
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering Ru nanoparticle size and metal–support interactions for enhanced catalytic hydrogen combustion†
Zohreh
Akbari
 *ab,
Mohammad Reza
Alizadeh Kiapi
c,
Thi Ha
My Pham
ab,
Loris
Lombardo
*ab,
David
Fairen-Jimenez
c and
Andreas
Zuttel
ab
*ab,
Mohammad Reza
Alizadeh Kiapi
c,
Thi Ha
My Pham
ab,
Loris
Lombardo
*ab,
David
Fairen-Jimenez
c and
Andreas
Zuttel
ab
aLaboratory of Materials for Renewable Energy (LMER), Institute of Chemical Sciences and Engineering (ISIC), Basic Science Faculty (SB), École Polytechnique Fedérale de Lausanne (EPFL) Valais/Wallis, Energypolis, Rue de l’Industrie 17, 1951 Sion, Switzerland. E-mail: zohreh.akbari@epfl.ch
bEmpa Materials Science & Technology, 8600 Dubendorf, Switzerland
cThe Adsorption & Advanced Materials Laboratory (A2ML), Department of Chemical Engineering & Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
First published on 2nd June 2025
Abstract
Catalytic hydrogen combustion (CHC) plays a crucial role in enhancing the safety and efficiency of fuel cells and electrolysers, thereby promoting the H2 economy. To increase the catalytic activity of supported metal particles for CHC, the active surface area can be increased through Ru fine dispersion, and intrinsic activity can be enhanced by optimising metal–support interactions (MSIs). In this study, we report the synthesis and CHC performance of highly dispersed Ru sub-nanoparticles on a γAl2O3 support with various Ru loadings. A clear correlation between Ru loading and CHC mass activity was identified. The highest mass activity is achieved at 1 wt% Ru, with a yield of 5.7 mmolH2 mol−1Ru s−1 at 80 °C. Lower Ru loadings lead to a strong MSI and subsequently to a lower Ru0/Ru–O ratio. Further, higher Ru loadings decrease metal dispersion, reducing CHC activity. Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and density functional theory (DFT) calculations confirmed the role of OH groups as key intermediates in the CHC mechanism over the Ru-γAl2O3 catalyst. Our findings highlight the impact of Ru nanoparticle size engineering on CHC mass activity and provide mechanistic insights and design principles for the development of highly active Ru catalysts, showing a way forward to achieve safer, integrated and efficient CHC utilisation.
1. Introduction
The ongoing energy and climate crises have accelerated the transition from fossil fuels to H2 as a sustainable, carbon-free energy carrier, enabling the large-scale integration of renewable energy sources. As an energy vector, H2 is crucial in driving decarbonization efforts across diverse sectors, particularly in energy conversion1–4 and power and heat production.5–7 To maintain safety standards, however, the H2 concentration in an exhaust must remain below the flammability limit of H2 in air (4 vol%).8,9 Although the controlled combustion of residual H2 is the most effective way to mitigate this risk while improving overall system efficiency, conventional, high-temperature combustion methods introduce several challenges, such as risks of flashback and NOx emissions.10,11Consequently, catalytic H2 combustion (CHC) has emerged as a promising alternative, enabling the conversion of H2 to water at temperatures below the autoignition point of H2.12,13 This approach is especially relevant in electrolysers and fuel cells, where H2 crossover significantly compromises safety and efficiency.14 Particularly in electrolysers operating at high H2 pressures, a thin layer of Pt catalyst coated on the Nafion membrane is often applied at the anode side.15 This layer catalyses H2 combustion in the presence of O2 and enhances both safety and stack lifetime. In fuel cells, CHC also contributes to improved energy efficiency by increasing the enthalpy of the gas stream before the expansion turbine and ultimately boosting the turbocharger efficiency.16
In a typical CHC process, the highly exothermic reaction between H2 and O2 takes place in the presence of a catalyst, yielding water as the sole product (eqn (1)).
| H2 + ½O2 → H2O ΔHH2fO(g) = −241.82 kJ mol−1 | (1) |
Pt and Pd catalysts are commonly employed in CHC owing to their unique ability to initiate H2 combustion at RT.17–20 Nevertheless, thermographic studies have revealed that at low flow rates, water formation below its evaporation temperatures extinguishes the reaction and subsequently decreases the reaction rate.21 Therefore, other candidates have been investigated to overcome the limitations associated with Pt and Pd-based catalysts.22–25 Despite these efforts, most alternative catalysts remain active only above 300 °C, limiting their practical applicability in low-temperature systems.
In addition to the metal catalyst, the support material plays a critical role in CHC activity.25–27 The Metal-support interaction (MSI) significantly influences the catalytic activity by modulating the charge transfer between the metal catalyst and the support, affecting the metal-O coordination number, the metal nanoparticle (NP) size and dispersion.26,28,29 Supported monometallic catalysts have shown strong size sensitivity in different catalytic processes.30 Recent advances in single-atom catalysis have further highlighted the importance of optimising MSIs to enhance catalytic activity and stability.31 Hence, it is essential to optimise the MSI to achieve high catalytic activity. However, tuning the catalytic activity through MSI regulation remains underexplored in CHC. Recently, it has been shown that by controlling the size of the Cu NPs using different Cu loadings, the Cu–O coordination can be tuned to achieve the highest mass activity at 5 wt% Cu on Al2O3 support.32 However, the mass activity only reached 0.009 molH2 mol−1Cu s−1, which is a relatively low mass conversion rate for temperatures as high as 300 °C. This highlights the critical role of MSI in determining catalytic performance, underscoring the need for a deeper understanding of this interaction to achieve high activity at lower temperatures.
Moreover, the rapid kinetics of the CHC process complicate the elucidation of the reaction mechanism. The widely accepted CHC mechanism for Pt and Pd catalysts involves the dissociative adsorption of O2 and H2 followed by the formation of H2O (eqn (2) and (3). If the adsorbed water reaches a sufficient concentration, it can dissociate back to OH (eqn (4)):33
| Oads + Hads → OHads | (2) |
| 2OHads + Hads → H2Oads → H2Ogas | (3) |
| H2Oads + Oads → 2OHads | (4) |
Building on these findings and limitations, Ru has emerged as a promising catalyst for initiating CHC at relatively low temperatures, with complete H2 conversion occurring just above 100 °C.35 This prevents water-induced inhibition and enhances system stability. Nonetheless, further insight into maximising mass activity by tuning the MSI is needed to optimise catalyst loading for practical applications.
In this paper, we describe the design and development of well-dispersed supported Ru NPs and the optimisation of their size to achieve the highest H2 mass conversion rate. We have synthesised a series of xRu-γAl2O3 (x = Ru loading in wt%) catalysts and examined the effect of Ru NPs size on MSI and CHC activity by varying the Ru loading (wt%). Moreover, we investigate the kinetics of the CHC reaction over the xRu-γAl2O3 catalyst, identifying a clear trend in the CHC activity and activation energies (Ea), that points to an optimum loading of Ru. To further elucidate the role of OH groups and the CHC mechanism on Ru-γAl2O3, operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is performed, complemented by density functional theory (DFT) calculations. Our findings provide guidance for the effective engineering of Ru NPs size and MSIs to achieve high CHC mass activity.
2. Experimental section
2.1. Synthesis of Ru-γAl2O3 catalysts
Commercially available γAl2O3 pellets (AlfaAesar) are crushed into powder. As described in our previous work, a support pretreatment is employed to enhance the MSI by introducing OH− groups.36,37 Briefly, the γAl2O3 is first heat-treated at 500 °C in air to remove any impurities and then immersed in a 3 M ammonia solution. After filtration, the powder is washed with hot DI water (80 °C) and dried at 120 °C overnight.The xRu-γAl2O3 (x = Ru wt%) catalysts are synthesised through the incipient wetness impregnation method. Ru chloride hydrate (RuCl3·xH2O) serves as the metal precursor, and DI water as the solvent. The obtained catalyst is heat-treated at 400 °C for 2 h under forming gas (FG, 5 vol% H2 in N2). This reduction temperature is chosen based on results obtained from the H2 temperature programmed reduction (H2-TPR) of RuCl3·xH2O (Fig. S1†).
Although the reduction process initiates the removal of the chlorine ions, complete removal is hindered by diffusion limitations and/or the strong adsorption of chlorine ions by Ru atoms. To address this, an additional chemical treatment step is performed to remove the chlorine ions using a 3 M ammonia solution, followed by a hot DI water washing procedure.38 Subsequently, pellets with the desired size (250–500 mesh sieve) are formed, and their CHC performance is investigated.
The heat-treatment atmosphere significantly impacts the resulting Ru phase and NP dispersion. An oxidising atmosphere leads to the agglomeration of Ru atoms and the formation of RuO2. In contrast, a reducing atmosphere stabilises the Ru NPs. Interested readers are referred to section 2 of the (ESI),† which describes the effects of the heat-treatment atmosphere on the phase (Fig. S2†) and particle size (Fig. S3†) of xRu-γAl2O3 catalysts.
2.2. Catalyst characterisation
The size and distribution of the Ru NPs are measured by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) using a Thermo Fisher Scientific Spectra200 system at 200 kV. Elemental maps are obtained by energy dispersive spectroscopy using Super-X detectors. The surface area, pore size, and pore size distribution are analysed by N2 adsorption/desorption isotherm at −196 °C performed using a Belsorp maxII instrument. H2 uptake measurements are also conducted using the same instrument at −196 °C. Powder X-ray diffraction (PXRD) is employed to characterise the crystal phases using a Bruker D8 Advance instrument with Cu Kα (λ = 1.54 Å) radiation at 40 kV and 40 mA. To study the reducibility of the catalysts, H2-TPR is conducted by implementing thermogravimetric analysis coupled with a mass spectrometer (TGA-MS, Netzsch). The surface chemistry of the catalysts is examined by X-ray photoelectron spectroscopy (XPS, Kratos Axis Supra with a monochromated Al Kα X-ray source). The binding energy of the samples is corrected by referencing the binding energy of C 1s to 284.8 eV. The Ru content is confirmed by inductively coupled plasma-mass spectrometry (ICP-QMS, nexION35010, PerkinElmer).Operando DRIFTS is used to investigate the mechanism of the CHC reaction over Ru-γAl2O3 catalysts. The spectra are recorded using a Bruker Tensor27 spectrometer. The setup is equipped with a Praying Mantis high-temperature chamber (Harrick Scientific) and a mercury–cadmium–telluride (MCT) detector. A pellet with the exact size of the chamber (6 mm) is made from the catalyst powder. The exhaust gas is analysed by MS. The same temperature procedure is used for reduction, and the CHC reaction condition is followed in the DRIFTS experiments. The DRIFTS spectra are collected by averaging 38 spectra with 2 cm−1 spectral resolution in the 800–4000 cm−1 range. The reference spectrum is collected on the reduced sample at RT under flowing He. Water and atmospheric compensation were applied for all spectra.
2.3. Catalytic activity tests
A schematic of the experimental setup is presented in Fig. S4.† The CHC performance is evaluated using a fixed-bed reactor. In each test, 100 mg of the catalyst pellets are placed in the middle of a 35 cm stainless steel tube with quartz wool, and the reactor is housed in a furnace (Carbolite Gero). The temperature of the reactor is controlled and monitored using two K-type thermocouples: one attached to the outside of the reactor and one positioned inside the reactor, just before the reaction bed. A gas flow of 20 ml. min−1 is supplied to the reactor using three mass flow controllers (Bronkhorst) for FG, N2, and synthesised air (80 vol% N2 and 20 vol% O2). The total gas hourly space velocity (GHSV) is maintained at 6000 h−1. The exhaust gas composition is analysed by MS (OmniStar 320, Pfeiffer Vacuum). Before introducing the reactive gas, the catalysts are activated (reduced) at 400 °C under FG. After reduction, the system is flushed with N2 and cooled to RT. Subsequently, the reactive gases are introduced to the reactor at a volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:96 (H2:O2:N2). The reactor temperature is then increased to 300 °C with a heating rate of 1 °C min−1. The H2 conversion and mass conversion rates are calculated using the following equations:25,32
:2:96 (H2:O2:N2). The reactor temperature is then increased to 300 °C with a heating rate of 1 °C min−1. The H2 conversion and mass conversion rates are calculated using the following equations:25,32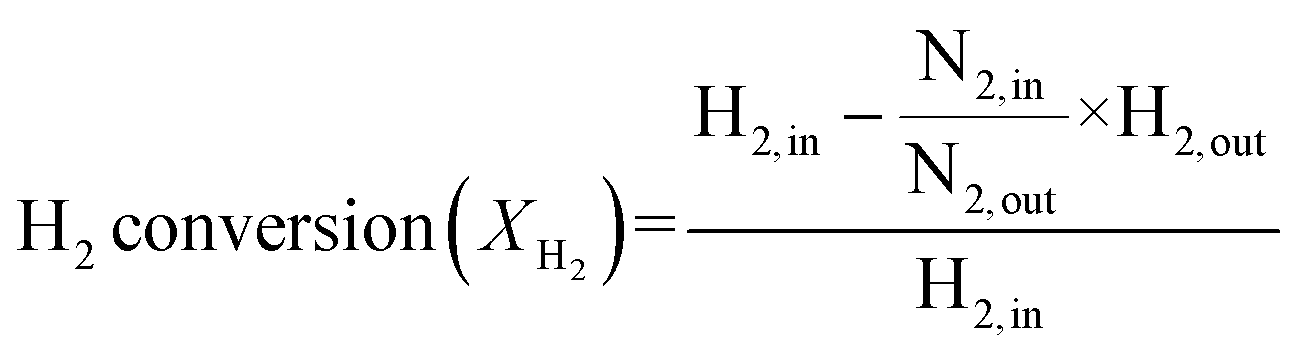 | (5) |
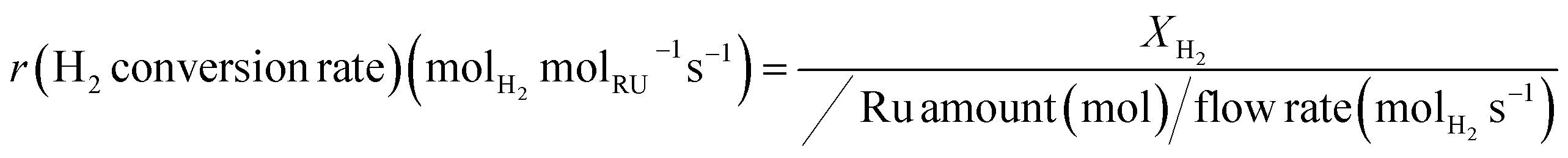 | (6) |
It is important to note that comparing the CHC activity of the catalysts using eqn (6) is only valid for low H2 conversions, where the axial change in the reactant concentration can be neglected throughout the catalyst bed. Consequently, we applied the Weisz-Prater Criteria39 to validate the temperature range in which internal and external mass transfers are not critical, and therefore, the calculated rates are not affected by mass transfer limitations.
2.4. DFT calculation
All spin-polarised DFT calculations in this study are performed using the Quantum ESPRESSO (QE) simulation package.40,41 The generalised gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional is employed,42 complemented by the D3BJ dispersion correction scheme to account for dispersion interactions.43 The core electrons are treated using the optimised norm-conserving Vanderbilt (ONCV) pseudopotentials,44 and a plane-wave energy cutoff of 480 eV is applied in all calculations. The Brillouin zone is sampled using a 4 × 4 × 1 Γ-centered Monkhorst–Pack grid, but for isolated H2, O2, and H2O molecules, only the Γ-point is used. Methfessel–Paxton smearing45 with a width of 0.2 eV is applied for surface/adsorbate systems, whereas a Gaussian smearing method with a narrow width of 0.001 eV is employed for isolated molecules. During geometry optimisations, structures are relaxed until the maximum force on any atom is less than 0.014 eV Å−1.Frequency calculations are conducted, using phonon calculations based on density functional perturbation theory (DFPT) as implemented in QE, for all atoms within the adsorbates and isolated molecules, and the frequencies of the Ru slab are excluded to improve computational efficiency. The results of these frequency calculations are used to compute thermochemical corrections to the electronic energy (EDFT) and obtain the Gibbs free energy (G) (eqn (7)).
| G = EDFT + EZPE + Ethermal(T) − T × S(T) | (7) |
The Ru (0001) surface is selected for modelling. A 4 × 2 supercell (cell a = 9.1709 Å, cell b = 10.5889 Å) is constructed from the optimised orthogonalized conventional cell of Ru, enabling the exploration of favourable adsorption configurations across various potential sites on the Ru (0001) surface. The slab model comprises three atomic layers because previous studies indicate that this thickness is sufficient to achieve energy convergence with minimal deviation from thicker slabs.46 The bottom layer is fixed to simulate the bulk structure, and a vacuum spacing of 15 Å is introduced perpendicular to the surface to minimise slab–slab interactions. Considering that adsorbates are placed on one side of the slab, resulting in an asymmetric unit cell, dipole corrections are applied along the surface-normal direction to account for asymmetry iRASPA software package is used47 to visualise the atomistic structures of different states.
3. Results and discussion
3.1. Effects of the Ru NP size and kinetics
The size and distribution of the synthesised xRu-γAl2O3 catalysts are presented in Fig. 1. The particle size was determined from the STEM images, and the analysis was conducted based on 100 Ru NPs for each catalyst, revealing well-dispersed Ru NPs in all samples. The results indicate that increasing the Ru loading leads to larger average particle sizes and broader particle size distributions. Among all the catalysts, 0.5Ru-γAl2O3 showed the smallest particle size, with a mean value of 0.42 ± 0.06 nm. Conversely, 10Ru-γAl2O3 showed the largest particle size, with a mean value of 1.60 ± 0.19 nm. The mean particle sizes of the different catalysts are summarised in Table S1.† | ||
| Fig. 1 AC-HAADF-STEM images of (a–e) xRu-γAl2O3 catalysts. The scale bar represents 20 nm. The histograms show the size distributions of each catalyst. (f) The mean particle size of xRu-γAl2O3 with respect to Ru loading (wt%). | ||
This trend in particle size is further illustrated in Fig. 1f, which shows the relationship between mean particle size and Ru loading. The results indicate that the particle size follows a geometrical correlation in which the size increases as a function of the (Ru loading)1/3. This trend aligns with the expected behaviour, where a geometric scaling law relates metal mass to particle size in a three-dimensional growth.
As the NP size increases, the surface-to-volume ratio decreases, leading to a corresponding drop in dispersion (D). Herein, D drops by 26.6% for the catalyst series from 0.5Ru-γAl2O3 to 10Ru-γAl2O3. The details of D calculations are provided in section 4 of the SI. D is later used to calculate the turnover frequency (TOF) over the xRu-γAl2O3 catalysts.
To evaluate and compare the catalytic performance of xRu-γAl2O3 catalysts during CHC, a reference test condition is established as follows: a 100 mg pellet, a stoichiometric ratio between H2 and O2, a gas composition of H2:O2:N2 = 4:2:94 vol%, and a total flow rate of 20 ml min−1 (GHSV = 6000 h−1). A typical H2 conversion vs. temperature curve obtained for the CHC reaction exhibits a sigmoidal shape. Note that chloride anions poison the catalyst surface (Fig. S5†). As such, this study only considers chlorine-washed catalysts. Section 5 in the ESI† is dedicated to investigating the effect of chlorine ions on the CHC performance and H2 uptake capacity of the Ru NPs.
Fig. 2a presents the effect of Ru loading on H2 conversion (XH2) at temperatures ranging from RT to 300 °C. The CHC activity enhancement is attributed to the increased number of active surface sites in samples with higher Ru loading. However, when H2 conversion is normalised to the mass of Ru, it becomes evident that the mass activity does not increase proportionally with Ru loading. Fig. 2b shows the H2 mass conversion rates over Ru, calculated at three different temperatures, 40, 60 and 80 °C. A clear trend in the Ru mass activity is observed for all temperatures, with 1Ru-γAl2O3 and 10Ru-γAl2O3 showing the maximum and minimum CHC activity, respectively. As the size of the Ru NPs changes, the number of active sites also varies. To investigate the effect of the fraction of surface atoms and include the size effect in the calculations, TOF is calculated using eqn (8) (square points in Fig. 2b).
 | (8) |
 | ||
| Fig. 2 (a) Conversion XH2, (b) mass activity (columns) and TOF (square points), and (c) kinetic parameters (columns as activation energy and square points as frequency factor) of the xRu-γAl2O3 catalysts versus Ru loading. | ||
The trend in TOF is similar to the mass activity for all temperatures, confirming that using 1 wt% Ru results in the optimal Ru NP size. Consequently, XH2 increases with higher Ru loadings at each temperature, whereas CHC mass activity decreases.
The kinetically controlled region is identified to study the kinetics of the CHC reaction over xRu-γAl2O3 catalysts. We have applied Weisz-Prater criteria to validate the absence of internal and external mass transfers in the temperature spans 40–80 °C. Using the Arrhenius model, the activation energy (Ea) and the frequency factor (A, pre-exponential factor) are calculated with results summarised in Fig. 2c. Two additional GHSVs (3000 and 12000 h−1) were tested for 2R-γAl2O3 and provided similar Ea, confirming the reliability of the used method to extract the kinetic information and that the kinetic parameters are independent of flow rate and GHSV. The resulting Ea values are summarised in Table S2.†
As can be seen from Fig. 2c, the Ea values are of the same order of magnitude for all xRu-γAl2O3 catalysts, with the minimum value of 17.2 kJ mol−1 observed for 1Ru-γAl2O3 catalyst.
Variations in the TOF and Ea values demonstrate that changes in the Ru loading influence the Ru NPs' size and their intrinsic catalytic activity, indicating the involvement of additional factors affecting intrinsic activity, such as size-dependent modifications in MSIs. These modifications can alter the xRu-γAl2O3 interface, impacting the nature and intrinsic properties of the Ru active sites.
3.2. Evaluation of the physicochemical properties
MSI significantly influences catalytic activity by altering electronic interactions and charge transfer.48 To assess changes in MSI with varying Ru particle sizes and determine the reduction properties of the catalysts, H2-TPR measurements are performed. Fig. 3a displays the normalised ion current of H2, detected by MS, for all the catalysts. Given that the Ru loading is low in 0.5 and 1Ru-γAl2O3, the signals exhibit some noise. Notably, the catalyst with the lowest Ru content, 0.5Ru-γAl2O3, displays the highest reduction temperature, indicating the presence of a strong MSI (SMSI). Such strong interactions result in reduced catalytic activity compared to 1Ru-γAl2O3, aligning with previous studies, where SMSI hinders the catalytic activity.49,50 The reason for diminished catalytic activity as a consequence of SMSI is the increase in the metal-O coordination number.32 The higher Ru–O compared to metallic Ru in the case of 0.5Ru-γAl2O3 catalyst is further confirmed by the lowest Ru0/RuOx ratio obtained from XPS analysis (Table 1). The SMSI in 0.5Ru-γAl2O3 catalyst is consistent with a study by Yan et al.,51 where they showed that an SMSI is expected at low Ru loadings (<1 wt%) on γAl2O3 supports. The origin of MSI is likely the adsorption of positively charged Ru ions on the hydroxylated γAl2O3 support during the synthesis procedure.52 | ||
| Fig. 3 (a) H2-TPR profiles and (b) PXRD patterns of the xRu-γAl2O3 catalysts. The H2-TPR tests are conducted under FG with a flow rate of 20 ml min−1 and a heating rate of 10 °C min−1. | ||
| Catalyst | 0.5Ru-γAl2O3 | 1Ru-γAl2O3 | 2Ru-γAl2O3 | 5Ru-γAl2O3 | 10Ru-γAl2O3 |
|---|---|---|---|---|---|
| Ru0/RuOx | 0.8 | 2.1 | 1 | 1.8 | 1.3 |
The minimum reduction temperature is achieved as the Ru loading increases to 2 wt%. This is an indication of Ru nanocluster formation with lower MSI.51 Although 2Ru-γAl2O3 exhibits the lowest reduction temperature, this does not necessarily correspond to higher mass activity, as seen in the CHC mass activity values (Fig. 2b). This discrepancy arises from the combined effects of weaker MSI and lower catalyst dispersion compared to 1Ru-γAl2O3 (Table S1†). Moreover, previous studies have also shown that achieving the highest catalytic activity requires an optimal MSI, which can be tuned by controlling the size of the NPs.53,54 Therefore, obtaining a high mass activity in CHC necessitates a balance between an optimal MSI and catalyst dispersion, which is achieved with 1Ru-γAl2O3 in our case.
Moreover, the TPR peak shifts to higher temperatures for the change in Ru loading from 2 to 10 wt%. This can be explained by the growth of the NPs as measured from the AC-STEM-HAADF images (Fig. 1). Also, the multiple peaks observed in the TPR profile of 5 and 10Ru-γAl2O3 are due to the broader size distribution of the Ru NPs.
PXRD measurements are performed to determine the crystallinity and phases present in the catalysts (Fig. 3b). The absence of any sharp metallic Ru peaks confirms the formation of well-dispersed Ru NPs.
To evaluate the influence of the surface area of the 0.5, 1, and 2Ru-γAl2O3 catalysts on their catalytic properties, N2 gas adsorption/desorption measurements are performed. The obtained N2 adsorption/desorption profiles are shown in Fig. S6a.† The obtained surface areas for the 0.5, 1, and 2Ru-γAl2O3 are 222, 216, and 215 m2 gr−1, respectively, whereas γAl2O3 has a BET surface area of 200 m2 gr−1. Similar values were expected because the Ru loading is low. Introducing Ru NPs onto γAl2O3 increases the number of pores below 5 nm, which is due to the impregnation of Ru. All three catalysts exhibit similar pore sizes and distributions; thus, the differences in CHC mass activity do not originate from catalyst porosity.
3.3. Evaluation of the electronic properties
XPS measurements are conducted on the reduced catalysts to elucidate the surface chemistry and the state of Ru. The XPS spectra are provided in Fig. 4, revealing the coexistence of metallic Ru (Ru0) and oxidised Ru (RuOx) states in all catalysts. As we later confirmed by DFT calculation, the dissociative adsorption of O2 on Ru presents no energy barrier. Hence, the presence of the oxide phase after reduction is expected owing to partial surface oxidation, considering that it was exposed to the air.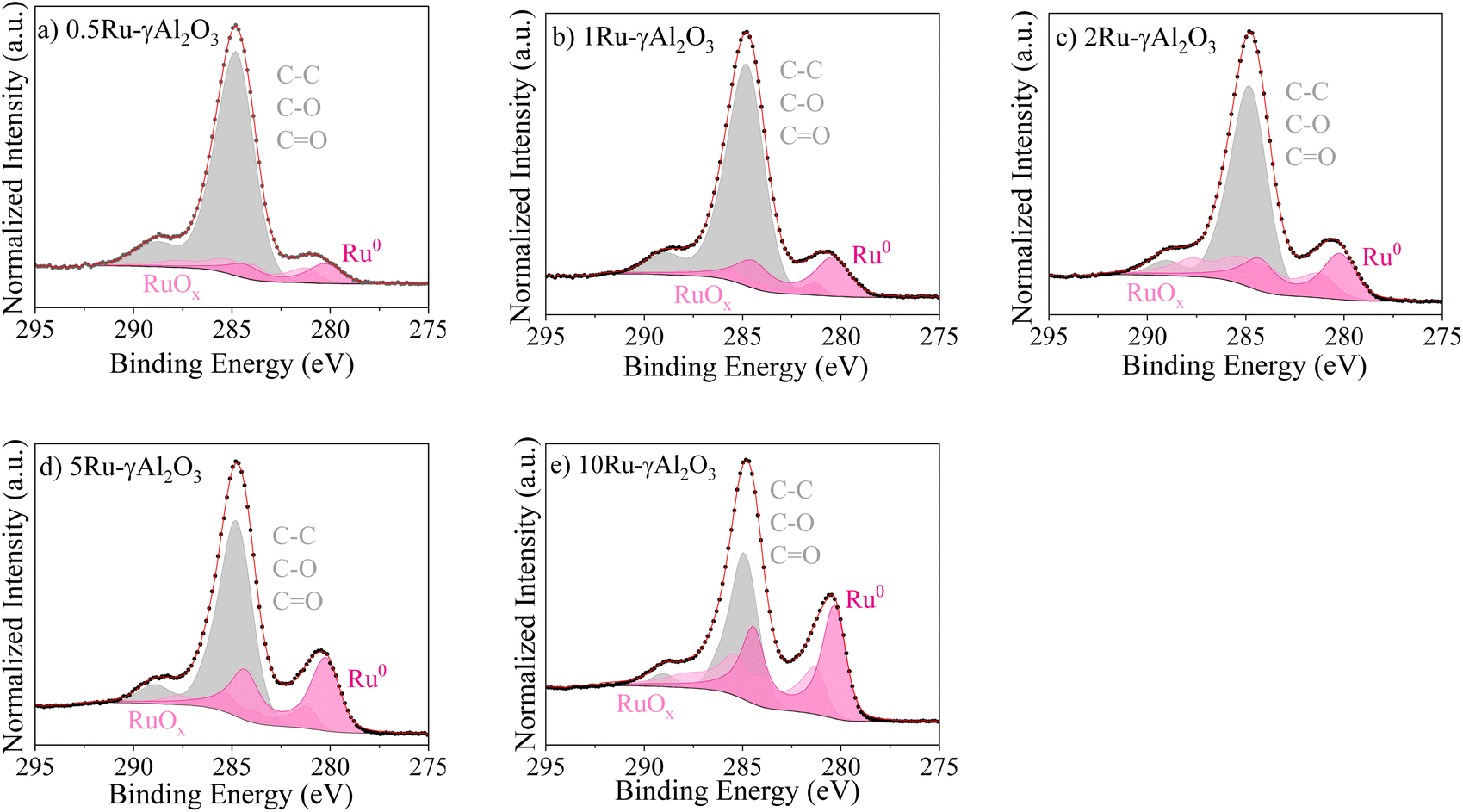 | ||
| Fig. 4 XPS Ru 3d spectra of the (a–e) 0.5, 1, 2, 5, and 10 γAl2O3 catalysts. The dotted points are experimental data, and the red lines are the fits. | ||
Table 1 shows the calculated ratio between the two Ru states (Ru0/RuOx). Notably, RuO2 is identified as the predominant Ru oxide phase. The lowest Ru0/RuOx ratio is obtained for 0.5Ru-γAl2O3, consistent with its smallest particle size. This results in the maximum number of exposed surface Ru atoms, which are more prone to oxidation upon environmental exposure. In contrast, the highest Ru0/RuOx ratio is observed for 1Ru-γAl2O3, consistent with our findings that a 1 wt% Ru loading provides the optimal MSI and Ru NP size, thereby achieving the highest CHC mass activity.
For 2Ru-γAl2O3, the weaker MSI facilitates easier oxidation upon exposure to the environment, resulting in a lower Ru0/RuOx ratio. Meanwhile, the 5Ru-γAl2O3 catalyst, which has a larger particle size than 2Ru-γAl2O3, exhibits fewer exposed Ru atoms and is less susceptible to oxidation. Consequently, a higher Ru0/RuOx ratio is observed owing to the increased proportion of bulk metallic Ru.
3.4. Mechanistic study
 | ||
| Fig. 5 DRIFTS spectra of 1Ru-γAl2O3 catalysts under reactive gas (4 vol% H2, 2 vol% O2, and 94 vol% He), from 20 to 300 °C. The reference spectrum (first spectrum from the bottom) is taken on the reduced catalyst at 20 °C. | ||
(1) The broad peaks at 3000–3750 cm−1 arise from the stretching of the different types of OH groups formed on the catalyst's surface.55,56 Based on previous studies,57–59 the peaks at 3630 and 3710 cm−1 do not correspond to the adsorbed OH on the surface of the γAl2O3. As a result, we assign these peaks to the formation of OH species on the Ru NPs.55 While the intensity of the peak at 3710 cm−1 increases as the temperature rises, the intensity at 3630 cm−1 decreases.
(2) The small peak around 1650 cm−1 arises from the OH bending of water molecules formed during the reaction.34
(3) A peak around 1830–1890 cm−1 appears at temperatures above 100 °C. It has been shown that oxide materials can give rise to overtone peaks in the 1800–2200 cm−1 range.60–62 Hadjiivanov et al.60 have assigned this peak to the oxidation of Run+ ions to Ru4+ in the presence of O2 in a hydrated RuOx phase. Thus, we assigned this peak to the formation of RuOx under the CHC reaction. To evaluate our assignment, we reduced the catalyst and found that this peak vanished as the temperature increased.
From points 1 and 2, we can conclude that even at RT, the catalyst is active toward the CHC reaction, as evidenced by the presence of the aforementioned OH evolution peaks. Moreover, Fig. 2a and 5 indicate that the CHC reaction over the 1Ru-γAl2O3 catalyst proceeds through an OH group-involved pathway. This conclusion is supported by the observed increase in peak intensity at 3630 and 3710 cm−1 with rising temperature. DFT calculations are used to further assess this observation and gain more mechanistic insights.
First, we examine the interactions between the Ru (0001) surface and key atomic and molecular species involved in the reaction process, including H, O, H2, O2, OH, and H2O. Adsorption studies are performed at typical sites - top, bridge, hollow hcp, and hollow fcc - using multiple possible orientations for the H2, O2, and H2O molecules. This enables us to identify the most energetically favourable adsorption configurations for the reactants, products, and intermediates (H, O, and OH) on the Ru (0001) surface. The resulting reaction pathway, Gibbs free energy profiles, and corresponding adsorption configurations for the CHC reaction are shown in Fig. 6. The top and side views of all the states are provided in Table S3.†
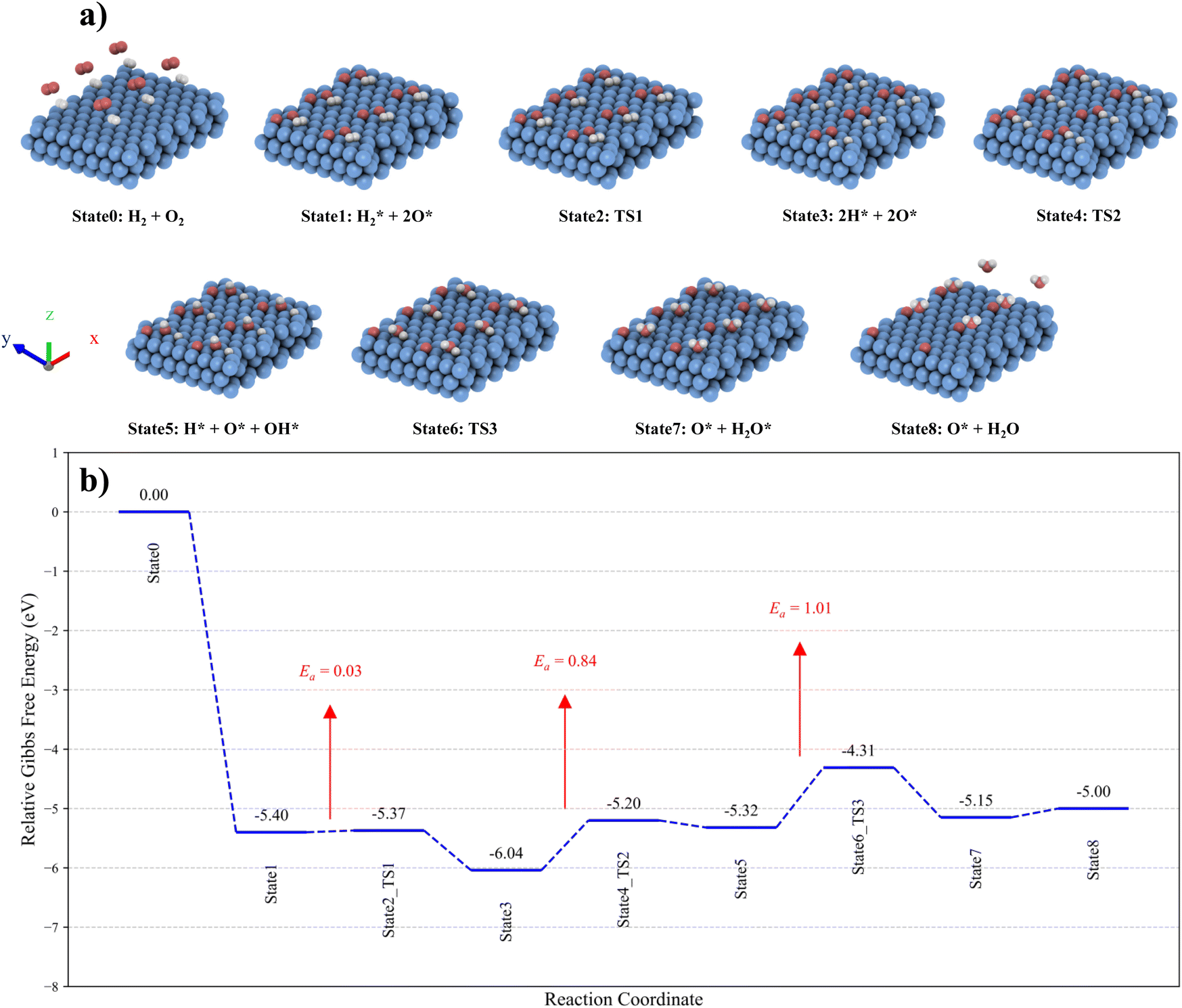 | ||
| Fig. 6 (a) The CHC reaction pathway over the Ru (0001) surface. The simulated cell is expanded 3 times in the x-direction and 2 times in the y-direction for better visualisation. (b) Gibbs free energy profiles and corresponding adsorption configurations at 120 °C. | ||
The interaction between the H2 molecule and the Ru (0001) surface was modelled as a two-step process involving adsorption and subsequent dissociation. In contrast, O2 dissociation was found to occur spontaneously, with no Gibbs free energy barrier, making direct dissociation highly favourable. This is consistent with the DRIFTS results, where the oxidation of Ru NPs is evidenced by the appearance of a peak around 1830–1890 cm−1.
Additionally, the Gibbs free energy change for the dissociation of O2 into oxygen adatoms, combined with the adsorption of H2 (State0 → State1), is calculated to be −5.4 eV, highlighting the strong binding affinity between oxygen adatoms and the Ru (0001) surface. This is significantly stronger than the Gibbs free energy change of −0.64 eV for the dissociation of H2 (State1 → State3). The H2 molecule preferentially adsorbs on the top site of the Ru(0001) surface, aligning parallel to the surface (State1). Once adsorbed, H2 readily dissociates into two H atoms (State3) with a very low Gibbs free energy barrier of 0.03 eV, demonstrating the surface's high catalytic efficiency for H2 dissociation. The most energetically favourable site for the dissociated oxygen adatoms is the hollow hcp site, whereas dissociated hydrogen atoms preferentially occupy the hollow fcc sites. These findings are consistent with results obtained in previous computational and experimental studies.46,66,67 Then, the reaction between oxygen and hydrogen species to form surface OH species (State3 → State5) on the Ru (0001) surface proceeds with an energy barrier of 0.84 eV.
The adsorbed OH species preferentially occupy the hollow fcc site (State5). Hence, during the second transition state (State4), the oxygen and hydrogen adatoms migrate to the nearest hollow fcc site to facilitate the reaction and take on the most stable configuration. Subsequently, H2O is formed (State5 → State7) through the reaction between OH and hydrogen species, with an energy barrier of 1.01 eV. Unlike OH, the H2O molecule favours adsorption at the top site. During the third transition state (State6), the OH and hydrogen species move to the nearest top site to form the adsorbed water molecule. This demonstrates the role of formed OH groups on the Ru NPs in the reaction pathway, as we observed by DRIFTS.
The desorption of the H2O molecule from the Ru (0001) surface (State7 → State8) is slightly endothermic, with an energy change of 0.15 eV, indicating that the water molecule can be readily removed. The calculated energy barriers for the transition states (State2, State4, and State6) reveal that the formation of the H2O molecule is the rate-determining step in the overall catalytic process on the Ru (0001) surface.
4. Conclusion
We synthesised well-dispersed sub-nano-sized xRu-γAl2O3 (x = 0.5, 1, 2, 5, and 10) catalysts through the incipient wetness impregnation method and evaluated their catalytic activity for the CHC reaction. By controlling the Ru particle size through varying Ru loadings, we optimised the MSI and Ru oxidation state to achieve the maximum CHC mass activity. A distinct relationship was identified between the CHC mass activity or turnover frequency (TOF) and Ru nanoparticle (NP) size. The highest CHC mass activity and TOF were achieved with an optimal Ru particle size of 0.75 ± 0.12 nm, corresponding to a 1 wt% Ru loading. At this loading, the catalyst exhibited a maximum mass activity of 5.7 mmolH2 mol−1Ru s−1 at 80 °C. Kinetic studies further revealed that the minimum activation energy for the CHC reaction (17.2 kJ mol−1) coincided with the optimal Ru NP size.Additionally, operando DRIFTS was conducted to investigate the CHC reaction mechanism using the 1Ru-γAl2O3 catalyst. The critical role of OH groups as intermediates in the CHC reaction was confirmed, as supported by DFT calculations. These insights provide a comprehensive understanding of the structure–performance relationship and the reaction mechanism, guiding the design of highly efficient Ru catalysts for CHC applications.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
We would like to thank Prof. Christophe Copéret for the discussions on DRIFTS data and Dr Mehrdad Asgari for assistance with DFT calculations. We also thank Interdisciplinary Center for Electron Microscopy (CIME) at EPFL for providing access to microscope facilities.References
- G. Anello, G. De Luna, G. De Felice, A. Saker, L. Di Felice and F. Gallucci, Int. J. Hydrogen Energy, 2024, 86, 922–930 Search PubMed.
- T. P. Minyukova and E. V. Dokuchits, Int. J. Hydrogen Energy, 2023, 48, 22462–22483 Search PubMed.
- S. Nuggehalli Sampathkumar, P. Aubin, K. Couturier, X. Sun, B. R. Sudireddy, S. Diethelm, M. Pérez-Fortes and J. Van herle, Int. J. Hydrogen Energy, 2022, 47, 10175–10193 Search PubMed.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 Search PubMed.
- G. Li, X. Zhang, Y. Zheng, Y. Zhu, W. Guo and Y. Tang, Int. J. Hydrogen Energy, 2023, 48, 22264–22276 Search PubMed.
- B. Fumey, S. Stoller, R. Fricker, R. Weber, V. Dorer and U. F. Vogt, Int. J. Hydrogen Energy, 2016, 41, 7494–7499 Search PubMed.
- L. V. Hoecke, L. Laffineur, R. Campe, P. Perreault, S. W. Verbruggen and S. Lenaerts, Energy Environ. Sci., 2021, 14, 815–843 Search PubMed.
- L. Gardner, Z. R. Liang, B. Ibeh, M. Hurley, S. Cotosman and J. Murphy, Hydrogen recombiners for non-nuclear hydrogen safety applications, International Conference on Hydrogen Safety, Canada, 2023 Search PubMed.
- W. Yu, X. Yu, S.-T. Tu and P. Tian, Int. J. Hydrogen Energy, 2017, 42, 14829–14840 Search PubMed.
- B. Fumey, T. Buetler and U. F. Vogt, Appl. Energy, 2018, 213, 334–342 Search PubMed.
- L. Hu, F. Chen, Y. Meng and Z. Sang, Int. J. Hydrogen Energy, 2024, 84, 334–343 Search PubMed.
- J. Kim, J. Yu, S. Lee, A. Tahmasebi, C.-H. Jeon and J. Lucas, Int. J. Hydrogen Energy, 2021, 46, 40073–40104 Search PubMed.
- M. van der Spek, C. Banet, C. Bauer, P. Gabrielli, W. Goldthorpe, M. Mazzotti, S. T. Munkejord, N. A. Røkke, N. Shah, N. Sunny, D. Sutter, J. M. Trusler and M. Gazzani, Energy Environ. Sci., 2022, 15, 1034–1077 Search PubMed.
- S. Choi, S.-H. Shin, D.-H. Lee, G. Doo, D. W. Lee, J. Hyun, J. Y. Lee and H.-T. Kim, J. Mater. Chem. A, 2022, 10, 789–798 Search PubMed.
- R.-T. Liu, Z.-L. Xu, F.-M. Li, F.-Y. Chen, J.-Y. Yu, Y. Yan, Y. Chen and B. Y. Xia, Chem. Soc. Rev., 2023, 52, 5652–5683 Search PubMed.
- G. Singer, P. Pertl and A. Trattner, Int. J. Hydrogen Energy, 2025, 106, 1158–1166 Search PubMed.
- G. M. Arzac, O. Montes and A. Fernández, Appl. Catal., B, 2017, 201, 391–399 Search PubMed.
- A. E. Kozhukhova, S. P. Du Preez and D. G. Bessarabov, Int. J. Hydrogen Energy, 2023, 51, 1079–1096 Search PubMed.
- J. Kim, A. Tahmasebi, S. Khoshk Rish, C.-H. Jeon and J. Yu, J. Energy Inst., 2023, 109, 101297 Search PubMed.
- F. Giarratano, G. M. Arzac, V. Godinho, D. Hufschmidt, M. C. Jiménez de Haro, O. Montes and A. Fernández, Appl. Catal., B, 2018, 235, 168–176 Search PubMed.
- I. H. Son, A. M. Lane and D. T. Johnson, J. Power Sources, 2003, 124, 415–419 Search PubMed.
- J. Zhang, Z. Hu, T. Man, Y. Zhao, M. Ding and Z. Guo, Nucl. Eng. Des., 2024, 428, 113481 Search PubMed.
- M. Haruta and H. Sano, Int. J. Hydrogen Energy, 1981, 6, 601–608 Search PubMed.
- Y. Varun, I. Sreedhar and S. A. Singh, J. Environ. Chem. Eng., 2022, 10, 108384 Search PubMed.
- V. M. Shinde and G. Madras, Catal. Today, 2012, 198, 270–279 Search PubMed.
- X. Zhang, N. Fei, K. Sang, W. Chen, G. Qian, J. Zhang, D. Chen, X. Zhou, X. Duan and W. Yuan, Cell Rep. Phys. Sci., 2024, 5(6), 102014 Search PubMed.
- E. Lalik, R. Kosydar, R. Tokarz-Sobieraj, M. Witko, T. Szumełda, M. Kołodziej, W. Rojek, T. Machej, E. Bielańska and A. Drelinkiewicz, Appl. Catal., A, 2015, 501, 27–40 Search PubMed.
- J. Chen, Y. Zhang, Z. Zhang, D. Hou, F. Bai, Y. Han, C. Zhang, Y. Zhang and J. Hu, J. Mater. Chem. A, 2023, 11, 8540–8572 Search PubMed.
- Z. Wang, Z. Mei, L. Wang, Q. Wu, C. Xia, S. Li, T. Wang and C. Liu, J. Mater. Chem. A, 2024, 12, 24802–24838 Search PubMed.
- X. He, F. Dong, W. Han, Z. Tang and Y. Ding, J. Mater. Chem. A, 2024, 12, 7470–7507 Search PubMed.
- H. Li, W. Wang, S. Xue, J. He, C. Liu, G. Gao, S. Di, S. Wang, J. Wang, Z. Yu and L. Li, J. Am. Chem. Soc., 2024, 146, 9124–9133 Search PubMed.
- X. Zhang, K. Sang, W. Chen, J. Zhang, G. Qian, D. Chen, X. Zhou, X. Duan and W. Yuan, Chem. Eng. J., 2024, 485, 149875 Search PubMed.
- S. Völkening, K. Bedürftig, K. Jacobi, J. Wintterlin and G. Ertl, Phys. Rev. Lett., 1999, 83, 2672–2675 Search PubMed.
- S. A. Singh, K. Vishwanath and G. Madras, ACS Appl. Mater. Interfaces, 2017, 9, 19380–19388 Search PubMed.
- Z. Akbari, M. D. Mensi, E. Oveisi, J. Ruffiner, W. Luo, C. D. Koolen and A. Züttel, Int. J. Hydrogen Energy, 2024 DOI:10.1016/j.ijhydene.2024.08.300.
- C. Li, Q. Sun, F. Cao, W. Ying and D. Fang, J. Nat. Gas Chem., 2007, 16, 308–315 Search PubMed.
- Z. Shahidian, K. Zare and S. M. Mousavi Safavi, Iran. J. Chem. Chem. Eng., 2020, 39, 61–69 Search PubMed.
- V. Mazzieri, F. Coloma-Pascual, A. Arcoya, P. C. L'Argentière and N. S. Fı́goli, Appl. Surf. Sci., 2003, 210, 222–230 Search PubMed.
- M. E. Davis and R. J. Davis, Fundamentals of Chemical Reaction Engineering, Courier Corporation, 2012 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 Search PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085117 Search PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 Search PubMed.
- F. Colasuonno and M. Lessio, J. Phys. Chem. C, 2023, 127, 23645–23653 Search PubMed.
- D. Dubbeldam, S. Calero and T. J. H. Vlugt, Mol. Simul., 2018, 44, 653–676 Search PubMed.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 Search PubMed.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16, 4528–4534 Search PubMed.
- Z. Wang, Z. Mei, L. Wang, Q. Wu, C. Xia, S. Li, T. Wang and C. Liu, J. Mater. Chem. A, 2024, 12, 24802–24838 Search PubMed.
- Y. Yan, Q. Wang, C. Jiang, Y. Yao, D. Lu, J. Zheng, Y. Dai, H. Wang and Y. Yang, J. Catal., 2018, 367, 194–205 Search PubMed.
- B. Yang, X. Chen, L. Guo and L. Zhang, J. Mater. Chem. A, 2024, 12, 19861–19884 Search PubMed.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 Search PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 Search PubMed.
- J. Zheng, C. Wang, W. Chu, Y. Zhou and K. Köhler, ChemistrySelect, 2016, 1, 3197–3203 Search PubMed.
- P. Liu, C. Zheng, W. Liu, X. Wu and S. Liu, ACS Catal., 2024, 14, 6028–6044 Search PubMed.
- X. Liu, J. Phys. Chem. C, 2008, 112, 5066–5073 Search PubMed.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 Search PubMed.
- W. Huang, X. Zhang, A.-C. Yang, E. D. Goodman, K.-C. Kao and M. Cargnello, ACS Catal., 2020, 10, 8157–8167 Search PubMed.
- K. Hadjiivanov, J.-C. Lavalley, J. Lamotte, F. Maugé, J. Saint-Just and M. Che, J. Catal., 1998, 176, 415–425 Search PubMed.
- G. Busca and J. C. Lavalley, Spectrochim. Acta, Part A, 1986, 42, 443–445 Search PubMed.
- Z. Opre, D. Ferri, F. Krumeich, T. Mallat and A. Baiker, J. Catal., 2007, 251, 48–58 Search PubMed.
- Y. Nanba, T. Ishimoto and M. Koyama, J. Phys. Chem. C, 2017, 121, 27445–27452 Search PubMed.
- M. Zhao and Y. Xia, Nat. Rev. Mater., 2020, 5, 440–459 Search PubMed.
- D. Ugur, A. J. Storm, R. Verberk, J. C. Brouwer and W. G. Sloof, J. Phys. Chem. C, 2012, 116, 26822–26828 Search PubMed.
- P. Ferrin, S. Kandoi, A. U. Nilekar and M. Mavrikakis, Surf. Sci., 2012, 606, 679–689 Search PubMed.
- F. Rose, M. Tatarkhanov, E. Fomin and M. Salmeron, J. Phys. Chem. C, 2007, 111, 19052–19057 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta03057a |
| This journal is © The Royal Society of Chemistry 2025 |