
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational discovery of a novel double transition metal nitride MXene and its applications as an anchoring and catalytic material in Li–Se batteries†
Rafiuzzaman
Pritom
 a,
Indrajit
Nandi
b,
Md Shahriar
Nahian
a,
Rahul
Jayan
ac,
Satyajit
Mojumder
d and
Md Mahbubul
Islam
*a
a,
Indrajit
Nandi
b,
Md Shahriar
Nahian
a,
Rahul
Jayan
ac,
Satyajit
Mojumder
d and
Md Mahbubul
Islam
*a
aDepartment of Mechanical Engineering, Wayne State University, Detroit, MI 48202, USA. E-mail: gy5553@wayne.edu
bDepartment of Mechanical and Industrial Engineering, University of Illinois at Chicago, Chicago, IL 60607, USA
cTheoretical Division, Los Alamos National Laboratory, Los Alamos, NM 87544, USA
dSchool of Mechanical and Materials Engineering, Washington State University, Pullman, WA 99164, USA
First published on 30th June 2025
Abstract
Double transition metal (DTM) nitride MXenes offer enhanced electrical conductivity and tunable catalytic properties compared to conventional carbide-based MXenes. In this study, we employed first-principles density functional theory (DFT) calculations to discover and computationally validate a novel DTM nitride MXene, Nb2TiN2, derived from its MAX phase precursor and investigated its potential as an anchoring material (AM) for Li–Se batteries. This newly proposed MXene expands the compositional landscape of DTM nitrides and opens new avenues for functional material design. We performed a comprehensive analysis of the thermodynamic and electronic properties of Nb2TiAlN2, and the MAX phase precursor to Nb2TiN2 to assess its structural stability and exfoliation potential. Exfoliation energy calculations confirmed the feasibility of synthesizing Nb2TiN2 from Nb2TiAlN2. We then explored the functionalized form, Nb2TiN2S2, evaluating its capability to serve as an effective anchoring material (AM) in Li–Se batteries by analyzing the reaction mechanisms and kinetics of the selenium reduction reaction (SeRR). Our results indicate that Nb2TiN2S2 exhibits a strong binding affinity for lithium polyselenides (Li2Sen), effectively suppressing the shuttle effect. Gibbs free energy calculations for the rate-limiting step of the SeRR reveal favorable kinetics and reduced reaction barriers. Overall, this study provides a detailed evaluation of the structural and electronic properties of a newly proposed DTM nitride MXene and its S-functionalized derivative and the catalyzing effect of Nb2TiN2S2 in accelerating the reaction kinetics in Li–Se batteries. These findings underscore the potential importance of the further exploration of MXenes to address current challenges in high-performance Li–Se batteries.
1. Introduction
Over the past decade, graphene has grabbed global attention as a groundbreaking two-dimensional (2D) material, demonstrating remarkable potential across diverse fields such as energy storage, electronics, and catalysis.1–4 This significant achievement has fueled a widespread interest in the search for alternative 2D materials that could further revolutionize these domains. Among these emerging materials, MXenes—a family of transition metal carbides, nitrides, and carbonitrides—have gained substantial recognition for their exceptional electrical conductivity, hydrophilic nature, mechanical flexibility, and dispersion ability.5,6 Their high specific surface area and the rich presence of surface functional groups make them highly adaptable for a broad range of applications, including supercapacitors, catalysis, sensors, energy storage systems, and optoelectronics.7–11 Today, MXenes stand as one of the most expansive and rapidly growing families of 2D materials, offering unmatched versatility and tunability for next-generation technological advancements. The unique combination of metallic conductivity, chemical stability, and functional surface chemistry has positioned MXenes as a promising class of functional materials.MXenes originate from a broader class of layered ternary carbides, nitrides, and carbonitrides known as MAX phases.6 These materials follow the general formula Mn+1AXn, where M represents a transition metal, A is a group 13 or 14 element, and X denotes carbon (C) or nitrogen (N). The unique structure of MAX phases allows them to be selectively etched to produce MXenes, which follow the formula Mn+1Xn. A key distinguishing feature of MXenes lies in their synthesis process, which involves the selective removal of the A-group element, such as aluminum, from MAX phases.12,13 This process is typically achieved using aggressive etching agents like hydrofluoric acid (HF) or ammonium bifluoride (NH4HF2), which effectively dissolve the A-layer while preserving the underlying Mn+1Xn layers.14 As a result, the final structure of MXenes can be represented by the formula Mn+1XnTx, where Tx denotes surface terminations (such as –OH, –F, or –O groups) that form during the etching process. The composition and surface chemistry of MXenes play a crucial role in determining their physical, chemical, and electronic properties.5,15 The ability to tune surface terminations and modify the structural composition opens up new opportunities for MXenes, particularly in electrochemical energy storage, catalysis, sensing, and electronic devices.
MXenes can be broadly categorized into two primary types based on their composition: carbide MXenes (c-MXenes) and nitride MXenes (n-MXenes).16 Among the broader family of MXenes, n-MXenes have attracted significant attention due to their superior electrical conductivity, enhanced mechanical strength, and improved catalytic performance compared to conventional c-MXenes.17–19 Previous studies have demonstrated that when comparing c-MXenes and n-MXenes with the same metal composition, n-MXenes exhibit superior electrical conductivity and enhanced capacitive properties. For example, Zhang et al.17 reported that n-MXenes exhibit higher in-plane Young's moduli and electrical conductivity compared to their c-MXene counterparts. This enhancement was attributed to the smaller lattice constants and reduced monolayer thickness of n-MXenes, which contribute to their superior mechanical and electronic properties. Another study indicated that substituting carbon with nitrogen in the same MXene composition results in a lower point of zero charge, which enhances the material's metallic characteristics and increases its capacitance.19 Molecular dynamics (MD) simulations further reinforced these findings, revealing that the Young's modulus of n-MXenes is significantly higher than that of c-MXenes.18 This enhanced mechanical strength, combined with their exceptional electrical conductivity and distinctive surface chemistry, enables n-MXenes to be utilized in a broad range of applications.20 Beyond mechanical and electrical performance, n-MXenes have also been explored for their magnetic properties. Studies on Mn2NTx suggest that 2D n-MXenes hold great promise for spintronic applications, making them potential candidates for next-generation magnetic devices.21 Additionally, Ti4N3Tx has been identified as a highly efficient electrocatalyst for the hydrogen evolution reaction (HER) as it exhibits a unique combination of metallic and semiconducting properties that result in enhanced catalytic activity.22 By employing density functional theory (DFT) calculations, Fan et al.23 demonstrated 2D V2N/V2NT2 monolayers functionalized with S as a potential S cathode host for Li–S batteries.
Recently, double transition metal MXenes (DTM-MXenes) have surfaced as another special class of 2D materials in the realms of advanced composites, energy storage, and catalysis, complementing the mono-metallic MXene counterparts.24–26 This focus can be attributed to their unique structure, which integrates two distinct transition metals instead of one. DTM-MXene has a formula of  , where M′ and M′′ are two different transition metals. A prior study has revealed that compared to single metal MXene Ti4C3, DTM-MXene Ti2Ta2C3 exhibits higher open circuit voltage and capacity to store lithium(Li) than Ti4C3.27 The mechanical properties of DTM-MXenes have also been investigated, with Mo2TiC2 showing an exceptionally high Young's modulus of 361 GPa, significantly surpassing that of Ti3C2 MXene.28 This enhanced mechanical strength reinforces their potential use in structural applications where durability and flexibility are crucial. Liu et al. studied the electrochemical characteristics of Ti2NbC2Tx as an anode material in Li-ion batteries.29 They reported a superior specific capacity and long cycling stability with Ti2NbC2Tx, which outperformed its single metal counterparts, Ti3C2Tx. Similar behavior was also noted for the TixTa4−xC3 MXene.26 All of these studies underscore the potential of DTM-MXene in various applications. Despite these advancements, most research on DTM-MXenes has primarily focused on c-MXene systems, with little to no studies investigating DTM n-MXenes. It can be hypothesized that DTM-n-MXene may exhibit superior performance by utilizing the characteristics of both n-MXene and DTM-MXene.
, where M′ and M′′ are two different transition metals. A prior study has revealed that compared to single metal MXene Ti4C3, DTM-MXene Ti2Ta2C3 exhibits higher open circuit voltage and capacity to store lithium(Li) than Ti4C3.27 The mechanical properties of DTM-MXenes have also been investigated, with Mo2TiC2 showing an exceptionally high Young's modulus of 361 GPa, significantly surpassing that of Ti3C2 MXene.28 This enhanced mechanical strength reinforces their potential use in structural applications where durability and flexibility are crucial. Liu et al. studied the electrochemical characteristics of Ti2NbC2Tx as an anode material in Li-ion batteries.29 They reported a superior specific capacity and long cycling stability with Ti2NbC2Tx, which outperformed its single metal counterparts, Ti3C2Tx. Similar behavior was also noted for the TixTa4−xC3 MXene.26 All of these studies underscore the potential of DTM-MXene in various applications. Despite these advancements, most research on DTM-MXenes has primarily focused on c-MXene systems, with little to no studies investigating DTM n-MXenes. It can be hypothesized that DTM-n-MXene may exhibit superior performance by utilizing the characteristics of both n-MXene and DTM-MXene.
The lithium selenium (Li–Se) batteries have recently emerged as an alternative to Li–S batteries.30,31 Se has a high theoretical gravimetric capacity (678 mA h g−1) and volumetric capacity (3268 mA h cm−3).32–35 Most significantly, Se has higher electrical conductivity 1 × 10−3 S m−1 (about 20 orders of magnitude than S), which can result in increased capacity utilization and rate capability.36 Unfortunately, the dissolution of soluble intermediate lithium polyselenides (Li2Sen) into the ether-based electrolyte during the electrochemical cycling results in a low battery capacity and inhibits the cycle performance of Li–Se batteries.37 Additional constraints like sluggish reaction kinetics and irreversibility also hinder their practical realization. To overcome these issues, for Li–Se systems, several strategies have been reported, such as confinement of Se in porous carbon, adsorption of polyselenides by metal oxide, insertion of carbon interlayer to adsorb soluble polyselenides, and design of nanoporous or nanofibrous Se structure.38–40 To achieve the desired performance in Li–Se batteries, the use of CoSe2-porous carbon composites,41 graphene-selenium hybrid microballs,42 and various metal organic frameworks (MOF) are found to mitigate, but not completely eliminate the dissolution of polyselenides. It has been observed that nitrogen-doped carbon structures, due to their porous character, facilitate an ion transport channel and better restrict Se inside the host.41,43 Han et al. demonstrated that the electrical and ionic conductivities can be enhanced, and the dissolution of polyselenides can be suppressed by probing the hierarchical structure created by the fusion of graphene and 3D porous carbon nanoparticles.44 The synthesized Fe-CNT/TiO2 catalyst through high-temperature sintering of a carbon nanotube-decorated MOF and MXene nanoarchitecture enhances Se hosting and demonstrates exceptional battery performance with high capacity retention and efficiency across various cycling rates.45 Se incorporation into the microporous carbon nanofibers via vacuum and heat treatment enhances the anchoring behavior and provides uniform distribution of the selenides on the carbon matrix.40 Despite extensive research on Li–Se batteries, challenges such as sluggish electrode kinetics and polyselenide shuttle effects remain unresolved. This highlights the need for further research into alternative anchoring materials (AMs) for Li–Se batteries to improve the polyselenides adsorption capacity and enhance the reaction kinetics.
This study focuses on the development of a novel DTM-n-MXene, Nb2TiN2, from its precursor MAX phase. To achieve this, we conducted a comprehensive computational study using first-principles DFT simulations to analyze the stability and electronic characteristics of MAX phases and the formation of Nb2TiN2 MXene. Our study began with an in-depth investigation of the stability of the Nb2TiAlN2 MAX phases, which was further extended to the evaluation of the exfoliation process by calculating the exfoliation energy. To assess the practical energy storage potential of the newly developed Nb2TiN2 MXene, we investigated its adsorption behavior and reaction kinetics in Li–Se batteries. Specifically, we studied sulfur-functionalized Nb2TiN2S2, evaluating its ability to stabilize Li2Sen and facilitate Se reduction reactions (SeRR). The adsorption mechanisms of Li2Sen on Nb2TiN2S2 and the kinetics of SeRR are all investigated using DFT simulations.
2. Calculation methodology
Spin polarized plane wave DFT calculations were carried out using the Vienna ab initio Simulation Package (VASP).46 The exchange–correlation energy was described using the Perdew–Burke–Ernzerhof (PBE) functional within the framework of the generalized gradient approximation (GGA).47 The PBE functional was selected for this study because of its demonstrated ability to accurately capture the electronic structure of MXenes and their interactions with lithium polychalcogenides in Li–chalcogen batteries.48–52 In addition, PBE offers a practical advantage with its significantly lower computational cost compared to hybrid functionals such as HSE06, which makes it well-suited for exploring larger system sizes and multiple adsorption configurations. The Projector Augmented Wave (PAW) method was employed to explicitly treat the interaction between valence and core electrons, enabling an accurate representation of the electronic structures by including the effect of core electrons on valence electron density. The plane-wave energy cutoff of 520 eV is used. The electronic structure minimization was performed using the blocked Davidson iteration scheme,46 and van der Waals (vdW) interactions were accounted for using the empirical DFT-D3 dispersion correction.53 We used the conjugate gradient method to optimize the geometry while letting the atoms relax until the residual force was less than 0.05 eV Å−1 and the energy convergence reached 1 × 10−4 eV. In order to sample the Brillouin zone integration for the electronic structure computations and atomic relaxations, the Monkhorst–Pack grid scheme with 8 × 8 × 1 k-point was used for the calculations related to MAX phase and MXene unit cell and 5 × 5 × 1 k-point was used for the Li2Sen adsorption calculations on the MXene substrate. To obtain the stable configuration of the MAX phase, the structure was fully relaxed, allowing for any adjustments in both shape and volume until the forces on atoms and the stress tensor components were minimized. The integration of the Brillouin zone was carried out using the tetrahedron method with Bloch corrections. Bader charge analysis was employed to quantify the charge population on the polyselenides and MXenes. We calculated the charge density difference based on the expression ρb = ρabsorbed state − (ρabsorbate + ρAM), where ρabsorbed state, ρabsorbate, and ρAM refer to the charge transfer of Li2Sen adsorbed AM, the isolated polyselenides, and the AM, respectively.3. Results and discussion
3.1 Stability analysis of Nb2TiAlN2 MAX phase
DFT simulations were employed to investigate the structural and thermodynamic stability of the Nb2TiAlN2 MAX phase. The study began with the structural optimization of the MAX phase, starting from its initial configuration obtained from the Materials Project database.54 The optimized structure, depicted in Fig. 1a, represents the energetically stable configuration of the Nb2TiAlN2 MAX phase. This converged structure served as the basis for subsequent analyses, including assessments of thermodynamic stability, electronic structure, and mechanical properties. We used the Open Quantum Materials Database (OQMD)55 to identify the competing phases into which Nb2TiAlN2 can decompose. OQMD contains DFT-calculated thermodynamic and structural properties for over 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 317811 structures. The Nb2TiAlN2 falls within the Nb–Ti–Al–N region of phase space. Our analysis revealed that Nb2TiAlN2 can decompose into AlN, TiN, Nb2N, and Nb3Al2N, as shown in Fig. 1b. Although these phases do not represent the complete set of competing phases in the Nb–Ti–Al–N quaternary system, they collectively represent the lowest-energy ground state configuration that accurately captures the stoichiometric composition of Nb2TiAlN2. The isometric view of the Gibbs tetrahedron of the 4-D (Nb–Ti–Al–N) phase diagram generated via the OQMD database is shown in Fig. S1.† The phase diagram shows the composition of stable and unstable phases in the phase space. To evaluate the thermodynamic stability, the formation energy of the Nb2TiAlN2 MAX phase was calculated and compared with those of competing phases in the Nb–Ti–Al–N system. We found that MAX phase possesses low formation energy (−114.90 eV) compared to the total formation energy of its competing phases (−109.90 eV), which suggests that the MAX phase is energetically more favorable and thus likely to be the stable phase under standard conditions. Additionally, phonon dispersion calculations were performed to assess the dynamic stability of the MAX phases. The calculation was carried out using Density Functional Perturbation Theory (DFPT) and analyzed with Phonopy.56 The absence of imaginary frequencies, illustrated in Fig. 1c, confirms the structural stability of the phase. To further investigate the material's stability, ab initio molecular dynamics (AIMD) simulations were conducted at elevated temperatures.
317811 structures. The Nb2TiAlN2 falls within the Nb–Ti–Al–N region of phase space. Our analysis revealed that Nb2TiAlN2 can decompose into AlN, TiN, Nb2N, and Nb3Al2N, as shown in Fig. 1b. Although these phases do not represent the complete set of competing phases in the Nb–Ti–Al–N quaternary system, they collectively represent the lowest-energy ground state configuration that accurately captures the stoichiometric composition of Nb2TiAlN2. The isometric view of the Gibbs tetrahedron of the 4-D (Nb–Ti–Al–N) phase diagram generated via the OQMD database is shown in Fig. S1.† The phase diagram shows the composition of stable and unstable phases in the phase space. To evaluate the thermodynamic stability, the formation energy of the Nb2TiAlN2 MAX phase was calculated and compared with those of competing phases in the Nb–Ti–Al–N system. We found that MAX phase possesses low formation energy (−114.90 eV) compared to the total formation energy of its competing phases (−109.90 eV), which suggests that the MAX phase is energetically more favorable and thus likely to be the stable phase under standard conditions. Additionally, phonon dispersion calculations were performed to assess the dynamic stability of the MAX phases. The calculation was carried out using Density Functional Perturbation Theory (DFPT) and analyzed with Phonopy.56 The absence of imaginary frequencies, illustrated in Fig. 1c, confirms the structural stability of the phase. To further investigate the material's stability, ab initio molecular dynamics (AIMD) simulations were conducted at elevated temperatures.
 | ||
| Fig. 1 The stable structures of (a) the Nb2TiAlN2 MAX phase; and (b) competing phases. (c) The phonon dispersion curve of the Nb2TiAlN2 MAX phase. | ||
The AIMD simulations were conducted using the canonical ensemble (NVT), where the system was maintained at a constant volume and temperature. The Nose–Hoover thermostat was applied to regulate the temperature at 1000 K. The AIMD simulation was run for 10 picoseconds (ps) with a time step of 1 femtosecond (fs). Fig. S2† presents the distribution of total energy and temperature over the entire simulation duration. The results indicate that both energy and temperature exhibit minimal fluctuations, suggesting that the atomic structure remains stable under high-temperature conditions. Additionally, the initial and final structures of Nb2TiAlN2 in AIMD simulation, shown in Fig. S3,† clearly depicts that the structure remains stable at high temperature even after 10 ps. The absence of significant energy deviations or structural distortions reinforces the high thermodynamic stability of Nb2TiAlN2 at elevated temperatures. All of these findings indicate that Nb2TiAlN2 MAX phase demonstrates favorable thermodynamic and dynamic stabilities.
3.2 Electronic conductivity of Nb2TiAlN2 MAX phase
MXene's metallic nature essentially originates from the d-localized electrons of the transition metal atoms. Previous studies showed that the metallic conductivity of MXenes and their functionalized forms allow electron diffusion between the MXene surface and the lithium polysulfides during the redox reactions to achieve high capacity in the Li–S batteries.50,57 This charge transfer also facilitates the chemical adsorption and catalytic conversion of higher-order polysulfides into lower-order polysulfides on the MXene surface. As a result, the metallic nature not only promotes efficient redox kinetics but also improves the polysulfides immobilization within the cathode scaffolds to mitigate the shuttle effect through electron-mediated interactions.58 Li–Se batteries with Nb2TiN2S2 are also expected to exhibit similar trends. To gain a deeper understanding of the electronic characteristics of the Nb2TiAlN2 MAX phase, we performed density of states (DOS) analysis. Fig. 2 presents the calculated total DOS for the Nb2TiAlN2 MAX phase. Our results reveal a significant density of states at the Fermi energy level (Ef = 0 eV), which is a clear indicator of the metallic nature and high electrical conductivity of Nb2TiAlN2.To further analyze the electronic contributions from different atoms and orbitals of Nb2TiAlN2, we performed projected density of states (PDOS) analysis which offers a breakdown of the contribution of individual atomic orbitals to the overall electronic states of the system. Our findings indicate that the d-orbitals of Nb and Ti play a dominant role in determining the electronic properties of Nb2TiAlN2. Both Nb-4d and Ti-3d states contribute significantly around the Fermi level. Additionally, the absence of an electronic band gap further confirms that Nb2TiAlN2 behaves as a metallic material rather than a semiconductor. The Al and N states contribute primarily to lower energy regions which indicates their minor role in direct conduction mechanisms but their potential influence on structural stability and bonding characteristics. The hybridization between the metal d-states and nonmetal p-states also suggests a complex bonding environment that supports both metallic and covalent interactions. | ||
| Fig. 2 Calculated band structure of the MAX phase. Total DOS is shown in the right plot whereas the contribution of individual orbital is presented in the left plot. | ||
3.3 Exfoliation of Nb2TiAlN2 MAX phase
The exfoliation of MAX phases into their corresponding 2D MXenes involves the selective removal of Al layers through chemical etching, typically using hydrofluoric acid (HF) or other etching agents. The elimination of these Al layers disrupts the strong bonding network within the MAX phase and leads to the formation of weak van der Waals interactions between the remaining MXene layers. These weak interlayer interactions allow for the subsequent intercalation process, in which specific molecules or ions are introduced between the layers. The intercalants expand the interlayer spacing and facilitate the mechanical or chemical separation of individual MXene flakes and ultimately yield a 2D structure with enhanced flexibility and high surface area.59To investigate the feasibility of exfoliating the Nb2TiAlN2 MAX phase into its 2D Nb2TiN2 MXene, we calculated the bond stiffness (k), which represents the resistance of a bond to deformation, and it is a crucial parameter for predicting the exfoliation behavior. It provides insight into which atomic bonds within the MAX phase are more susceptible to breaking during the exfoliation process. The calculated bond stiffness values for various atomic bonds in Nb2TiAlN2 are presented in Table 1. Our results suggest that the bond stiffness values for Nb–Al and Al–N bonds are significantly lower than those of Ti–N, N–N, and Nb–N bonds. This indicates that Al atoms are weakly bonded to their neighboring atoms. The relatively strong bonding of Ti and Nb with nitrogen further supports the structural integrity of the remaining MXene layers after Al removal, allowing the formation of a stable 2D Nb2TiAlN2 MXene. To quantitatively assess the thermodynamic feasibility of exfoliation, we further calculated the exfoliation energy (Eexf), which provides a measure of the energy required to separate the 2D MXene from the bulk MAX phase. The exfoliation energy was calculated using the equation Eexf = (Ebulk MAX phase − E2D-MXene − EAl)/A, where Ebulk MAX phase, E2D-MXene, EAl, and A denote the energy of bulk Nb2TiAlN2 MAX phase, the energy of 2D Nb2TiN2 MXene, the energy of Al atom in its bulk fcc phase and the surface area of Nb2TiN2 MXene flake, respectively. Our calculations yielded an Eexf value of approximately 0.20 eV Å−2. Previous studies have reported that the critical exfoliation energy threshold is ∼0.25 eV Å−2, with values below this threshold indicating a high probability of successful exfoliation.60 Since our calculated exfoliation energy is well below this critical limit, this strongly suggests that the exfoliation of Nb2TiAlN2 MAX phase into 2D Nb2TiN2 MXene is thermodynamically favorable.
| Nb–Al | Ti–N | N–N | Nb–N | Al–N | |
|---|---|---|---|---|---|
| Bond stiffness, k (N m−1) | 609 | 1103 | 1111 | 1401 | 77 |
3.4 Nb2TiN2 MXene and sulfur functionalization
The optimized geometry of the Nb2TiN2 MXene after the exfoliation from its MAX phase is illustrated in Fig. 3a and c. The Nb2TiN2 monolayer adopts a quint-layer configuration, where a central Ti atomic layer is symmetrically enclosed between two N layers, with Nb layers exposed on both the top and bottom surfaces. This structural arrangement follows the sequence of Nb–N–Ti–N–Nb. After the relaxation of the Nb2TiN2 monolayer, the optimized lattice parameters are found as a = b = 3.01 Å with a layer thickness of 5.08 Å. The bond lengths of Ti–N and Nb–N are observed as approximately 2.11 Å and 2.16 Å, respectively. The incorporation of functional groups provides additional knobs to tune the structural and electronic properties of Nb2TiN2 MXene. The introduction of functional groups such as –S, and –O can improve its hydrophilicity, conductivity, and interaction with other materials.61,62 Additionally, controlled surface functionalization allows for tailoring the work function and charge transfer properties. Here, we incorporated S functionalization into the studied MXene and systematically evaluated its stability in comparison to the pristine phase. However, for functionalization, there are 3 possible adsorption sites (i) above the N atom (ii) above the Ti atom, and (iii) above the Nb atom. In our previous study, conducted for Mo2TiC2-MXene, we found that the C top site is energetically favorable and dynamically stable compared to the other two sites.63 Following that, we applied the S-functionalization at all three sites, shown in Fig. S4,† and calculated their formation energies to determine the most favorable site for the functionalization. Interestingly, our result indicates that the N-top site of Nb2TiN2 MXene possesses relatively more negative formation energy (∼−6.0 eV) compared to the Ti-top site(∼−5.4 eV) and is the most stable configuration. However, it was found that the Nb-top site was structurally unstable for S-functionalization. These findings are in good agreement with our previous study.63Fig. 3b and d depict the optimized configurations of the S-terminated Nb2TiN2 MXene. Notably, the bond length of Nb–S is measured at approximately 2.45 Å. | ||
| Fig. 3 Structure of optimized geometries of Nb2TiN2 MXene and its S-functionalized counterparts. (a–d) Represent the top view and the side view, respectively. | ||
To gain deeper insights into the bonding characteristics and the effects of functionalization, we analyzed the electron localization function (ELF) for both the pristine and S-functionalized Nb2TiN2 MXenes. ELF provides a quantitative measure of charge distribution within the material, ranging from 0 to 1, where ELF = 0 represents complete electron delocalization, characteristic of metallic or weakly bonded systems, while ELF = 1 corresponds to strong electron localization, typically observed in covalent or ionic bonding scenarios. Fig. 4a presents a comparative visualization of the charge density in Nb2TiN2 MXene before and after functionalization. In the pristine structure, a high electron density is observed around the N atoms, which highlights substantial charge transfer from Nb and Ti atoms to N. As a result, N plays a crucial role in governing the charge distribution, leading to strong Nb–N and Ti–N interactions. Upon S functionalization, notable changes in charge localization are observed. Due to increased charge transfer from the metal atoms, the S atoms exhibit an enriched electron cloud, and this charge transfer is stronger than the one observed on N in the pristine MXene. Due to its higher electronegativity and polarizability, S interacts more effectively with the underlying Nb and Ti atoms, forming bonds with a more pronounced covalent character. Additionally, the redistribution of charge density upon functionalization may influence the electronic properties, catalytic activity, and mechanical integrity, as electron-rich surface terminations can alter the material's work function and interaction with external species.
 | ||
| Fig. 4 (a) Electron localization function (ELF) and (b) phonon dispersion analysis of Nb2TiN2 MXene before functionalization and after functionalization. | ||
To assess the dynamic stability of pristine and S-functionalized Nb2TiN2 MXenes, we performed a phonon dispersion calculation, as presented in Fig. 4b. The phonon dispersion plot reveals that all vibrational modes exhibit positive frequencies across the Brillouin zone which confirms the dynamical stability of both the pristine and functionalized MXenes. Although a minor negative frequency is observed for functionalized case, it is insignificant and likely arises from numerical artifacts or computational approximations rather than indicating a true structural instability. We also performed AIMD calculations using an NVT ensemble with a Nose–Hoover thermostat at a temperature of 1000 K for 10 ps to evaluate the thermodynamic stability at high temperatures. The energy and temperature evolution with time shows insignificant fluctuations (Fig. S5 and S7†) and the identical initial and final structures were found to be highly stable for both pristine and S-functionalized MXene (Fig. S6 and S8†). The absence of substantial imaginary frequencies and rapid fluctuations of energy and temperature suggest that both materials maintain their structural integrity and thermodynamic stability in adverse conditions, making them promising candidates for further investigation in applications requiring stable MXene structures. As our simulations establish the stability of the Nb2TiN2S2 MXene, we further explore its potential application in Li–Se batteries, specifically as an anchoring and catalyzing material to enhance battery performance.
3.5 Polysulfide adsorption on Nb2TiN2S2
We first investigated the ability of the Nb2TiN2S2 MXene to immobilize the otherwise soluble higher order polyselenides. Typically, the metal exposed surfaces of bare MXenes exhibit extremely high adsorption strength, which results in the decomposition of polyselenides. To ensure reversible M–X battery operation, we incorporated surface modifications with S and studied their impact on the electrochemical performance as AMs.The fundamental electrochemical processes in Li–Se and Li–S batteries share significant similarities.64 During discharge, Li ions migrate from the anode and react with the Se cathode, forming intermediate Li2Sen. These intermediates undergo further reduction and ultimately yield the final discharge products, Li2Se2 and Li2Se. To understand the anchoring behavior of Nb2TiN2S2 for potential applications in metal–chalcogenide batteries, we employed DFT simulations to study the adsorption of Se8 and Li2Sen species. The interaction between the polyselenides and electrode surface plays a critical role in determining battery performance, particularly in mitigating the shuttling effect and improving the stability of Li–Se systems. In general, polychalcogenides interact with substrates through covalent bonding between metal sites and the surface S atoms of the MXene material. To identify the most energetically favorable adsorption configurations, we initially placed Li2Sen species in various positions on the Nb2TiN2S2 surface and performed structural optimizations. To quantify the anchoring strength of polyselenides on the pristine Nb2TiN2S2 surface, we calculated the adsorption energies (Eads) by employing the formula Eads = ELi2Sen + EAM − ELi2Sen+AM, where ELi2Sen, EAM, and ELi2Sen+AM denote the DFT energies of Li2Sen, AMs, and the polyselenide-adsorbed substrate. Additionally, we compared the adsorption characteristics of Nb2TiN2S2 MXene with graphene which was reported in a previous study on Li–Se battery.62Fig. 5a presents the calculated adsorption energies of different Li2Sen species on Nb2TiN2S2 and graphene. Our results indicate a progressive increase in the adsorption energy with increasing lithiation for Nb2TiN2S2, demonstrating enhanced polyselenide anchoring as more Li atoms are incorporated. We compared the polyselenides absorption strengths on Nb2TiN2S2 with graphene as a representative carbonaceous material. The data for Li2Sen adsorption on graphene has been taken from our previous study.62Fig. 5a exhibits a significant improvement in binding characteristics of Nb2TiN2S2 over graphene substrate. Fig. 5b presents the relaxed atomic configurations of Li2Sen species adsorbed on the Nb2TiN2S2 surface. The optimized structures reveal that the Li2Sen species remain well-conserved during the adsorption process without undergoing significant structural or chemical degradation. The Se8 molecules tend to adopt a nearly parallel orientation relative to the Nb2TiN2S2 surface, with a minimum spacing of approximately 3.06 Å. For the other Li2Sen species the adsorption predominantly occurs at the bridging sites, where each Li atom establishes strong Li–S interactions with the nearby S atoms on the Nb2TiN2S2 surface. This bridging configuration enhances the anchoring stability of the polyselenides, effectively preventing their dissolution and unwanted migration, which are critical challenges in Li–Se battery systems. The interaction with the Nb2TiN2S2 substrate provides a strong anchoring performance while maintaining the structural integrity of the Li2Sen. To evaluate the possibility of polyselenide dissolution into the electrolytes, we compared our calculated adsorption energies with the adsorption energies obtained for higher order polyselenides (Li2Se8, Li2Se6, and Li2Se4) bound with the most common electrolytes solvents 1,3-dioxalane (DOL) and 1,2-dimethoxyethane (DME) (shown in Fig. 5a), which were already reported in our previous work.62 Our findings suggest that Nb2TiN2S2 substrate exhibits stronger binding characteristics for the polyselenides than DME and DOL, which indicates the capability of Nb2TiN2S2 substrate to inhibit the dissolution of polyselenides into electrolytes, thereby limiting the shuttle effect in the Li–Se batteries.
 | ||
| Fig. 5 (a) The calculated adsorption energies of Se8 and Li2Sen adsorbed on graphene and Nb2TiN2S2 (b) Optimized geometric configurations of Se8 and Li2Sen adsorbed on Nb2TiN2S2. Color code for atoms: Nb: forest green; Ti: sky blue; N: violate; S: yellow; Se: green; Na: navy blue; Li: purple. The data for graphene is taken from ref. 62. | ||
To further understand the adsorption mechanism and establish a connection between the adsorption energies and underlying chemical interactions, we performed Bader charge analysis to quantify the charge transfer between Li2Sen species and the Nb2TiN2S2 substrate. Our calculations reveal that Se8 exhibited the lowest charge transfer, which aligns with its lower binding energy. The computed charge transfer values for different Li2Sen species are presented in Fig. 6a, where it has been observed that as the discharge process progresses, the quantity of electrons transferred from the polyselenides to the substrate increases. Positive charge transfer values indicate that charge is getting transferred from the polyselenides to the Nb2TiN2S2 substrate. As a result, the lower-order polyselenides (Li2Se4, Li2Se2, and Li2Se) are significantly prone to donate more charge to the AMs compared to higher-order species, which corroborates with their stronger interaction with Nb2TiN2S2. Higher-order polyselenides, which exhibit weaker charge transfer, also demonstrate lower binding energies, while lower-order polyselenides show higher charge transfer and stronger adsorption.
 | ||
| Fig. 6 (a) Computed charge transfer of Li2Sen adsorbed Nb2TiN2S2. (b) Differential charge density (DCD) calculation of Se8 and Li2Sen on Nb2TiN2S2 substrate. The blue and pink colors define the charge accumulation and depletion, respectively. | ||
To gain further insight into the nature of charge redistribution during adsorption, we also computed the average change in atomic charge of Li and Se atoms before and after adsorption. By comparing the atomic charge values in the gas phase (isolated Li2Sen molecules) and adsorbed phase (Li2Sen on Nb2TiN2S2), we observed a notable trend: the charge transfer primarily originates from Se atoms rather than Li atoms. For example, the charge transfers of Li and Se were found −0.0168|e| and −1.317|e|, respectively, for Li2Se adsorption. This finding is consistent with our previous research on Na–S battery systems, where we utilized Mo2TiC2S2 MXene as an AM and observed a similar charge transfer mechanism.65 In both cases, the chalcogenide-substrate interaction is primarily facilitated by chalcogen atoms (S or Se), which play a crucial role in stabilizing the polychalcogenide species and preventing their dissolution.63 We also conducted the differential charge density (DCD) analysis to explain the charge transfer between the substrate and the adsorbates. The DCD analysis, shown in Fig. 6b, revealed a significant charge depletion around the Se atoms, confirming that Se acts as the primary electron donor in the adsorption process. Correspondingly, charge accumulation was observed on the Nb2TiN2S2 substrate. This charge redistribution strengthens the interaction between the chalcogenide species and the substrate and develops the anchoring effect that prevents the dissolution of higher order polyselenides.
To understand the contribution between chemisorption and physisorption in the interactions between Li2Sen species and the substrates, we conducted further analysis to isolate the effect of vdW interactions. This analysis quantifies the contribution of vdW forces to adsorption by calculating the relative vdW ratio, which is determined by comparing the adsorption energy with and without vdW interactions. This was achieved by computing the adsorption energy for each polyselenide species using the formula,
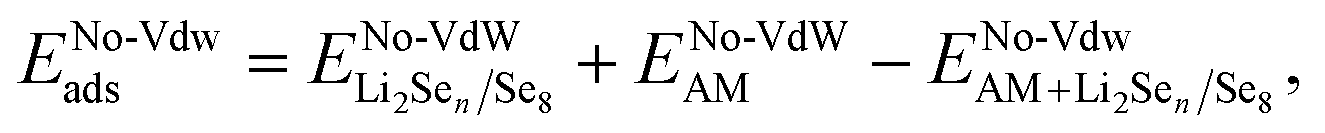
 , ENo-VdWAM and
, ENo-VdWAM and  represent the DFT energies of isolated Li2Sen/Se8 molecules, the AM, and the polyselenide-adsorbed system without vdW interactions, respectively. The relative contribution of vdW interactions was then calculated using the expression:
represent the DFT energies of isolated Li2Sen/Se8 molecules, the AM, and the polyselenide-adsorbed system without vdW interactions, respectively. The relative contribution of vdW interactions was then calculated using the expression:
The calculated VdW ratio values, as shown in Fig. 7, reveal a strong dependence of adsorption behavior on the degree of lithiation. Notably, Se8 exhibits an exceptionally high vdW ratio, approaching nearly 100%, indicating that Se8 adsorption is predominantly driven by vdW forces with minimal chemical interaction. This suggests that Se8 remains weakly bonded to the substrate through non-covalent interactions, well corroborated with the observed weaker adsorption behavior on Nb2TiN2S2. However, as lithiation progresses, the vdW ratio decreases significantly, indicating that chemical interactions become dominant with increasing Li content in the polyselenides. This trend is inversely related to the adsorption energy, further supporting the notion that as Li atoms interact with Se, they enhance the chemical binding by reducing the relative contribution of vdW interactions. Additionally, our analysis reveals that polyselenides exhibit stronger vdW interactions with graphene compared to Nb2TiN2S2. This finding indicates that while graphene exhibits weak physisorption behavior toward Li2Sen due to the non-polar nature of carbon, Nb2TiN2S2 shows stronger chemical interactions with Li-polyselenides, making it a more effective material for anchoring Li2Sen species.
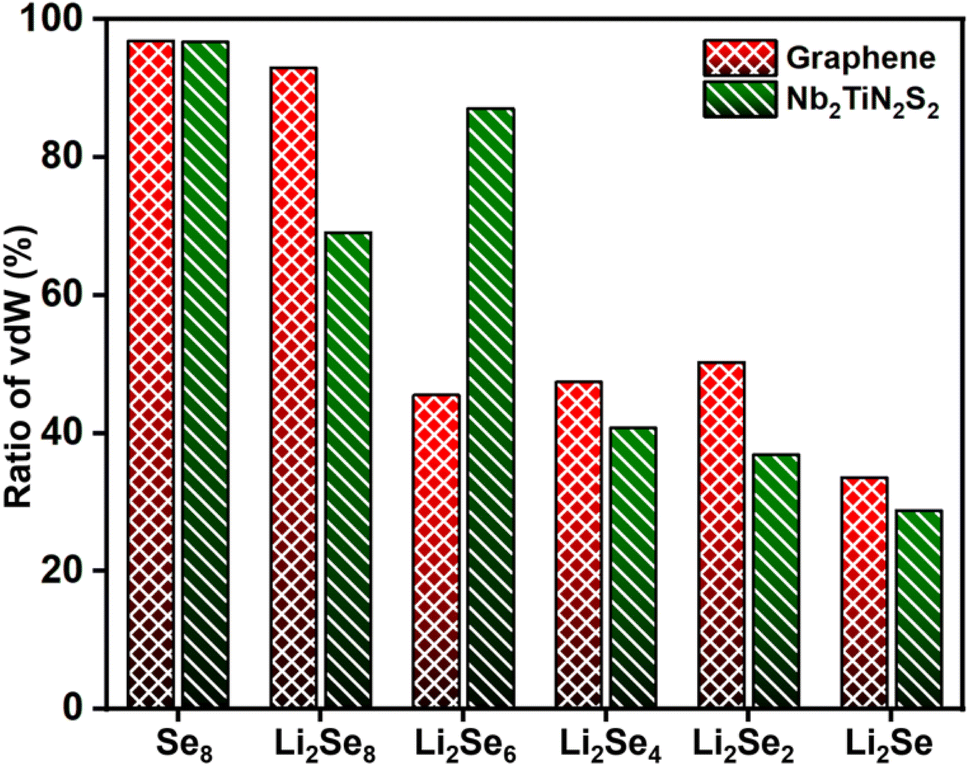 | ||
| Fig. 7 The ratio of van-der Waals interactions of Se8 and Li2Sen absorbed on graphene and Nb2TiN2S2. The data for graphene is taken from ref. 62. | ||
3.6 Electronic density of states of adsorbed Li2Sen
To ensure efficient redox reactions in Li–Se batteries, it is crucial that AMs maintain their intrinsic electronic conductivity following Li2Sen adsorption. A loss of conductivity upon adsorption could hinder electron transport, limiting the charge transfer kinetics and negatively impacting the overall electrochemical performance. To investigate this, we conducted DOS calculations for Nb2TiN2S2 before and after Li2Sen adsorption to assess how adsorption affects the electronic characteristics of the substrate. The TDOS curve (Fig. S9†) confirms that pristine Nb2TiN2S2 is metallic in nature, which aligns well with prior studies on functionalized MXenes.62,63,66 The presence of a significant density of states at the Fermi level (EF) in the pristine system indicates high electronic conductivity which suggests Nb2TiN2S2 a potential host material for electrochemical applications. To further understand the contributions of different atomic orbitals, we analyzed the PDOS. As shown in Fig. S9,† the Ti-3d orbitals contribute the most to the electronic states near the Fermi level, while Nb-4d, S-3p, and N-2p states also make noticeable but relatively smaller contributions.After the adsorption of Li2Sen, the PDOS plots (Fig. 8) reveal that a significant number of electronic states are still present at the Fermi level, confirming that Nb2TiN2S2 retains its metallic conductivity even after polyselenides adsorption. This is particularly important because Se8 is inherently insulating, and a conductive host material is essential to mitigate this limitation. In contrast to Se8 adsorption, where the insulating characteristics are more pronounced, Li2Sen adsorption induces the emergence of adsorption peaks below the Fermi level, which correspond to charge transfer interactions between Li2Sen and the substrate. The PDOS further demonstrates that the contribution from Se-derived states is higher than that of S near the Fermi level, although this increase is negligible and does not significantly impact the overall conductivity of the system. This suggests that the electronic properties of Nb2TiN2S2 remain largely unaffected by Se-rich environments, further supporting its role as a stable, conductive AM.
 | ||
| Fig. 8 PDOS of Se8 and Li2Sen adsorbed on Nb2TiN2S2. The Fermi level is denoted by vertical lines. | ||
3.7 Selenium reduction reaction (SeRR)
To further elucidate the SeRR kinetics occurring on the Nb2TiN2S2 monolayer during the Li–Se battery discharge process, we investigated the overall reaction mechanisms based on the formation of Li2Se from Se8 and bulk Li. Understanding the thermodynamic feasibility of these transformations is crucial in assessing the electrochemical performance of Nb2TiN2S2 as an AM for Li2Sen. To quantify the driving forces of each step in the SeRR pathway, we computed the Gibbs free energy (ΔG) for each reaction step on the Nb2TiN2S2 substrate and compared the results with gas-phase data (Fig. 9). These free energy calculations allow us to determine the rate-determining step of the entire conversion process. Calculation details for this analysis were provided in our previous work.67 | ||
| Fig. 9 Gibbs free energy diagram of selenium reduction reaction in vacuum and on Nb2TiN2S2 for Li–Se systems. | ||
The reduction of Se8 to Li2Se8 marks the first step in the Li–Se battery discharge process. Our calculations indicate that this initial reduction step is a spontaneous exothermic reaction for both the gas phase and the Nb2TiN2S2-anchored systems. The exothermic nature of this step suggests that Li incorporation into Se occurs readily. The Li2Se8 → Li2Se6 conversion is also found to be exothermic on both vacuum and AM. The third step, which involves the conversion of Li2Se6 to Li2Se4, is found to be endothermic, however the studied AM reduced the energy barrier for the formation of soluble Li2Se4 compared to gas phase conversion. Notably, in M–X (M = Li, Na, or K; X = S, Se) batteries, the M2X4 to M2X2 conversion is widely recognized as the rate-determining step in the overall discharge process.65,68–70 Our results reveal that in gas phase, the Li2Se4 → Li2Se2 transformation exhibits a significant energy barrier of 1.25 eV, which could hinder the reaction kinetics and slow down the discharge process. However, when the reaction occurs on Nb2TiN2S2, the energy barrier is dramatically reduced to 0.201 eV, marking an 83.94% reduction in the reaction barrier compared to vacuum conditions. This substantial decrease in energy requirement evidence that the Nb2TiN2S2 substrate effectively accelerates the rate-determining step, enabling faster polyselenide conversion and enhancing the discharge reaction kinetics of Li–Se batteries. The improved Se reduction kinetics arises from the strong adsorption of polyselenides on Nb2TiN2S2, which facilitates the reduction of higher order polyselenides to lower order polyselenides, owing to the electron transfer at the electrode–electrolyte interface. The sulfur functionalized Nb2TiN2 MXene provides abundant adsorption sites for polyselenides to suppress the shuttle effect by immobilizing the lithium polyselenides species (Fig. 5). At the same time, its high electrical conductivity enhances electron transfer during the adsorption process (Fig. 6a). The strong binding between polyselenides and the Nb2TiN2S2 surface, coupled with charge depletion from the polyselenide species toward Nb2TiN2S2 (Fig. 6b), accelerates the Se reduction reaction and lowers the energy barriers of intermediate steps. This effect is particularly pronounced in the Li2Se4 → Li2Se2 conversion step compared to other stages (Fig. 9). Overall, these findings suggest that Nb2TiN2S2 can effectively serve as both a catalytic and anchoring host to accelerate the conversion kinetics and improve the cycling performance of Li–Se batteries. A similar trend of acceleration in reduction kinetics was observed in our previous studies, where surface modification-based metal–chalcogenides batteries exhibited significantly improved discharge reaction rates due to substrate-induced energy barrier reduction.63,65,67,71 In conclusion, during discharge, the use Nb2TiN2S2 as an AM proved to be effective for Li–Se batteries where Li–Se exhibited faster reduction reaction kinetics.
4. Conclusion
In this study, we discovered a new MXene, Nb2TiN2, and evaluated its structural and electronic properties, along with its potential as a cathode catalyst for Li–Se batteries. We started our investigation by analyzing the precursor Nb2TiAlN2 MAX phase. With the relaxed MAX phase structure, we employed phonon dispersion and AIMD simulation to examine the dynamic stability and thermodynamic stability at high temperatures, respectively. Our finding suggests that the MAX phase exhibits excellent dynamic stability with zero negative frequency and thermodynamic stability even at 1000 K. The DOS analysis provides evidence of the presence of states at the Fermi level with the conducting property of the catalyst majorly arising from the Nb and Ti ‘d’ orbitals. The transformation of 2D MXene from the MAX phase was also examined with the calculation of exfoliation energy and bond stiffness of various bonds. Al–N and Nb–Al bonds are found to be significantly weaker than the other bonds, which is in favor of the exfoliation process. On the other hand, the exfoliation energy was also found within the critical value. We investigated the binding mechanism and reaction kinetics of Li2Se on the Nb2TiN2S2 substrate. Simulations revealed that Nb2TiN2S2 is effective in anchoring the Li2Sen to mitigate the shuttle effect. During the anchoring process, the charge transfer occurs from the Se atoms of the polyselenides to the AM material and the charge transfer increases as the order of polyselenides decreases. The DOS analysis of the polyselenides adsorption on AM indicates the presence of the metallic nature even after adsorption. According to the Gibbs free energy profile, Li2Se4 to Li2Se2 conversion is found to be the rate-determining step, and the inclusion of Nb2TiN2S2 lowers the energy barrier and is expected to accelerate the reaction kinetics significantly. In conclusion, the results demonstrate that Nb2TiN2S2 MXene possesses significant potential to advance the development of high-performance and sustainable Li–Se batteries, as well as broader catalytic systems. This investigation can be extended to other chalcogen-based systems such as Li–S, Na–S, and Na–Se batteries. Although the fundamental mechanisms share common features, there are also distinct differences depending on the chalcogen species and ion involved. On the other hand, the influence of different surface terminations such as –O, –F, and –OH on the catalytic performance of Nb2TiN2 MXene could offer another promising direction to explore. In our future work, we plan to utilize this MXene with various functional groups in these alternative battery chemistries to explore further insights into its catalytic functionality.Data availability
The authors confirm that the data required to reproduce the findings of this study are available within the article and can be reproduced by density functional theory calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by the National Science Foundation (Award No. CBET-2400109) and the Ralph E. Powe Junior Faculty Enhancement Award.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- H. Hu, J. H. Xin, H. Hu, X. Wang and Y. Kong, Appl. Catal., A, 2015, 492, 1–9 CrossRef CAS.
- M. Sang, J. Shin, K. Kim and K. J. Yu, Nanomaterials, 2019, 9, 374 CrossRef CAS PubMed.
- A. G. Olabi, M. A. Abdelkareem, T. Wilberforce and E. T. Sayed, Renewable Sustainable Energy Rev., 2021, 135, 110026 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- K. A. Papadopoulou, A. Chroneos, D. Parfitt and S.-R. G. Christopoulos, J. Appl. Phys., 2020, 128, 170902 CrossRef CAS.
- R. Akhter and S. S. Maktedar, J. Materiomics, 2023, 9, 1196–1241 CrossRef.
- Á. Morales-García, F. Calle-Vallejo and F. Illas, ACS Catal., 2020, 10, 13487–13503 CrossRef.
- Y. Pei, X. Zhang, Z. Hui, J. Zhou, X. Huang, G. Sun and W. Huang, ACS Nano, 2021, 15, 3996–4017 CrossRef CAS PubMed.
- X. Li, Z. Huang, C. E. Shuck, G. Liang, Y. Gogotsi and C. Zhi, Nat. Rev. Chem., 2022, 6, 389–404 CrossRef PubMed.
- H. Vadakke Neelamana, S. M. Rekha and S. V. Bhat, Chem. Mater., 2023, 35, 7386–7405 CrossRef CAS.
- M. Alhabeb, K. Maleski, T. S. Mathis, A. Sarycheva, C. B. Hatter, S. Uzun, A. Levitt and Y. Gogotsi, Angew. Chem., Int. Ed., 2018, 57, 5444–5448 CrossRef CAS PubMed.
- S. Bagheri, A. Lipatov, N. S. Vorobeva and A. Sinitskii, ACS Nano, 2023, 17, 18747–18757 CrossRef CAS PubMed.
- C. Zhou, X. Zhao, Y. Xiong, Y. Tang, X. Ma, Q. Tao, C. Sun and W. Xu, Eur. Polym. J., 2022, 167, 111063 CrossRef CAS.
- H. Riazi, M. Anayee, K. Hantanasirisakul, A. A. Shamsabadi, B. Anasori, Y. Gogotsi and M. Soroush, Adv. Mater. Interfaces, 2020, 7, 1902008 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- N. Zhang, Y. Hong, S. Yazdanparast and M. A. Zaeem, 2D Mater., 2018, 5, 045004 CrossRef CAS.
- S. M. Hatam-Lee, A. Esfandiar and A. Rajabpour, Appl. Surf. Sci., 2021, 566, 150633 CrossRef CAS.
- C. Zhan, W. Sun, P. R. C. Kent, M. Naguib, Y. Gogotsi and D. Jiang, J. Phys. Chem. C, 2019, 123, 315–321 CrossRef CAS.
- D. Johnson, Z. Qiao, E. Uwadiunor and A. Djire, Small, 2022, 18, 2106129 CrossRef CAS PubMed.
- H. Kumar, N. C. Frey, L. Dong, B. Anasori, Y. Gogotsi and V. B. Shenoy, ACS Nano, 2017, 11, 7648–7655 CrossRef CAS PubMed.
- A. Djire, H. Zhang, J. Liu, E. M. Miller and N. R. Neale, ACS Appl. Mater. Interfaces, 2019, 11, 11812–11823 CrossRef CAS PubMed.
- K. Fan, Y. Ying, X. Luo and H. Huang, J. Mater. Chem. A, 2021, 9, 25391–25398 RSC.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- I. Hussain, U. Sajjad, O. J. Kewate, U. Amara, F. Bibi, A. Hanan, D. Potphode, M. Ahmad, M. S. Javed, P. Rosaiah, S. Hussain, K. Khan, Z. Ajmal, S. Punniyakoti, S. S. Alarfaji, J.-H. Kang, W. Al Zoubi, S. Sahoo and K. Zhang, Mater. Today Phys., 2024, 42, 101382 CrossRef CAS.
- R. Syamsai, J. R. Rodriguez, V. G. Pol, Q. Van Le, K. M. Batoo, S. F. Adil, S. Pandiaraj, M. R. Muthumareeswaran, E. H. Raslan and A. N. Grace, Sci. Rep., 2021, 11, 688 CrossRef CAS PubMed.
- D. Maldonado-Lopez, J. R. Rodriguez, V. G. Pol, R. Syamsai, N. G. Andrews, S. J. Gutiérrez-Ojeda, R. Ponce-Pérez, M. G. Moreno-Armenta and J. Guerrero-Sánchez, ACS Appl. Energy Mater., 2022, 5, 1801–1809 CrossRef CAS.
- R. Jayan, A. Vashisth and M. M. Islam, J. Am. Ceram. Soc., 2022, 105, 4400–4413 CrossRef CAS.
- W. Liu, J. Cao, F. Song, D.-D. Zhang, M. Okhawilai, J. Yi, J.-Q. Qin and X.-Y. Zhang, Rare Met., 2023, 42, 100–110 CrossRef CAS.
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 14–29 RSC.
- J. Sun, Z. Du, Y. Liu, W. Ai, K. Wang, T. Wang, H. Du, L. Liu and W. Huang, Adv. Mater., 2021, 33, 2003845 CrossRef CAS PubMed.
- C.-P. Yang, Y.-X. Yin and Y.-G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef CAS PubMed.
- S.-F. Zhang, W.-P. Wang, S. Xin, H. Ye, Y.-X. Yin and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 8759–8765 CrossRef CAS PubMed.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed.
- C.-P. Yang, S. Xin, Y.-X. Yin, H. Ye, J. Zhang and Y.-G. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8367 CrossRef CAS PubMed.
- J. T. Lee, H. Kim, M. Oschatz, D.-C. Lee, F. Wu, H.-T. Lin, B. Zdyrko, W. I. Cho, S. Kaskel and G. Yushin, Adv. Energy Mater., 2015, 5, 1400981 CrossRef.
- K. Sun, H. Zhao, S. Zhang, J. Yao and J. Xu, Ionics, 2015, 21, 2477–2484 CrossRef CAS.
- T. Liu, Y. Zhang, J. Hou, S. Lu, J. Jiang and M. Xu, RSC Adv., 2015, 5, 84038–84043 RSC.
- Y. Liu, L. Si, Y. Du, X. Zhou, Z. Dai and J. Bao, J. Phys. Chem. C, 2015, 119, 27316–27321 CrossRef CAS.
- J. Yang, H. Gao, D. Ma, J. Zou, Z. Lin, X. Kang and S. Chen, Electrochim. Acta, 2018, 264, 341–349 CrossRef CAS.
- H.-C. Youn, J. H. Jeong, K. C. Roh and K.-B. Kim, Sci. Rep., 2016, 6, 30865 CrossRef CAS PubMed.
- Z. Li and L. Yin, Nanoscale, 2015, 7, 9597–9606 RSC.
- K. Han, Z. Liu, J. Shen, Y. Lin, F. Dai and H. Ye, Adv. Funct. Mater., 2015, 25, 455–463 CrossRef CAS.
- S. Vallem, S. Song, Y. Oh, J. Kim, M. Li, Y. Li, X. Cheng and J. Bae, J. Colloid Interface Sci., 2024, 665, 1017–1028 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- N. Li, J. Fan and J. Jia, J. Power Sources, 2025, 640, 236783 CrossRef CAS.
- J. Sun, R. Yu, D. Legut, J. S. Francisco and R. Zhang, Adv. Mater., 2025, 2501523 CrossRef PubMed.
- D. Rao, L. Zhang, Y. Wang, Z. Meng, X. Qian, J. Liu, X. Shen, G. Qiao and R. Lu, J. Phys. Chem. C, 2017, 121, 11047–11054 CrossRef CAS.
- M. Fang, J. Han, S. He, J.-C. Ren, S. Li and W. Liu, J. Am. Chem. Soc., 2023, 145, 12601–12608 CrossRef CAS PubMed.
- T. Boteju, S. Ponnurangam and V. Thangadurai, J. Phys. Chem. C, 2024, 128, 3652–3661 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 1–15 Search PubMed.
- A. Togo, J. Phys. Soc. Jpn., 2023, 92, 012001 CrossRef.
- F. Li, S. Mei, X. Ye, H. Yuan, X. Li, J. Tan, X. Zhao, T. Wu, X. Chen, F. Wu, Y. Xiang, H. Pan, M. Huang and Z. Xue, Adv. Sci., 2024, 11, 2404328 CrossRef CAS PubMed.
- Y. C. Jeong, J. H. Kim, S. H. Kwon, J. Y. Oh, J. Park, Y. Jung, S. G. Lee, S. J. Yang and C. R. Park, J. Mater. Chem. A, 2017, 5, 23909 RSC.
- J. Björk, J. Halim, J. Zhou and J. Rosen, npj 2D Mater. Appl., 2023, 7, 1–7 CrossRef.
- Y. Cheng, L. Wang, Y. Song and Y. Zhang, J. Mater. Chem. A, 2019, 7, 15862–15870 RSC.
- L. Zhu, J. Wang, J. Liu, R. Wang, M. Lin, T. Wang, Y. Zhen, J. Xu and L. Zhao, Nanomaterials, 2024, 14, 666 CrossRef CAS PubMed.
- R. Jayan and M. M. Islam, Nanoscale, 2020, 12, 14087–14095 RSC.
- M. S. Nahian, R. Jayan, T. Kaewmaraya, T. Hussain and M. M. Islam, ACS Appl. Mater. Interfaces, 2022, 14, 10298–10307 CrossRef CAS PubMed.
- L.-C. Zeng, W.-H. Li, Y. Jiang and Y. Yu, Rare Met., 2017, 36, 339–364 CrossRef CAS.
- M. S. Nahian, R. Jayan and M. M. Islam, ACS Catal., 2022, 12, 7664–7676 CrossRef CAS.
- Y. Wang, J. Shen, L.-C. Xu, Z. Yang, R. Li, R. Liu and X. Li, Phys. Chem. Chem. Phys., 2019, 21, 18559–18568 RSC.
- R. Jayan and M. M. Islam, ACS Appl. Mater. Interfaces, 2021, 13, 35848–35855 CrossRef CAS PubMed.
- T. Li, Z. Wang, J. Hu, H. Song, Y. Shi, Y. Jiang, D. Zhang and S. Huang, Chem. Eng. J., 2023, 471, 144736 CrossRef CAS.
- D.-R. Deng, H.-J. Xiong, Y.-L. Luo, K.-M. Yu, J.-C. Weng, G.-F. Li, J. Lei, Y. Li, M.-S. Zheng and Q.-H. Wu, Adv. Mater., 2024, 36, 2406135 CrossRef CAS PubMed.
- Y. Zhong, Q. Wang, S.-M. Bak, S. Hwang, Y. Du and H. Wang, J. Am. Chem. Soc., 2023, 145, 7390–7396 CrossRef CAS PubMed.
- R. Jayan and M. M. Islam, J. Phys. Chem. C, 2021, 125, 4458–4467 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta03099d |
| This journal is © The Royal Society of Chemistry 2025 |