DOI:
10.1039/D5TA02142A
(Paper)
J. Mater. Chem. A, 2025,
13, 20868-20883
Network capture effect-driven enhanced activation of peroxymonosulfate by iron-doped carbon quantum dots derived from ferrous gluconate for efficient ciprofloxacin degradation: DFT calculations and mechanism analysis†
Received
16th March 2025
, Accepted 20th May 2025
First published on 6th June 2025
Abstract
Carbon quantum dots (CQDs) have gained extensive application in advanced oxidation processes (AOPs) due to the advantages of their unique carbon network structure. However, CQDs still face issues such as uneven electron distribution and low utilization of active sites during application, which have limited their further use in peroxymonosulfate (PMS)-based AOPs. In this study, ferrous gluconate derived iron-doped CQDs (Fe-CQDs) with a unique structure of Fe doping sites were synthesized by a one-step method to activate PMS. The degradation of ciprofloxacin in the Fe-CQDs/PMS system reached 98.19% within 30 minutes, surpassing that in the CQDs/PMS system (30.39%) and PMS system (25.30%), with approximately 10.74 times and 12.92 times higher reaction rates, respectively. Mechanistic studies have revealed that the Fe3+ sites on Fe-CQDs can optimize the electronic structure of CQDs and enhance the utilization of active sites within the carbon network structure, improving the electron transfer ability from Fe-CQDs to PMS. This optimization strengthens the contact and activation of PMS by Fe-CQDs through the network capture effect. Simultaneously, the carbon network on Fe-CQDs provides abundant reactive sites for PMS activation and pollutant degradation reactions. DFT calculations were utilized to explore the correlation between pollutants and active species by computing their corresponding Fukui functions. This work presents novel insights into the network capture effect on the Fe-CQD-based PMS activation system.
1 Introduction
The high-biological toxicity, low-biodegradability, and potential carcinogenicity of refractory organics in water have attracted significant attention.1 Traditional treatments such as adsorption2 and biodegradation3 cannot effectively degrade emerging organic pollutants. In addition, traditional treatments face problems such as severe secondary pollution, long treatment cycles, and poor shock-load resistance.4–6 Effective treatments are urgently needed to improve the oxidation efficiency. Advanced oxidation processes (AOPs) utilizing peroxymonosulfate (PMS) have garnered significant interest due to their high treatment efficiency and low energy consumption.7 The asymmetric structure of PMS (HO–O–SO3) and the elongated superoxide O–O bond length (IO–O = 1.326 Å) facilitate its activation.1 Researchers have developed various catalysts to effectively activate PMS.8 Among them, carbon quantum dots (CQDs) have drawn considerable attention due to the advantages of widely sourced raw materials, low preparation cost, low toxicity, high chemical stability and biocompatibility. CQDs have nanoscale dimensions, high specific surface area, and abundant defect structures and active sites, which can significantly improve the interaction efficiency with PMS.9,10 However, the uneven electron density distribution on the surface of CQDs leads to low PMS activation efficiency, making it difficult to efficiently generate reactive oxygen species.11 The carbon network structure of CQDs exhibits low density and underutilization of effective active sites, hindering sufficient contact and activation of PMS.12 Furthermore, pollutants or intermediate products adsorbed on the CQD surface may cover these active sites, resulting in a site-blocking effect that deactivates the CQDs and prevents further PMS activation.13 Therefore, there is an urgent need to modify CQDs to enhance their PMS activation capability, along with further investigation of the key mechanism in the CQDs/PMS system.
Doping with heteroatoms is a commonly used method for modifying CQDs.14 Based on the nature of the doped atoms, doping can be divided into metal and non-metal types.15 Among them, metal doping can modulate the electronic structure of CQDs through the incorporation of metals with different valence states and unoccupied orbits.16 Iron-based doping of CQDs is the most common modification method.17 Fe atom doping can enhance the electron accepting ability of CQDs, thereby further strengthening the electron transfer processes (ETP) and providing more active centers, enabling effective activation of PMS.18,19 However, the traditional synthesis method of exogenous iron-doped carbon quantum dots (Fe-CQDs) has some drawbacks that affect their catalytic performance. Exogenous iron doping requires the introduction of iron into CQD structures through high-temperature carbonization or chemical post modification, and precise control of iron loading and dispersion is necessary.19 However, poor compatibility between iron precursors and carbon substrates can lead to aggregation and reduce the density of active sites.16 At the same time, externally doped iron atoms have weak binding with the carbon matrix and are prone to dissolution during the reaction (e.g., Fe2+/Fe3+), leading to secondary pollution and catalyst deactivation.20 In addition, exogenous methods require the additional introduction of iron salts (e.g., FeCl3 and Fe(NO3)3), which increases raw material costs and potential toxicity.21 In the activated PMS system of exogenous Fe-CQDs, heterogeneous interfaces are easily formed by exogenous iron doping, which hinders the rapid transfer of electrons from Fe-CQDs to PMS, resulting in lower activation efficiency than that of homogeneous systems doped with endogenous iron.22 In terms of the selectivity of active species, exogenous iron doped systems tend to generate free radicals, which are easily quenched by anions such as Cl− and HCO3− in water.18 Exogenous Fe-CQDs rely on specific active species for the degradation of some pollutants, and have low mineralization rates for complex organic compounds.23 Compared to exogenous methods, endogenous doping can achieve low-cost green synthesis using natural iron precursors.14 In addition, endogenous iron has a more stable structure and lower dissolution rate due to the bonds between iron and the carbon skeleton.13 In PMS activation systems, endogenous iron doping significantly improves stability and broad-spectrum performance due to its better structural stability and synergistic effect.24 Meanwhile, endogenous Fe-CQDs can effectively improve the electron transfer efficiency between Fe-CQDs and PMS, and enhance anti-interference ability through non-radical pathways,25–27 making endogenous Fe-CQDs a more promising method for PMS activation.
In addition, the specific mechanism of PMS activation by CQDs remains incompletely elucidated. In most studies, researchers have focused on the mediating role of doped active sites in generating reactive species, while neglecting the potential contributions and mechanistic influences of the intrinsic carbon network structure of CQDs on reactive species formation. The carbon network structure of CQDs is a nanoscale framework formed by carbon atoms through a combination of sp2 and sp3 hybridization.28 This structure offers high chemical stability, excellent electrical conductivity, and sp2-hybridized domains that provide electron transport channels to facilitate interfacial charge transfer.29 Defect sites and surface functional groups (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH) within the carbon network can directly adsorb and activate PMS, offering abundant reactive interfaces.30 More importantly, the carbon network structure not only generates reactive radicals via surface functional groups such as carbonyl groups, but also enables electron transfer mediated by the carbon skeleton.31 In addition, existing studies on CQD-mediated PMS activation predominantly rely on free radical pathways, with insufficient exploration of non-radical pathways such as singlet oxygen and surface-mediated electron transfer.32 Therefore, it is important to conduct a further in-depth investigation into the behavior of the CQDs' carbon network in capturing PMS and pollutants, as well as its contribution to reactive species generation mechanisms.
O and –COOH) within the carbon network can directly adsorb and activate PMS, offering abundant reactive interfaces.30 More importantly, the carbon network structure not only generates reactive radicals via surface functional groups such as carbonyl groups, but also enables electron transfer mediated by the carbon skeleton.31 In addition, existing studies on CQD-mediated PMS activation predominantly rely on free radical pathways, with insufficient exploration of non-radical pathways such as singlet oxygen and surface-mediated electron transfer.32 Therefore, it is important to conduct a further in-depth investigation into the behavior of the CQDs' carbon network in capturing PMS and pollutants, as well as its contribution to reactive species generation mechanisms.
Herein, we synthesized Fe-CQDs using ferrous gluconate (FG, Fe(C6H11O7)2·2H2O) as an endogenous precursor for activating PMS and selected ciprofloxacin (CIP) to investigate the performance of the Fe-CQDs/PMS system. In addition, the possible mechanism was also studied and proposed. Various characterization methods were used to study Fe-CQDs and investigate their degradation ability under different influencing conditions. In addition, the mechanism by which Fe-CQDs/PMS forms a hybrid oxidation system was analyzed through electrochemical and quenching methods. The generation mechanism was further explored using density functional theory (DFT). Moreover, factors affecting the hybrid systems have also been studied. We also refined models for different pollutants through DFT computations and evaluated attack sites, degradation pathways, and the toxicity of intermediates. This work offers novel insights into the network capture effect in Fe-CQD-based PMS activation systems.
2 Materials and methods
2.1 Chemicals
Ferrous gluconate (FG, 98%, Aladdin Industrial Corporation), acetaminophen (ACE, 99%, Yien Chemical Technology Co., Ltd), peroxymonosulfate (PMS, 42–46% KHSO5 basis, Yien Chemical Technology Co., Ltd), glucose (99%, Sinopharm Chemical Reagent Co., Ltd), and sodium thiosulfate pentahydrate (Na2S2O3·5H2O, 99%, Sinopharm Chemical Reagent Co., Ltd). Ultrapure water was used throughout the experiment. The chemicals were used without further purification; the detailed information is listed in Text S1.†
2.2 Synthesis of CQDs and Fe-CQDs
Fe-CQDs were synthesized via a hydrothermal process. First, 3.00 g of FG was dissolved in 60 mL of ultrapure water to form the precursor solution. The precursor solution was transferred to a 100 mL Teflon-lined autoclave and kept at 200 °C for 10 h. The reactor was gradually cooled down to room temperature. The product was further purified using 0.22 μm aqueous filter membranes. Then the Fe-CQDs were obtained through a dialysis method (MWCO = 500 Da, 24 h). For characterization, the brown powders were obtained through a freeze-drying method. CQDs were synthesized by the same method except that FG was replaced with glucose.
2.3 Characterization
Transmission electron microscopy (TEM), X-ray diffraction spectrometry (XRD), Raman spectra, X-ray photoelectron spectroscopy (XPS), Fourier transformed infrared spectra (FT-IR), Brunauer–Emmett–Teller (BET) surface area, electron paramagnetic resonance (EPR) and an electrochemical workstation were employed to characterize the obtained catalysts. Please refer to Text S2† for a detailed description.
2.4 Experimental procedures
The catalytic performance of CQDs and Fe-CQDs was measured through catalytic degradation experiments of CIP. First, the catalyst was added to 100 mL of CIP (2.5 mg L−1, 5 mg L−1, 7.5 mg L−1, and 10 mg L−1). The rotor speed was 500 rpm, and the catalyst (3 μg L−1, 6 μg L−1 and 9 μg L−1) was stirred for 30 minutes to ensure full dispersion in the solution and to achieve adsorption and desorption equilibrium. Then, PMS (0.1 mM, 0.2 mM, 0.3 mM, 0.4 mM, and 0.5 mM) was added to the solution to trigger the reaction and samples were collected at specific time points. The sampling time points were −30, 0, 5, 10, 15, 20, 25, and 30 minutes, respectively. A sample volume of 1 mL was collected each time and thoroughly mixed with 20 μL of 0.1 mmol L−1 sodium thiosulfate (Na2S2O3), added dropwise to quench the reaction. The sample was filtered through a 0.22 μm aqueous filter membrane. All experiments were conducted at room temperature (25 °C).
2.5 Analytical methods
The target contaminants' concentrations and detailed conditions of the detection method are displayed in Table S1.† The detailed detection conditions for CIP degradation intermediates are displayed in Text S3.† The conditions of the PMS concentration detection method are displayed in Text S4.†
2.6 Theoretical calculations
Density functional theory (DFT) calculations for ACE were performed using the revised version A.031 of the Gaussian16 software package. Multiwfn was used for auxiliary computing.33 The optimized models of CQDs and Fe-CQDs and the first principles calculations of the PMS activation pathway are detailed in Text S5.†
3 Results and discussion
3.1 Characterization of CQDs and Fe-CQDs
Fig. 1A–D show the TEM images of Fe-CQDs and CQDs. As shown in TEM images, Fe-CQDs and CQDs exhibited spherical morphology. The lattice spacing for both Fe-CQDs and CQDs was measured to be 0.21 nm, with average particle sizes of 6.11 nm and 6.52 nm (Fig. 1E and F), respectively. A lattice spacing of 0.21 nm corresponded to the graphite layer.34 The element mapping of Fe-CQDs is shown in Fig. S1,† where C, O, and Fe elements are evenly distributed on Fe-CQDs. The element mapping of CQDs (Fig. S2†) also showed the even distribution of C and O elements. Fig. 1G shows the XRD pattern of Fe-CQDs and CQDs. The unclear peak near 21° corresponds to the graphitized structure.35 The low Fe doping level may contribute to the unclear peak observed in Fe-CQDs.27 Graphitization was also confirmed by Raman spectroscopy (Fig. 1H). The ratio (ID/IG) of the characteristic peaks of sp2 and sp3 hybridized carbon appearing near 1578 nm (G-band) and 1346 nm (D-band) was calculated to be 2.324. Compared with CQDs, a higher ID/IG value (ID/IG = 2.418) indicated a higher degree of graphitization and more defects in Fe-CQDs.25,28 FT-IR analysis was utilized to investigate the surface functional groups found in CQDs and Fe-CQDs (Fig. 1I).36 The absorption peaks identified at 2971 cm−1, 2935 cm−1, and 1423 cm−1 were associated with the stretching vibrations of saturated –CH and –CH2 groups, as well as the symmetric deformation vibration of –CH3.37 The peaks observed at 1636 cm−1 and 1066 cm−1 were likely attributed to the stretching vibrations of CO and C–O, respectively.38 It is worth noting that a broad and strong –OH absorption peak appeared at 3403 cm−1, which is mainly attributed to the abundant –OH groups of FG and coordinating water.39 Comparing CQDs and Fe-CQDs, the surface of Fe-CQDs contained more –OH groups than CQDs, indicating that –OH may contribute to the enhanced catalytic ability of Fe-CQDs.40 Meanwhile, the red-shift of the vibration absorption peak of the –OH group may result from the significant association of hydroxyl compounds.41 Additionally, the peak detected at 565 cm−1 in Fe-CQDs corresponded to the Fe–O bond.42 Furthermore, the BET surface area and desorption average pore diameter of Fe-CQDs were 17.04 m2 g−1 and 8.39 nm, respectively (Fig. 1J and K).
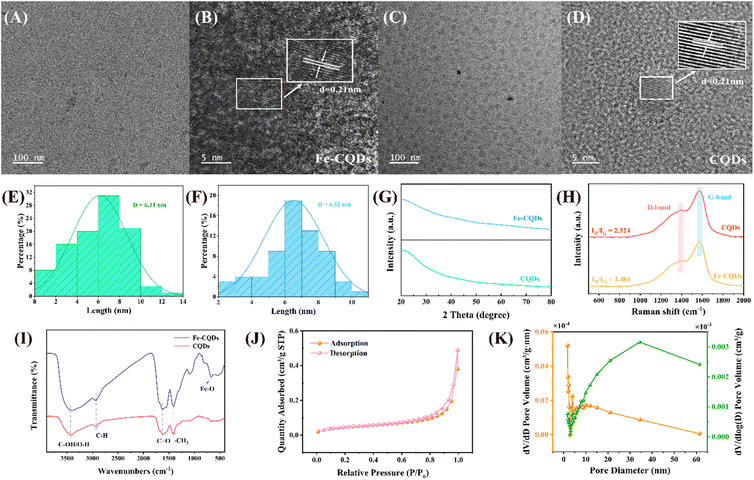 |
| | Fig. 1 (A) TEM images and (B) lattice fringe of Fe-CQDs. (C) TEM images and (D) lattice fringe of CQDs. The average particle size of (E) Fe-CQDs and (F) CQDs. (G) X-ray diffraction (XRD) patterns of CQDs and Fe-CQDs. (H) Raman spectra of CQDs and Fe-CQDs. (I) Fourier transform infrared (FT-IR) images of CQDs and Fe-CQDs. (J) The N2 adsorption–desorption isotherm and (K) corresponding pore size distribution of Fe-CQDs. | |
The elemental composition and valence states of CQDs and Fe-CQDs were investigated by XPS spectroscopy.43 The XPS survey spectra of CQDs and Fe-CQDs are shown in Fig. S3.† CQDs mainly contain C (66.44%) and O (33.56%). Fe-CQDs are composed of C (66.44%), O (28.40%), and Fe (2.87%). This indicated that Fe was present in Fe-CQDs at lower doping levels. Energy dispersive spectroscopy (EDS) also showed that the Fe content was less than 1% (Fig. S4†), confirming the low doping amount of Fe in Fe-CQDs. The C 1s spectrum (Fig. 2A) could be deconvoluted into three peaks (284.80 eV, 286.70 eV, and 287.47 eV), designated as CC/C–C, C–O, and CO, respectively.44,45 A higher concentration of C–O was observed in Fe-CQDs. In the O 1s spectrum (Fig. 2B), peaks at 531.62 eV, 533.30 eV, and 529.80 eV were attributed to CO, C–O, and hydroxyl groups, respectively.5,46 In the Fe 2p spectrum (Fig. 2C), the main peaks for Fe 2p1/2 and Fe 2p3/2 appeared at 724.61 eV and 711.26 eV, respectively, belonging to Fe3+.47 The typical Fe 2p1/2 and Fe 2p3/2 peaks appeared at 721.46 eV and 709.03 eV, respectively, belonging to Fe2+.48 In particular, Fe3+ had the highest content in Fe-CQDs, indicating that Fe3+ is the dominant state in Fe-CQDs. To further understand the electron transfer abilities, electrochemical experiments were conducted. Electrochemical impedance spectroscopy (EIS) was utilized to examine the e− transfer characteristics of CQDs and Fe-CQDs.49 As shown in Fig. 2D, the arc radius of the EIS Nyquist curve for Fe-CQDs was smaller than that of CQDs. This indicated that the resistance of Fe-CQDs was lower than that of CQDs, suggesting that the addition of Fe was beneficial for reducing resistance and enhancing the e− transfer ability of Fe-CQDs.43 Meanwhile, the Tafel slope (Fig. 2E) was also calculated, and the slope value of Fe-CQDs (23.69 mV dec−1) was 15 times smaller than that of CQDs (310.94 mV dec−1), indicating that Fe-CQDs had excellent catalytic performance.50 Linear sweep voltammetry (LSV) (Fig. 2F) showed that Fe-CQDs had an obviously higher current density than CQDs, which further demonstrated that Fe-CQDs can promote electron separation and utilization, making the ETP between Fe-CQDs and PMS more favorable.51
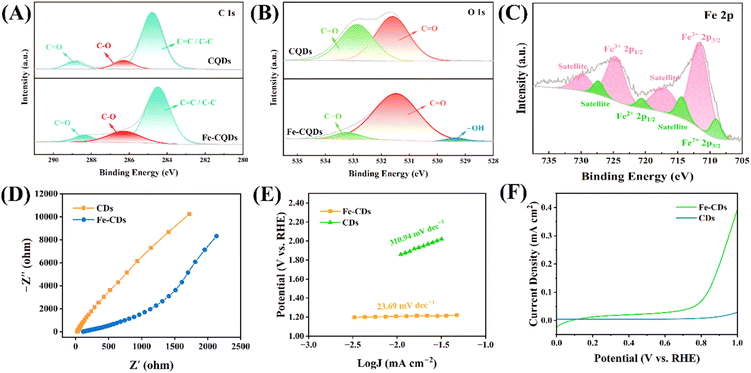 |
| | Fig. 2 High-resolution X-ray photoelectron spectroscopy (XPS) spectra of (A) C 1s and (B) O 1s of CQDs and Fe-CQDs. High-resolution XPS spectrum of (C) Fe 2p of Fe-CQDs. (D) Electrochemical impedance spectroscopy (EIS) spectra of CQDs and Fe-CQDs. (E) Tafel slopes of CQDs and Fe-CQDs. (F) Linear sweep voltammetry (LSV) curves of CQDs and Fe-CQDs. | |
3.2 Catalytic performance of the Fe-CQDs/PMS system
The effectiveness of Fe-CQDs in activating PMS was assessed through the degradation process of ciprofloxacin (CIP) (Fig. 3A). The degradation efficiency of Fe-CQDs/PMS, CQDs/PMS and PMS was 98.19%, 30.39% and 25.30%, respectively (reaction conditions: [Fe-CQDs]0 = 6 μg L−1, [PMS]0 = 0.3 mM, [CIP]0 = 5 mg L−1). Correspondingly, the pseudo-first-order kinetic constants were 0.1085 min−1, 0.0101 min−1 and 0.0084 min−1, respectively (Fig. 3B). To determine the adsorption–desorption capacity of Fe-CQDs and CQDs, we conducted adsorption experiments (Fig. S5†). Fe-CQDs and CQDs can achieve adsorption–desorption equilibrium with adsorbed CIP percentages of 23.94% and 4.65%, respectively. This indicates that adsorption by Fe-CQDs and CQDs was not the main contributor to CIP removal. The reaction rate of the Fe-CQDs/PMS system increased by 12.92 times compared to PMS system, indicating that the incorporation of Fe can effectively enhance the activation of PMS. In addition, with the doping of Fe into Fe-CQDs, the catalytic performance of CQDs improved significantly. This suggests that Fe serves as an important active site in the system. XPS indicated that Fe3+ is the main valence state in Fe-CQDs, while Fe2+ is only present only in small amounts. To investigate the effect of doped Fe2+ and Fe3+ on PMS activation, the effect of dissociative Fe3+ and Fe2+ on CIP degradation was also examined. The addition of dissociative Fe3+ or Fe2+ did not effectively activate PMS, resulting in only 34.69% and 27.26% degradation, respectively. This suggests that free Fe3+ and Fe2+ may not play an important role in CIP degradation. Considering that Fe3+ is the main oxidation state of Fe-CQDs, the doped Fe3+ may be the important sites in activating PMS. The effect of differential initial CIP concentrations on degradation is demonstrated in Fig. 3C. The pseudo-first-order kinetic constant is shown in Fig. S5.† At a concentration of 2.5 mg L−1, the system achieved 100% removal in 10 minutes with a high reaction rate (0.4485 min−1). However, as the concentration increased (7.5 mg L−1 to 10 mg L−1), the degradation marginally declined to 91.75% and 84.56%. This indicated that even at higher concentrations of CIP, the Fe-CQDs/PMS system still retained excellent degradation ability. The effect of the dosage of Fe-CQDs is shown in Fig. 3D. The degradation efficiency increased as the dosage of Fe-CQDs rose from 3 μg L−1 to 9 μg L−1 (91.08% to 100%). However, the degradation rates at concentrations of 6 μg L−1 and 9 μg L−1 were comparable, suggesting that a higher concentration of Fe-CQDs (do not significantly enhance degradation ability). Certain studies have also proposed that H+ can inhibit the process by reducing the concentration of radicals involved in degradation.52,53 Under higher pH conditions, CIP also exists in the form of CIP–O−, which is difficult to degrade.54 The significant inhibition is also attributed to the rapid reaction and consumption of ˙OH and SO4˙− in alkaline environments.45 In addition, PMS is unstable and easily decomposes under alkaline conditions, which further inhibits the degradation of CIP.46
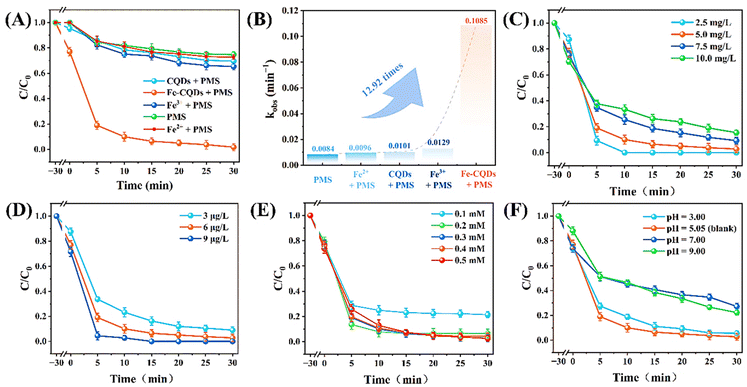 |
| | Fig. 3 (A) CIP degradation and (B) pseudo-first-order kinetic constant of Fe-CQDs and CQDs in different systems. Effect of different concentrations of (C) CIP, (D) Fe-CQDs, and (E) PMS. (F) Effect of different initial pH values on CIP degradation (experimental conditions: [Fe-CQDs]0 = 6 μg L−1, [PMS]0 = 0.3 mM, [CIP]0 = 5 mg L−1, initial pH = 5.05; except indicated). | |
3.3 Identification of reactive oxygen species (ROS)
Quenching experiments were performed to verify the relative contributions of different ROS. Fig. 4A and B show the results of quenching experiments and corresponding kobs values, respectively. Methanol (MeOH, 300 mM), isopropanol (IPA, 200 mM), p-benzoquinone (p-BQ, 10 mM), and sodium azide (NaN3, 10 mM) were used as scavengers for SO4˙− and ˙OH, ˙OH, O2˙−, and 1O2, respectively.55 DMSO was further used as a scavenger for high-valent iron species, ˙OH and SO4˙−. As shown in Fig. 4A, the degradation efficiency decreased to 29.69% after the introduction of DMSO, with the highest inhibition compared to other active species. Due to the fact that DMSO can quench both high-valent iron species and ˙OH and SO4˙−, and high-valent iron species act as transient active species with fast generation and transformation rates in the system, the contribution of high-valent iron species is not clear and should be further investigated. The introduction of MeOH and IPA resulted in a decrease in degradation efficiency to 36.69% and 40.85%, respectively, indicating that ˙OH plays a more significant role as an active species than SO4˙− (Fig. S6†). The presence of 1O2 and O2˙− was also detected. NaN3 and p-BQ reduced the degradation efficiency to 47.95% and 54.18%, indicating that the certain contribution of 1O2 and O2˙−. In addition, the contribution rate of e− was investigated using potassium dichromate (K2Cr2O7) (Fig. S7†). After introducing 1 mM of K2Cr2O7, the degradation efficiency was 85.47%, indicating a certain contribution of ETP. However, with the continuous increase in concentration, the degradation efficiency exhibited obvious improvement. This was attributed to the suppression of the ETP. After the ETP was suppressed, the generation active species through non-ETP pathways was enhanced, improving the activation and utilization of PMS. In addition, higher concentration of Cr served as co-catalysts for Fe3+ sites on Fe-CQDs and promoted the activation of PMS.35
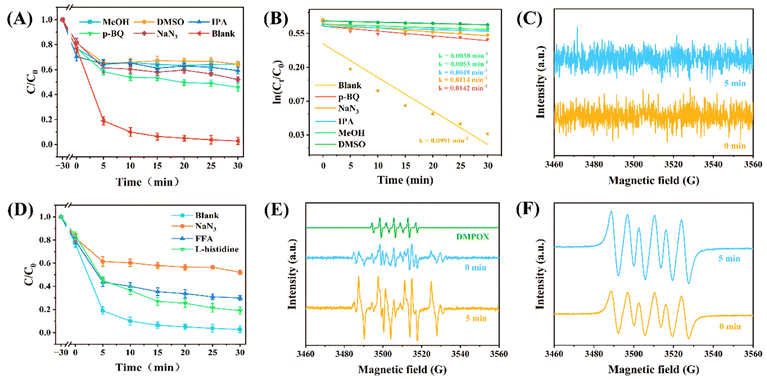 |
| | Fig. 4 (A) Quenching experiment. (experimental conditions: [MeOH]0 = 300 mM, [IPA]0 = 200 mM, [NaN3]0 = [p-BQ]0 = 10 mM, [DMSO]0 = 30 mM). (B) Pseudo-first-order kinetic constant of the quenching experiment. (C) EPR spectra of TEMP–1O2. (D) Degradation efficiency of different 1O2 quenchers in the Fe-CQDs/PMS system. (Experiment condition: [NaN3]0 = [FFA]0 = [L-histidine]0 = 10 mM). EPR spectra of (E) DMPO–˙OH/SO4˙− and (F) DMPO–O2˙− adducts in the Fe-CQDs/PMS system. | |
EPR was used to detect ROS generated in Fe-CQDs/PMS systems. 2,2,6,6-Tetramethyl-4-piperidinol (TEMP) was used as a spin trapping agent for 1O2.56 It is worth noting that no obvious 1O2 characteristic peak was observed. Previous studies have indicated that upon activation, PMS generates non-radical pathways represented by 1O2.39 To further determine the signal of 1O2, we attempted filtering and testing (Fig. 4C) but did not find obvious typical peaks. This indicated that 1O2 may not have been generated. The quenching effect observed with NaN3 may be due to the presence of ˙OH and SO4˙− radicals. To further determine the contribution of 1O2, furfuryl alcohol (FFA) and L-histidine were used, showing similar results as NaN3 (Fig. 4D).57 A recent work reported that quenching agents commonly used to detect 1O2 may significantly shield ˙OH and SO4˙−, leading to an overestimation of the contribution of 1O2.58 We further measured the concentration of 1O2 by a probe method. As expected, only 0.96 μM 1O2 was detected, which indicated that the concentrations of 1O2 in the system were relatively low. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was used as a spin trapping agent for ˙OH and SO4˙−, as shown in Fig. 4E.59 In addition to ˙OH and SO4˙− signals, a DMPOX signal was also present. The generation of DMPOX suggested a strong ETP.60 This indicated that the Fe-CQDs/PMS system had strong oxidizing properties, and it further oxidized DMPO into DMPOX. This also suggested that Fe-CQDs had excellent activation ability for PMS. Meanwhile, due to the higher SO42− concentration than OH− in the system, the overall peak shape was different from the conventional DMPO–˙OH/SO4˙− peak shape. As shown in Fig. 4F, a typical quartet signal with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 was detected, suggesting the formation of the characteristic DMPO–O2˙− adducts.61
:1:1:1 was detected, suggesting the formation of the characteristic DMPO–O2˙− adducts.61
3.4 Mechanism investigation
In this study, the contributions of high-valent iron, ˙OH, and O2˙− were similar. To account for the contributing effects, we proposed an oxidation system dominated by the “Dual-Pathway,” namely the hybrid oxidation system. High-valent iron species and Fe-CQDs-PMS* dominated the non-radical pathway, while ˙OH and O2˙− jointly dominated the radical pathway.
Non-radical pathway: Fe(IV) and Fe-CQDs-PMS*. High-valent iron species are often utilized as selective oxidizing agents, capable of reacting with organic compounds that have a high electron density while remaining unaffected by the presence of chlorides or carbonates in the environment. Based on the previous results, high-valent iron species exhibited the highest contribution in the Fe-CQDs/PMS system, indicating that the Fe3+ sites on Fe-CQDs served as important reactive sites on Fe(IV) generation. Fe3+ reacted with PMS to form Fe2+ and SO5˙− (eqn (1)).50 Fe2+ was oxidized to Fe(IV) again by PMS (eqn (2)).62
| | | Fe3+ + HSO5− → Fe2+ + SO5−˙ + H+ | (1) |
| | Fe2+ + HSO5− → ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeIVO + SO42− + H+ FeIVO + SO42− + H+ | (2) |
To investigate the generation of Fe(IV), PMSO was adopted as a probe for determining its concentration. As depicted in Fig. 5A, changes occurred in the concentrations of PMSO and PMSO2, with the peak generation of PMSO2 reaching 9.19 μM. This implies that Fe(IV) was produced via the cycling of iron. The utilization efficiency of CQDs and Fe-CQDs on PMS was measured (Fig. 5B). Compared with CQDs, Fe3+ sites effectively promoted iron cycling, making the utilization of PMS more efficient. The efficient iron cycle not only generated high-valent iron species to participate in oxidation, but also mediated the generation of other active species. The enhanced efficiency observed in this microenvironment could be attributed to the larger specific surface area of Fe-CQDs, or it may be associated with the strong interaction between Fe-CQDs and PMS. Fe-CQDs can also activate PMS to give rise to highly reactive surface complex species, permitting PMS to adhere to the catalyst surface and form metastable peroxides. The catalyst–PMS* metastable reaction complex has been identified as the most probable binding form.63 To determine the presence of catalyst–PMS*, electrochemical experiments were conducted for further investigation. The platform potential corresponding to the catalyst–PMS* was measured.64 As shown in Fig. 5C, the addition of PMS significant enhanced the potential of Fe-CQDs, while CQDs exhibited a more modest increase. When CIP was introduced into the reaction, there was a notable reduction in the potential for both Fe-CQDs and CQDs. This finding suggests that Fe-CQDs-PMS* and CQDs-PMS* are formed, indicating that Fe-CQDs and PMS are closely associated, creating an effective microenvironment for iron cycling. Furthermore, it is believed that carbon-based materials facilitate non-radical oxidation pathways more effectively. The electron-deficient C–O groups present on Fe-CQDs can act as mediators to enhance electron transfer and serve as key active sites within these non-radical mechanisms. Additionally, electrons can function as conductive links through Fe-CQDs, improving the transfer of e− from pollutants (e− donors) to reactive complex intermediates (e− acceptors).
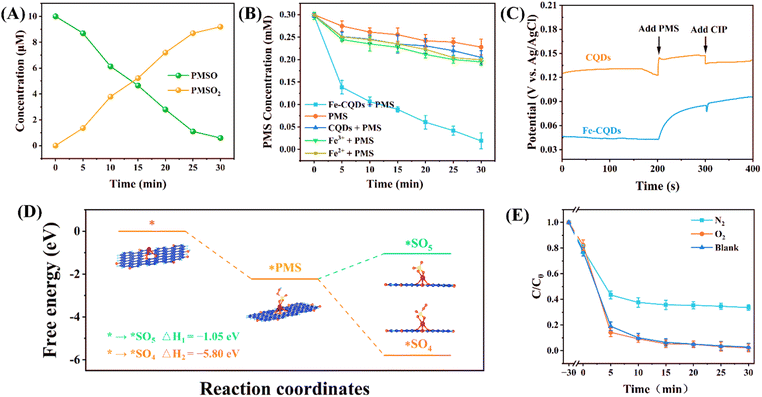 |
| | Fig. 5 (A) Variation in the concentrations of PMSO and PMSO2 within the Fe-CQDs/PMS system. (B) Changes in PMS concentration across different systems. (C) Open-circuit potential (OCP) measurements for both Fe-CQDs/PMS and CQDs/PMS systems. (D) Free energy associated with the decomposition of PMS into *SO4 and *SO5. (E) Assessment of degradation efficiency in the Fe-CQDs/PMS system under N2, O2, and air conditions. | |
Radical pathway: ˙OH and O2˙−.
In the Fe-CQD/PMS system, ˙OH was the most important free radical active species. Generally, ˙OH is mainly generated through two different pathways. In the first pathway, PMS generates SO4˙− by receiving e− (eqn (3)).65 The generated SO4˙− reacts with H2O (eqn (4)) and OH− (eqn (5)) to generate ˙OH, which is also an important reason why SO4˙− cannot effectively participate in the reaction in the free radical pathway.66 The second pathway is to activate PMS through Fe-CQDs. PMS combines with Fe-CQDs to form Fe-CQDs–PMS* complexes. Fe-CQDs can activate PMS through dehydrogenation (–H) or dehydroxylation (–OH).67 In most catalyst activated PMS systems, pathways involving dehydrogenation have been widely reported.1,44,53 This pathway generally involves dehydrogenation to form SO5−˙, which then undergoes self-coupling (eqn (6)) and reacts with other species (H2O, OH−, etc.) to form a system dominated by 1O2. However, the free radical pathway was dominated by ˙OH, which was distinct from the majority of reported studies. To further understand why ˙OH is the main free radical within the system, we carried out first principles analyses on the two aforementioned activation pathways and computed the activation energies for the two distinct pathways. The optimized modeling of Fe-CQDs is shown in Fig. S8.† From Fig. 5D, it can be seen that the activation energy of *SO4 was lower than that of *SO5. This indicates that Fe-CQDs tend to promote the dehydroxylation of PMS, leading to the generation of *SO4 when combined with PMS. In addition, Fe-CQDs can reduce the activation energy of pathway 2, making the generation of ˙OH more favorable, which further explains why 1O2 has low contribution to degradation in this system.
The sp2 hybridized carbon atoms found in the plentiful carbon rings of Fe-CQDs, along with the presence of C–O and CO functional groups, can act as e− carriers that facilitate the activation of PMS through an ETP. These sp2 hybridized carbon atoms can lead to the generation of ˙OH. Furthermore, the enhanced movement of π electrons within Fe-CQDs aids in transferring unpaired electrons and breaking O–O bonds in PMS. The CC bond within Fe-CQDs, in conjunction with the freely flowing π electrons, enables the activation of PMS. The carbon atom in the CO group and the C–O bond can receive e− from oxygen, which facilitates the activation of PMS. CO can also extend the length of the O–O bonds in PMS, thereby facilitating the activation of PMS to produce ˙OH. A recent study indicated that persistent free radicals (C–H groups) at the carbon center can act as e− donors by transferring e− to PMS for its activation, after which they are oxidized to CO.68 This process also accelerates the redox cycling of PMS activated by Fe3+ sites. In addition, the plentiful –OH groups on the surface of Fe-CQDs are capable of forming hydrogen bonds with PMS, facilitating activation of PMS to generate ˙OH through redox reactions. The resulting ˙OH can subsequently oxidize CIP via hydrogen capture or hydrogenation processes. The generated e− also reacted with dissolved oxygen to generate O2˙− (eqn (7)). To clarify this pathway, nitrogen gas (N2) and O2 were used to investigate the generation of O2˙−. As shown in Fig. 5E, the degradation was attributed to O2˙−, which can accelerate the recycling of iron, and activate PMS under low redox potential (EO2˙−/O2 = −0.33 V).3
| | | e− + HSO5− → SO4−˙ + OH− | (3) |
| | | H2O + SO4−˙ → ˙OH + H+ + SO42− | (4) |
| | | OH− + SO4−˙ → ˙OH + SO42− | (5) |
| | | SO5−˙ + SO5−˙ → 2SO42− + 1O2 | (6) |
Network capture effect.
In the Fe-CQDs/PMS system, the trace dosage of Fe-CQDs has impressively achieved the efficient activation of PMS and the efficient removal of CIP. We believe that the unique network structure of Fe-CQDs can effectively capture PMS, enhanced by Fe3+ sites, and thus effectively remove CIP at low concentrations. This process is termed the network capture effect of Fe-CQDs (Fig. 6A). In CQDs, the charge distribution on the catalyst surface is non-uniform, leading to an insufficient adsorption capacity for PMS. Reinforced by Fe3+ sites, the charge of Fe-CQDs was redistributed, and Fe3+ acted as the main active site for capturing PMS. The O atoms in the Fe–O bonds of Fe-CQDs possessed considerable electronegativity, strongly attracting electrons from Fe3+ and making it an electron-deficient Fe site. As an electron-rich species, PMS has a strong propensity for combination and reaction with the electron-deficient Fe sites, thereby enabling Fe-CQDs to capture and activate PMS efficiently and accurately. Simultaneously, the Fe3+ sites reduced the adsorption energy of PMS (ΔE = 1.98 eV), further indicating that the enhanced capture ability of these electron-deficient Fe3+ sites can effectively activate PMS, maintaining excellent degradation performance even at low concentrations of Fe-CQDs. Furthermore, the reticular structure of Fe-CQDs can provide active sites for the further generation of radicals and non-radicals, enhancing the decomposition and utilization of PMS. With abundant sp2 hybridized carbon, electron rich functional groups, and iron active sites, Fe-CQDs have a structural basis that triggers a hybrid oxidation system simultaneously. Typically, the metal and non-metal sites on Fe-CQDs function as the site to generate active species.67 First, sp2 hybridized carbon and electron rich groups (C–O, CO, etc.) can serve as interfacial sites for PMS activation in the free radical pathway.59 The rapid transfer of π electrons, along with the numerous defect structures present in a highly graphitized carbon framework, can promote the breaking of O–O bonds in PMS. The breaking of O–O bonds can generate radicals (˙OH, SO4˙−etc.) to degrade pollutants. In terms of non-radical mechanisms, groups that are rich in electrons may serve as intermediates to enhance e− transfer and trigger non-radical pathways.68 Metal sites can also trigger the activation of peroxide bonds within PMS, giving rise to metastable peroxide intermediates, thereby achieving the generation of non-radical pathways.67 The enhanced network capture effect can also capture CIP, facilitating its reaction with active species generated on Fe-CQDs. This also shortened the contact time between Fe-CQDs and CIP, allowing the generated active species to quickly react with CIP and be consumed, further promoting the decomposition and activation of PMS. To further elucidate the mechanism of the network capture effect on PMS activation through the electron transfer pathway, differential charge density and charge transfer quantities were calculated for both Fe-CQDs and CQDs after PMS adsorption. As shown in Fig. 6B, before PMS adsorption, the Bader charge value of the Fe3+ site on Fe-CDs was 0.482e, which increased to 1.157e after PMS adsorption. This demonstrates that the Fe3+ site at the network capture end of Fe-CQDs can transfer more electrons to PMS for activation, thereby endowing Fe-CQDs with superior PMS capture and activation capabilities. Meanwhile, the number of electrons transferred from the substrate portion of Fe-CQDs (the carbon quantum dot region excluding the Fe3+ site) to PMS was calculated to be 0.734 e−, whereas CQDs transferred only 0.634 e− to PMS. This indicates that Fe3+ effectively reinforces the network capture effect, enhancing electron transfer from the “network” portion of Fe-CQDs to PMS, thus improving both contact efficiency and activation performance. Notably, after PMS adsorption, the O–O bond length in PMS increased from 1.448 Å (when adsorbed on CQDs) to 1.468 Å (when adsorbed on Fe-CQDs). This elongation facilitates easier cleavage of HSO5− into free radicals, further confirming the superior catalytic activity of Fe-CDs.69 The differential charge density spectra further confirm that the Fe3+ sites on Fe-CQDs have higher electron density. When Fe-CQDs adsorb PMS, the electron density near the Fe3+ site further increases, thereby enhancing the electron transfer process from the substrate to PMS. However, no significant electron accumulation process occurs after CQDs adsorb PMS, which results in a very limited number of electrons that can be transferred to PMS, limiting the activation of PMS. This also explains the possible reason for the significant catalytic performance of Fe-CQDs for CQDs. Based on the effective capture stage and active platform, Fe-CQDs can activate PMS and generate abundant active species at low dosage, maintain high efficiency across a wide range of environmental conditions.
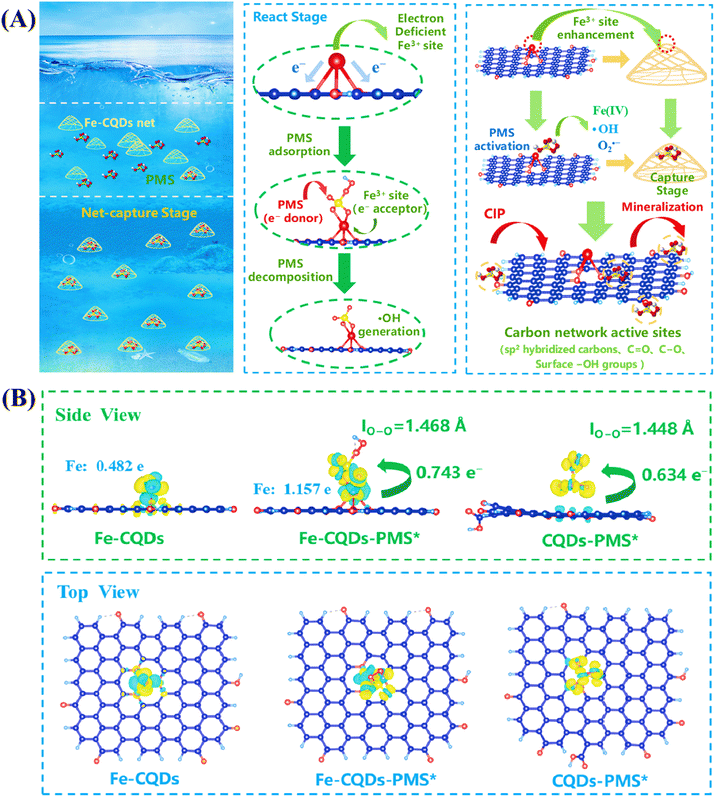 |
| | Fig. 6 (A) Possible mechanism of the network capture effect in the Fe-CQDs/PMS system. (B) Bader charge value and differential charge density spectra of Fe-CQDs, Fe-CQDs-PMS* and CQDs-PMS*. | |
Possible mechanism of the Fe-CQDs/PMS system.
Based on the discussion above, the Fe-CQD/PMS system discussed in this study functions as a hybrid oxidation system primarily driven by radicals (˙OH and O2˙−) and non-radicals (Fe(IV)). The possible mechanism of the Fe-CQDs/PMS system is shown in Fig. 7. Previous analyses of the mechanisms of non-radical and radical pathways have proved that Fe3+ sites are key to the generation of this hybrid oxidation system. The Fe3+ site acted as the active center in the iron cycling process to activate PMS, producing high-valent iron species and functioning as the primary source for the non-radical pathway. Through the network capture effect, Fe-CQDs can effectively capture PMS and CIP, and activate PMS to generate active species. The Fe3+ sites on Fe-CQDs significantly enhance the electron transfer from Fe-CQDs to PMS, with a higher Bader charge value to activate PMS and generate radicals. The metastable complexes formed by the binding of Fe-CQDs with PMS and the C–O bond induced ETP in Fe-CQDs are also important non-radical pathways. In the free radical pathway, Fe-CQDs promote PMS to preferentially remove –OH, making ˙OH one of the dominant free radicals. The sp2 hybridized carbon, C–O, and CO in Fe-CQDs can also promote further activation of PMS, thereby generating more reactive oxygen species. The unique structural characteristics of Fe-CQDs are the reason why the Fe-CQDs/PMS system has evolved into a hybrid oxidation mechanism that includes both free radical and non-radical processes, demonstrating its effectiveness in promoting pollutant degradation.
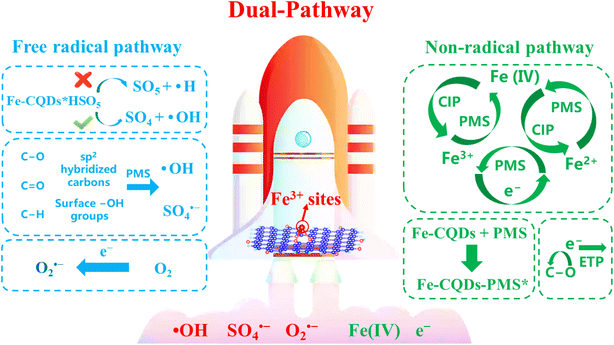 |
| | Fig. 7 Possible mechanism of the Fe-CQDs/PMS system. | |
3.5 Practical application of the Fe-CQDs/PMS system
In actual water bodies, many inorganic ions have been reported to react quickly with free radicals. Reaction between inorganic ions and free radicals caused changes in conditions, inhibiting the degradation efficiency. In addition, complex organic pollutant matrices exist in actual water bodies. The performance of the Fe-CQDs/PMS system was evaluated. Different anions were introduced into the Fe-CQDs/PMS system, and significant differences were observed in the inhibitory effects of some anions on degradation (Fig. 8A). NO3− and SO42− both showed slight inhibitory effects, with degradation efficiencies of 95.65% and 95.37%, respectively. NO3− can react with ˙OH and SO4˙− to generate NO3˙ which has weaker oxidation ability (eqn (8) and (9)), slightly inhibiting the degradation process.43 In addition, excessive SO42− can make the decomposition and activation of PMS difficult, thereby affecting the electron transfer of PMS.54 With the introduction of Cl−, the degradation efficiency decreased to 88.50%, showing a stronger inhibitory effect than NO3− and SO42−. This is because Cl− can react with HSO5−, which is decomposed from PMS to generate HOCl (eqn (10)), and also react with ˙OH.46 The reaction between SO4˙− and Cl− also consumed free radicals (eqn (11)).44 HCO3− emerged as the most influential anion, leading to a notable reduction in degradation efficiency to 59.22%. This may be due to the role of HCO3− as a scavenger for free radicals, which decreases the concentration ˙OH and SO4˙− (eqn (12) and (13)).45 In addition, HCO3− at higher concentrations competes with CIP for the adsorption sites of the catalyst, leading to a decrease in degradation efficiency.7 CO32− exhibits strong inhibition with the degradation efficiency dropping to 78.00%. This is attributed to CO32− suppressing the generation and concentration of free radicals.64
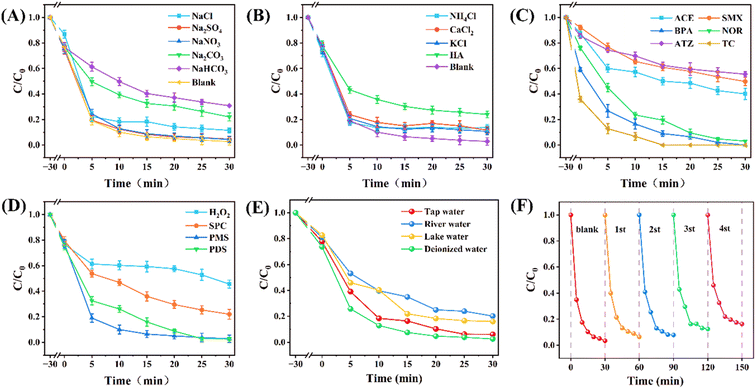 |
| | Fig. 8 Effect of different (A) anions, (B) cations and humic acid (HA) on CIP degradation. ([Initial concentration]0 = 10 mM, [Fe-CQDs]0 = 6 μg L−1, [PMS]0 = 0.3 mM); (C) degradation of various contaminants by the Fe-CQDs/PMS system. (Experimental conditions: [Fe-CQDs]0 = 6 μg L−1, [PMS]0 = 0.3 mM, [pollutants]0 = 5 mg L−1; except indicated). (D) Effect of different oxidants on CIP degradation. ([PMS]0 = [H2O2]0 = [PDS]0 = [SPC]0 = 0.3 mM). (E) The CIP degradation efficiency of the Fe-CQDs/PMS system in different water bodies (river water from the Xiangjiang river and lake water from Taozihu lake). (F) Recyclability test of the Fe-CQDs/PMS system over 4 cycles. | |
Unlike anions, the inhibitory effect of different cations on the system was generally low (Fig. 8B), indicating that the Fe-CQDs/PMS system had excellent adaptability in cationic environments.
| | | NO3− + SO4˙− → SO42− + NO3˙ | (8) |
| | | NO3− + ˙OH → NO3˙ + OH− | (9) |
| | | HSO5− + Cl− → HOCl + SO42− | (10) |
| | | SO4˙− + Cl− → Cl˙+ SO42− | (11) |
| | | HCO3− + ˙OH → H2O + CO3˙− | (12) |
| | | HCO3− + SO4˙− → SO42− + HCO3˙ | (13) |
The Fe-CQDs/PMS system has satisfactory degradation effects on different pollutants (Fig. 8C). The degradation efficiency of ACE, SMX, BPA, NOR, ATZ, and TC by the Fe-CQDs/PMS system was 59.95%, 50.22%, 100%, 96.96%, 44.52%, and 100%, respectively. The kobs values (Fig. S9†) were 0.0229 min−1, 0.0193 min−1, 0.1237 min−1, 0.1075 min−1, 0.0141 min−1 and 0.1664 min−1, respectively. It is evident that Fe-CQDs/PMS demonstrated effective degradation of various pollutants. In addition, different oxidants (H2O2, SPC and PDS) were used to study the potential of Fe-CQDs in activating different oxidation systems (Fig. 8D). Various oxidants achieved degradation rates of over 50%, indicating that Fe-CQDs have excellent activation potential for different commonly used oxidants. PDS and PMS have similar activation modes and energies, exhibiting degradation capabilities similar to those observed during PMS activation. For H2O2 and SPC, the Fe3+ sites on Fe-CQDs have difficulty undergoing the Fenton reaction with H2O2; they must first accept electrons to be converted to Fe2+ before further activating the oxidant, which significantly inhibits the activation efficiency.50 The degradation capacity of the Fe-CQDs/PMS system for CIP was investigated in different water body environments (Fig. 8E). Under deionized water conditions, the degradation efficiency of CIP was the highest (97.40%). However, in tap water, the degradation efficiency was slightly inhibited (93.92%), which might be due to the residual chlorine in the tap water network competing for the active species in the system. Meanwhile, the degradation efficiency of CIP in river water (79.80%) and lake water (83.92%) was still satisfying. The slight inhibition is mainly attributed to the complex water matrix in river water and lake water. Additionally, the reusability of the Fe-CQDs/PMS system was also tested (Fig. 8F). No significant catalyst deactivation was observed over four cycles, and the degradation efficiency in the fourth cycle still reached 83.65%, indicating that the Fe-CQDs/PMS system exhibits good degradation performance over multiple cycles. The above results indicated that Fe-CQDs are an efficient catalyst with low dosage, promising performance, and universality. Fe-CQDs can activate different oxidants and achieve effective removal of different pollutants, showing promising application potential in wastewater treatment.
3.6 DFT calculations, degradation pathways and toxicity
DFT calculations of different pollutants.
DFT calculations were utilized to explore the correlation between pollutants and active species. In the present study, molecular models of seven pollutants were optimized, and their corresponding Fukui HOMO–LUMO, and electrostatic potentials (ESP) were also computed. Tables S2–S8† provide the Fukui function values corresponding to each pollutant. f0, f+, and f− correspond to free radical attacks (˙OH, SO4˙−, O2˙−), nucleophilic attacks, and electrophilic attacks (1O2), respectively.70 As the value of f increases, the likelihood of an attack also increases. The calculation results of different pollutants are shown in Fig. 9. The 20O atom in CIP has the highest f value (f− = 0.0873, f+ = 0.0878, and f0 = 0.0875), rendering the 20O site the most vulnerable to attack by active species (Fig. 9A). The benzene rings in BPA have higher f0 values at 1C, 13C, 15C, and 17C, making them more susceptible to free radical attacks (Fig. 9B). The 16O atom on NOR has the highest f value (f− = 0.0873, f+ = 0.0912, and f0 = 0.0872), making it vulnerable to attacks (Fig. 9C). Both 2C and 6C of SMX have high f− values, indicating that both 2C and 6C atoms are susceptible to attack (Fig. 9D). It is worth noting that 6C has the highest f− value (f− = 0.0572, f+ = 0.0707, and f0 = 0.0639). This makes 6C the most susceptible to attack. The 24O atom of TC has the highest f− and f0 values, which increases its vulnerability to attacks from free radicals and electrophiles (Fig. 9E). The f− values of 25N, 26N, and 28Cl in ATZ were all relatively high, with 25N having the highest f− value (f− = 0.1254), making it the most susceptible to electrophilic attack (Fig. 9F). 28Cl has the highest f+ and f0 values (f+ = 0.1603 and f0 = 0.1308), making it the most susceptible to nucleophilic and radical attacks. The 5C atom of ACE has the highest f− value (f− = 0.0703, f+ = 0.1374, and f0 = 0.1039), making it the most vulnerable site for attack (Fig. 9G). The HOMO–LUMO isosurface results further confirmed the analysis of Fukui's function. It is also notable that the HOMO–LUMO outcomes suggest that for the majority of pollutants, the carbon atoms on the benzene ring are more prone to oxidation, followed by the heteroatoms (N, Cl, etc.) attached to it. This is consistent with most literature reports and intermediate product results from mass spectrometry analysis. In addition, we analyzed the surface charge distribution of various pollutants through ESP. Regions with higher ESP values (red deeper) are more prone to nucleophilic attacks; in contrast (blue deeper), regions with lower ESP values are prone to electrophilic attacks.59 This indicated that the –OH group attached to the benzene ring in BPA is more vulnerable to attack. The ternary and pentagonal structures of CIP are more susceptible to nucleophilic attacks, while functional groups containing oxygen are more likely to undergo electrophilic reactions. Additionally, the oxygen-containing functional groups found in NOR, SMX, and TC also showed susceptibility to nucleophilic attacks, whereas ACE was more prone to electrophilic attacks.
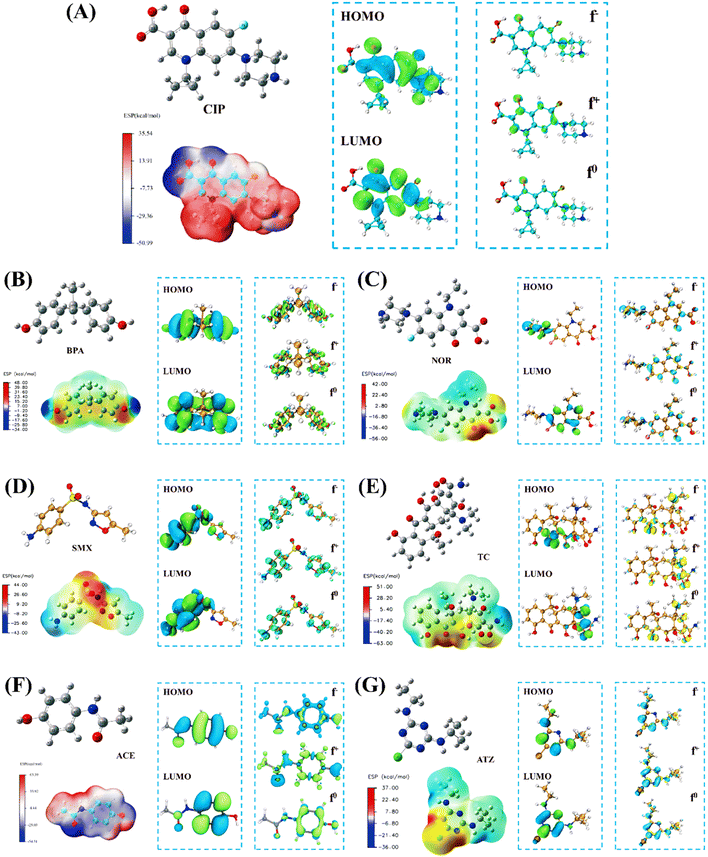 |
| | Fig. 9 Optimized modeling, HOMO–LUMO, f−, f+, f0 and ESP isosurface of (A) CIP, (B) BPA, (C) NOR, (D) SMX, (E) TC, (F) ACE and (G) ATZ. | |
Possible degradation pathway of CIP.
HPLC-MS was used to detect the intermediates in the system. Twenty intermediates were identified, and their detailed structural analysis information is presented in Table S9.† Five possible reaction pathways (pathway I to V) have been proposed (Fig. 10A). In pathway I, P1 (m/z = 362) is initially generated via the opening of the piperazine ring within CIP, and subsequently, P2 (m/z = 334) and P3 (m/z = 306) are generated as a consequence of successive losses of CO.71 The –NH2 group on P3 was attacked and removed by OH and SO4˙− in the system, resulting in the formation of P4 (m/z = 291). P4 then underwent oxidation, leading to the loss of CO and producing P5 (m/z = 263).54 P5 can generate P6 (m/z = 245) and P7 (m/z = 219) through processes such as defluorination or decarboxylation. Additionally, it is capable of producing P8 (m/z = 211) by breaking the pyridine ring. Ultimately, P8 transforms into P11 (m/z = 109) via a series of reactions including decarboxylation, defluorination, and oxidation.72 In pathway II, CIP first undergoes decarboxylation to form P12 (m/z = 288). Upon cleavage of the CC bond within the pyridine ring, P12 undergoes further oxidation to produce P13 (m/z = 181). In pathway III, the opening of the quinolone ring of CIP results in the generation of P14 (m/z = 282), which is subsequently oxidized to form P15 (m/z = 200). Notably, both P13 and P15 from pathways II and III can undergo further oxidation to generate P9, which can then be further oxidized into smaller molecular species. In pathway IV, the fluorine atoms in CIP are replaced by hydroxyl groups, leading to the generation of P16 (m/z = 330). P16 undergoes decarboxylation to generate P17 (m/z = 285). In pathway V, CIP is first subjected to hydroxylation resulting in P18 (m/z = 348), followed by decarboxylation and additional hydroxylation, ultimately yielding P19 (m/z = 320). Finally, through the replacement of fluorine atoms and the opening of the quinolone ring structure, P20 (m/z = 318) is generated. Ultimately, reactive species facilitate the mineralization of intermediates from various pathways into CO2, H2O, or other small molecular compounds.
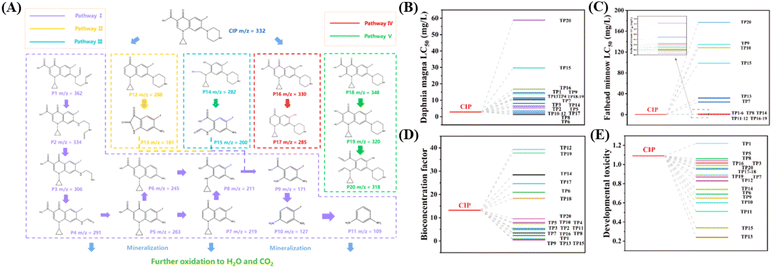 |
| | Fig. 10 (A) Possible degradation pathways in the Fe-CQDs/PMS system. Estimated toxicity with T.E.S.T. for CIP and the transformation products in the Fe-CQDs/PMS process on (B) Daphnia magna LC50 (48 h). (C) Fathead minnow LC50 (96 h). (D) Bioconcentration factor. (E) Developmental toxicity. | |
Toxicity assessment of the Fe-CQDs/PMS system.
The toxicity of CIP and its degradation intermediates was also evaluated through the Toxicity Estimation Software Tool (T.E.S.T.). We calculated the toxicity of the Fe-CQDs/PMS system during degradation using daphnia magna LC50 (48 h), fathead minnow LC50 (96 h), bioconcentration factor, and developmental toxicity. As shown in Fig. 10B–E, excluding certain intermediates, the LC50 values for Daphnia magna and Fathead minnow showed varying degrees of increase for intermediates produced during the degradation of CIP. Simultaneously, the bioconcentration factor and developmental toxicity presented a similar tendency, revealing a significant reduction in the overall toxicity to organisms. This implies that with as the degradation process progresses, CIP and the highly toxic intermediates generated during the degradation can eventually be oxidized into products of lower toxicity and further mineralized into substances of even lower toxicity, thereby making Fe-CQDs/PMS more environmentally benign.
4 Conclusions
In this study, Fe-CQDs with FG as the precursor were synthesized to efficiently activate PMS in a hybrid oxidation system. At low catalyst addition (6 μg L−1) and PMS concentration (0.3 mM), the Fe-CQDs/PMS system achieved a degradation efficiency of 98.19% on CIP in 30 minutes with a high reaction rate (0.1085 min−1). The Fe-CQDs/PMS system also exhibited excellent degradation ability under different conditions, and effectively degraded various organic pollutants (CIP, BPA, NOR and TC). Mechanism investigation suggested that Fe-CQDs can enhance the activation of PMS and effectively react with CIP through the network capture effect. The network capture effect effectively enhances the utilization efficiency of active sites in Fe-CQDs and the electron transfer from Fe-CQDs to PMS, promotes the generation of active species, and ultimately leads to the formation of hybrid oxidation systems. The non-radical pathway primarily involves the formation of high-valent iron species, generated through metal redox reactions that were enabled by Fe3+ sites on Fe-CQDs. In addition, the Fe-CQDs–PMS* metastable peroxide intermediates formed during the reaction and the C–H enhanced ETP on Fe-CQDs also contribute to the non-radical pathway. The radical pathway was mainly driven by the catalytic dehydroxylation of PMS into –OH facilitated by Fe-CQDs. The C–O, CO, and C–H bonds on Fe-CQDs, in conjunction with the sp2 hybridized carbons on the carbon ring and surface –OH groups, further promote the activation of PMS into ˙OH and SO4˙−. Under the combined influence of metal sites and the carbon ring structure of Fe-CQDs, the generation of the hybrid oxidation system achieved directional induction. This work provides an efficient AOP and offers new insights into the formation mechanism and induction methods of hybrid oxidation systems.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Songru Xie: investigation, conceptualization, data curation, formal analysis, visualization, writing – original draft, validation. Longbo Jiang: funding acquisition, investigation, formal analysis, validation, writing – review & editing. Wei Liu: project administration, supervision, writing – review & editing. Qiaomei Lu: supervision, writing – review & editing. Guanjun Zeng: investigation, formal analysis. Hou Wang: investigation, formal analysis. Jiajia Wang: investigation, formal analysis, software. Xingzhong Yuan: funding acquisition, project administration, supervision. Haiwei Jiang: funding acquisition, project administration, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support provided by the Major Program of Xiangjiang Laboratory (24XJ01003), the National Natural Science Foundation of China (U23A2055258, 52100008, and 22308136), the Natural Science Foundation of Hunan Province, China (2023JJ30136 and 2023JJ10012), the Program of Xiangjiang Laboratory (25XJ03015), the Natural Science Foundation of Changsha, China (kq2402189), the Environmental research project of the Hunan Department of Ecological Environment (HBKYXM-2023011), the Open project of the Jiangxi Provincial Key Laboratory of Greenhouse Gas Accounting and Carbon Reduction (No. 2024JXKLCOP010), the Open Funding of the Key Laboratory of Monitoring for Heavy Metal Pollutants, Ministry of Ecology and Environment (KLMHM202419), the Scientific Research Project of the Hunan Education Department (No. 23B0039 and No. 22B0670), the Jiangxi Provincial Natural Science Foundation (No. 20232BAB214063), and the Jiangxi Province Ganpo Juncai Support Program—Training Program for Academic and Technical Leaders in Major Disciplines (20243BCE51126).
Notes and references
- F. Chen, G.-X. Huang, F.-B. Yao, Q. Yang, Y.-M. Zheng, Q.-B. Zhao and H.-Q. Yu, Water Res., 2020, 173, 115559 CrossRef CAS PubMed.
- Y. Núñez-de La Rosa, L. G. C. Durango, M. R. Forim, O. R. Nascimento, P. Hammer and J. M. Aquino, Appl. Catal., B, 2023, 327, 122439 CrossRef.
- S. Zhu, Z. Wang, C. Ye, J. Deng, X. Ma, Y. Xu, L. Wang, Z. Tang, H. Luo and X. Li, Chem. Eng. J., 2022, 432, 134180 CrossRef CAS.
- M.-M. Chen, H.-Y. Niu, C.-G. Niu, H. Guo, S. Liang and Y.-Y. Yang, J. Hazard. Mater., 2022, 424, 127196 CrossRef CAS PubMed.
- Y. Deng, J. Wang, J. Wang, H. Zhang, H. Xiao, C. Zhang and W. Wang, Appl. Catal., B, 2023, 338, 123041 CrossRef CAS.
- J. Kim, G. N. Coulibaly, S. Yoon, A. A. Assadi, K. Hanna and S. Bae, Water Res., 2020, 184, 116171 CrossRef CAS PubMed.
- Q. Li, M. Wang, J.-Q. Chen, X. Liu, J. Wang and Y. Mu, Water Res., 2023, 233, 119729 Search PubMed.
- Z. Zhu, X. Li, M. Luo, M. Chen, W. Chen, P. Yang and X. Zhou, J. Colloid Interface Sci., 2022, 605, 330–341 Search PubMed.
- W. Han, D. Li, M. Zhang, H. Ximin, X. Duan, S. Liu and S. Wang, J. Hazard. Mater., 2020, 395, 122695 CrossRef CAS PubMed.
- A. Hassani, P. Eghbali, F. Mahdipour, S. Wacławek, K.-Y. A. Lin and F. Ghanbari, Chem. Eng. J., 2023, 453, 139556 CrossRef CAS.
- J. Yan, Y. Zhou, J. Shen, N. Zhang and X. Liu, Spectrochim. Acta, Part A, 2023, 302, 123105 CrossRef CAS PubMed.
- Y. Gao, Q. Wang, G. Ji and A. Li, Chem. Eng. J., 2022, 429, 132387 Search PubMed.
- X. Li, S. Ding, Z. Lyu, P. Tieu, M. Wang, Z. Feng, X. Pan, Y. Zhou, X. Niu, D. Du, W. Zhu and Y. Lin, Small, 2022, 18, 2203001 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, L. Wang, R. Fu, L. Sui, H. Song, Y. Hu and S. Lu, Adv. Mater., 2023, 35, 2302536 CrossRef CAS PubMed.
- J. Li, J. Yao, Q. Yu, X. Zhang, S. A. C. Carabineiro, X. Xiong, C. Wu and K. Lv, Appl. Catal. B Environ. Energy, 2024, 351, 123950 CrossRef CAS.
- A. Annamalai, K. Annamalai, R. Ravichandran, A. K. Anilkumar, G. M R and S. Elumalai, Mater. Today Chem., 2023, 30, 101536 CrossRef CAS.
- S. Adil, W. S. Kim, T. H. Kim, S. Lee, S. W. Hong and E.-J. Kim, J. Hazard. Mater., 2020, 396, 122757 CrossRef CAS PubMed.
- Z. Yang, Y. Cui, B. Pan and J. J. Pignatello, Environ. Sci. Technol., 2023, 57, 18918–18928 CrossRef CAS PubMed.
- Z. Zhu, X. Li, M. Luo, M. Chen, W. Chen, P. Yang and X. Zhou, J. Colloid Interface Sci., 2022, 605, 330–341 CrossRef CAS PubMed.
- J. Lei, N. Zhou, S. Sang, S. Meng, J. Low and Y. Li, Chin. J. Catal., 2024, 65, 163–173 CrossRef CAS.
- N. Tejwan, A. K. Saini, A. Sharma, Th. A. Singh, N. Kumar and J. Das, J. Controlled Release, 2021, 330, 132–150 CrossRef CAS PubMed.
- C. Liu, S. Mao, H. Wang, Y. Wu, F. Wang, M. Xia and Q. Chen, Chem. Eng. J., 2022, 430, 132806 CrossRef CAS.
- M. Shao, Q. Yu, N. Jing, Y. Cheng, D. Wang, Y.-D. Wang and J.-H. Xu, Lab Chip, 2019, 19, 3974–3978 RSC.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- P. Zhang, X. Cao, L. Gu, H. Yu, F. Wu, Y. Liu, X. Liu, Y. Gao and H. Zhang, Sep. Purif. Technol., 2024, 342, 126975 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- A. Gholipour, M. Jahanshahi and H. Emadi, J. Cluster Sci., 2024, 35, 237–251 CrossRef CAS.
- C. Huang, M. Duan, Y. Shi, H. Liu, P. Zhang, Y. Zuo, L. Yan, Y. Xu and Y. Niu, J. Colloid Interface Sci., 2023, 645, 933–942 CrossRef CAS PubMed.
- P. Muhammad, S. Hanif, J. Li, A. Guller, F. U. Rehman, M. Ismail, D. Zhang, X. Yan, K. Fan and B. Shi, Nano Today, 2022, 45, 101530 CrossRef CAS.
- X. Yue, X. Miao, X. Shen, Z. Ji, H. Zhou, Y. Sun, K. Xu, G. Zhu, L. Kong, Q. Chen, N. Li and X. He, J. Colloid Interface Sci., 2019, 540, 167–176 Search PubMed.
- T. Luo, Y. Nie, J. Lu, Q. Bi, Z. Cai, X. Song, H. Ai and R. Jin, Mater. Des., 2021, 208, 109878 CrossRef CAS.
- L. Jiang, S. Zhou, J. Yang, H. Wang, H. Yu, H. Chen, Y. Zhao, X. Yuan, W. Chu and H. Li, Adv. Funct. Mater., 2022, 32, 2108977 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Jiang, S. Xie, H. Chen, J. Yang, X. Wang, W. Li, X. Peng, Z. Wu, H. Wang, J. Wang and X. Yuan, Appl. Catal. B Environ. Energy, 2025, 365, 124881 Search PubMed.
- T. Yu, H. Chen, T. Hu, J. Feng, W. Xing, L. Tang and W. Tang, Appl. Catal., B, 2024, 342, 123401 Search PubMed.
- B. Ning, Z. Chen, Y. Cai, F.-X. Xiao, P. Xu, G. Xiao, Y. He, L. Zhan and J. Zhang, Langmuir, 2024, 40, 9144–9154 Search PubMed.
- Y. Wei, Y. Wu, J. Wang, Y.-H. Wu, Z. Weng, W.-Y. Huang, K. Yang, J.-L. Zhang, Q. Li, K.-Q. Lu and B. Han, J. Mater. Chem. A, 2024, 12, 18986–18992 RSC.
- Z. Zheng, J. Min, X. Wang, C. W. Lung, K. Shih and I. M. C. Lo, Environ. Sci. Technol., 2023, 57, 8414–8425 CrossRef CAS PubMed.
- Y. Wang, Y. Bai, C. Han, Z. Li, X. Lun and C. Zhang, Environ. Sci. Pollut. Res., 2023, 30, 108538–108552 CrossRef CAS PubMed.
- L. Jiang, J. Yang, S. Zhou, H. Yu, J. Liang, W. Chu, H. Li, H. Wang, Z. Wu and X. Yuan, Coord. Chem. Rev., 2021, 439, 213947 Search PubMed.
- J. Zhang, L. Zhan, B. Ning, Y. He, G. Xiao, Z. Chen and F.-X. Xiao, Inorg. Chem. Front., 2024, 11, 6970–6980 Search PubMed.
- Q. Ci, Y. Wang, B. Wu, E. Coy, J. J. Li, D. Jiang, P. Zhang and G. Wang, Advanced Science, 2023, 10, 2206271 Search PubMed.
- J. Chen, R. Ren, Y. Liu, C. Li, Z. Wang and F. Qi, Water, 2023, 15, 4241 CrossRef CAS.
- X. Huang, X. Wang, H. Xu, Y. Zhang, G. Zheng, Z. Yang, Q. Ye, Y. Wang and J. Zhang, Process Saf. Environ. Prot., 2024, 181, 75–86 CrossRef CAS.
- N. Li, R. Li, X. Duan, B. Yan, W. Liu, Z. Cheng, G. Chen, L. Hou and S. Wang, Environ. Sci. Technol., 2021, 55, 16163–16174 CrossRef CAS PubMed.
- J. Dai, Z. Wang, K. Chen, D. Ding, S. Yang and T. Cai, Chem. Eng. J., 2023, 453, 139889 CrossRef CAS.
- Y. Bao, C. Lian, K. Huang, H. Yu, W. Liu, J. Zhang and M. Xing, Angew. Chem., Int. Ed., 2022, 61, e202209542 Search PubMed.
- H. Fang, C. Zhou, S. Xu, J. Shi, Y. Hu and G. Liu, J. Alloys Compd., 2023, 958, 170547 Search PubMed.
- Y. Liu, C. Gao, L. Liu and H. Wang, Chem. Eng. J., 2023, 464, 142577 Search PubMed.
- C. Cheng, W. Ren, F. Miao, X. Chen, X. Chen and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218510 CrossRef CAS PubMed.
- Y. Wei, J. Miao, J. Ge, J. Lang, C. Yu, L. Zhang, P. J. J. Alvarez and M. Long, Environ. Sci. Technol., 2022, 56, 8984–8992 Search PubMed.
- Q. Zhou, C. Song, P. Wang, Z. Zhao, Y. Li and S. Zhan, Proc. Natl. Acad. Sci. U. S. A, 2023, 120, e2300085120 Search PubMed.
- Z. Wang, J. Bao, H. He, S. Mukherji, L. Luo and J. Du, Chem. Eng. J., 2023, 458, 141513 CrossRef CAS.
- W. Chen, X. Dai, Z. Liu, B. Du, X. Zheng, D. Ma and X. Huang, Chem. Eng. J., 2023, 469, 143922 CrossRef CAS.
- S. Zhou, L. Jiang, H. Wang, J. Yang, X. Yuan, H. Wang, J. Liang, X. Li, H. Li and Y. Bu, Adv. Funct. Mater., 2023, 33, 2307702 CrossRef CAS.
- S. Wang, J. Li, W. Wang, C. Zhou, Y. Chi, J. Wang, Y. Li and Q. Zhang, J. Environ. Manage., 2023, 342, 118192 CrossRef CAS PubMed.
- X. Wang, W. Tang, Q. Li, W. Li, H. Chen, W. Liu, J. Yang, X. Yuan, H. Wang and L. Jiang, Sep. Purif. Technol., 2024, 342, 127056 CrossRef CAS.
- H. Wang, L. Gao, Y. Xie, G. Yu and Y. Wang, Water Res., 2023, 244, 120480 CrossRef CAS PubMed.
- Y. Wu, X. Li, H. Zhao, F. Yao, J. Cao, Z. Chen, F. Ma, D. Wang and Q. Yang, Chemosphere, 2022, 287, 132061 CrossRef CAS PubMed.
- C. Wang, S. Jia, Y. Zhang, Y. Nian, Y. Wang, Y. Han, Y. Liu, H. Ren, S. Wu, K. Yao and X. Han, Appl. Catal., B, 2020, 270, 118819 CrossRef CAS.
- W. Ren, C. Cheng, P. Shao, X. Luo, H. Zhang, S. Wang and X. Duan, Environ. Sci. Technol., 2022, 56, 78–97 CrossRef CAS PubMed.
- J. Zhang, D. Wang, F. Zhao, J. Feng, H. Feng, J. Luo and W. Tang, Water Res., 2022, 227, 119324 CrossRef CAS PubMed.
- G. Liu, W. Guan, D. Chen, W. Liu, H. Mi, Y. Liu and J. Xiong, Environ. Sci. Pollut. Res., 2022, 30, 33133–33141 CrossRef PubMed.
- P. Zhou, J. Xu, J. Guo, X. Hou, L. Dai, X. Xiao and K. Huo, Green Chem., 2024, 26, 6005–6018 RSC.
- L. Jiang, W. Li, H. Wang, J. Yang, H. Chen, X. Wang, X. Yuan and H. Wang, J. Hazard. Mater., 2024, 469, 133806 CrossRef CAS PubMed.
- H. Luo, E. Almatrafi, W. Wang, Y. Yang, D. Huang, W. Xiong, M. Cheng, C. Zhou, Y. Zhou, Q. Lin, G. Fang, G. Zeng and C. Zhang, Sci. Total Environ., 2023, 871, 162048 CrossRef CAS PubMed.
- H. Ming, P. Zhang, Y. Yang, Y. Zou, C. Yang, Y. Hou, K. Ding, J. Zhang and X. Wang, Appl. Catal., B, 2022, 311, 121341 Search PubMed.
- L. Jiang, Q. Lu, H. Chen, Y. Su, G. Zeng, M. Li, S. Xie, X. Wang, W. Li, X. Yuan, J. Wang, H. Wang and Y. Xie, Sep. Purif. Technol., 2025, 362, 131937 Search PubMed.
- T. Chen, G. Zhang, H. Sun, Y. Hua, S. Yang, D. Zhou, H. Di, Y. Xiong, S. Hou, H. Xu and L. Zhang, Nat. Commun., 2025, 16, 2402 CrossRef CAS PubMed.
- Q. Zhou, C. Song, P. Wang, Z. Zhao, Y. Li and S. Zhan, Proc. Natl. Acad. Sci. U. S. A, 2023, 120, e2300085120 CrossRef CAS PubMed.
- H. Yan, C. Lai, S. Liu, D. Wang, X. Zhou, M. Zhang, L. Li, D. Ma, F. Xu, X. Huo, L. Tang, M. Yan, J. Nie and X. Fan, Chem. Eng. J., 2023, 473, 145253 CrossRef CAS.
- S. Zhu, H. Li, L. Wang, Z. Cai, Q. Wang, S. Shen, X. Li and J. Deng, Chem. Eng. J., 2023, 458, 141415 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abc,
Wei
Liu
d,
Qiaomei
Lu
a,
Guanjun
Zeng
e,
Hou
Wang
ab,
Jiajia
Wang
a,
Xingzhong
Yuan
*abc,
Wei
Liu
d,
Qiaomei
Lu
a,
Guanjun
Zeng
e,
Hou
Wang
ab,
Jiajia
Wang
a,
Xingzhong
Yuan









