
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazine oxidation-assisted electrocatalytic water splitting with Prussian blue analog-derived V-doped CoFe-layered double hydroxide nanosheets†
Baghendra
Singh
,
Toufik
Ansari
and
Arindam
Indra
 *
*
Department of Chemistry, Indian Institute of Technology (BHU), Varanasi, UP-221005, India. E-mail: arindam.chy@iitbhu.ac.in
First published on 21st June 2025
Abstract
Efficient and sustainable hydrogen production through electrocatalytic water splitting remains a critical challenge, hindered primarily by the sluggish oxygen evolution reaction (OER). In this regard, leveraging the hydrazine oxidation reaction (HzOR) as an anodic alternative significantly lowers the overall cell voltage, promoting energy-efficient hydrogen evolution. In this study, we report Prussian blue analog (PBA)-derived vanadium-doped cobalt-iron layered double hydroxide (V-CoFe-LDH) nanosheets as an efficient electrocatalyst for the HzOR in the alkaline medium. The PBA-derived V-CoFe-LDH offered a high surface area, large porosity, and coordination unsaturation, and produced 2D nanosheets. The introduction of mixed-valence V4+/V5+-species modulated the electronic structure and enhanced the active site density, offering facile access to the higher oxidation states of Co and Fe-ions to improve the catalytic performance. The V-CoFe-LDH exhibited superior HzOR activity, achieving a significant reduction in the potential requirement (0.70 V in 3-electrode and 0.42 V in 2-electrode systems) compared to the anodic OER. Moreover, the structural modification in PBA-derived V-CoFe-LDH led to an improved HzOR compared to the hydrothermally prepared V-CoFe-LDH-HT. The operando Raman studies elucidated the formation of the *NH2 intermediate on the V-CoFe-LDH surface, and further confirmed the breaking of the N–N bond during the HzOR.
Introduction
Electrochemical hydrogen production by water splitting requires a high operational voltage (>1.23 V), leading to significant consumption of electricity and associated costs, which hinder its feasibility for large-scale implementation.1–4 Therefore, developing efficient, energy-saving, and low-cost catalysts for hydrogen production has gained considerable importance.1 In water splitting, the anodic oxygen evolution reaction (OER) works as the bottleneck because of its sluggish kinetics and high energy demand.5–8 To overcome this, anodic oxidation reactions (AORs) of inorganic and organic substrates have been explored to lower the overall voltage requirement, increase the efficiency of the cell, and improve the hydrogen production rate.9–13Among different AORs, the anodic hydrazine oxidation reaction (HzOR) is particularly important as it can largely improve hydrogen production.14–16 As the HzOR requires a very low theoretical oxidation potential of −0.33 V vs. RHE, the replacement of the OER (theoretical potential is 1.23 V vs. RHE) by the HzOR can significantly reduce the operating voltage and, consequently, the energy demand, making it a highly attractive alternative for practical applications (Fig. 1).17,18 Different research groups have explored HzOR-assisted H2 production and analyzed the techno economical aspects.19–24 For example, Badreldin et al. showed the economic feasibility of the HzOR over other anodic oxidation reactions.19 The H2 production cost (0.30 USD per kg H2) by hydrazine oxidation was found to be economically viable compared to other anodic reactions like urea, methanol, ethanol oxidation, and the OER.20 However, the high cost of hydrazine still poses a challenge for the commercial use of the HzOR.20–24
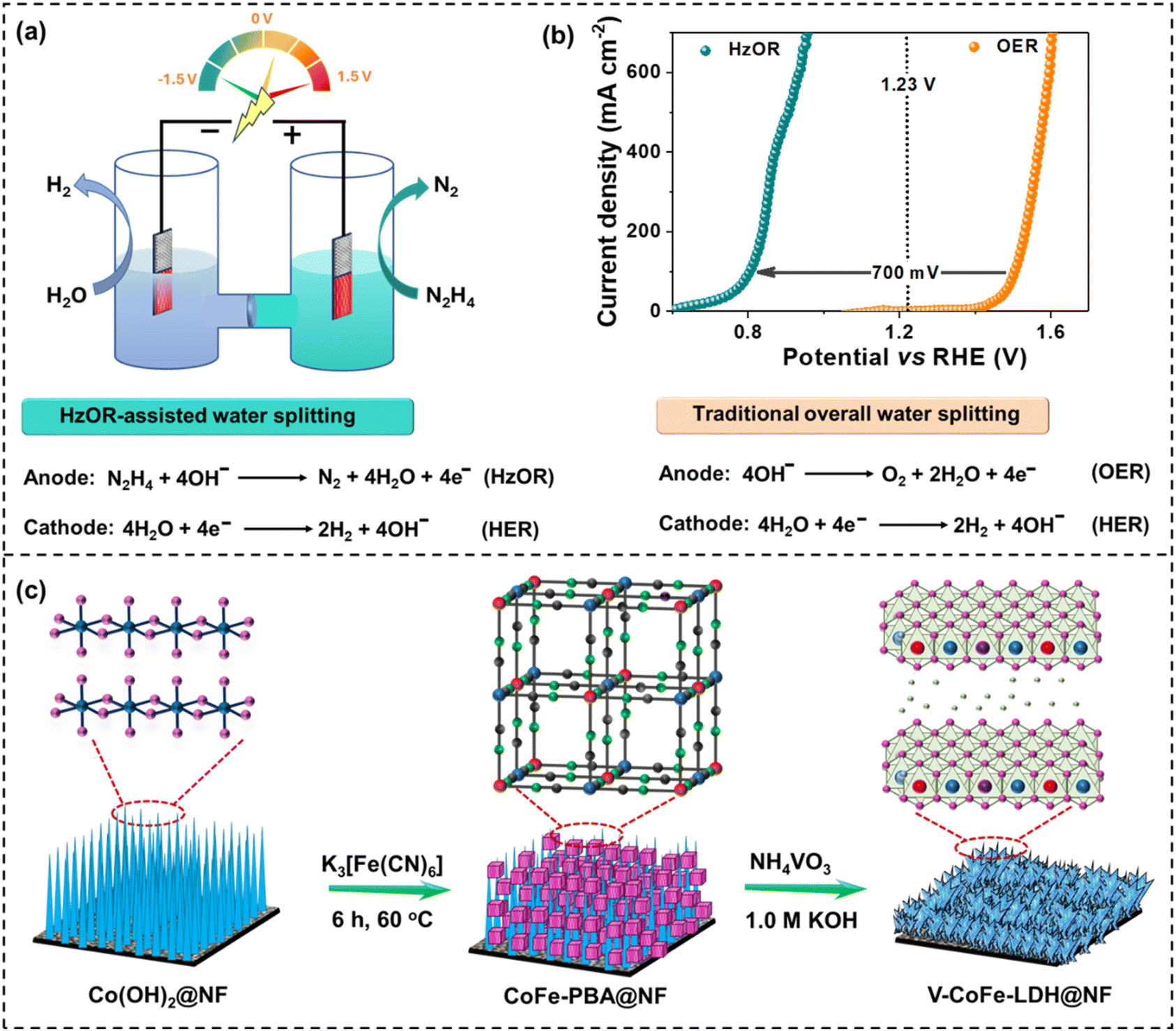 | ||
| Fig. 1 (a) Schematic illustration showing hydrazine oxidation-assisted water splitting; (b) the advantages of hydrazine oxidation-assisted water splitting over traditional water splitting; and (c) alkaline etching of CoFe-PBA in the presence of NH4VO3 to produce V-CoFe-LDH. Color code of the atoms – Co: blue, O: pink, Fe: red, C: green, N: gray, V: purple. | ||
In recent years, substantial research has been directed toward the design of advanced electrocatalysts for catalyzing both the cathodic and anodic reactions in HzOR-assisted hydrogen production systems.15,16,25–32 In this context, numerous electrocatalysts based on noble metals have been reported for electrocatalytic HzOR activity. For example, the RhRu0.5-alloy,33 AuPt alloy,34 Rh2P,35 and so on have been developed for efficient HzOR coupled with the hydrogen evolution reaction (HER). Furthermore, transition metal-based catalysts such as Ni(OH)2/Ni2P,36 P, W-Co3N,37 NiCoP,38 NiCo-MOF/Mxene,39 NiS2,40 and Co/MoO2 (ref. 41) demonstrated excellent activity and stability for the HzOR.42–45 In further advancement, researchers have extensively utilized transition metal-based hydroxides and layered double hydroxides (LDHs) for efficient HzOR.36,46,47
Interestingly, the use of a metal–organic framework (MOF) as the precursor of LDH was found to be beneficial for electrocatalytic performance.48–51 MOF-derived LDHs provide a high surface area, atomic level thickness, tuned porosity, and optimized electronic properties for improved electrocatalytic activity and stability.52–56 The electrocatalytic anodic oxidation activity of LDHs is further enhanced by the addition of high-valent metal ions.56–58
In this work, we have synthesized V-CoFe-LDH using CoFe-PBA as the precursor with simultaneous incorporation of V4+/V5+-ions during alkaline etching (Fig. 1). Previous work has demonstrated the synthesis of various hydroxides by the alkaline etching of PBAs.59 Additionally, we have seen that PBAs undergo electrochemical conversion into M(O)(OH) nanosheets during the alkaline water splitting.52–56 Nevertheless, it has not been investigated yet, how high-valent metal ions like V4+/V5+ can be incorporated into PBA-derived LDH nanosheets to improve the electrocatalytic activity.
Previous studies indicate that the HzOR requires a highly efficient catalyst with excellent electronic conductivity and robust stability.33–36,38,40 Moreover, the presence of high-valent metal ions significantly boosts the catalytic performance due to optimized electronic properties.33–35,37,41,46,47 Herein, we combined multiple strategies to enhance the efficiency of the HzOR using V-doped CoFe-LDH catalysts: (i) the self-supported catalyst not only improves catalyst–support interactions but also facilitates charge transfer, improving the electrochemical activity and stability.60 (ii) The introduction of V4+/V5+-ions in the structure of CoFe-LDH optimized the electronic structure, resulting in high catalytic efficiency.61 (iii) The atomic-scale thin V-CoFe-LDH nanosheets offer coordination and electronic unsaturation at the metal centers with a high density of active binding sites for reactant molecules. (iv) The use of PBA as the precursor results in a large surface area, increased porosity, and modulated electronic properties in V-CoFe-LDH.62
As a result, V-CoFe-LDH nanosheets exhibited significantly improved HzOR activity compared to CoFe-LDH. Additionally, the incorporation of V4+/V5+-ions in CoFe-LDH was found to boost the efficacy of water splitting when the OER was substituted with the HzOR. The PBA-derived V-CoFe-LDH nanosheets achieved a current density of 100 mA cm−2 for the OER at the voltage demand of 1.50 V vs. RHE, while only 0.80 V vs. RHE was required for the HzOR. Additionally, V-CoFe-LDH manifested a cell potential of 1.22 V for 100 mA cm−2 current density for the HzOR, significantly lower than that of water splitting involving the anodic OER (1.64 V) in a two-electrode system. Furthermore, the efficiency of this newly developed catalyst was demonstrated by its remarkable HzOR activity compared to V-CoFe-LDH-HT, prepared by the hydrothermal method.
Results and discussion
Synthesis and characterization of the catalysts
Nickel foam-supported CoFe-PBA was synthesized following a methodology developed in our laboratory.54,55 The formation of CoFe-PBA@NF was verified using powder X-ray diffraction (PXRD) and infrared (IR) spectroscopy and is outlined in the ESI (Fig. S1 and S2†). CoFe-PBA@NF was treated with ammonium metavanadate in an aqueous KOH solution, resulting in the formation of V-CoFe-LDH nanosheets (Fig. 1). Further, the amount of ammonium metavanadate was optimized by preparing two more vanadium-doped CoFe-layered double hydroxides (denoted as V-CoFe-LDH-1 and V-CoFe-LDH-2) to attain the highest HzOR activity. For comparison purposes, CoFe-LDH was also prepared by treating CoFe-PBA in an aqueous KOH solution without ammonium metavanadate. Additionally, a comparative V-CoFe-LDH-HT catalyst was synthesized by a hydrothermal method (Table S1†).The V-incorporation in CoFe-LDH could happen in three possible ways; (i) introduction of V in the lattices of LDH replacing Co and Fe sites; (ii) intercalation of VOxn− between the layers of LDH; and/or (iii) both. To understand this process, we have carried out powder X-ray diffraction (PXRD). The PXRD pattern of both catalysts showed the characteristic peaks of CoFe-LDH (JCPDS no. 50-0235) (Fig. S3†). A positive shift of 0.12° in the two-theta angle of (003) and (015) peaks was observed for V-CoFe-LDH in contrast to CoFe-LDH. The incorporation of V4+/V5+, with smaller ionic radii than Co2+ and Fe3+, into the lattice of CoFe-LDH led to lattice contraction and hence a positive shift in the PXRD peak of V-CoFe-LDH was observed.50 This analysis confirmed the successful incorporation of V into the catalyst structure (Fig, S3†). On the other hand, intercalation of VOxn− between the layers of LDH has been ruled out as it leads to the expansion of the inter-planar distance.
Fourier transform infrared (FTIR) spectroscopy provided evidence for the formation of LDH. The FTIR spectrum of V-CoFe-LDH and CoFe-LDH revealed the absence of –CN bridges, characteristic of the PBA framework, indicating its decomposition and the formation of a new catalyst structure (Fig. S4†).48,50 IR peaks of V-CoFe-LDH corresponding to the stretching and bending vibrations of the –OH group were observed at 3490 cm−1 and 1645 cm−1, respectively. The FTIR spectrum of CoFe-LDH exhibited identical peaks with a positive shift of 6 cm−1 compared to V-CoFe-LDH (Fig. S4†). The combined PXRD and FTIR analyses validate the successful synthesis of V-CoFe-LDH.
Further, the electronic environment of the elements in the catalysts was examined by X-ray photoelectron spectroscopy (XPS). The major peaks in the Co 2p XPS of V-CoFe-LDH were observed at 782.08 eV and 797.74 eV for Co 2p3/2 and Co 2p1/2, respectively (Fig. 2a).50,51,54 Deconvolution of Co 2p3/2 revealed the peaks for Co2+ and Co3+ at 784.26 eV and 781.91 eV, respectively. In comparison to CoFe-LDH, the Co 2p3/2 peak in V-CoFe-LDH was shifted by 0.57 eV toward higher binding energy. The Co3+/Co2+ peak area ratio was higher for V-CoFe-LDH (1.66) in contrast to CoFe-LDH (0.47) (Fig. 2a).50,51,54
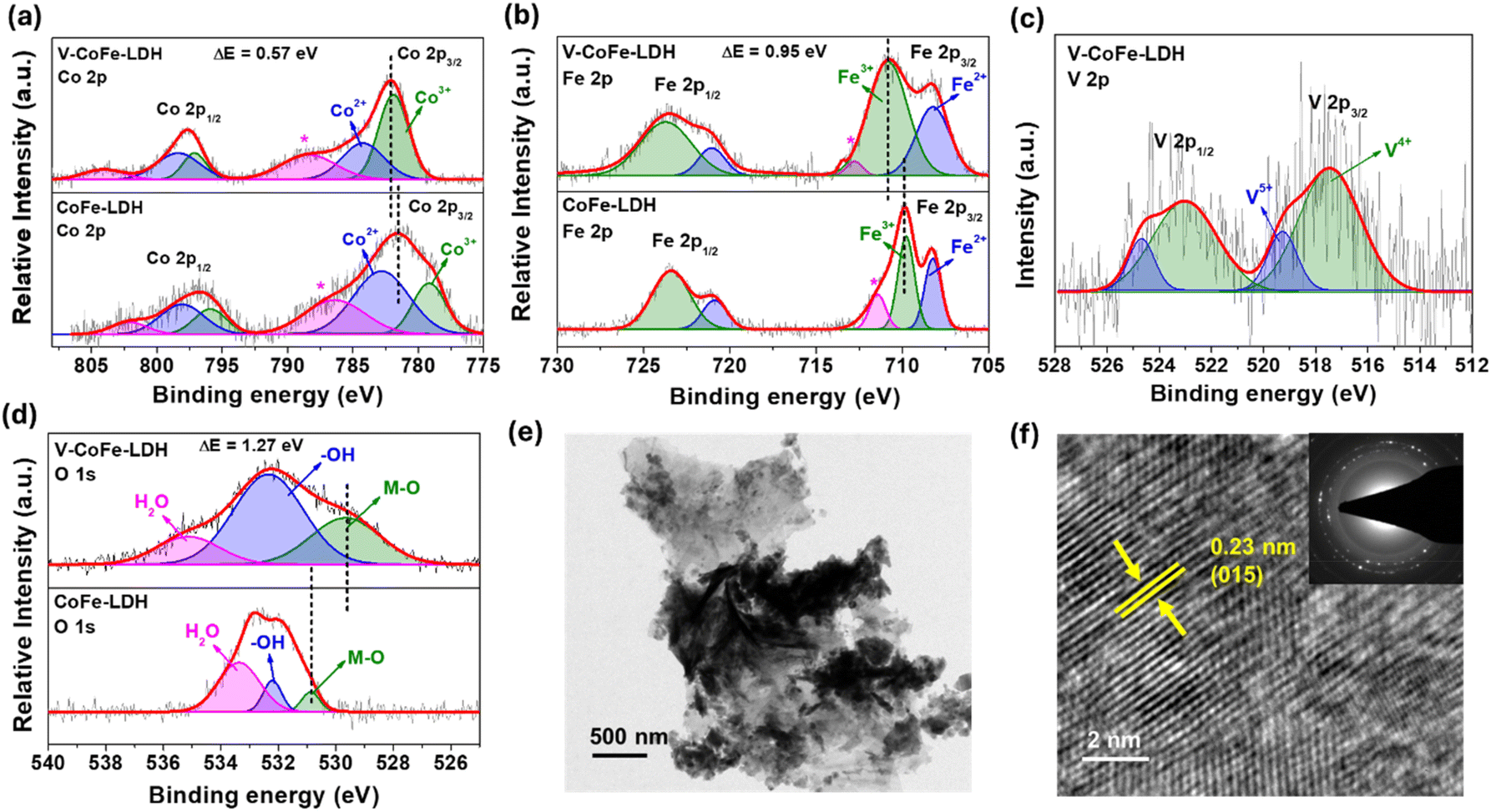 | ||
| Fig. 2 (a) Co 2p XPS of CoFe-LDH and V-CoFe-LDH depicting a positive shift of the Co 2p3/2 binding energy in V-CoFe-LDH; (b) Fe 2p XPS of CoFe-LDH and V-CoFe-LDH showing a positive shift of the Fe 2p3/2 peak in V-CoFe-LDH; (c) V 2p XPS of V-CoFe-LDH showing the peaks for V4+ and V5+; (d) O 1s XPS data of CoFe-LDH and V-CoFe-LDH showing the negative shift of the peak for the M–O bond in V-CoFe-LDH; (e) TEM image of V-CoFe-LDH; and (f) HRTEM image of V-CoFe-LDH showing the lattice spacing with a d-value of 0.23 nm for the (015) plane of LDH (SAED inset). | ||
V-CoFe-LDH was found to have a Co 2p3/2-Co 2p1/2 spin–orbit coupling energy difference of 15.66 eV, indicating the presence of mixed-valent Co2+ and Co3+ species (Fig. 2a). The high Lewis acidic nature of V4+/V5+ facilitated electron transfer from Co2+ to V4+/V5+, enhancing the electrophilicity of Co sites.61
In the Fe 2p XPS spectra of V-CoFe-LDH, peaks corresponding to Fe 2p3/2 and Fe 2p1/2 were observed at 710.84 eV and 723.57 eV, respectively (Fig. 2b).50,51,54 Further fitting of the Fe 2p3/2 peak identified Fe2+ and Fe3+ species at 708.20 eV and 710.80 eV, respectively. Similar to Co 2p XPS, the Fe 2p3/2 peak of V-CoFe-LDH was shifted to a higher binding energy by 0.95 eV than CoFe-LDH, showing higher positive charge density on Fe in V-CoFe-LDH (Fig. 2b).50,51,54 The Fe3+/Fe2+ ratio in V-CoFe-LDH was found to be 1.94 while 1.30 was observed for CoFe-LDH.
Two peaks, representing V 2p3/2 and V 2p1/2 were deconvoluted from the V 2p XPS spectra of V-CoFe-LDH at 517.47 eV and 523.03 eV, respectively (Fig. 2c).56,61 The signals at 517.50 eV and 519.29 eV validated the existence of V4+ and V5+ in V-CoFe-LDH. The binding energy of the V 2p spectrum of V-CoFe-LDH was observed between VO2 and V2O5, further suggesting the presence of V4+ and V5+.63 The coexistence of V4+ and V5+ observed in the V 2p XPS can be attributed to the strong electronic interaction of vanadium with Co and Fe atoms. The strong Lewis acid V5+ ion withdraws electron density from Co and Fe-centers and gets reduced to V4+.56,61
The O 1s XPS spectrum of V-CoFe-LDH displayed peaks at 529.62 eV, 532.35 eV, and 535.13 eV, corresponding to M–O bonds, surface –OH, and adsorbed H2O, respectively (Fig. 2d).56,61 The M–O peak in V-CoFe-LDH shifted negatively by 1.27 eV compared to CoFe-LDH, confirming electronic modifications due to V-doping (Fig. 2d).56
The morphological features of the catalysts were obtained from transmission electron microscopy (TEM) and scanning electron microscopy (SEM). SEM images showed that both V-CoFe-LDH and CoFe-LDH had a nanosheet morphology, with the nanosheets assembling into a flower-like structure (Fig. S5 and S6†). TEM studies further confirmed the nanosheet morphology of V-CoFe-LDH (Fig. 2e). High-resolution TEM (HRTEM) detected a lattice spacing of 0.23 nm, assigned to the (015) plane of CoFe-LDH (Fig. 2f).64 Furthermore, the polycrystalline nature of V-CoFe-LDH was detected in the selected area electron diffraction (SAED) pattern (Fig. 2f inset). Atomic force microscopy analysis also showed 2–8 nm thickness of V-CoFe-LDH nanosheets (Fig. S7†).
Energy dispersive X-ray (EDX) spectroscopy confirmed the presence of Co, Fe, V, and O in V-CoFe-LDH and Co, Fe, and O in CoFe-LDH (Fig. S8 and S9†). EDX analysis also provided information about the elemental composition, specifically the Co and Fe ratios in V-CoFe-LDH and CoFe-LDH (Table S1†).
The EDX and XPS analyses showed a notable reduction in Fe content following vanadium incorporation in CoFe-LDH, suggesting the partial substitution of Fe by V in the catalyst structure. Further, XPS supports this observation by revealing shifts in the binding energies. More conclusive results were obtained from the PXRD studies (Fig. S3†). A positive shift in the two-theta value was observed for V-CoFe-LDH compared to CoFe-LDH. This shift can be explained by the lattice contraction, resulting from the substitution of Fe3+ ions in the LDH by V4+/V5+ ions with smaller ionic radii. These results confirm the successful substitution of vanadium into the metallic sites of LDH.
Electrochemical characterization
The redox behavior of the metal sites in the catalysts was evaluated by cyclic voltammetry (CV). An oxidation peak at 1.31 V vs. RHE was visible in the CV of CoFe-LDH, corresponding to the Co2+/Co3+ oxidation couple (Fig. 3a).65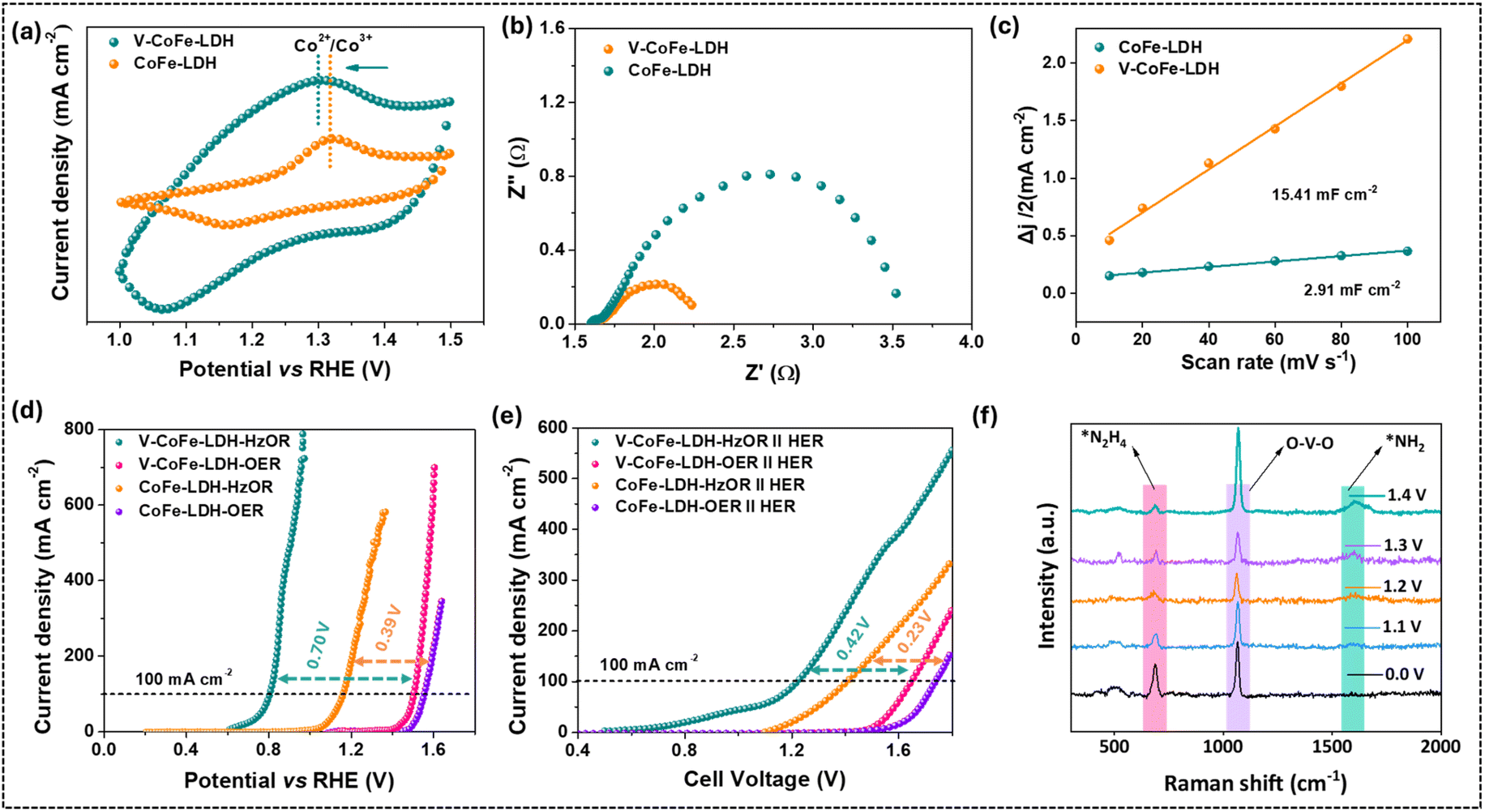 | ||
Fig. 3 (a) CV plots of CoFe-LDH and V-CoFe-LDH manifesting the oxidation peak of Co2+/Co3+; (b) EIS data of CoFe-LDH and V-CoFe-LDH showing the lower Rct of V-CoFe-LDH; (c) Cdl profiles of CoFe-LDH and V-CoFe-LDH manifesting the larger Cdl of V-CoFe-LDH; (d) LSV profiles for the OER and HzOR activity of CoFe-LDH and V-CoFe-LDH verifying the excellent activity of V-CoFe-LDH; (e) LSV profiles for the overall water splitting activity of CoFe-LDH and V-CoFe-LDH in a two-electrode system with or without hydrazine; and (f) operando Raman spectroscopic studies of hydrazine oxidation at different cell voltages in a two-electrode set-up using V-CoFe-LDH as the anode and cathode (reaction conditions: 1.0 M KOH +![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.1 M N2H4, 25 °C). 0.1 M N2H4, 25 °C). | ||
V-CoFe-LDH manifested the Co2+/Co3+ oxidation wave at a lower potential (1.29 V vs. RHE) than CoFe-LDH, suggesting that the inclusion of V facilitated the oxidation of Co2+ to Co3+, enhancing O–O bond formation more effectively. In order to determine the quantity of active Co sites in the prepared catalysts, the redox peak area in the CV profile was integrated (Fig. S10†).17,48 A larger number of active sites were observed in V-CoFe-LDH (14.24 × 1018 sites) compared to CoFe-LDH (3.71 × 1018 sites). The existence of a larger number of active sites in V-CoFe-LDH promoted facile binding of substrates and improved the catalytic activity.
Electrochemical impedance spectroscopy (EIS) showed the lower charge-transfer resistance (Rct) of V-CoFe-LDH (0.59 Ω) than that of CoFe-LDH (1.90 Ω), indicating improved electron transfer due to V-introduction in the catalyst (Fig. 3b). The double-layer capacitance (Cdl) of V-CoFe-LDH was calculated to be 15.41 mF cm−2, higher than that of CoFe-LDH (2.91 mF cm−2) (Fig. 3c and S11†). Further, the electrochemical surface area (ECSA) values were calculated to be 385.25 cm2 for V-CoFe-LDH and 72.75 cm2 for CoFe-LDH (Fig. S11†).
Electrocatalytic water oxidation
The electrocatalytic OER performance of the catalysts was evaluated in 1.0 M KOH solution. The linear sweep voltammetry (LSV) revealed that CoFe-LDH achieved a current density of 100 mA cm−2 for the OER at a potential of 1.55 V vs. RHE (Fig. 3d). The introduction of V in CoFe-LDH lowered the required potential for the OER, reducing it to 1.50 V vs. RHE to deliver the same current density. Further, Tafel plots were utilized to assess the reaction kinetics of the developed catalysts. The Tafel slopes for V-CoFe-LDH and CoFe-LDH during the OER were determined to be 63 mV dec−1 and 128 mV dec−1, respectively (Fig. S12†). These results confirmed that both the required potential and reaction kinetics were improved significantly when vanadium was introduced into the structure of CoFe-LDH.66 As discussed earlier, V-incorporation in CoFe-LDH also improved the charge transfer kinetics (Fig. 3b).Electrocatalytic hydrazine oxidation
The HzOR activity of the synthesized LDHs was demonstrated in 1.0 M KOH containing 0.1 M hydrazine. To achieve a high faradaic efficiency for the HzOR, it should be oxidized at a lower potential than the OER onset potential.4 Interestingly, V-CoFe-LDH required 0.80 V vs. RHE to achieve 100 mA cm−2 current density when hydrazine (0.1 M) was added to the electrolyte (Fig. 3d). Even the HzOR takes place before the theoretical potential of water oxidation (1.23 V vs. RHE). This led to a marked reduction in the potential requirement (by 0.70 V) when the OER was substituted by the HzOR (Fig. 3d). The V-CoFe-LDH also avoided OER interference by achieving an industrial-level current density of 400 mA cm−2 at 0.87 V vs. RHE for the HzOR, which was below the onset potential for the OER (1.42 V vs. RHE) (Fig. 3d).In contrast, the potential of CoFe-LDH for the HzOR was recorded to be 1.16 V vs. RHE with a reduction of 0.39 V compared to the OER with the same catalyst (Fig. 3d). Interestingly, V-CoFe-LDH manifested a significant potential reduction (0.70 V) for the HzOR in contrast to CoFe-LDH (0.39 V). This result established that the HzOR activity was significantly improved due to the incorporation of high valent V4+/V5+ in CoFe-LDH. Further, the performance of V-CoFe-LDH surpassed that of V-CoFe-LDH-HT, V-CoFe-LDH-1, and V-CoFe-LDH-2 (Fig. S13†). The catalytic activity of V-CoFe-LDH for the HzOR was also found to be comparable to that of catalysts reported in the literature (Table S2†).
The OER and HzOR activities of V-CoFe-LDH were also evaluated in 0.1 M (pH: 12.12), 0.5 M (pH: 13.24), and 2.0 M (pH: 14.00) aqueous KOH solutions (Fig. S14 and S15†). The OER performances deteriorated in 0.1 M and 0.5 M KOH solutions while the activity was enhanced in 2.0 M KOH solution. Similarly, the HzOR performance declined in 0.1 M and 0.5 M KOH solutions, whereas in 2.0 M KOH, the activity remained nearly similar to that observed in 1.0 M KOH.
In further evaluation, V-CoFe-LDH was used as the anode and cathode to construct a two-electrode water electrolyzer. This electrolyzer manifested 100 mA cm−2 for overall water splitting at a cell voltage of 1.64 V (Fig. 3e). The cell voltage dropped to 1.22 V for the same current density when hydrazine was added to the electrolyte (Fig. 3e). This leads to a 0.42 V reduction in cell voltage for the HzOR compared to the OER. Moreover, V-CoFe-LDH also required a low cell voltage when compared with CoFe-LDH (Fig. 3e).
We have also carried out repeated chronoamperometric measurements for the HzOR for 5 cycles in a three-electrode system, demonstrating the excellent stability of V-CoFe-LDH for hydrazine oxidation (Fig. S16†). Furthermore, hydrogen production increased 3.9-fold during hydrazine oxidation relative to the OER (Fig. S17†).
In order to realize the reaction mechanism, the electrocatalytic hydrazine oxidation activity of V-CoFe-LDH was evaluated in a typical two-electrode system using an aqueous electrolyte containing 1.0 M KOH and 0.1 M N2H4 (Fig. 3f). After the introduction of N2H4 into the electrolyte, a strong Raman band appeared at 683 cm−1, corresponding to the N–H stretching mode of adsorbed *NH2NH2.38 As the potential was applied, the intensity of the *NH2NH2 peak decreased, while a new peak emerged at 1595 cm−1, attributed to the *NH2 intermediate.38,67,68 Notably, the *NH2 peak was not detectable at 0.0 V, but the intensity of the peak increased with increasing potential. This observation suggests that NH2NH2 is consumed during the reaction, leading to the formation of *NH2 species. Based on these findings, the proposed reaction mechanism involves the initial adsorption of N2H4 on the V-CoFe-LDH surface to form *NH2NH2, followed by cleavage of the N–N bond to yield the *NH2 intermediate. The *NH2 species then undergoes stepwise dehydrogenation to produce N2.38,67
The high HzOR activity of V-CoFe-LDH is attributed to strong Co 3d–O 2p–V 2p/Fe 2p orbital interactions and partial electron transfer from Co and Fe to V (Fig. S18†).17,50 The Co3+ in its low-spin state (t2g6eg0) experiences significant electron–electron repulsion with the bridging O2− ligand, while Fe3+ in its high-spin state (t2g3eg2) shows a poor electron–electron repulsion with the O2− ligand.17,50 The 3d0 outer electronic configuration of V5+, characterized by empty t2g and eg orbitals, facilitates π-donation from the bridging oxygen atoms to the vanadium center.17,50 This electron redistribution optimizes the electron density around the metal ions, stabilizing high-valent states and promoting catalytic activity.
The effect of V-doping on the electronic structure and catalytic activity of LDHs based on density functional theory (DFT) calculations were reported in the literature.69–72 The DFT calculations revealed that the formation of O* from HO* is the rate-determining step on the NiFe-LDH surface.70 After V-doping, the Gibbs free energy change of the rate determining step is reduced, facilitating the formation of O* to promote the O–O bond formation. Li et al. showed that the bandgap was significantly reduced after the doping of V in NiFe-LDH, improving the electronic conductivity.70 Similarly, Jiang et al. explored V-doped nickel hydroxide for the OER and explained the improved activity by the DFT calculations.71 The DFT calculations also revealed a higher water adsorption energy for V-Co(OH)2 (1.14 eV) compared to Co(OH)2 (0.60 eV).73
The crucial role of V in enhancing the catalytic HzOR activity of Ni3N was also previously reported.74 The V-doping in Ni3N promotes the dehydrogenation of N2H4, thereby improving the efficiency of hydrazine oxidation. V-doping also modulates the reaction kinetics of the HzOR.74 The DFT calculations revealed stepwise dehydrogenation of hydrazine involving intermediates *N2H4, *N2H3, *N2H2, *N2H, and *N2. Importantly, the dehydrogenation of N2H4 is the rate-determining step in the HzOR and it has a lower energy barrier on V-Ni3N (0.27 eV) compared to Ni3N (0.49 eV), indicating faster kinetics with the V-doped catalyst. Further, Bader charge analysis revealed weaker interaction between N2H3 and V-Ni3N, consistent with the lower free energy barrier for this intermediate than that of Ni3N. Overall, V-doping in the catalyst modulates the electronic structure, reduces the energy barrier and enhances HzOR activity.
Post-catalytic characterization
The anodic oxidation of an electrocatalyst leads to a change in the electronic as well as morphological features of the catalyst.75,76 Therefore, post-catalytic characterization was conducted to detect the changes in the electronic as well as morphological features after anodic oxidation (120 h of HzOR CA).A shift of 0.41 eV in the Co 2p3/2 peak towards higher binding energy was observed in the Co 2p XPS spectra of V-CoFe-LDH after CA, suggesting a higher concentration of Co3+ as a result of anodic oxidation (Fig. S19†).50,51,54 The Co 2p XPS of CoFe-LDH after CA also manifested a positive shift of 0.16 eV in the Co 2p3/2 peaks in contrast to the fresh catalyst (Fig. S20†).28,29,32 The Fe 2p XPS also showed a similar type of effect after anodic oxidation (Fig. S21 and S22†).28,29,32 According to these findings, both catalysts produce high-valent metal ions during anodic oxidation. Likewise, upon CA, the V4+ and V5+ peaks in the V 2p XPS spectra of V-CoFe-LDH shifted negatively when compared with the fresh catalyst (Fig. S23†).56
The O 1s XPS analysis for V-CoFe-LDH after CA displayed three peaks with a positive shift of 0.88 eV in the M–O bond (Fig. S24†).50,51,54 The O 1s XPS of CoFe-LDH also demonstrated a negative shift of 0.84 eV when compared with the fresh CoFe-LDH catalyst (Fig. S25†). The presence of V4+/V5+ was confirmed from the V 2p XPS spectra, but with a decreased intensity, suggesting partial V leaching from the V-CoFe-LDH during anodic oxidation (Table S1†).56
The leaching of vanadium, as well as iron from the electrocatalysts during anodic oxidation, has also been previously reported by us and other groups.56 Consequently, the surface analysis indicated a lower V content in the V-CoFe-LDH after CA in contrast to the fresh sample (Table S1†). In addition, we have performed ICP-AES analysis to understand the leaching of the metal ions during the catalytic process. A significant amount of V and Fe leaching was detected in the solution during the HzOR (Table S3†). However, the leaching of Fe in V-CoFe-LDH after CA was not as significant as previously reported in the case of the OER (Table S1†).51,53,54
The morphological features of V-CoFe-LDH after CA were investigated using SEM and TEM. SEM images showed the agglomeration of the nanosheets (Fig. S26†). The TEM images also confirmed the slight agglomeration after CA while HRTEM detected the (012) plane of CoFe-LDH with a lattice spacing of 0.25 nm (Fig. S27†).76 Post-catalytic characterization verified that anodic oxidation produced more high-valent Co3+/Fe3+species with a slight variation in the catalyst's morphology.
Conclusions
In summary, we have developed high-valent V-doped CoFe-LDH nanosheets by the alkaline etching of a CoFe-PBA precursor. The incorporation of V4+/V5+-ions promotes electron withdrawal from Co and Fe sites. This electronic interaction speeds up reaction kinetics, enhances charge transfer, and raises the density of active sites. Consequently, V-CoFe-LDH achieved a current density of 100 mA cm−2 at 0.80 V vs. RHE for the HzOR, significantly lower than the 1.50 V required for the OER. It further reached an industrial-scale current density of 400 mA cm−2 at 0.87 V vs. RHE for the HzOR. In a two-electrode setup, substituting the OER with the HzOR reduced the cell voltage by 0.42 V, leading to a 3.9-fold increase in hydrogen production relative to the OER. The operando Raman analysis confirmed the cleavage of the N–N bond of hydrazine during the HzOR. This work thus presents a rational design approach for high-valent V-doped LDH nanosheets with enhanced HzOR activity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
B. Singh: methodology, electrochemical measurement, characterization, data acquisition and formal analysis, writing the original draft; T. Ansari: electrochemical measurement, product analysis, Raman studies; A. Indra: project design, review and editing, supervision, conceptualization, resources and project administration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A. I. acknowledges the core research grant (CRG/2023/002395). T. A. is grateful to PMRF, Govt. of India (ID: 1103080) for a fellowship.References
- Z. Li, L. Sun, Y. Zhang, Y. Han, W. Zhuang, L. Tian and W. Tan, Coord. Chem. Rev., 2024, 510, 215837 CrossRef CAS.
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC.
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS.
- J. Li and G. Zheng, Adv. Sci., 2017, 4, 1600380 CrossRef PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- Z. Kou, X. Li, L. Zhang, W. Zang, X. Gao and J. Wang, Small Sci., 2021, 1, 2100011 CrossRef CAS PubMed.
- D. Liu, Y. Cai, X. Wang, Y. Zhuo, X. Sui, H. Pan and Z. Wang, Energy Environ. Sci., 2024, 17, 6897–6942 RSC.
- A. K. Singh, D. Kumar, B. Singh and A. Indra, Synlett, 2022, 34, 552–560 Search PubMed.
- D. Yao, Y. Zhang, S. Zhang, J. Wan, H. Yu and H. Jin, J. Mater. Chem. A, 2023, 11, 16433–16457 RSC.
- C. Deng, C. Y. Toe, X. Li, J. Tan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2201047 CrossRef CAS.
- X. Liu, Y. Han, Y. Guo, X. Zhao, D. Pan, K. Li and Z. Wen, Adv. Energy Sustainability Res., 2022, 3, 2200005 CrossRef CAS.
- B. Singh and H. Gupta, Chem. Commun., 2024, 60, 8020–8038 RSC.
- T. Y. Burshtein, Y. Yasman, L. Muñoz-Moene, J. H. Zagal and D. Eisenberg, ACS Catal., 2024, 14, 2264–2283 CrossRef CAS.
- K. Ojha, E. M. Farber, T. Y. Burshtein and D. Eisenberg, Angew. Chem., Int. Ed., 2018, 57, 17168–17172 CrossRef CAS PubMed.
- B. Singh, R. Kumar, T. Ansari, A. Indra and A. Draksharapu, Chem. Commun., 2024, 60, 9432–9435 RSC.
- X. Xu, H. C. Chen, L. Li, M. Humayun, X. Zhang, H. Sun, D. P. Debecker, W. Zhang, L. Dai and C. Wang, ACS Nano, 2023, 17, 10906–10917 CrossRef CAS PubMed.
- A. Badreldin, E. Youssef, A. Djire, A. Abdala and A. Wahab, Cell Rep. Phys. Sci., 2023, 04, 101427 CrossRef CAS.
- S. Lia, Y. Houa, L. Jiang, G. Fenga, Y. Gec and Z. Huang, Energy Rev., 2025, 04, 100105 CrossRef.
- L. Zhu, J. Huang, G. Meng, T. Wu, C. Chen, H. Tian, Y. Chen, F. Kong, Z. Chang, X. Cui and J. Shi, Nat. Commun., 2023, 25, 1997 CrossRef PubMed.
- S. Hu, H. Wu, C. Feng and Y. Ding, Int. J. Hydrogen Energy, 2020, 45, 21040–21050 CrossRef CAS.
- A. Caravaca, A. de Lucas-Consuegra, A. B. Calcerrada, J. Lobato, J. L. Valverde and F. Dorado, Appl. Catal., B, 2013, 135, 302–309 CrossRef.
- C. R. Cloutier and D. P. Wilkinson, Int. J. Hydrogen Energy, 2010, 35, 3967–3984 CrossRef CAS.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li and G. Zhang, Adv. Funct. Mater., 2021, 31, 2103673 CrossRef CAS.
- S. Singh, M. Yadav, D. K. Singh, D. K. Yadav, P. K. Sonkar and V. Ganesan, New J. Chem., 2022, 46, 13422–13430 RSC.
- S. Zhang, C. Zhang, X. Zheng, G. Su, H. Wang and M. Huang, Appl. Catal., B, 2023, 324, 122207 CrossRef CAS.
- J. Zhang, Y. Liu, J. Li, X. Jin, Y. Li, Q. Qian, Y. Wang, A. El-Harairy, Z. Li, Y. Zhu, H. Zhang, M. Cheng, S. Zeng and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 3881–3890 CrossRef CAS PubMed.
- Q. Sun, L. Wang, Y. Shen, M. Zhou, Y. Ma, Z. Wang and C. Zhao, ACS Sustain. Chem. Eng., 2018, 6, 12746–12754 CrossRef CAS.
- X. Zhai, Q. Yu, G. Liu, J. Bi, Y. Zhang, J. Chi, J. Lai, B. Yang and L. Wang, J. Mater. Chem. A, 2021, 9, 27424–27433 RSC.
- Q. Sun, M. Zhou, Y. Shen, L. Wang, Y. Ma, Y. Li, X. Bo, Z. Wang and C. Zhao, J. Catal., 2019, 373, 180–189 CrossRef CAS.
- T. Shi, B. Gao, H. Meng, Y. Fu, D. Kong, P. Ren, H. Fu and Z. Feng, Green Chem., 2024, 26, 4209–4220 RSC.
- X. Fu, D. Cheng, C. Wan, S. Kumari, H. Zhang, A. Zhang, H. Huyan, J. Zhou, H. Ren, S. Wang, Z. Zhao, X. Zhao, J. Chen, X. Pan, P. Sautet, Y. Huang and X. Duan, Adv. Mater., 2023, 35, 2301533 CrossRef CAS PubMed.
- Y. Yu, S. J. Lee, J. Theerthagiri, Y. Lee and M. Y. Choi, Appl. Catal., B, 2022, 316, 121603 CrossRef CAS.
- Y. Zhao, N. Jia, X.-R. Wu, F.-M. Li, P. Chen, P.-J. Jin, S. Yin and Y. Chen, Appl. Catal., B, 2020, 270, 118880 CrossRef CAS.
- H.-M. Yang, H.-Y. Wang, M.-L. Sun and Z.-Y. Yuan, Chem. Eng. J., 2023, 475, 146134 CrossRef CAS.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- L. Zhu, J. Huang, G. Meng, T. Wu, C. Chen, H. Tian, Y. Chen, F. Kong, Z. Chang, X. Cui and J. Shi, Nat. Commun., 2023, 14, 1997 CrossRef CAS PubMed.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- J. Wang, X. Ma, T. Liu, D. Liu, S. Hao, G. Du, R. Kong, A. M. Asiri and X. Sun, Mater. Today Energy, 2017, 3, 9–14 CrossRef.
- Y. Guo, X. Liu, Y. Zang, Y. Wu, Q. Zhang, Z. Wang, Y. Liu, Z. Zheng, H. Cheng, B. Huang, Y. Dai and P. Wang, J. Mater. Chem. A, 2022, 10, 17297–17306 RSC.
- G. Meng, Z. Chang, L. Zhu, C. Chen, Y. Chen, H. Tian, W. Luo, W. Sun, X. Cui and J. Shi, Nanomicro Lett., 2023, 15, 212 CAS.
- M. Du, H. Sun, J. Li, X. Ye, F. Yue, J. Yang, Y. Liu and F. Guo, Chemelectrochem, 2019, 6, 5581–5587 CrossRef CAS.
- B. Li, K. Wang, J. Ren and P. Qu, New J. Chem., 2022, 46, 7615–7625 RSC.
- Y. Zhao, Y. Sun, H. Li, S. Zeng, R. Li, Q. Yao, H. Chen, Y. Zheng and K. Qu, J. Colloid Interface Sci., 2023, 652, 1848–1856 CrossRef CAS PubMed.
- G. Liu, Z. Wang, T. Shen, X. Zheng, Y. Zhao and Y.-F. Song, Nanoscale, 2021, 13, 1869–1874 RSC.
- Z. Li, M. Shao, H. An, Z. Wang, S. Xu, M. Wei, D. G. Evans and X. Duan, Chem. Sci., 2015, 6, 6624–6631 RSC.
- B. Singh, T. Ansari and A. Indra, ACS Appl. Nano Mater., 2024, 7, 15754–15762 CrossRef CAS.
- B. Singh, A. Yadav and A. Indra, J. Mater. Chem. A, 2022, 10, 3443–3468 RSC.
- B. Singh, A. K. Patel and A. Indra, Mater. Today Chem., 2022, 25, 100930 CrossRef CAS.
- B. Singh, O. Prakash, P. Maiti, P. W. Menezes and A. Indra, Chem. Commun., 2020, 56, 15036–15039 RSC.
- P. Mukherjee, K. Sathiyan, R. B. -Ziv and T. Zidki, Inorg. Chem., 2023, 62, 14484–14493 CrossRef CAS PubMed.
- T. Ansari, D. Bagchi, S. Ghosh, J. Niklas Hausmann, A. Indra and P. W. Menezes, Chem.–Eur. J., 2025, 31, e202404174 CrossRef CAS PubMed.
- B. Singh, P. Mannu, Y. C. Huang, R. Prakash, S. Shen, C. L. Dong and A. Indra, Angew. Chem., Int. Ed., 2022, 61, e202211585 Search PubMed.
- B. Singh, T. Ansari, N. Verma, Y.-C. Huang, P. M. Mannu, C.-L. Dong and A. Indra, J. Mater. Chem. A, 2024, 12, 19321–19330 RSC.
- B. Singh, Y.-C. Huang, A. Priyadarsini, P. Mannu, S. Dey, G. K. Lahiri, B. S. Mallik, C.-L. Dong and A. Indra, J. Mater. Chem. A, 2023, 11, 15906–15914 RSC.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W. F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- W.-D. Zhang, X. Yan, T. Li, Y. Liu, Q.-T. Fu and Z.-G. Gu, Chem. Commun., 2019, 55, 5467–5470 Search PubMed.
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS.
- B. Singh and A. Indra, Dalton Trans., 2021, 50, 2359–2363 Search PubMed.
- B. Singh, O. Prakash, P. Maiti and A. Indra, ACS Appl. Nano Mater., 2020, 3, 6693–6701 CrossRef CAS.
- J. Mendialdua, R. Casanova and Y. Barbaux, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- A. K. Singh, S. Ji, B. Singh, C. Das, H. Choi, P. W. Menezes and A. Indra, Mater. Today Chem., 2022, 23, 100668 CrossRef CAS.
- Y. Bi, Z. Cai, D. Zhou, Y. Tian, Q. Zhang, Q. Zhang, Y. Kuang, Y. Li, X. Sun and X. Duan, J. Catal., 2018, 358, 100–107 CrossRef CAS.
- Y. Fu, T. Li, G. Zhou, J. Guo, Y. Ao, Y. Hu, J. Shen, L. Liu and X. Wu, Nano Lett., 2020, 20, 4960–4967 CrossRef CAS PubMed.
- J. Lan, M. Luo, J. Han, M. Peng, Hu. Duan and Y. Tan, Small, 2021, 17, 2102814 Search PubMed.
- K. Bera, A. Karmakar, S. Kumaravel, S. S. Sankar, R. Madhu, H. N. Dhandapani, S. Nagappan and S. Kundu, Inorg. Chem., 2022, 61, 4502–4512 CrossRef CAS PubMed.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 Search PubMed.
- J. Liang, H. Shen, Y. Ma, D. Liu, M. Li, J. Kong, Y. Tang and S. Ding, Dalton Trans., 2020, 49, 11217–11225 RSC.
- N. Wu and X. Du, Int. J. Hydrogen Energy, 2023, 48, 23877–23884 Search PubMed.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- J. Zhang, Y. Liu, J. Li, X. Jin, Y. Li, Q. Qian, Y. Wang, A. El-Harairy, Z. Li, Y. Zhu, H. Zhang, M. Cheng, S. Zeng and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 3881–3890 CrossRef CAS PubMed.
- Y. Li, X. Du, J. Huang, C. Wu, Y. Sun, G. Zou, C. Yang and J. Xiong, Small, 2019, 15, 1901980 CrossRef PubMed.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- Z. Kong, J. Chen, X. Wang, X. Long, X. She, D. Li and D. Yang, Mater. Adv., 2021, 2, 7932–7938 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst synthesis, characterization, and optimization of the catalytic reactions. See DOI: https://doi.org/10.1039/d5ta02480c |
| This journal is © The Royal Society of Chemistry 2025 |