DOI:
10.1039/D5TA02013A
(Paper)
J. Mater. Chem. A, 2025,
13, 33365-33384
Shape anisotropy in iron oxide nanocrystals: ligand field and photocatalytic efficiency under tropical sunlight
Received
11th March 2025
, Accepted 18th August 2025
First published on 18th August 2025
Abstract
Recent efforts in structure–activity-based sustainable photocatalyst design for solar energy harvesting highlight the need for a deeper understanding of the photocatalytic process and structural control in synthetic methods. Also, the control over the nanocrystal structure and the shape anisotropy in Earth-abundant materials like iron (mineral) oxide (α-Fe2O3), particularly during bottom-up synthetic approaches, is of multiple significance yet poorly understood. Furthermore, the functional correlation of shape anisotropy with photocatalytic efficiency and its fundamental relevance in geochemical processes remains largely unexplored. This work investigated the comparative role of naturally relevant organic ligands from sugar press mud (PM) with chemical surfactants to induce shape anisotropy among iron oxide nanocrystals in an aqueous sol–gel (bottom-up) synthetic approach. Using electroanalytical tools, we further examine the dynamic link between the structure and activity of these nanocrystals during photocatalysis. Our results revealed that the transformation of hematite (α-Fe2O3) nanocrystals from spherical to sheet-and rod-like morphologies (∼24–44 nm) is broadly consistent with non-classical crystallization theory (NCCT), even in the presence of biogenic ligands (PM) as additives. Moreover, the differences in photocatalytic efficiency (rate constants, k ∼0.014–0.038 min−1) are better explained by using a combined framework of ‘Langmuir–Hinshelwood (L–H) kinetic model and Marcus–Gerischer charge (e−/h+) transfer theory’ than solely by the traditional band gap (Eg ∼ 2.0 eV) and charge carrier (e−/h+) dynamics approach. These findings may provide insight into the rational design of sustainable photocatalysts for solar energy harvesting and contribute to understanding the fundamental geochemical (light-mineral interaction) processes in nature.
1 Introduction
The recent spurt in low-dimensional and Earth-abundant inorganic materials for sustainable photo- and electrocatalytic applications has renewed our interest in long-known iron oxides, particularly hematite (α-Fe2O3).1 This is primarily due to their remarkable magnetic, optical, and redox properties—attributes that have intricately linked them to modern catalytic applications and the history of life on Earth.1–3 Moreover, suitable methods for designing and synthesizing nanoscale iron oxides with efficient catalytic properties are now being extensively studied experimentally and computationally for their catalytic applications.4 The bottom-up soft synthetic routes often lack fine control over material design.5 However, there are efforts to learn to control the design of hematite (α-Fe2O3) nanocrystals from biological (biomimetic) and geochemical (mineral formation) processes.6 Also, methods are being developed using data-driven artificial intelligence (AI) based machine learning and computational calculations for suitable catalyst design.7 Nevertheless, the success of these methods depends highly on the theoretical understanding of solution-based nucleation and crystallisation processes.7 Similarly, there is also a requirement for a deeper understanding of photo- and electrocatalytic processes, particularly regarding the interfacial charge (h+/e−) transfer processes.8 This theoretical understanding will help with error-free algorithm design. Also, the high-quality experimental data on the structure and activity will enable AI model training on catalyst design.4,7
Moreover, the efforts to replace commercial surfactants for sustainable photocatalyst design using wet chemical (bottom-up) synthesis remain limited due to the poor control over the catalyst's structural and functional properties.6 However, our previous study reported an appreciable degree of control over the wurtzite phase (ZnO, 63mc) and morphology (sheet-like) of a nanoscale (15–20 nm) photocatalyst in a press mud (PM) based biogenically controlled wet chemical process.9,10 Nevertheless, there is a lack of mechanistic understanding regarding this.8 Also, with the use of electroanalytical tools, particularly capacitance based (impedance spectroscopy) measurements, the calculated electrochemically active surface area (ESCA) and the low resistance to charge transfer (Rct) showed a significant correlation between the shape anisotropy of the photocatalyst (ZnO sheets, 15–20 nm) and photocatalytic dye degradation rate constant (k).9 Since the theoretical framework for nucleation and crystallisation is constantly evolving and the ligand (solvent perturbation) controlled process shows evidence of non-classical crystallisation, there is a need for extensive investigation in this direction.11,12 Also, there are reports of shifting attention to the mechanistic exploration of photocatalysis from traditional band gap engineering and charge carrier dynamics-led approaches to interfacial charge transfer processes.13–15 Theories, like Marcus–Gerischer for interfacial charge (e−/h+) transfer at the semiconductor–electrolyte interface, are gaining prominence in photocatalytic applications.8 Recently, many studies of various hematite (α Fe2O3) based photocatalysts have been reported for interfacial charge transfer using electroanalytical tools.16
We, in this work, investigated the origin and evolution of shape anisotropy in iron oxide nanocrystals using the same biogenic ligand (PM), i.e., lignocellulosic agro-industrial waste, as a solvent perturbation or capping agent in comparison with fine chemical-based ligands (MC and CT) in an aqueous sol–gel (bottom-up) based wet chemical process. Also, beyond the traditionally known band gap engineering and charge carrier dynamics-based approach, we utilize electroanalytical techniques and Marcus–Gerischer type adiabatic (e−–h+) transfer theory to explain the observed differences in the overall photocatalytic behaviour of the morphologically different and nano-sized (25–45 nm) hematite (α-Fe2O3) photocatalysts. Thus, this work offers a fresh insight into biogenic ligand-induced anisotropic control over mineral oxides and a mechanistic understanding of structure–activity relationships.
2 Experimental
2.1 Materials
All the chemicals were of analytical grade and were used as obtained. Detailed information on the materials and their uses is available in Sections S2.1–S2.3 in the SI.
2.2 Procedure
A summary is provided below. Detailed synthesis steps and complete characterization protocols are available in Sections S2.1–S2.3 in the SI. We first prepared the aqueous extract of dry sugar press mud (PM) following the methods described in our previous study, Verma et al. (2023)9 (for details, refer to Section S2.1 in the SI). This PM extract was used in the synthesis as a biogenic capping agent source compared to other commercially available surfactants like cetyltrimethylammonium bromide (CTAB) and methyl cellulose (MC). A sol–gel (aqueous) synthesis experiment was designed to study the role of these ligands in the physico-chemical characteristics of iron oxide (α-Fe2O3) nanoparticles in general and the morphological variation in particular.9 To study the absolute effect of ligands on nanoparticles, we first undertook acid (FeAH) and base hydrolysis (FeBH) pathways separately without using any ligands in the system. Then, by choosing the base hydrolysis pathway, we used all three ligands one by one separately in three experiments. The steps involved in synthesis are described in Scheme 1 (for a detailed procedure of synthesis, refer to Section 2.5 in the SI).
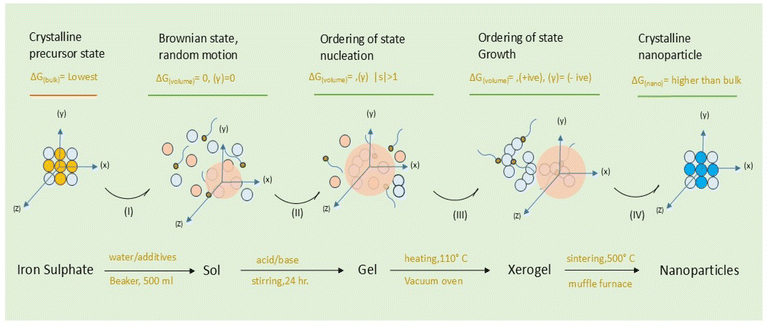 |
| | Scheme 1 Formation of iron oxide (α-Fe2O3) nanoparticles in an aqueous sol–gel acid (H2SO4)–base (NaOH) hydrolysis process and the evolution of the state through reaction stages (light blue circle indicates hydrated iron; the red circle indicates the water molecule; the head–tail entity indicates the additive/ligand in solution; the yellow circle indicates sulfate ions; dark blue indicates the oxygen atom; (γ) surface energy of nuclei, |s| = (C/Ceq)T the supersaturation level). | |
A summary of the synthesis is also given in Table 1. The sintered samples were characterised using advanced surface, electronic, diffraction, and microscopic techniques (Section 2.4 SI).9 The microscopic catalyst dye-interface characterisation was done using advanced electroanalytical techniques (Section 2.7 in the SI). Furthermore, a dilute aqueous solution of rhodamine 6G dye (0.0596 × 10−3 M) in a beaker with the characterised five iron oxide (α-Fe2O3) nanoparticles of different shapes and sizes, a catalyst (100 mg L−1), was exposed to the direct sunlight of tropical summer for photocatalytic study with the dye. The photocatalytic activity was spectrophotometrically followed using the absorption coefficient (ε) corresponding to λmax = 540 nm (Sections 2.5–2.6 SI).9 The macroscopic rate constant (kp) was calculated using the Langmuir–Hinshelwood (L–H) kinetic model (eqn (12)) and percentage degradation of the dye using eqn (S1) (SI). To rationalize the variation in the macroscopic rate constant of dye degradation (Kp), the charge transfer rate was analysed by electrochemical investigation of the catalyst–dye interface. The capacitive and faradaic response (for details see Section 2.7 in the SI) of the interface yielded parameters like flat band potential (Efb), electrochemically active surface area (ECSA), low resistance to charge transfer (Rct), electrochemical work function (ϕ), Epzc, and redox potential (E).9 The complete methods and procedures of electrochemical investigation and calculation of parameters are described in Section 2.7 (SI). The observed variation in the macroscopic photocatalytic rate constant (Kp) was rationalized using electrochemical parameters, particularly the flat band potential (Efb) based charge (e−/h+) transfer rate (microscopic rate constant ke) at the catalyst–dye interface using Marcus–Gerischer charge transfer theory for semiconductor electrolyte interfaces.
Table 1 Summary of synthesis (sol–gel) experiments at 35 °C with stirring at 750 rpm
| Sr. no. |
Precursor/salt |
Additives/ligands |
Form/lattice |
Average size (nm) |
Shape |
Surface charge (ζ) (mV) |
| 1 |
FeSO4·7H2O |
No (BH) |
α-Fe2O3 rhombohedral (hexagonal), distorted cubic γ-Fe2O3 |
∼44 |
Aggregate |
−4.3 |
| 2 |
FeSO4·7H2O |
No (AH) |
α-Fe2O3 rhombohedral (hexagonal), γ-Fe2O3/Fe3O4 |
∼38 |
Aggregate |
−10.5 |
| 3 |
FeSO4·7H2O |
CTAB |
α-Fe2O3 rhombohedral (hexagonal), γ-Fe2O3 |
∼24 |
Rod-like |
−17.8 |
| 4 |
FeSO4·7H2O |
MC |
α-Fe2O3 rhombohedral, (hexagonal), γ-Fe2O3 |
∼27 |
Sheet-like |
−18.1 |
| 5 |
FeSO4·7H2O |
PM |
α-Fe2O3 rhombohedral, (hexagonal), γ-Fe2O3 |
∼31 |
Sheet-like |
−28.7 |
3 Results and discussion
3.1 Synthesis and characterization of iron oxide nanoparticles
The five iron oxide samples have been characterized as hematite (α-Fe2O3), a form of iron oxide. However, there have been traces of other crystalline forms, particularly γ-Fe2O3 & Fe3O4, in all samples along with a noticeable Fe3O4 impurity in the FeAH sample (Fig. 1a, b, and1a–d, SI). These variations in the forms and phases are primarily due to the difference in the synthesis protocols, i.e., acid and base hydrolysis and the varying ligand environment. Moreover, to elucidate the absolute effect of capping ligands on nanoparticles, we first used acid (FeAH) and base (FeBH) hydrolysis without any external capping agent in the first attempt. Thereafter, to compare the role of a lignocellulosic ligand from biogenic sources (PM) with those of analytical grade commercial surfactants, we followed the base hydrolysis protocol and varied the additives (ligands), namely methylcellulose (MC), cetyltrimethylammonium bromide (CTAB), and press mud (PM) extract in sequence. The process in all cases was room temperature (35°) based aqueous sol–gel synthesis with iron sulphate (FeSO4·7H2O) as precursor salt and NaOH as a base (for details refer to Table 1 & Section S2.2, SI). A schematic and mechanistic overview of the reaction protocol is also provided in Scheme 1. The choice of commercial surfactants (CTAB and MC) and biogenic extract (PM) was made to investigate the comparative effect of ligand-based perturbation on the overall synthetic outcome of the reaction in general and the shape anisotropy of the nanoparticles in particular. Moreover, PM was also used as a ligand along with CTAB, and MC to assess a sustainable synthetic method for the future replacement of these commercial surfactants. This was also to understand the possible role of humic substances during mineral crystallization in the natural environment. For instance, secondary mineral formation occurring in geological and soil environments in the presence of degraded organic substances from vegetable sources bears similarity to this biogenic (PM) case. Nevertheless, well-defined laboratory-scale systems do not often correlate with natural systems. Thus, the overall rationale for the design of the synthetic part of the experiment was to understand the evolution of the shape and forms of iron oxides (α-Fe2O3) in the presence of external ligands relevant to sustainable synthetic protocols and geochemical processes.
 |
| | Fig. 1 (a and b) (a) X-ray diffractograms indexed with reference JCPDS cards. (b) Raman spectra of various iron oxide nanoparticle samples from FeAH to FePM, showing phase and polymorphic variations (peaks not indexed in Fig. 2a show impurities in the samples); the (*) at XRD peaks corresponds to α-Fe2O3; (**) corresponds to Fe3O4; (***) corresponds to γ-Fe2O3; and (****) corresponds to ε-Fe2O3. The unindexed peaks belong to other additional peaks. | |
3.1.1 Crystal lattice, morphology, surface, and size.
In the absence of external ligands as additives, the observation revealed that the simple acid hydrolysis resulted in predominantly hematite formation, i.e., α-Fe2O3, a red-brown solid (Fig. S2, SI). Similarly, relatively facile base hydrolysis also resulted in hematite (α-Fe2O3). The formation of the same oxide irrespective of the synthesis route (acid/base) is due to the final oxidation of various oxyhydroxides (α/γ-FeOOH) in the sol–gel state to the thermodynamically stable form, i.e., hematite (α-Fe2O3). However, traces of impurities (γ-Fe2O3/Fe3O4), particularly in FeAH and FePM, are most likely due to the incomplete oxidation of oxyhydroxides during sintering under limited oxygen. The slight variation in the sample's color (Fig. S2, SI) is due to the combined effects of size, surface, defects, and impurities. Reddish-brown and blackish-red are often associated with the hematite (α-Fe2O3) and magnetite (Fe3O4) forms, respectively. Moreover, the presence of α-Fe2O3 as the dominant form in both pathways (acid/base) is due to the same sintering conditions (500 °C) under which only the most stable form (α-Fe2O3) dominates. Furthermore, the perturbation of the same system by changing the ligands, i.e., surfactants and biogenic extract, significantly affected the nanocrystal shape, size, and the polymorphic forms (Fig. 5a–t). For instance, the average crystallite size calculated using the Debye–Scherrer eqn (S1), (SI), ranges from ∼20.0 nm to ∼55.0 nm. This shows the ligands' differential effect on the nanocrystals' size. For instance, the FeCT (∼24 nm) sample shows the smallest average particle size, followed by FeMC (∼27 nm) and then FePM (∼31 nm) with respect to FeBH (∼44 nm) and FeAH (∼38 nm) (for details, refer to Table 1). This indicates that the PM ligands remain in the third place in controlling the size of the iron oxide (α-Fe2O3) nanoparticles (FePM). The indexing of the XRD (Fig. 1a, b and S1a–d, SI) peaks (2θ = 10–70°) and considering the Raman shift modes (280–620 cm−1) (Fig. 1b, S2d & Table S2) differentiated that, unlike the other four cases, FeAH has trace amounts of γ-Fe2O3 (maghemite) and (magnetite) (Fe3O4) impurities.17–19 Nevertheless, the indexing of the diffraction pattern (Fig. 1a) with reference to JCPDS cards indicated the overall predominance of the α-Fe2O3 form in all samples.19 As a matter of fact, pre- and post-annealed XRD diffraction (Fig. 2a and b) patterns of samples with ligands (FeCT, FeMC &FePM) showed a topotactic phase transition of the different oxyhydroxides, particularly α-goethite (α-FeOOH), with γ-lepidocrocite (γ-FeOOH) in traces, of iron to the most stable hematite (α-Fe2O3) form in the pre- and post-annealed samples. The maghemite (γ-Fe2O3) and magnetite impurities can be assigned to the limited oxygen supply and relatively low temperature (500 °C) of sintering, which led to the incomplete conversion of α/γ-FeOOH to hematite (Fig. 3a, b, and 4a, b). Also, the relative intensity ratio of the most intense XRD peaks corresponding to different Miller index (hkl) values (Fig. 1a and S1a–c, SI) indicated the morphological differences in the iron oxide nanoparticles.20–22 A closer analysis (Fig. S1d, SI) of Raman spectra of the samples, along with the resolved region of the XRD (Fig. S1a–c, SI) pattern, shows the samples to be anisotropic in composition while containing impurities of different polymorphic forms of hematite (Fe2O3), particularly maghemite (γ-Fe2O3) and magnetite.17,18 The observed trace of ε-Fe2O3 form (Fig. 1a and S1a–d), particularly in the FeAH sample, was not seen (Fig. 2a and b) in repeated experiments, explaining its transient and rare existence. Furthermore, according to Bragg's diffraction eqn (S1), the variation in the internal crystal features of the samples has been observed. For instance, the lattice constants are for rhombohedral (hexagonal) structures {a = 5.051–5.06 Å and c = 13.81–13.87 Å} in cases of FeMC, FeCT and FePM and inverse cubic in the case of FeBH {a = 8.46 Å} vis-à-vis interplanar spacing in the range of d(104) 2.75–2.76 Å and d(311) 2.56 Å, respectively (Table S1, SI). This clearly showed the changes in the lattices of the samples. Similarly, the asymmetric FTIR peaks (Fig. 3, 4a, b, S3a and b, SI) for FeCT, FeMC, and FePM are at around 400–600 cm−1 with the varying degrees of the axial ratios (a/b ≠ 1) of nano-oscillators, suggesting the morphological differences in iron oxide nanoparticles in varying ligand environments.21 Also, the FTIR analysis of the samples (Fig. 3, 4a, b and S3a, SI) shows that along with the surface hydroxyl (Fe–O–H) group (∼3500 cm−1 broad) due to moisture, the traces of carbonyl and carboxylic functionalities (1600–1400 cm−1 symmetric and asymmetric stretching modes of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) attached to hydrocarbon residues (1400–1200 cm−1 bending modes of –C–H) from ligands are present on the surface of nanoparticles.23,24 Nonetheless, their intensities reflect their residual presence on the surface. However, incomplete decomposition of capping ligands during sintering was observed in pre- and post-analysis of the samples with ligands (Fig. 3, 4a and b).19,24 The loss of organic ligands and of water is also evident from the decrease of characteristic (C–H) stretching frequencies (Fig. 3a and b) and weight loss in TGA analysis (Fig. S8).19 The bands in the ∼470–490 cm−1 and ∼540–570 cm−1 regions are commonly attributed to Fe3+–O asymmetric (Eu) and (A2u) modes of iron oxide (α-Fe2O3) in an octahedral environment. Also, the band peaks at ∼450–550 cm−1 and ∼570–590 cm−1 can be assigned to T1u mode of (Fe3+–O) in a tetrahedral (Td) environment and T1u mode of Fe3+/Fe2+–O, a mixed valence state in an octahedral (Oh) environment, respectively.25 However, the spectra of magnetite (γ-Fe2O3) overlap with those of both magnetite and hematite and thus are often not clearly distinguishable.25 The asymmetries and distortions in the characteristic FTIR peak (Fig. 3, 4a and b) reflect the samples' polymorphic and anisotropic variation.26 The two bands at around 380–450 cm−1 and 570–700 cm−1 indicate the maghemite impurity in the samples (Fig. 3, 4a and b). Previous studies have demonstrated similar results, but with chemically pure surfactants and different methods.19,26–29 Also, the FESEM images of pre- (Fig. S4a–c, SI) and post-annealed (Fig. 4a–e, SI) samples indicate the relative variations in the morphology of these samples. The high-resolution TEM images of annealed samples (Fig. 5a–t) confirmed the ligand effect and the change of morphology from more regular shapes (axial ratios a/b ∼ 1) with an irregular matrix for FeAH and FeBH to more irregular shapes (axial ratios a/b ≠ 1) with a regular matrix for FeCT, FeMC, and FePM samples.19,26 The shape of Fe-AH (Fig. 5a–c) and Fe–BH (Fig. 5e–g) nanoparticles is symmetrical with irregular matrices. However, the samples with capping ligands like Fe-MC (m–o) and FePM (q–s) show the emergence of sheet-like nanoparticles with an inclination to regular matrices. Likewise, FeCT (Fig. 5i–k) shows the evolution
of rod-like nanoparticles.19,24,30,31 This collectively shows that experiments without external ligands in solution (FeAH and FeBH) yielded nanoparticles with a random matrix of sphere-like nanoparticles, whereas systems with capping ligands have shown ordering. Also, the pre- and post-FESEM analysis (Fig. S4a–c and S5a–e, SI) of resynthesized samples with ligands (FeMC, FeCT, and FePM) shows the corresponding precursor morphology of the nanoparticles. This variation in the morphology of nanoparticles due to different ligand environments observed in pre- and post-FESEM analysis appears to have originated in solutions due to the ligand's effect and has developed based on this ligand-guided template shape during the sintering process in a topotactic phase transition (Fig. 2a and b). This is quite apparent from the needle- and plate-shaped template morphology in pre-annealed samples (Fig. S4a–c, SI) and rod- and sheet-like morphology in annealed samples (Fig. S5a–e) for FeCT, FeMC, and FePM samples, respectively. However, it is also arguable that due to the relatively low temperature (500 °C) of annealing, where complete atomic diffusion-led thermodynamic shape-equilibration is less favoured, the initially (pre-annealed) defined kinetic shapes remain statistically dominant and preserved. However, the mechanism behind this shape differentiation is likely to be the diffusion-controlled differential growth rates of specific crystallite faces in both the solution and sintering stages. This has resulted in nanoparticles of different shapes.23,30,32 Furthermore, the SAED pattern of the samples shows a slight shift from the polycrystalline (Fig. 5d and h) to monocrystalline (Fig. 5(l, p and t)) nature of the material when capping ligands are introduced as additives in the solution. This confirms the relative control of the additives over material lattice growth, with FeCT being the most effective. Moreover, the relatively high impurity level in the FePM sample is indicated by the XRD (Fig. 1a, b & S1a–d, SI) and an SEM/EDX analysis (Fig. S9a–j, S10(i–v) and Table S3, SI) of the samples.24 The SEM/EDX analysis, i.e., micrographs (Fig. S10(i–iii), inset) and colourmaps (Fig. S9a–j) of the various regions of all five iron oxide NP samples represented the nearly uniform distribution of metals with varying degrees of impurities in the respective samples.24 The samples with analytical grade commercial capping agents like CTAB and simple acid and base hydrolysis (FeBH) have shown the least impurity (Table S3) level. Generally, the sulfur from the precursor side, i.e., in the range of (0.5–7%) w/w, is visible as an impurity. As expected, the FePM sample showed the largest impurity due to the source of the ligand (PM). However, in some places, the exposed carbon tape (encircled area with arrow) has contributed to the ‘C’ as an impurity, and the same has been omitted.33 In summary, the comprehensive characterization and analysis confirm that all the samples produced predominantly hematite (α-Fe2O3) nanoparticles with varying size and morphology. The other polymorphic forms are due to incomplete conversion of oxyhydroxide during sintering.19,34
O) attached to hydrocarbon residues (1400–1200 cm−1 bending modes of –C–H) from ligands are present on the surface of nanoparticles.23,24 Nonetheless, their intensities reflect their residual presence on the surface. However, incomplete decomposition of capping ligands during sintering was observed in pre- and post-analysis of the samples with ligands (Fig. 3, 4a and b).19,24 The loss of organic ligands and of water is also evident from the decrease of characteristic (C–H) stretching frequencies (Fig. 3a and b) and weight loss in TGA analysis (Fig. S8).19 The bands in the ∼470–490 cm−1 and ∼540–570 cm−1 regions are commonly attributed to Fe3+–O asymmetric (Eu) and (A2u) modes of iron oxide (α-Fe2O3) in an octahedral environment. Also, the band peaks at ∼450–550 cm−1 and ∼570–590 cm−1 can be assigned to T1u mode of (Fe3+–O) in a tetrahedral (Td) environment and T1u mode of Fe3+/Fe2+–O, a mixed valence state in an octahedral (Oh) environment, respectively.25 However, the spectra of magnetite (γ-Fe2O3) overlap with those of both magnetite and hematite and thus are often not clearly distinguishable.25 The asymmetries and distortions in the characteristic FTIR peak (Fig. 3, 4a and b) reflect the samples' polymorphic and anisotropic variation.26 The two bands at around 380–450 cm−1 and 570–700 cm−1 indicate the maghemite impurity in the samples (Fig. 3, 4a and b). Previous studies have demonstrated similar results, but with chemically pure surfactants and different methods.19,26–29 Also, the FESEM images of pre- (Fig. S4a–c, SI) and post-annealed (Fig. 4a–e, SI) samples indicate the relative variations in the morphology of these samples. The high-resolution TEM images of annealed samples (Fig. 5a–t) confirmed the ligand effect and the change of morphology from more regular shapes (axial ratios a/b ∼ 1) with an irregular matrix for FeAH and FeBH to more irregular shapes (axial ratios a/b ≠ 1) with a regular matrix for FeCT, FeMC, and FePM samples.19,26 The shape of Fe-AH (Fig. 5a–c) and Fe–BH (Fig. 5e–g) nanoparticles is symmetrical with irregular matrices. However, the samples with capping ligands like Fe-MC (m–o) and FePM (q–s) show the emergence of sheet-like nanoparticles with an inclination to regular matrices. Likewise, FeCT (Fig. 5i–k) shows the evolution
of rod-like nanoparticles.19,24,30,31 This collectively shows that experiments without external ligands in solution (FeAH and FeBH) yielded nanoparticles with a random matrix of sphere-like nanoparticles, whereas systems with capping ligands have shown ordering. Also, the pre- and post-FESEM analysis (Fig. S4a–c and S5a–e, SI) of resynthesized samples with ligands (FeMC, FeCT, and FePM) shows the corresponding precursor morphology of the nanoparticles. This variation in the morphology of nanoparticles due to different ligand environments observed in pre- and post-FESEM analysis appears to have originated in solutions due to the ligand's effect and has developed based on this ligand-guided template shape during the sintering process in a topotactic phase transition (Fig. 2a and b). This is quite apparent from the needle- and plate-shaped template morphology in pre-annealed samples (Fig. S4a–c, SI) and rod- and sheet-like morphology in annealed samples (Fig. S5a–e) for FeCT, FeMC, and FePM samples, respectively. However, it is also arguable that due to the relatively low temperature (500 °C) of annealing, where complete atomic diffusion-led thermodynamic shape-equilibration is less favoured, the initially (pre-annealed) defined kinetic shapes remain statistically dominant and preserved. However, the mechanism behind this shape differentiation is likely to be the diffusion-controlled differential growth rates of specific crystallite faces in both the solution and sintering stages. This has resulted in nanoparticles of different shapes.23,30,32 Furthermore, the SAED pattern of the samples shows a slight shift from the polycrystalline (Fig. 5d and h) to monocrystalline (Fig. 5(l, p and t)) nature of the material when capping ligands are introduced as additives in the solution. This confirms the relative control of the additives over material lattice growth, with FeCT being the most effective. Moreover, the relatively high impurity level in the FePM sample is indicated by the XRD (Fig. 1a, b & S1a–d, SI) and an SEM/EDX analysis (Fig. S9a–j, S10(i–v) and Table S3, SI) of the samples.24 The SEM/EDX analysis, i.e., micrographs (Fig. S10(i–iii), inset) and colourmaps (Fig. S9a–j) of the various regions of all five iron oxide NP samples represented the nearly uniform distribution of metals with varying degrees of impurities in the respective samples.24 The samples with analytical grade commercial capping agents like CTAB and simple acid and base hydrolysis (FeBH) have shown the least impurity (Table S3) level. Generally, the sulfur from the precursor side, i.e., in the range of (0.5–7%) w/w, is visible as an impurity. As expected, the FePM sample showed the largest impurity due to the source of the ligand (PM). However, in some places, the exposed carbon tape (encircled area with arrow) has contributed to the ‘C’ as an impurity, and the same has been omitted.33 In summary, the comprehensive characterization and analysis confirm that all the samples produced predominantly hematite (α-Fe2O3) nanoparticles with varying size and morphology. The other polymorphic forms are due to incomplete conversion of oxyhydroxide during sintering.19,34
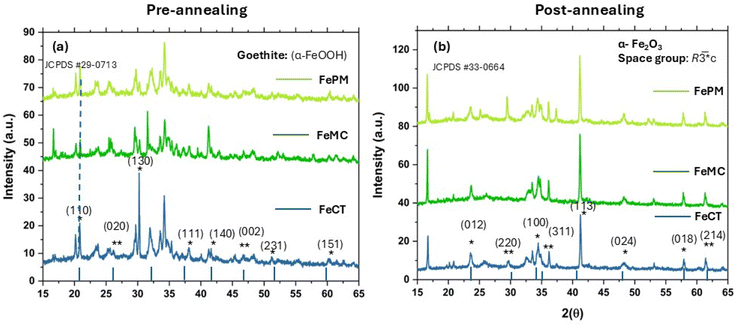 |
| | Fig. 2 (a and b) (a) X-ray diffractograms of pre-annealed samples showing dominant oxyhydroxides form goethite (α-FeOOH); (b) annealed samples showing topotactic phase transformation of α-FeOOH into α-Fe2O3; (peaks not indexed in (a) show impurities of other forms in the samples); (*) at XRD peaks indicates α-FeOOH in (a) and corresponding α-Fe2O3 in (b); (**) indicates lepidocrocite (γ-FeOOH) in (a) and the corresponding γ-Fe2O3 impurity in (b). | |
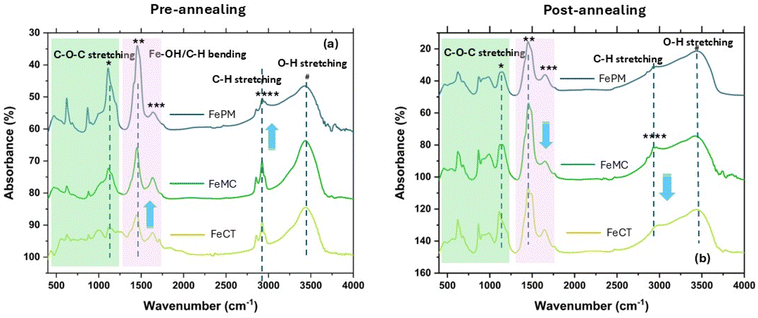 |
| | Fig. 3 (a and b) Fourier transform infrared spectroscopy (FTIR) spectrum of pre-annealed (a) and annealed (b) samples with capping ligands (FeCT, FeMC & FePM), showing the intensity changes of characteristic ligands (C–H/O) and Fe–O frequencies of goethite and hematite forms. | |
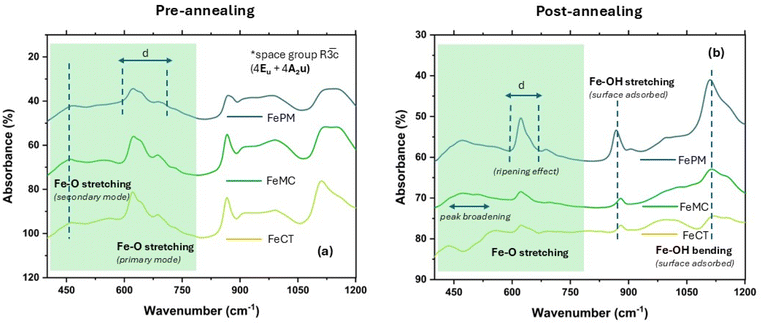 |
| | Fig. 4 (a and b) Fourier transforms infrared spectroscopy (FTIR) spectrum of pre-annealed (a) and annealed (b) samples with capping ligands (FeCT, FeMC & FePM), showing characteristic variation/distortion in the shape of the peaks assigned to metal oxide bonds, indicating shape anisotropy and lattice distortions aspect ratio (a/b ≥ 1) in the system. | |
 |
| | Fig. 5 (a–t) Transmission electron microscopy (TEM) images and corresponding SAED-patterns for the various iron oxide nanocrystals (annealed samples) with their aspect ratio (a/b ≥ 1), showing shape (aggregate, rod & sheet) anisotropy (a/b ≥ 1) in the system. | |
3.1.2 Electronic symmetry, surface charge, and band gap.
The lattice defects, shape anisotropy, and polymorphic impurities are also visible in the room temperature (27 °C) X-band (ν = 9.5 GHz) EPR spectrum of the samples. The two bands in the EPR spectrum (Fig. Sa and b, SI) at around 2000–2500 G and 3200–3600 G indicate hematite, with traces of maghemite and magnetite impurities. Also, the FeBH sample has a distinct broad signal indicating its mixed (Fe2+/Fe3+) valence in the inverse spinel structure of magnetite. The signal with a high line width (ΔG > 100) and ‘g’ ≈ 1.939 for high spin Fe3+ (S = 5/2) ions in an octahedral environment shows strong exchange interaction due to mixed valence states. The sharp signal of FeAH is due to Fe3+ and the more regular structure of hematite. However, other than these two cases (FeAH & FeBH) without capping ligands, the peak distortion in different samples (with capping ligands) indicates the shape anisotropy and defects in the electronic environment around the Fe3+ center.35,36 Furthermore, the zeta potential (ζ) measurement shows the negative surface charge (Fig. S11, SI & Table 1) in the range of −4.03–28.07 mV in ethanol dispersion. Ethanol, a solvent having a lower dielectric constant than water, minimizes the effect of pH on ζ; thus, the measured ζ fairly reflects the charge on the surface. From a low and negative value, in the case of no capping ligands (FeBH −4.03 mV), the zeta potential increases to a high negative value in the case of capping ligands (FePM −28.7 mV). However, the negative ζ in all cases demonstrates the effect of anionic capping ligands modifying the surface charges of the nanoparticles. Nonetheless, along with the residual presence of surface ligands, such a large variation of ζ, may also be due to the polymorphic and anisotropic variation of nanoparticles. Noticeably, this ζ also shows a correlation with the observed potential of zero charge (Epzc) discussed below.9 The diffuse reflectance UV-vis absorption spectra with the absorption edge at 565 nm to 515 nm (Fig. S7a), along with Tauc analysis (Fig. S7b and c), eqn (S4), show that the indirect band gap (Eg) of the samples lies in the range of 1.9 to 2.1 eV. This variation in the band gap is primarily due to the variation in size and crystalline defects, or due to the mid-gap and surface states introduced by the phase (Fe3O4 & γ-Fe2O3) impurities and residual capping ligands on the surface of the samples.24,37,38 This agrees with the observed varying levels of magnetite (Fe3O4) and maghemite (γ-Fe2O3) impurities, in the respective samples. However, the heterogeneity-led noise in the spectrum hampers clear analysis, thus inviting further investigations. Moreover, such impurity-based variation in the band gap has been extensively studied and reported in the literature.37,38
3.1.3 Nanoparticle size distribution.
Following a method reported by Foreman-Ortiz et al. (2022),39 a low concentration (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) ethanol-based dispersion (viscosity; ethanol 12.0 millipoise) of the samples (FeAH, FeBH, FeCT, FeMC, and FePM), when studied using an NTA probe (Fig. S12.0(a–c)–S12.9(a–l), SI), revealed crucial information (Fig. S13, SI) on system evolution, settling, and pathways by means of various distribution curves. These distribution curves, namely, the particle concentration (particle per mL) vs. diameter (nm), surface area concentration (nm2 mL−1) vs. diameter, volume concentration (nm3 mL−1) vs. diameter of nanoparticles, and the intensity distribution solid curve of the diameter, describe the statistical distribution of the system.39 These plots give insights into the number, surface area, and volume contributions of particles of various sizes and reaction outcomes and the probable evolutionary pathways of the nanoparticles. For instance, the comparative mean particle diameter (nm) distribution (Fig. S13, SI) for the various iron oxide nanoparticle samples synthesized using varying ligands leads to a distinct picture of the variation in the diameter distribution (size contribution) of the nanoparticles. This is clearly due to the different behaviours of the additives in the solution during system evolution, i.e., seed formation, crystal growth, and the Ostwald ripening stage of the nanoparticles. This can be understood considering the fact (Fig. S13, SI) that in iron oxide synthesis, D90, the distribution pattern of 90% of the nanoparticles in each sample, shows the highest particle diameter for Fe–BH and the lowest particle diameter for Fe–MC. This is logically expected because of the absence of a capping agent, which allows unrestrained crystal growth in the case of Fe–AH (Fig. S12.1(a–l), SI) and Fe–BH (Fig. S12.3(a–l)), whereas effective capping ability tapered this growth by minimizing the surface free energy (ΔG) in the case of surfactants.40 Also, the surface area concentration (nm2 mL−1) vs. diameter curve of sample FeCT (Fig. S12.5a–d) shows high surface area at the smallest size, indicative of early stage nucleation in the FeCT system, while an increase in surface area for FeCT, FeMC, and FePM (Fig. S12.5–S12.9(e−h)) without a proportional increase in volume (Fig. S3.5–S3.9(i–l)) indicates shape anisotropy in the system. Nevertheless, the overall control of the particle size diameter remains similar for FeCT, FeMC, and FePM, as indicated by the mean distribution of NPs (Fig. S13 red curve line). Thus, this shows the competitive capping ability of PM-based ligands. The modal distribution of NPs, the D10 distribution pattern of 10% of nanoparticles in each sample, can be ascribed to the mixing error or the handling error.39
:1000) ethanol-based dispersion (viscosity; ethanol 12.0 millipoise) of the samples (FeAH, FeBH, FeCT, FeMC, and FePM), when studied using an NTA probe (Fig. S12.0(a–c)–S12.9(a–l), SI), revealed crucial information (Fig. S13, SI) on system evolution, settling, and pathways by means of various distribution curves. These distribution curves, namely, the particle concentration (particle per mL) vs. diameter (nm), surface area concentration (nm2 mL−1) vs. diameter, volume concentration (nm3 mL−1) vs. diameter of nanoparticles, and the intensity distribution solid curve of the diameter, describe the statistical distribution of the system.39 These plots give insights into the number, surface area, and volume contributions of particles of various sizes and reaction outcomes and the probable evolutionary pathways of the nanoparticles. For instance, the comparative mean particle diameter (nm) distribution (Fig. S13, SI) for the various iron oxide nanoparticle samples synthesized using varying ligands leads to a distinct picture of the variation in the diameter distribution (size contribution) of the nanoparticles. This is clearly due to the different behaviours of the additives in the solution during system evolution, i.e., seed formation, crystal growth, and the Ostwald ripening stage of the nanoparticles. This can be understood considering the fact (Fig. S13, SI) that in iron oxide synthesis, D90, the distribution pattern of 90% of the nanoparticles in each sample, shows the highest particle diameter for Fe–BH and the lowest particle diameter for Fe–MC. This is logically expected because of the absence of a capping agent, which allows unrestrained crystal growth in the case of Fe–AH (Fig. S12.1(a–l), SI) and Fe–BH (Fig. S12.3(a–l)), whereas effective capping ability tapered this growth by minimizing the surface free energy (ΔG) in the case of surfactants.40 Also, the surface area concentration (nm2 mL−1) vs. diameter curve of sample FeCT (Fig. S12.5a–d) shows high surface area at the smallest size, indicative of early stage nucleation in the FeCT system, while an increase in surface area for FeCT, FeMC, and FePM (Fig. S12.5–S12.9(e−h)) without a proportional increase in volume (Fig. S3.5–S3.9(i–l)) indicates shape anisotropy in the system. Nevertheless, the overall control of the particle size diameter remains similar for FeCT, FeMC, and FePM, as indicated by the mean distribution of NPs (Fig. S13 red curve line). Thus, this shows the competitive capping ability of PM-based ligands. The modal distribution of NPs, the D10 distribution pattern of 10% of nanoparticles in each sample, can be ascribed to the mixing error or the handling error.39
3.2 Nucleation, crystal growth, and shape anisotropy
The crystallization behavior of iron oxide (α-Fe2O3) nanoparticles synthesized via a sol–gel route using FeSO4·7H2O in varied ligand environments (CTAB, methyl cellulose (MC), biogenic ligand (PM), and no ligand (AH and BH)) demonstrates the influence of both classical and non-classical nucleation-growth mechanisms (Fig. 6). The process began with hydrolysis of iron salt solution, a pale green solution, [Fe(H2O)x], to FeOH2 (blue color), to its instant oxidation of Fe2+ to Fe3+ forming (FeOH)3 (green color solution) upon NaOH addition (eqn (2) and (3)), followed by gelation and sintering at 500 °C. The chemical transformation can be represented as eqn (1) and (2):| | Fe2+ + ¼O2 + ![[/]](https://www.rsc.org/images/entities/char_e11f.gif) H2O → Fe(OH)3↓ + 2H+ H2O → Fe(OH)3↓ + 2H+ | (1) |
| | | Fe(OH)3 → α-FeOOH → α-Fe2O3 + 3H2O | (2) |
 |
| | Fig. 6 Schematic diagram involving distinct stages and pathways available to systems for ligand-controlled crystallisation and Ostwald ripening; the two pathways show classical crystallisation and a prenuclear cluster (PNC) led non-classical pathway, indicating the loci and the pathway for the origin and growth of shape anisotropy in iron oxide nanocrystals during system evolution and phase separation processes in a sol–gel scheme. | |
The initiation of nucleation is thermodynamically governed by the solubility product (Ksp) of Fe(OH)3, typically around 10−38 at 25 °C. Upon base addition, the ionic product (IP) eqn (3) becomes
Supersaturation arises when IP >
Ksp, and its extent is described by the degree of supersaturation (
S) (
eqn (4) and
(5)):
| | 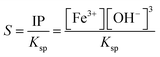 | (4) |
| | 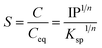 | (5) |
where ‘
C’ is the concentration of the solute,
Ceq is the equilibrium concentration,
Ksp is the solubility product, and ‘
n’ is the number of ions. This ratio quantifies the chemical potential driving nucleation. A higher (
S =
IP/
Ksp > 1) ratio enhances the driving force for phase separation but also alters the thermodynamics and kinetics of nucleus formation.
41,42 This supersaturation (
S > 1) is the driving force for nucleation and crystal growth. According to Classical Nucleation Theory (CNT), the formation of a stable nucleus requires overcoming a free energy barrier (Δ
G) (
eqn (6) and
(7)) given by
43| | 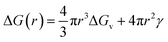 | (6) |
| | | ΔGv = −KTlnS | (7) |
where Δ
Gv is the volume free energy (negative under supersaturation),
γ is the interfacial energy,
Vm is the molar volume of the solid, ‘
K’ is the Boltzmann constant,
T is the temperature, and
S is the supersaturation. The formation of a stable nucleus requires overcoming a critical free energy barrier (Δ
G*) and the critical nucleus radius (
r*), which are given by
eqn (8) and
(9) (ref.
43):
| | 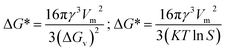 | (8) |
| | 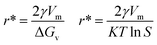 | (9) |
In the ligand-free system, as observed, the morphologies of the iron oxide NPs (
Fig. 5a–t) in the absence of any additives as external perturbation in solution remain inclined to the random distribution of more regular (
a/
b ∼ 1) shapes.
43 This is in line with the absence of a surface-passivating agent leading to high
γ and unstable supersaturation, resulting in high Δ
G* and low nucleation density. This promoted uncontrolled aggregation and yielded poorly resolved, isotropic morphologies. Thus, irrespective of the pathways (acid or base hydrolysis), no significant shape resolution (
Fig. 5a–h) occurs in the case of processes where there is no external ligand present (FeAH and FeBH) in the system. However, in other cases with ligands, more pronounced shape resolution, as reflected in axial ratios (
a/
b ≠ 1) diverging significantly from unity, was found.
29 In particular, the inclusion of CTAB, a cationic surfactant, significantly altered interfacial dynamics. CTAB molecules preferentially adsorbed onto specific crystal facets, selectively lowering
γ and stabilizing certain planes. This facilitated anisotropic, facet-directed growth
via a diffusion-controlled regime, described by
eqn (13) (ref.
43):
| | 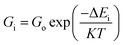 | (10) |
where Δ
Ei corresponds to the activation barrier for ion attachment at the crystallite facet. This effect resulted in nanoplates or rod-like hematite structures (
Fig. 5i–l) with enhanced shape anisotropy. Methyl cellulose, a high molecular weight non-ionic polymer, introduced steric hindrance and formed a viscous sol network during the experiment.
11,43–45 This constrained ion mobility and suppressed aggregation, enabling a controlled growth regime, likely through elongation along the
c-axis due to directional sol crowding, resulting in sheet-like structures (
Fig. 5m–p). In the case of the biogenic ligand, composed of polyfunctional groups (carboxyl, hydroxyl, and phenolic), strong chelation with Fe
3+ and hydrogen bonding to particle surfaces altered both
γ and local ion activity, also resulting in sheet-like morphology (
Fig. 5q–t). Beyond lowering the nucleation barrier, this facilitated non-classical nucleation pathways, particularly oriented attachment and cluster aggregation, where crystalline building blocks align and fuse based on lattice compatibility (
Fig. 6). This is typically seen in cluster aggregation (or Smoluchowski-type) kinetics, which is used to describe how the number of clusters of a specific size ‘
i’ changes over time as described by
eqn (11) (ref.
43):
| | 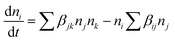 | (11) |
Here,
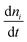
is the rate of change of the number concentration of clusters (or particles) of size ‘
i’ with respect to time, Σ
βJknJnk represents the formation of clusters of size ‘
i’ by binary collisions (or aggregations) of smaller clusters. Furthermore,
nj and
nk represent the number concentrations of clusters of sizes
j and
k, respectively. The term
βjk represents the rate constant (or the collision kernel) for the aggregation of clusters of sizes
j and
k. The summation runs over all pairs such that
j +
k =
ij +
k =
ij +
k =
i. So only those combinations that lead to size ‘
i’ are considered. The second term
niΣ
βijnj similarly indicates the loss of clusters of size ‘
i’ as they collide with other clusters of size ‘
j’ to form larger clusters.
43 These dynamics bypass the traditional atom-by-atom growth of classical models, explaining the formation of highly oriented, anisotropic hematite structures. Moreover, the current discussion on solution phase classical
vs. non-classical nucleation and growth suggests that additive-controlled processes are generally inclined to a non-classical path.
42,46 However, a similar shape anisotropy in zinc oxide nanoparticles was also reported in our previous study (L. M. Verma
et al. 2024).
9 In summary, the shape and phase outcomes across ligand systems are rationalized by considering both thermodynamic and kinetic aspects,
i.e.,
Ksp-defined supersaturation (
S), ligand-induced modulation of
γ, and the coexistence of classical nucleation with non-classical assembly routes. This framework explains how surface-active ligands steer iron oxide crystallization during sol–gel synthesis. However, beyond these differences in solution phase evolutionary pathways, the sintering of resynthesized samples (FeCT, FeMC, and FePM) at a fixed temperature (500 °C) in a muffle furnace showed the retention of the relative effect of the ligands as observed in this experiment. The resynthesized pre-annealed (vacuum oven drying at 150 °C) iron oxide samples analysed by XRD (
Fig. 2a and b), FTIR (
Fig. 3a and b), and TGA (Fig. S8, SI) confirm the definitive pre- and post-annealing effect of the ligands on the shape and morphology of the final iron oxide (α-Fe
2O
3) nanocrystals. Also, FESEM images of pre- and post-annealed (Fig. S4a and S5a–d, SI) samples show template morphologies of the samples developing into the corresponding final shape. For instance, the needle-shaped goethite (α-FeOOH) samples in the case of FeCT grow into rod-like morphologies after annealing. This suggests the template-based Ostwald ripening of the needle-shaped crystal – a precise mechanism for non-classical kinetic crystallization pathways. As the complete thermodynamic equilibration of shape in the case of α-Fe
2O
3 is reported at 700–900 °C, the result in this case is largely kinetically controlled.
26 Furthermore, the previous work shows that most of such studies are concerned with completely defined cases, involving fine chemical surfactants as additives, yet still lack complete mechanistic understanding and control over nucleation and crystallization.
43 Among the reported biogenic ligand-based approaches, using the extract of lignocellulosic waste (PM) presents a novel case of ligand-modulated size and morphology control. However, these findings align with the recent efforts in these directions. Nevertheless, during this study, it remained challenging to conclude the exact role of the PM extract in shaping and controlling the crystallisation process primarily due to the complexity of the degraded lignocellulosic substance (PM) based aqueous extract. However, given its importance in sustainable catalysis and relevance to the mineralisation processes in the natural environment vis-à-vis the complexity of iron oxide polymorphism, this needs to be further investigated on a case-by-case basis and in far greater detail under more suitable conditions.
11,47
3.3 Photocatalytic activity under tropical sunlight
The variation of photocatalytic activity (rate constant Kp) of morphologically different iron oxide (FeAH, FeBH, FeCT, FeMC, and FePM) nanoparticles was investigated to understand the role of the catalyst structure and interface-based differences, for their correlation with activity (Fig. 7a–f). The overall experiment involves nanoscale (∼20–55 nm) metal oxide (α-Fe2O3) based aqueous phase dye degradation in the presence of tropical sunlight. The designed setup facilitated the interaction of broad-spectrum tropical sunlight with the rhodamine dye in an aqueous solution, offering near steady-state illumination.9,48 The variation in the dye degradation rate (Kp) was followed spectrophotometrically. As a matter of fact, metal oxide-based photocatalysis, particularly its rate and mechanism, has been studied extensively during the past few decades.49,50 However, greater emphasis has been placed on selected materials like TiO2, RuO2, and ZnO. Also, the observed photocatalytic rate constants are often macroscopic (bulk).50 In the mechanisms part, aspects like light absorption, charge carrier dynamics, surface states, and quantum efficiency have received much attention.49 Nevertheless, crucial elements like interfacial charge (e−/h+) transfer and microscopic kinetics still require rigorous investigations. In this study, we have attempted to understand the variation in the macroscopic (bulk) rate constant (Kp) of dye degradation using the Langmuir–Hinshelwood (L–H) kinetic model. Also, with the help of Marcus–Gerischer's theory for semiconductor–electrolyte interfaces, the microscopic variation in the interfacial charge transfer rate (kET) at the catalyst dye interface was investigated. These models are described below in greater detail. To handle this non-equilibrium photocatalytic system electrochemically and to allow the system to be in the Langmuir–Hinshelwood (L–H) kinetics regime, the dye concentration was kept low, i.e., 0.596 × 10−3 M, along with a moderate catalyst load of 0.1 g L−1. This low dye concentration helped decrease the possibility of complete surface coverage and multilayer adsorption of the dye on the catalyst surface after the adsorption-desorption equilibrium. Thus, a reasonable approximation of this photocatalytic system to pseudo-first order kinetics is possible (Fig. 9b &S15, SI). Furthermore, this helped the weakening of the non-linearities in the photocatalytic activity of the catalyst due to the crowding effect.9 In addition, the setup with this configuration allows its proximity to the real cases in nature where mineral oxides, humic substances, or pollutants interact under sunlight.9 The result showed that after completion of 4.0 hours, the total degradation of the dye under the same conditions but with different photocatalysts was found to be clearly different. This was quantified as (%) degradation due to the photocatalytic activity (PA) of the catalysts (eqn (S5)) and is shown in Fig. S14 (SI). This showed the highest degradation, 86.1%, by FeAH, followed by 76.9% by FePM and 52.8% by FeMC, and close to 43.5% of FeCT systems under light conditions. Also, the lowest degradation of 29.3% was observed in the case of the FeBH system (Fig. S14, SI). However, 14.6% degradation of the dye under dark conditions was shown by the FePM catalyst, vis-à-vis 10.2% of degradation without any catalyst under the same conditions (Fig. S14, SI). This could be attributed to the residual absorption of light during the whole experiment or the dark catalysis. Nevertheless, 16.9% degradation without any catalyst under light conditions shows the fundamental role of light in such processes (Fig. S14, SI). This variation in degradation (%) after the same period of 4.0 h is clearly due to the different photocatalytic rates shown by the catalysts. Heterogeneous dye degradation at low concentration generally follows the Langmuir–Hinshelwood (L–H) kinetic model as described by eqn (12) (ref. 51):| | 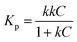 | (12) |
where ‘Kp’ is the rate constant for the overall photocatalytic dye degradation; ‘k’ is the surface reaction rate constant; ‘K’ is the adsorption equilibrium constant; and ‘C’ is the dye concentration. In the case of low concentrations of the dye, KC ≪ 1. eqn (12) simplifies into a pseudo-first-order reaction eqn (13) (ref. 51):| | 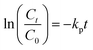 | (13) |
where ‘Kp’ is the apparent and overall rate constant of the degradation reaction, ‘C0’ is the initial concentration of the dye, and ‘Ct’ is the concentration of the dye at time ‘t’. The rate of degradation, kp, was calculated using eqn (13) for all catalytic systems. The curve fitting (R2 = 0.95–0.98) of dye concentration (lnC/C0) vs. time (Fig. 10b) suggested a pseudo-first-order rate constant ‘Kp’ (min−1) with its value in the range of 0.0139 to 0.0338 min−1. This agrees with the described (L–H) kinetic model in eqn (12). However, the apparent variation in Kp led to a significant difference in the degradation percentage by different catalytic systems after 4.0 hours. As a matter of fact, for steady state illumination with moderate to high light intensity, the rate is independent of light intensity. Also, in pseudo-first-order kinetics, the rate constant ‘Kp’ is not an actual rate constant. Thus, this difference in ‘Kp’ is more complex and may stem from the various sequential stages in photocatalysis, particularly the rate-determining step. Since the significant key steps in photocatalysis are the light absorption, generation of charge carriers (e−/h+), and the charge transfer (Fig. 8) at the catalyst–dye interface, any of these can be the rate-determining step.52 Thus, factors like efficiency of the photon absorption coefficient (α), efficiency of charge (h+/e−) carrier dynamics, and interfacial charge transfer barrier can affect the rate constant (Kp) accordingly. As a matter of fact, in most cases, the interfacial charge transfer step is rate-determining (k) in such processes.8,51 Thus, the factors affecting the interfacial charge transfer rate constant (kET) should correlate with the overall rate constant (Kp). Interestingly, in this experiment, the observed Kp appears to be intricately related to the morphology and interface structure of the catalyst, which in turn is related to the interfacial charge transfer barriers.9,48 Here, it has been observed that the morphologically similar sheet-like structures of the α-Fe2O3 catalyst systems FeMC (Kp = 0.0245 min−1) and FePM (Kp = 0.0338 min−1) have identical catalytic activity. This is different in terms of the catalytic rate constant (Kp) compared to the other set of morphologically similar sphere-like structures, i.e., FeAH (Kp = 0.0379 min−1) and FeBH (Kp = 0.0139 min−1) systems. However, as a deviation from this trend, the FeBH (sphere-like) system shows the lowest photocatalytic activity. This underlines the inevitable role of the lattice and interface structure beyond morphology.9,24 Thus, such comparisons must factor in other parameters like lattice and interface descriptors. Also, the highest activity of FeAH cannot be explained due to the possible effect of shape anisotropy alone – this requires deeper investigations.52 We utilised the role of interfacial parameters like electrochemically active surface area (ESCA), electrochemical work function (ø), resistance to interfacial charge transfer (Rct), and the flat band potential (Efb) for structure–activity relationship.31,53,54 Moreover, the interplay of several factors with a complex rate law for photocatalytic activity makes its rate often elusive to many degrees. Thus, a proper explanation for this variation in rate requires a macroscopic (bulk) yet comprehensive rate law vis-à-vis its correlation with the microscopic detail of the factors affecting the overall rate.52 Based on this, a comprehensive rate law that factors in all the geometric, electronic, and surface electrical properties of the interface can be written as| | 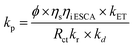 | (14) |
where ‘kp’ is the overall photocatalytic rate constant, ‘ϕ’ is the photon absorption efficiency, ηs is the charge separation efficiency, ‘ηi’ is the charge injection efficiency, Rct is the resistance to charge transfer, kr is the rate of recombination, ‘kd’ is the surface desorption rate, and ‘kET’ is the rate of interfacial charge transfer dealt with using Marcus–Gerischer theory. On expanding eqn (14) by substituting kET with the Marcus term and ‘Kr’ and ‘Kd’ with recombination and Langmuir and Hinshelwood (L–H) terms, respectively, further multiplied with the band bending term f(Δϕ), we get eqn (15):| | 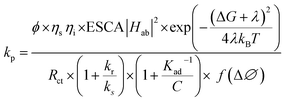 | (15) |
 |
| | Fig. 7 (a–f) UV-visible absorption spectrum of the dye–catalyst suspension under direct tropical sunlight exposure showing a continuous decrease in the absorption coefficient (ε) at the λmax (540 nm); (a) control experiment (dye–catalyst system in the dark); (b) FeBH catalyst system; (c) FeMC; (d) FeAH system. (e) FePM catalyst system; (f) FeCT catalyst system. | |
 |
| | Fig. 8 Proposed simplistic view of shape anisotropy and surface electrical property led modulation of the energy gap (ΔE), standard reduction potential (E0), and band bending (Efb) at the nanoparticle and dye interface – affecting the electrochemical work function Φ0ad, charge transfer barriers (Rct), driving force (ΔGo), and dynamics (kET) during sunlight driven charge transfer. | |
Since the Marcus equation is given as eqn (16),
| | 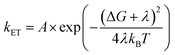 | (16) |
Also, A can be written as eqn (17):
| | 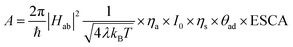 | (17) |
where pre-exponential factor A includes all the surface, geometrical and electronic factors, and the squared absolute value of ‘
H’ describes the electronic coupling of the energy levels (electronic states) during electron transfer, which has its quantum mechanical origin. Moreover, the fundamental Marcus equation is for homogeneous outer sphere mechanism (OSM) based
e-transfer reactions.
8 The modified form of the Marcus equation for semiconductor–electrolyte interfaces (OSM-type charge transfer),
i.e., the Marcus–Gerischer equation, is given as below
eqn (18):
| | 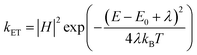 | (18) |
where
E − E0 is the energy gap between the catalyst and receptor. Taking this equation into account, an integrated rate law equation can be written as
eqn (19):
| | 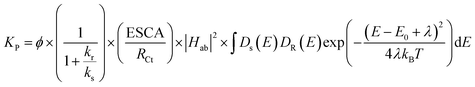 | (19) |
where
ϕ is dependent on light intensity, absorption coefficient, and particle size,
kr: recombination rate constant,
ks: charge separation rate, and
E −
E0 is the energy of the redox couple in solution (relative to vacuum). Band edge positions (
Efb) are embedded in the
Ds (
E) shift at the solution interface. Also, considering the complexity and importance of the interfacial charge transfer step in the photocatalytic process in heterogeneous photocatalytic systems, Ye
et al. (2024)
8 summarized the current relevant framework and photophysical process with a future need and possible treatments of the problem. It highlights the phenomenological success of Butler–Vilmer theory for the metal electrolyte interface with electrical potential as bias, yet recognizes its unsuitability to deal with the semiconductor electrolyte interface with light as bias.
8
3.4 Interfacial charge transfer and Marcus–Gerischer theory
Various authors have previously studied photocatalysis using an electro-analytical approach. However, it was generally limited to TiO2 systems, and the kinetics were studied in macroscopic terms.55–57 For instance, Baram et al. (2010)55 studied mesoporous TiO2 for photocatalytic degradation of methylene blue using EIS. They showed from the Mott–Schottky analysis and polarization curves that a high surface area of tubular TiO2 does not translate into good photocatalytic activity. Our previous study also reported a similar result for ZnO sheets and Rhodamine dye.9 Likewise, Baumanis et al. (2008)56 studied TiO2 thin film electrodes using Mott–Schottky measurements for photocatalytic reduction and oxidation of methyl viologen and methylene blue, respectively. They reported a correlation of photocatalytic activity with flat donor density and flat band potential.56 In this study, we also observed, using electrochemical measurements, a promising correlation between the shape anisotropy and the photocatalytic rate constant (Kp). The anisotropy-dependent catalyst surface electrical properties effectively influenced the interface structure and function in terms of the charge (e−/h+) transfer barrier. For instance, the sphere-like α-Fe2O3 (FeAH) nanocatalyst (∼38 nm) with high electrochemically active surface area (ECSA = 6.2), low resistance to charge transfer (Rct), and a low electrochemical work function (Φ0ad = 4.44) outperforms (‘k’ = 0.0379 min−1) the other morphologically different (sheet-like) α-Fe2O3 (FePM, FeCT, and FeMC) nanocatalysts (Fig. 9a &Table 2). Furthermore, it also outperformed a morphologically similar (sphere-like) catalyst (FeBH = 0.139 min−1) with the highest electrochemical active surface area (ECSA = 9.2). In general, sheet-like morphologies are considered relatively more suitable for surface charge transfer-based catalysis. Also, the high ESCA value and low Rct correlate with the high photocatalytic rate constant as observed in this study and were also reported in our previous work. However, the deviation in the photocatalytic rate constant (k = 0.0139 min−1) from the predicted ECSA value in the case of FeBH can be attributed to the importance of other electronic and surface factors (Fig. 9a &Table 2). Moreover, the measured negative flat band potential (Efb) in the case of FeAH appears to be a reasonable descriptor for photocatalytic activity (Fig. 10a). However, the comprehensive explanation for the correlation among structural, electronic and surface properties with the observed photocatalytic rate constant ‘Kp’ lies in the overall process and the complete mechanism of photocatalysis itself. Fundamentally, as described above, heterogeneous photocatalysis is a surface-based light (hv) driven interfacial charge transfer (h+/e−) process and thus fairly obeys the laws of thermodynamics. However, it has been found to fall in a mixed regime of surface reaction-controlled (macroscopic) & charge transfer-controlled (microscopic) kinetics. We can explain this by broadly separating the whole process into multiple sequential and parallel phases. As described above, these are, namely, light absorption and charge carrier (h+/e−) generation, charge migration, and charge transfer at the solid–liquid interface, followed by surface reactions. Generally, as described above in eqn (12) and (13), at low dye concentrations and moderate to high light intensity, the overall dye degradation follows pseudo-first-order kinetics (Fig. 10b) as a simplified Langmuir–Hinshelwood model (KC ≫ 1). This is why the photocatalytic rate (macroscopic) often correlates with the Brunauer–Emmett–Teller (BET) surface area. Also, the charge carrier dynamics (recombination) are often described by second-order or trap-assisted Shockley–Read–Hall models. However, interfacial charge transfer is best described by the extended Marcus-type kinetics described by the Marcus–Gerischer theory. Considering all these factors together, a simplified overall rate (eqn (14)) for photocatalytic dye degradation can involve descriptors associated with macroscopic rate constant Kp (adsorption and surface reactions) and microscopic rate constant kET (interfacial charge transfer). The latter will include the electrochemical descriptors related to the structure of the interface at the solid–liquid dye junction (Fig. 8). In particular, the flat-band potential (Efb) may parametrize the geometry and surface functional group-led structural variation in all five catalyst systems and can give the energy difference between the donor and acceptor (E − E0). The electrochemically active surface area (ECSA) may factor in the surface density of the states in the given catalyst. Similarly, the measured frequency-dependent (Rct), i.e., resistance to charge transfer through the solution phase, can describe the solvent reorganization energies (λ). Also, the electrochemical work function (ϕ) may describe the electronic coupling in the donor and receptor energy states. Thus, combining all these factors, a general rate law (eqn (15)) for charge transfer at the interface can give the variation in the microscopic rate constant (kET). However, as per eqn (15), this requires a foregoing correlation between shape anisotropy and the electronic structure of the catalyst. Since anisotropy refers to the direction-dependent variation in properties, such as conductivity, surface energy, and defect distribution, which becomes even more significant in nanostructured or oriented materials.58 Also, the surface conduction band edge ECB(S) is modified by well-known band bending (Fig. 10a) of semiconductors at the electrolyte interface as described by eqn (20):where ECB(S) is the conduction band edge at the semiconductor (α-Fe2O3) surface and Δϕbb is the band bending due to the aqueous dye solution contact. This modifies the energy gap (E − E0) between the donor (E) and acceptor (E0), and this further modifies the driving force ΔG (Fig. 11) for the charge transfer as per eqn (21):| | | ΔG = −e(ECB,surf − Eacceptor) = −e(EFb − Δϕbb − Eacceptor) | (21) |
where ECB,surf =E(donor) and Acceptor (E0). This band-bending-led variation in the driving force of the reaction (ΔG) leads to different charge transfer rate constants (kET), thereby affecting the photochemical behaviour of these systems (Fig. 11).59 The substitution of ΔG in eqn (18) can be further rationalized by the fact that different crystal surface planes exhibit distinct band structures, leading to varying degrees of valence band (VB) and conduction band (CB) bending. For instance, Lei et al. via STEM profile-view imaging and electronic structure analysis revealed that the photodegradation activity of CeO2 is surface structure-sensitive following the order {110} ≫ {100}.60 Similarly, Meng et al. showed that facet engineering in cubic In2O3 (001) controls its electron band structure and thus its PEC activity.61 Furthermore, it is also established that SrTiO3 {100} surfaces are highly active as photons execute many more transitions along the (100) directions than in the other directions due to the weakly dispersive bands along this direction.62 Here, in this case, shape anisotropy in semiconductors, hematite (α-Fe2O3), plays a crucial role in modulating their photocatalytic behavior, by modulating the flat band potential (EFb), potential of zero charge (EPZC), and band bending (Fig. 10a and Table 3).63 The possible distinct crystallographic facets exposed by rod-like, sheet-like, and sphere-shaped hematite (α-Fe2O3) particles exhibit varied surface energies, resulting in facet-specific band edge positions and work functions (Fig. 10a and Table 3). These variations govern the extent of band bending at the semiconductor/electrolyte interface (Fig. 10a), influencing charge separation and interfacial redox kinetics (Fig. 10b and 11). For instance, a previous study showed that the different crystallographic planes, such as (110) and (012), of α-Fe2O3, exhibit varying surface state densities and carrier mobility, leading to facet-dependent shifts in flat band potential.64 Also, a study on Fe2O3 nanorods decorated with a thin ZnO layer observed a negative change of approximately 20 mV in the EFb, attributed to forming a heterojunction that facilitates upward band bending and enhances charge separation efficiency.65 These shifts critically influence charge separation efficiency and the ability of photogenerated electrons and holes to participate in redox reactions (Fig. 8). Moreover, anisotropy also alters the distribution of surface states, which affects the EFb by shifting the balance between surface donor and acceptor-like states. For instance, α-Fe2O3 nanocubes, exposing {012} and {104} facets, were found to facilitate the IO3− reduction, while α-Fe2O3 octahedra, exposing {101} and {111} facets, on the other hand, are found to be very inactive, owing to their crystallographic orientations.59 Such changes also lead to non-uniform band bending across different facets, impacting the space-charge layer formation and ultimately controlling photovoltage generation and charge transfer kinetics. Furthermore, internal electric fields arising from facet-dependent space-charge distributions enhance directional charge transport, as demonstrated for TiO2 single crystals.66 Also, in highly confined anisotropic systems, quantum size effects further modify the density of states near the band edges, thereby altering optical and catalytic properties. These phenomena provide a fundamental basis for the observed correlation between shape anisotropy and photocatalytic efficiency. Furthermore, the observed differences in surface redox potentials (E0), differential capacitive behaviour (Cdl), charge transfer resistance (Rct), and flat band potential (Efb) of this interface can be attributed to the anisotropy or surface-based electrical properties (Tables 2, 3, Fig. S16a–f, S17a–e and S19, SI). Also, the significant variation in the electrochemically active surface area (ECSA), electrochemical work function (Φ0ad), potential of zero charge (Epzc) is functionally correlated with the differences in photochemical activity (Fig. 10b and Table 2). Moreover, to separate the effect of ligand-induced surface electrical properties from anisotropy, further investigations are needed. Also, the ECSA calculations may overestimate active sites if redox processes interfere with Cdl measurements, which may oversimplify the case. To validate the same, Mott–Schottky (MS) analysis was done using EIS at 1000 Hz within the potential range of 1 to −1 V vs. RHE (Fig. 11a). The slope of MS plots suggests that all the samples are n-type semiconductors.67 It is observed that the negative values of VFb follow the order FeAH > FePM > FeBH > FeMC > FeCT, indicating the reduction ability of the surfaces, while from the slope, the donor density was found to be the highest for FeBH, followed by the FeAH system.67 Moreover, to validate the dominant contribution from the capacitive nature and the least impact of the faradaic contributions, the MS of the best FeAH catalyst was investigated at multiple frequencies (Fig. S20, SI), which shows that there is no change in the slope and VFb of the system as a function of frequency.67 This independence of the MS plots over a wide range of frequencies indicates capacitive dominance (Fig. S20, SI).67 However, the highest upward band bending of the FeAH system allows better charge separation and shorter carrier diffusion length (τd), enabling it to outperform other catalysts, including FeBH, with higher ESCA (Fig. 10b). Nevertheless, band bending does not linearly translate into photocatalytic activity in the case of FeBH, highlighting the role of other factors (Fig. 10a and b). Furthermore, the observed variation in surface redox behaviour of the catalysts confirms the differences in the charge transfer efficiency at the catalyst surface (Fig. S17a–e, SI). For instance, in the anodic (0.3 to 2.5 vs. RHE) scans (Fig. S17, SI) of all the interface systems, two redox peaks were observed at ca. 0.45 V and 0.9 V vs. RHE, which are attributed to Fe+/Fe2+ and Fe2+/Fe3+ redox species. Interestingly, the peak current (jp,a) for Fe2+/Fe3+ transformation for the FeAH system is higher than that for the other samples, which agrees with its high Kp value (Fig. 10b). Interestingly, the redox peaks in FeCT (Fig. S17c, SI) are absent in this potential window, probably due to surface modification. Besides this, the reduction peak corresponding to Fe3+/Fe2+ is much more dominant in the FeAH sample (Fig. S17a, SI). This showed the catalyst systems' clear electronic and surface modification due to synthetic conditions altering the energy barrier for the redox processes at the interface. Likewise, a similar correlation is observed in the other cases, excluding FeBH, possibly due to the other electronic factors. However, a weak redox peak at higher potential (1.7–2.1 V) may be associated with some higher oxidation states of iron, since there are no such oxidation states other than Fe3+ and Fe2+ that are visible in the XPS spectrum (Fig. S18, SI). Also, an examination of the capacitive region (0.0 to 0.9 vs. RHE) of the catalyst systems (FeCT, FeMC, and FePM) shows almost similar CV responses and current densities, which are much higher than those of the rest of the samples (Fig. S16b–e). Furthermore, using the slope jcapvs. ϑ and Cdl, the corresponding ECSA values were found to be in the order of FeBH > FeAH > FeMC > FePM > FeCT, as shown in Fig. S16a–f and mentioned in Table 2. Remarkably, the ESCA values of the FeAH, FeMC, FePM, and FeCT catalyst systems also align closely with the order of the Kp.9 This highlights the functional role of ESCA over the total geometrical area (BET isotherm) for surface adsorption. A similar observation was also reported in our previous study of ZnO sheets.9 However,
the higher current density and the highest ECSA value for FeBH do not agree with its observed Kp. This disagreement in the case of FeBH highlights the role of band alignment as observed from Efb. Also, from the Nyquist plots (Fig. 9a), a close observation suggests that the lower Rct values are associated with a higher Kp, reflecting the ease of charge (e−) transfer with the low Rct.9,57 Moreover, the significant deviation from this in the case of FeBH can be ascribed to its unfavourable band alignment (Efb) for charge transfer. Furthermore, from Fig. 9b, presenting the Bode plot, it can be observed that all the samples other than FeBH in the high-frequency region behave almost similarly (capacitive) with the phase angle in the 80–90° range. This also indicates a similar and better lifetime τ = 1/2πfc of charge carriers, which promotes good charge dynamics. However, FeBH shows that it acts as a resistor (phase angle zero) and involves only charge transfer in the high-frequency domain, which agrees with its observed Kp. Moreover, in a low-frequency region, the phase angle is minimum for the FeAH interface, showing charge leakage and explaining its higher Kp. The clear antagonism of FeAH and FeBH in Bode's plot explains their inverse photocatalytic activity (Fig. 9b). Nonetheless, to gain further insights into the surface electrical behaviour of the FeX interface (X= AH, BH, CT, PM, and MC), the potential of zero charge was estimated from the differential capacitance plots (Fig. S16a–d, SI). The potential of zero charge EPZC is the applied potential at which the net charge on the surface is almost zero (σsurface = 0 at E = EPZC) and the double-layer capacitance is minimum.68 Thus, it helps understand the catalyst and adsorbate interaction in the solution and the possibility of band bending. Also, EPZC plays a central role in catalytic performance, as it defines the degree of polarizability and Φ0ad of a system. Systems with low EPZC are more susceptible to anodic polarization, while lower electrochemical work functions favour more feasible electron transfer across the interface.69 From Fig. S19a–d (SI), all the samples show positive EPZC, indicating that surfaces are negatively charged, as inferred by the zeta potential measurements as well, and the EPZC values follow the order FeCT > FePM ≫ FeMC > FeAH > FeBH. The correlation between the EPZC and Φ0ad is given by eqn (22) and (23):70–73| | | EPZC = Φ0ad + EReference | (22) |
where Φ0ad represents the electrochemical work function, EPZC is the potential of zero charge and EReference is the potential at the reference electrode, which is assumed to be −3.44 V vs. RHE.74
 |
| | Fig. 9 (a and b) Nyquist (a) and Bode plots (b) obtained at OCP for all the samples, (a) showing the differences in resistance to charge transfer (Rct) at various catalyst dye-interfaces, due to the shape anisotropy and surface-based differences at the catalyst-dye interface. (b) The changes in the phase angle (θ) over different frequency ranges (region-I/II/III: low, medium and high), indicating the variation in the dominance of the charge transfer, capacitive, and resistive processes at the various catalyst (FeAH-FePM) based interfaces. | |
Table 2 Interfacial parameters (calculated from cyclic voltammetry measurements) and the observed rate constants (Kp) for the various catalyst systems
| Catalyst |
C
dl (μF cm−2) |
ECSA |
R
f (cm−2) |
Rate constant (Kp) |
| FeBH |
374.3 |
9.4 |
300 |
0.0139 min−1 |
| FeAH |
248.4 |
6.2 |
197 |
0.0379 min−1 |
| FeCT |
45.8 |
1.2 |
38 |
0.0211 min−1 |
| FeMC |
91.6 |
2.1 |
73 |
0.0245 min−1 |
| FePM |
73.1 |
1.8 |
57 |
0.0338 min−1 |
 |
| | Fig. 10 (a and b) Mott–Schottky plots (a) for all samples, obtained at 1000 Hz frequency in the potential range of −1 to 1 V vs. RHE, showing the estimated variation in the flat band potential (Efb) and donor density in the samples. (b) LnC/C0vs. t showing the variation in the photocatalytic rate constant Kp and its correlation with the trend in the variation of flat band potential (Efb). | |
 |
| | Fig. 11 The proposed shift in Marcus potential curves and their modification due to different extents of band bending (Efb) affecting charge transfer driving force (ΔGo) and reorganisation energy (λ) leading to different charge transfer rates (kET) at the semiconductor-based catalyst (α-Fe2O3) dye (electrolyte) interface. | |
Table 3 The electrochemical work function, Φ0ad (eV), and potential of zero charges, EPZC, estimated from differential capacitive plots (Fig. S16a–f), along with the observed rate constants (Kp) for the various catalyst systems
| Catalyst |
E
PZC (V) vs. RHE |
Φ
0ad (eV) |
Rate constant (Kp) |
| FeBH |
0.71 |
4.15 |
0.0139 min−1 |
| FeAH |
1.09 |
4.44 |
0.0379 min−1 |
| FeCT |
1.34 |
4.78 |
0.0211 min−1 |
| FeMC |
1.03 |
4.47 |
0.0245 min−1 |
| FePM |
1.16 |
4.60 |
0.0338 min−1 |
The solvent-modified work function (Φ = Evac − EF) quantifies the energy penalty for electron shuttling across the catalyst surface–dye interface. The Φ0ad was calculated from eqn (12) (Table 3), and it was found that FeAH shows the second lowest Φ0ad after FeBH, indicating high solution-state polarizability of the catalyst, which also agrees closely with the observed Kp value for the FeAH system. Noticeably, this lowering of Φ0ad could be attributed to the influence of the ligands on the Fe surface. Moreover, this significant agreement between these electrochemical descriptors, particularly the ESCA values and macroscopic (bulk) photocatalytic rate constant (Kp), highlights the roles of the number of active sites in achieving higher rate constants. Furthermore, combining this with band alignment due to Efb also suggests the interfacial charge transfer step to be the rate-determining step for at least the charge transfer process (kET). Furthermore, the alignment of the energy levels (E − E0) of donors and acceptors correlates with the macroscopic rate constant (Kp), suggesting this to be a rate-determining step in the whole process. In fact, the factors that affect charger transfer at the interface affect the macroscopic rate constant (Kp). This indicates that the process is essentially controlled by the interfacial charge transfer (Fig. 11). Since heterogeneous photocatalytic processes are inherently non-equilibrium and adiabatic charge transfer processes, they can be approximated to an OSM-based charge transfer process at the hematite dye interface. Thus, this can be rationalised by using the Marcus–Gerischer electron transfer theory. However, to combine the macroscopic rate constant (Kp) from the Langmuir–Hinshelwood (L–H) model with the microscopic charge transfer rate constant (kET), a unified form can be presented from eqn (14) and (15), where pre-exponential factors can be regarded as representing everything other than the exponential Marcus term. If non-interfacial (surface adsorption, light absorption, and charge recombination dynamics) factors are taken as a constant pre-exponential (A) term in all the cases, the band alignment due to band bending (Efb) directly relates to driving force ΔG° (eqn (21)) of the charge transfer as given in the Marcus term (Fig. 11). It is observed that a catalyst system like FeAH with a high ESCA value and favourable (Efb) can show a faster rate for charge transfer (kET), which translates further into corresponding higher Kp values. Likewise, FeBH, having the highest ESCA value but unfavourable (Efb), shows the lowest driving force for the microscopic rate constant (kET), thus the lowest overall macroscopic rate constant (Kp) for photocatalytic degradation. This is likely due to the domination of the pre-exponential factor in the case of FeBH. In contrast, the domination of Marcus' term in the case of FeAH may shift the rate-determining step from charge transfer in the case of FeAH to charge separation in the case of FeBH (Fig. 11). This example highlights the role of interfacial charge transfer as well as preceding steps in the photocatalytic process. Moreover, various authors have reported similar effects of interface structures (energy level Ef − Ea alignment, charge transfer distance, and surface electrostatics) on the interfacial charge transfer rate.13–15,75 For instance, Ding et al. 2015 (ref. 14) studied photoexcited CdSe–CdS core–shell quantum dots (QDs) covalently linked with alkyl thiol substituted ferrocene and reported the decrease of the hole transfer rate (kht) with an increase in CdS shell thickness and alkyl length (β) in the framework of Marcus theory.14 The same group of scientists, in another study, Tarafder et al. 2014,15 reported the dependence of the rate (Marcus inverted region) of hole (h+) transfer on electronic coupling factor |H| and driving force of the reaction in a CdSe/CdS quantum rod (QR) tethered ferrocene system.15 They, in another consecutive investigation, showed the relation between driving force of charge transfer and its rate using a modulation of energy gap (Edonor − Eacceptor) between the donor (CdSe/CdS, QDs) and acceptor (alkyl thiol linked substituted ferrocene), in the inverted region of Marcus theory.13 However, the present case, α-Fe2O3/Rh6G dye, though resembling such cases, lacks rigorous empirical evidence and thus should be further investigated in greater detail. Moreover, in general, it can be concluded from the electrochemical investigations that the FeAH system, a sphere-like α-Fe2O3 structure with the highest ESCA (6.2), low Rct, and low work function (Φ0ad = 4.4) with less positive band bending (Efb) and better charge dynamics (LD & τ), favours the best charge transfer (kET) and, hence, the highest photocatalytic rate constant (Kp). However, despite the highest (ESCA) value of the FeBH system, the lowest Kp underlines the role of band alignment (Efb) and other electronic and optical parameters in a photocatalyst design.
4 Conclusion
In summary, this study provides a deeper understanding of the shape anisotropy evolution in iron oxide nanocrystals during the synthesis of hematite (α-Fe2O3) nanoparticles from iron(II) sulfate (Fe2SO4·7H2O) via a sol–gel route in various ligand environments. The varying ligand conditions included CTAB, a cationic surfactant, methyl cellulose, a non-ionic polymer, a biogenic ligand (PM) (poly functional groups), and ligand-free conditions. This demonstrates the intricate interplay of classical and non-classical crystallization pathways. The observed shift from spherical to sheet- and rod-like structures highlights the role of molecular identities of ligands in directing nucleation and crystal growth. The experiment has shown that the similarities in molecular identity of MC and PM have led to the sheet-like morphologies of the nanoparticles of similar size (∼35–45 nm) in the cases of FePM and FeMC. This study suggests the possible replacement of commercial surfactants with suitable biogenic sources without adversely affecting their activity. Also, this method can be used in sustainable materials design and catalysis. Furthermore, the use of an electroanalytical approach to deal with the photocatalytic (α-Fe2O3/Rh6G) interface revealed that the variations in photocatalytic efficiency (rate constant Kp) are generally due to the differential rate of interfacial charge transfer (kET). This can be described by combining the Marcus–Gerischer electron transfer theory with the Langmuir–Hinselwood kinetic model, with the help of electrochemical descriptors, particularly Efb, along with ESCA, EPZC, and Φ0ad. The plausible correlation among shape anisotropy, interface structure, and the charge transfer kinetics underlines its role in efficient catalyst design. Rather than merely relying on conventional approaches of band gap engineering and charge carrier dynamics, these electrochemical descriptors appear to be more significant for efficient catalyst design. For instance, the best catalyst in this case has shown high ESCA, low Rct & low Φ0ad vis-à-vis a favourable band alignment (EFb). Also, the electrochemical approach offers a more direct and quantitative means for evaluating interfacial charge transfer, adsorption characteristics, and catalytic activity than conventional optical or spectroscopic techniques. By capturing real-time changes in electronic states and surface reactivity, this method may provide an effective tool to correlate the characteristics of the catalyst with its performance, offering an improved approach for structure–activity relationship studies. Furthermore, despite many differences, the overall research highlights the unique and fundamental similarity in electrochemical and photochemical interfaces due to the key and common step of interfacial charge transfer in both processes. Moreover, beyond its immediate implications for photocatalysis, this work offers valuable insights into the dynamic aspects of nanoscale interfaces, particularly the semiconductor–aqueous interface and similar interfaces in geochemical environments. By establishing a direct link between shape anisotropy, interfacial electrochemical properties, and catalytic performance, our findings contribute to the broader understanding of photocatalysis, which is crucial in fields ranging from environmental remediation to sustainable energy conversion. Additionally, the study underscores the potential of biogenic ligands in tailoring nanostructures, opening new avenues for green synthesis approaches in materials science. These insights pave the way for future research into the rational design of functional nanomaterials, particularly for applications in energy harvesting, catalysis, and environmental sustainability.
Author contributions
All the work, including ideation, experimentation, analysis, and writing, was done by author Dr Lahur Mani Verma. Aejaz Ul Basir helped during electrochemical experiments and analysis, and Prof. Pravin P. Ingloe and Prof. Satyawati Sharma supervised the work.
Conflicts of interest
There are no conflicts to declare.
Data availability
All the experimental data related to the article are provided in the manuscript and SI, and the same can be provided if required at any time.
The supplementary information (SI) includes details of synthesis protocols, elctrochemical and photoctalytic investigations, additional structural and surface characterization (XRD patterns, SEM images, FTIR spectra), SEM-EDX and NTA analysis, EPR spetrum, UV-DRS spectra, electrochemical data (CV, pZC), theoretical calculations, and supporting figures/tables referenced in the article. See DOI: https://doi.org/10.1039/d5ta02013a.
Acknowledgements
The authors are thankful to the University Grant Commission (UGC) and the Indian Institute of Technology Delhi (CRF and NRF) for supporting this research. AUB is thankful to the CSIR-HRDG for the fellowship. The authors are also grateful to Professor Jai Deo Singh, Department of Chemistry, IIT Delhi, for his constant and sincere motivation.
References
- N. Arif, M. N. Zafar, M. Batool, M. Humayun, M. A. Iqbal, M. Younis, L. Li, K. Li and Y. J. Zeng, J. Mater. Chem. C, 2024, 12, 12653–12691 RSC.
- D. S. Chaudhari, R. P. Upadhyay, G. Y. Shinde, M. B. Gawande, J. Filip, R. S. Varma and R. Zbořil, Green Chem., 2024, 26, 7579–7655 RSC.
- H. J. Cleaves, A. M. Scott, F. C. Hill, J. Leszczynski, N. Sahai and R. Hazen, Chem. Soc. Rev., 2012, 41, 5502–5525 RSC.
- F. T. Geldasa, M. A. Kebede, M. W. Shura and F. G. Hone, RSC Adv., 2023, 13, 18404–18442 RSC.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119(8), 4819–4880 CrossRef CAS PubMed.
- V. Singh, S. Patra, N. A. Murugan, D. C. Toncu and A. Tiwari, Mater. Adv., 2022, 3, 4069–4087 RSC.
- C. Ye, D. S. Zhang, B. Chen, C. H. Tung and L. Z. Wu, ACS Cent. Sci., 2024, 10(3), 529–542 CrossRef CAS PubMed.
- L. M. Verma, A. Kumar, A. U. Bashir, U. Gangwar, P. Ingole and S. Sharma, Nanoscale Adv., 2024, 6, 155–169 RSC.
- L. M. Verma, A. Kumar, A. Kumar, G. Singh, U. Singh, S. Chaudhary, S. Kumar, A. R. Sanwaria, P. P. Ingole and S. Sharma, Sci. Rep., 2024, 14, 4074 CrossRef CAS.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54(1), 57–93 CrossRef CAS.
- J. H. Olshansky, T. X. Ding, Y. V. Lee, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 15567–15575 CrossRef CAS PubMed.
- T. X. Ding, J. H. Olshansky, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 2021–2029 CrossRef CAS PubMed.
- K. Tarafder, Y. Surendranath, J. H. Olshansky, A. P. Alivisatos and L. W. Wang, J. Am. Chem. Soc., 2014, 136, 5121–5131 CrossRef CAS PubMed.
- B. K. Jha, S. Chaule and J. H. Jang, Mater. Chem. Front., 2024, 8, 2197–2226 RSC.
- J. López-Sánchez, A. Serrano, A. Del Campo, M. Abuín, O. Rodríguez De La Fuente and N. Carmona, Chem. Mater., 2016, 28, 511–518 CrossRef.
- F. S. Yen, W. C. Chen, J. M. Yang and C. T. Hong, Nano Lett., 2002, 2, 245–252 CrossRef CAS.
- F. N. Sayed and V. Polshettiwar, Sci. Rep., 2015, 5, 9733 CrossRef CAS PubMed.
- M. Andres, A. Mifsud and C. J. Serna, J. Chem. Soc., Faraday Trans., 1990, 86, 959–963 RSC.
- M. Andrés-Vergés and C. J. Serna, J. Mater. Sci. Lett., 1988, 7, 970–972 CrossRef.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13(7), 7359–7365 CrossRef CAS.
- C. Cheng, F. Xu and H. Gu, New J. Chem., 2011, 35, 1072–1079 RSC.
- S. Meneceur, H. Hemmami, A. Bouafia, S. E. Laouini, M. L. Tedjani, D. Berra and M. S. Mahboub, Biomass Convers. Biorefin., 2024, 14, 5357–5372 CrossRef CAS.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CrossRef CAS.
- L. MacHala, J. Tuček and R. Zbořil, Chem. Mater., 2011, 23(14), 3255–3272 CrossRef CAS.
- J. Scheck, B. Wu, M. Drechsler, R. Rosenberg, A. E. S. Van Driessche, T. M. Stawski and D. Gebauer, J. Phys. Chem. Lett., 2016, 7, 3123–3130 CrossRef CAS.
- Z. Zhou, X. Zhu, D. Wu, Q. Chen, D. Huang, C. Sun, J. Xin, K. Ni and J. Gao, Chem. Mater., 2015, 27, 3505–3515 CrossRef CAS.
- D. Maiti, U. Manju, S. Velaga and P. S. Devi, Cryst. Growth Des., 2013, 13, 3637–3644 CrossRef CAS.
- J. Cheon, N. J. Kang, S. M. Lee, J. H. Lee, J. H. Yoon and S. J. Oh, J. Am. Chem. Soc., 2004, 126, 1950–1951 CrossRef CAS.
- M. Alagiri and S. B. A. Hamid, J. Sol-Gel Sci. Technol., 2015, 74, 783–789 CrossRef CAS.
- P. K. Narnaware and C. Ravikumar, J. Phys. Chem. C, 2020, 124, 25010–25027 CrossRef CAS.
- A. Rajendran, M. Alsawalha and T. Alomayri, J. Saudi Chem. Soc., 2021, 25, 101307 CrossRef CAS.
- L. MacHala, J. Tuček and R. Zbořil, Chem. Mater., 2011, 23(14), 3255–3272 CrossRef CAS.
- K. Dyrek and M. Che, Chem. Rev., 1997, 97(1), 305–332 CrossRef CAS.
- M. P. Herring, L. Khachatryan and B. Dellinger, World Acad. Sci. Eng. Technol., 2015, 9(7), 804–812 Search PubMed.
- H. Wan, L. Hu, X. Liu, Y. Zhang, G. Chen, N. Zhang and R. Ma, Chem. Sci., 2023, 14, 2776–2798 RSC.
- J. Zhang and S. Eslava, Sustainable Energy Fuels, 2019, 3, 1351–1364 RSC.
- I. U. Foreman-Ortiz, T. Fung Ma, B. M. Hoover, M. Wu, C. J. Murphy, R. M. Murphy and J. A. Pedersen, J. Colloid Interface Sci., 2022, 615, 50–58 CrossRef CAS PubMed.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- R. Demichelis, P. Raiteri, J. D. Gale, D. Quigley and D. Gebauer, Nat. Commun., 2011, 2, 590 Search PubMed.
- D. Gebauer, P. Raiteri, J. D. Gale and H. Cölfen, Am. J. Sci., 2018, 318, 969–988 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114(15), 7610–7630 CrossRef CAS PubMed.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- P. J. M. Smeets, A. R. Finney, W. J. E. M. Habraken, F. Nudelman, H. Friedrich, J. Laven, J. J. De Yoreo, P. M. Rodger and N. A. J. M. Sommerdijk, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7882–E7890 CrossRef CAS PubMed.
- D. Gebauer, Minerals, 2018, 8, 179 CrossRef.
- D. Gebauer, J. D. Gale and H. Cölfen, Small, 2022, 18(28), 2107735 CrossRef CAS.
- L. M. Verma, U. Singh, H. Arora, A. Kumar, V. Dhayal, P. P. Ingole, S. Sharma and A. R. Sanwaria, Mater. Chem. Phys., 2023, 306, 128050 CrossRef CAS.
- H. L. Tan, F. F. Abdi and Y. H. Ng, Chem. Soc. Rev., 2019, 48, 1255–1271 RSC.
- S. T. Kochuveedu, Y. H. Jang and D. H. Kim, Chem. Soc. Rev., 2013, 42, 8467–8493 RSC.
- H. D. Tran, D. Q., P. T. Do and U. N. P. Tran, RSC Adv., 2023, 13, 16915–16925 RSC.
- R. Yanagi, T. Zhao, D. Solanki, Z. Pan and S. Hu, ACS Energy Lett., 2022, 7(1), 432–452 CrossRef CAS.
- B. Ahmmad, K. Leonard, M. Shariful Islam, J. Kurawaki, M. Muruganandham, T. Ohkubo and Y. Kuroda, Adv. Powder Technol., 2013, 24, 160–167 CrossRef CAS.
- G. Kasivelu, T. Selvaraj, K. Malaichamy, D. Kathickeyan, D. Shkolnik and S. Chaturvedi, New J. Chem., 2020, 44, 11373–11383 RSC.
- N. Baram and Y. Ein-Eli, J. Phys. Chem. C, 2010, 114, 9781–9790 Search PubMed.
- C. Baumanis and D. W. Bahnemann, J. Phys. Chem. C, 2008, 112, 19097–19101 Search PubMed.
- W. H. Leng, Z. Zhang, J. Q. Zhang and C. N. Cao, J. Phys. Chem. B, 2005, 109, 15008–15023 CrossRef CAS.
- Y. Xia, Q. Chen and U. Banin, Chem. Rev., 2023, 123, 3325–3328 CrossRef CAS.
- M. H. Huang, G. Naresh and H. S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4–15 CrossRef CAS PubMed.
- W. Lei, T. Zhang, L. Gu, P. Liu, J. A. Rodriguez, G. Liu and M. Liu, ACS Catal., 2015, 5, 4385–4393 CrossRef CAS.
- M. Meng, X. Wu, X. Zhu, L. Yang, Z. Gan, X. Zhu, L. Liu and P. K. Chu, J. Phys. Chem. Lett., 2014, 5, 4298–4304 CrossRef CAS PubMed.
- C. Ricca and U. Aschauer, J. Chem. Phys., 2022, 156, 154703 CrossRef CAS PubMed.
- J. Sun, W. Xia, Q. Zheng, X. Zeng, X. Zeng, W. Liu, G. Liu and P. Wang, ACS Omega, 2020, 5, 12339–12345 CrossRef CAS PubMed.
- Q. Xiang, G. Chen and T. C. Lau, RSC Adv., 2015, 5, 52210–52216 RSC.
- Y. C. Liang and C. S. Hung, ACS Omega, 2020, 5, 16272–16283 CrossRef CAS PubMed.
- P. Mikrut, D. Mitoraj, R. Beranek and W. Macyk, Appl. Surf. Sci., 2021, 566, 150662 CrossRef CAS.
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 RSC.
- A. S. Shatla, M. Landstorfer and H. Baltruschat, ChemElectroChem, 2021, 8, 1817–1835 CrossRef CAS.
- T. Li, S. Ciampi and N. Darwish, ChemElectroChem, 2022, 9(17), e202200255 CrossRef CAS.
- A. N. Frumkin, J. Electroanal. Chem., 1965, 9, 173–183 CAS.
- S. Trasatti, J. Electroanal. Chem., 1971, 33, 351–378 CrossRef CAS.
- A. U. Bashir and P. P. Ingole, ACS Electrochem., 2025, 1, 1055–1065 CrossRef CAS.
- A. U. Bashir and P. P. Ingole, Small, 2025, 21, 2409457 CrossRef CAS PubMed.
- R. Kant, J. Kaur and G. K. Mishra, J. Phys. Chem. C, 2020, 124, 2273–2288 CrossRef CAS.
- R. Chen, D. Zhang, Z. Wang, D. Li, L. Zhang, X. Wang, F. Fan and C. Li, J. Am. Chem. Soc., 2023, 145, 4667–4674 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Aejaz Ul
Bashir
a,
Pravin P.
Ingole
ab,
Aejaz Ul
Bashir
a,
Pravin P.
Ingole














 is the rate of change of the number concentration of clusters (or particles) of size ‘i’ with respect to time, ΣβJknJnk represents the formation of clusters of size ‘i’ by binary collisions (or aggregations) of smaller clusters. Furthermore, nj and nk represent the number concentrations of clusters of sizes j and k, respectively. The term βjk represents the rate constant (or the collision kernel) for the aggregation of clusters of sizes j and k. The summation runs over all pairs such that j + k = ij + k = ij + k = i. So only those combinations that lead to size ‘i’ are considered. The second term niΣβijnj similarly indicates the loss of clusters of size ‘i’ as they collide with other clusters of size ‘j’ to form larger clusters.43 These dynamics bypass the traditional atom-by-atom growth of classical models, explaining the formation of highly oriented, anisotropic hematite structures. Moreover, the current discussion on solution phase classical vs. non-classical nucleation and growth suggests that additive-controlled processes are generally inclined to a non-classical path.42,46 However, a similar shape anisotropy in zinc oxide nanoparticles was also reported in our previous study (L. M. Verma et al. 2024).9 In summary, the shape and phase outcomes across ligand systems are rationalized by considering both thermodynamic and kinetic aspects, i.e., Ksp-defined supersaturation (S), ligand-induced modulation of γ, and the coexistence of classical nucleation with non-classical assembly routes. This framework explains how surface-active ligands steer iron oxide crystallization during sol–gel synthesis. However, beyond these differences in solution phase evolutionary pathways, the sintering of resynthesized samples (FeCT, FeMC, and FePM) at a fixed temperature (500 °C) in a muffle furnace showed the retention of the relative effect of the ligands as observed in this experiment. The resynthesized pre-annealed (vacuum oven drying at 150 °C) iron oxide samples analysed by XRD (Fig. 2a and b), FTIR (Fig. 3a and b), and TGA (Fig. S8, SI) confirm the definitive pre- and post-annealing effect of the ligands on the shape and morphology of the final iron oxide (α-Fe2O3) nanocrystals. Also, FESEM images of pre- and post-annealed (Fig. S4a and S5a–d, SI) samples show template morphologies of the samples developing into the corresponding final shape. For instance, the needle-shaped goethite (α-FeOOH) samples in the case of FeCT grow into rod-like morphologies after annealing. This suggests the template-based Ostwald ripening of the needle-shaped crystal – a precise mechanism for non-classical kinetic crystallization pathways. As the complete thermodynamic equilibration of shape in the case of α-Fe2O3 is reported at 700–900 °C, the result in this case is largely kinetically controlled.26 Furthermore, the previous work shows that most of such studies are concerned with completely defined cases, involving fine chemical surfactants as additives, yet still lack complete mechanistic understanding and control over nucleation and crystallization.43 Among the reported biogenic ligand-based approaches, using the extract of lignocellulosic waste (PM) presents a novel case of ligand-modulated size and morphology control. However, these findings align with the recent efforts in these directions. Nevertheless, during this study, it remained challenging to conclude the exact role of the PM extract in shaping and controlling the crystallisation process primarily due to the complexity of the degraded lignocellulosic substance (PM) based aqueous extract. However, given its importance in sustainable catalysis and relevance to the mineralisation processes in the natural environment vis-à-vis the complexity of iron oxide polymorphism, this needs to be further investigated on a case-by-case basis and in far greater detail under more suitable conditions.11,47
is the rate of change of the number concentration of clusters (or particles) of size ‘i’ with respect to time, ΣβJknJnk represents the formation of clusters of size ‘i’ by binary collisions (or aggregations) of smaller clusters. Furthermore, nj and nk represent the number concentrations of clusters of sizes j and k, respectively. The term βjk represents the rate constant (or the collision kernel) for the aggregation of clusters of sizes j and k. The summation runs over all pairs such that j + k = ij + k = ij + k = i. So only those combinations that lead to size ‘i’ are considered. The second term niΣβijnj similarly indicates the loss of clusters of size ‘i’ as they collide with other clusters of size ‘j’ to form larger clusters.43 These dynamics bypass the traditional atom-by-atom growth of classical models, explaining the formation of highly oriented, anisotropic hematite structures. Moreover, the current discussion on solution phase classical vs. non-classical nucleation and growth suggests that additive-controlled processes are generally inclined to a non-classical path.42,46 However, a similar shape anisotropy in zinc oxide nanoparticles was also reported in our previous study (L. M. Verma et al. 2024).9 In summary, the shape and phase outcomes across ligand systems are rationalized by considering both thermodynamic and kinetic aspects, i.e., Ksp-defined supersaturation (S), ligand-induced modulation of γ, and the coexistence of classical nucleation with non-classical assembly routes. This framework explains how surface-active ligands steer iron oxide crystallization during sol–gel synthesis. However, beyond these differences in solution phase evolutionary pathways, the sintering of resynthesized samples (FeCT, FeMC, and FePM) at a fixed temperature (500 °C) in a muffle furnace showed the retention of the relative effect of the ligands as observed in this experiment. The resynthesized pre-annealed (vacuum oven drying at 150 °C) iron oxide samples analysed by XRD (Fig. 2a and b), FTIR (Fig. 3a and b), and TGA (Fig. S8, SI) confirm the definitive pre- and post-annealing effect of the ligands on the shape and morphology of the final iron oxide (α-Fe2O3) nanocrystals. Also, FESEM images of pre- and post-annealed (Fig. S4a and S5a–d, SI) samples show template morphologies of the samples developing into the corresponding final shape. For instance, the needle-shaped goethite (α-FeOOH) samples in the case of FeCT grow into rod-like morphologies after annealing. This suggests the template-based Ostwald ripening of the needle-shaped crystal – a precise mechanism for non-classical kinetic crystallization pathways. As the complete thermodynamic equilibration of shape in the case of α-Fe2O3 is reported at 700–900 °C, the result in this case is largely kinetically controlled.26 Furthermore, the previous work shows that most of such studies are concerned with completely defined cases, involving fine chemical surfactants as additives, yet still lack complete mechanistic understanding and control over nucleation and crystallization.43 Among the reported biogenic ligand-based approaches, using the extract of lignocellulosic waste (PM) presents a novel case of ligand-modulated size and morphology control. However, these findings align with the recent efforts in these directions. Nevertheless, during this study, it remained challenging to conclude the exact role of the PM extract in shaping and controlling the crystallisation process primarily due to the complexity of the degraded lignocellulosic substance (PM) based aqueous extract. However, given its importance in sustainable catalysis and relevance to the mineralisation processes in the natural environment vis-à-vis the complexity of iron oxide polymorphism, this needs to be further investigated on a case-by-case basis and in far greater detail under more suitable conditions.11,47












