
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface amorphization advances activity and stability for acidic oxygen evolution†
Muhammad Bilal
Hussain‡
 ab,
Munir
Ahmad‡
ab,
Shabab
Hussain
c,
Rida
Javed
ab,
Zulakha
Zafar
ab,
Dinghua
Zhou
ab,
Arunpandiyan
Surulinathan
ab,
Renfei
Feng
d,
Xian-Zhu
Fu
b,
Shao-Qing
Liu
*b and
Jing-Li
Luo
*b
ab,
Munir
Ahmad‡
ab,
Shabab
Hussain
c,
Rida
Javed
ab,
Zulakha
Zafar
ab,
Dinghua
Zhou
ab,
Arunpandiyan
Surulinathan
ab,
Renfei
Feng
d,
Xian-Zhu
Fu
b,
Shao-Qing
Liu
*b and
Jing-Li
Luo
*b
aCollege of Civil and Transportation Engineering, Shenzhen University, Shenzhen, 518055, Guangdong, China
bShenzhen Key Laboratory of Energy Electrocatalytic Materials, Guangdong Research Center for Interfacial Engineering of Functional Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: sq.liu@szu.edu.cn; jll@szu.edu.cn
cShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, P. R. China
dCanadian Light-Source-Inc 44 Innovation-Blvd, Saskatoon, Saskatchewan S7N-0X4, Canada
First published on 16th June 2025
Abstract
The oxygen evolution reaction (OER) in acidic environments is crucial for various energy storage/conversion technologies. Enhancing acidic OER efficiency through precise optimization of the electronic structure at active metal sites remains a significant challenge. Due to structural flexibility and accessible active sites, amorphization offers a knob to enhance the activity of Ru-based catalysts. Here, we developed a RuO2/IrO2 composite catalyst characterized by a crystalline core and an amorphous surface structure. Density functional theory results indicate that surface amorphization can effectively lower the reaction energy barrier and accelerate electron transfer than its fully crystalline counterpart. Operando differential electrochemical mass spectrometry proved that surface amorphization inhibits the leaching of lattice oxygen. As a result, the RuO2/IrO2 composite demonstrates superior OER performance, achieving a low overpotential of 174 mV at 10 mA cm−2 and remarkable stability for 95 hours in a 0.5 M H2SO4 electrolyte. This work inspires new design ideas for high-performance acid electrocatalysts, providing insights for developing robust, efficient materials for sustainable energy applications.
1. Introduction
The oxygen evolution reaction (OER) plays an undoubtedly vital role in the energy conversion system, including hydrogen production through water electrolysis, CO2 reduction for generating clean fuels, and its integration with energy conversion devices such as metal–air batteries.1–5 Compared to alkaline media, the OER in acidic electrolytes offers a higher reaction rate and fewer side reactions, making it essential for efficient electrolytic cell design.2,6–8 However, the OER requires high energy to overcome the kinetic barrier of the four-electron transfer reaction, necessitating the development of efficient electrocatalysts.8–13 While many transition metal-based electrocatalysts perform well under alkaline conditions, only a few are effective for acidic OER.14–20Ruthenium (Ru)-based oxides (e.g., RuO2) and iridium (Ir)-based oxides (e.g., IrO2) are currently the most widely used catalysts for acidic OER due to their remarkable activity and corrosion resistance.21–24 However, IrO2, despite its superior stability, is limited by its scarcity and relatively lower catalytic activity. RuO2, in particular, is favored for its higher activity and lower cost compared to IrO2. However, it suffers from poor durability due to active site collapse and Ru dissolution under acidic conditions.13,25,26 During the oxygen evolution reaction (OER), ruthenium (Ru) is oxidized to higher valence states, including Ru4+ and Ru6+. These states are less stable, especially under acidic conditions, making Ru prone to dissolution. Specifically, Ru6+ is highly reactive and dissolves into the electrolyte, often forming soluble species such as RuO4, which contributes to catalyst degradation.27–30 While numerous efforts have been made to address the stability and activity challenges of RuO2 and IrO2 through RuIr bimetallic oxides, these advancements have predominantly relied on crystalline materials. Amorphous materials, with their lack of long-range order, enable substantial structural reorganization during catalytic reactions. This disorder creates numerous dangling bonds and defects, offering abundant active sites. Their flexibility enhances active site exposure and efficient charge transfer, significantly boosting catalytic performance.31,32 Moreover, amorphous materials exhibit unique isotropic properties and strong corrosion resistance, making them promising candidates for stable catalysts.33,34 However, the potential of amorphous RuO2/IrO2 composites remains largely unexplored. Designing an amorphous RuO2/IrO2 composite is a promising approach to achieving synergistic activity and stability for acidic OER demands.
Herein, we reported a RuO2/IrO2 composite catalyst with a crystalline core and an amorphous surface. The RuO2/IrO2 catalyst displays a significantly reduced overpotential of 174 mV and exhibits sustained stability for 95 hours at 10 mA cm−2 in a 0.5 M H2SO4 electrolyte. Amorphous materials excel in catalytic applications by providing more active sites and better charge transfer than their crystalline counterparts, enhancing overall efficiency. The DFT calculations demonstrate that the IrO2/RuO2 composite with an amorphous surface outperforms its full crystalline counterpart in catalytic activity by reducing the reaction energy barrier and improving electron transfer. In addition, (Fig. 1–4) operando differential electrochemical mass spectrometry (DEMS) proved that surface amorphization prevents the loss of lattice oxygen. These findings underscore the significant potential of amorphous materials in acidic OER.
 | ||
| Fig. 1 Morphology and composition profile analysis. (a) Schematic illustration of the synthesis of IrO2/RuO2-CC; (b–d) TEM, HRTEM and HAADF-STEM images of IrO2/RuO2-CC; (e) SAED pattern of IrO2/RuO2-CC; (f) STEM-EDS elemental mapping of IrO2/RuO2-CC. | ||
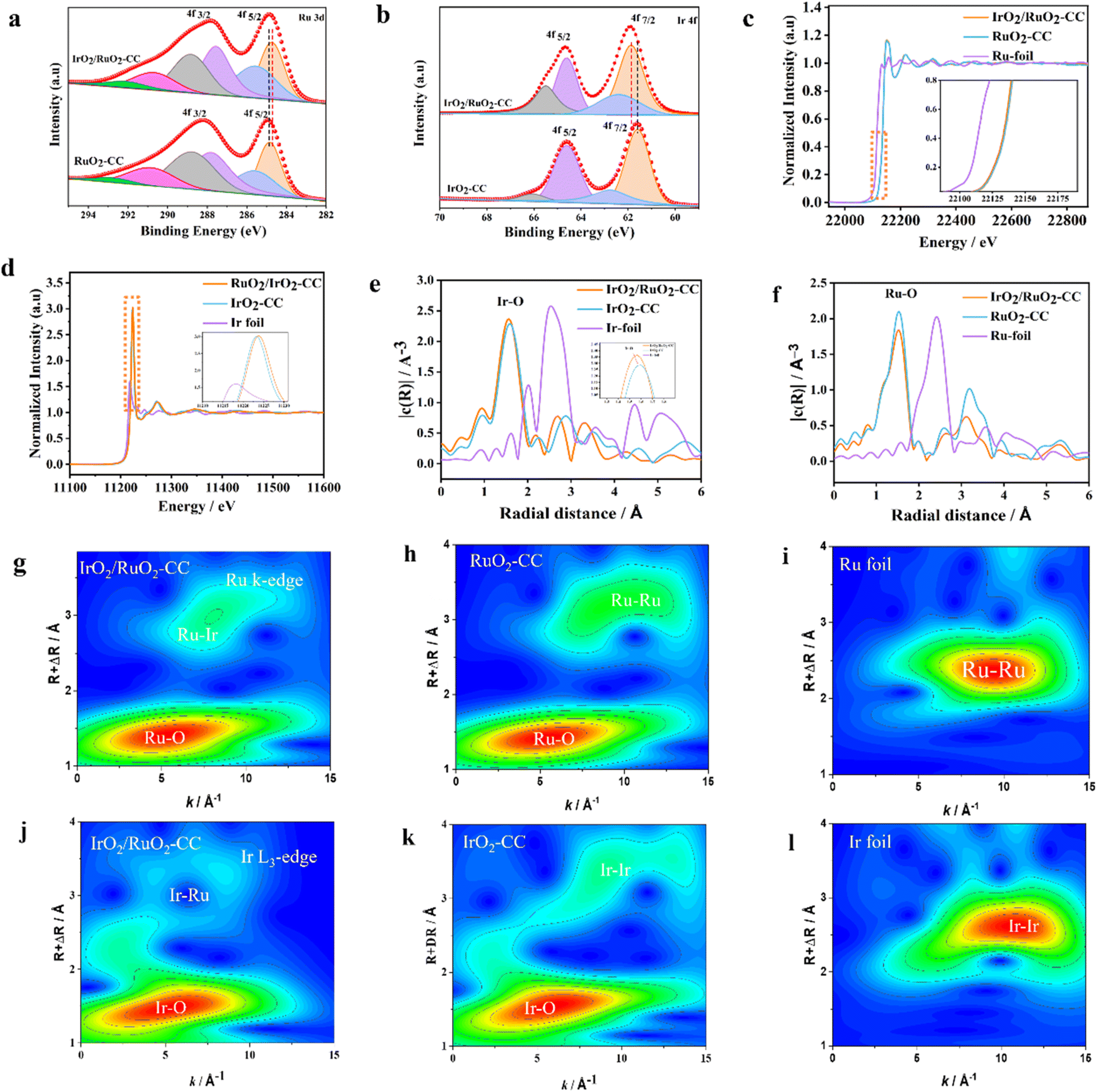 | ||
| Fig. 2 XPS and XAS characterization studies of electrocatalysts. (a) Ru 3d XPS spectra of IrO2/RuO2-CC and RuO2-CC; (b) Ir 4f XPS spectra of IrO2/RuO2-CC and IrO2-CC; (c) Ru K-edge XANES spectra of IrO2/RuO2-CC, RuO2-CC, and Ru foil; (d) Ir L3-edge XANES spectra of IrO2/RuO2-CC, IrO2-CC, and Ir foil; (e) Fourier-transformed EXAFS spectra at the Ru K-edge for IrO2/RuO2-CC, RuO2-CC, and Ru foil; (f) Fourier-transformed EXAFS spectra at the Ir L3-edge collected for IrO2/RuO2-CC, IrO2-CC, and Ir foil; (g–i) Ru K-edge WT-EXAFS spectra of IrO2/RuO2-CC, C–RuO2, and Ru foil; (j–l) Ir L3-edge WT-EXAFS spectra of IrO2/RuO2-CC, IrO2-CC, and Ir foil, respectively. | ||
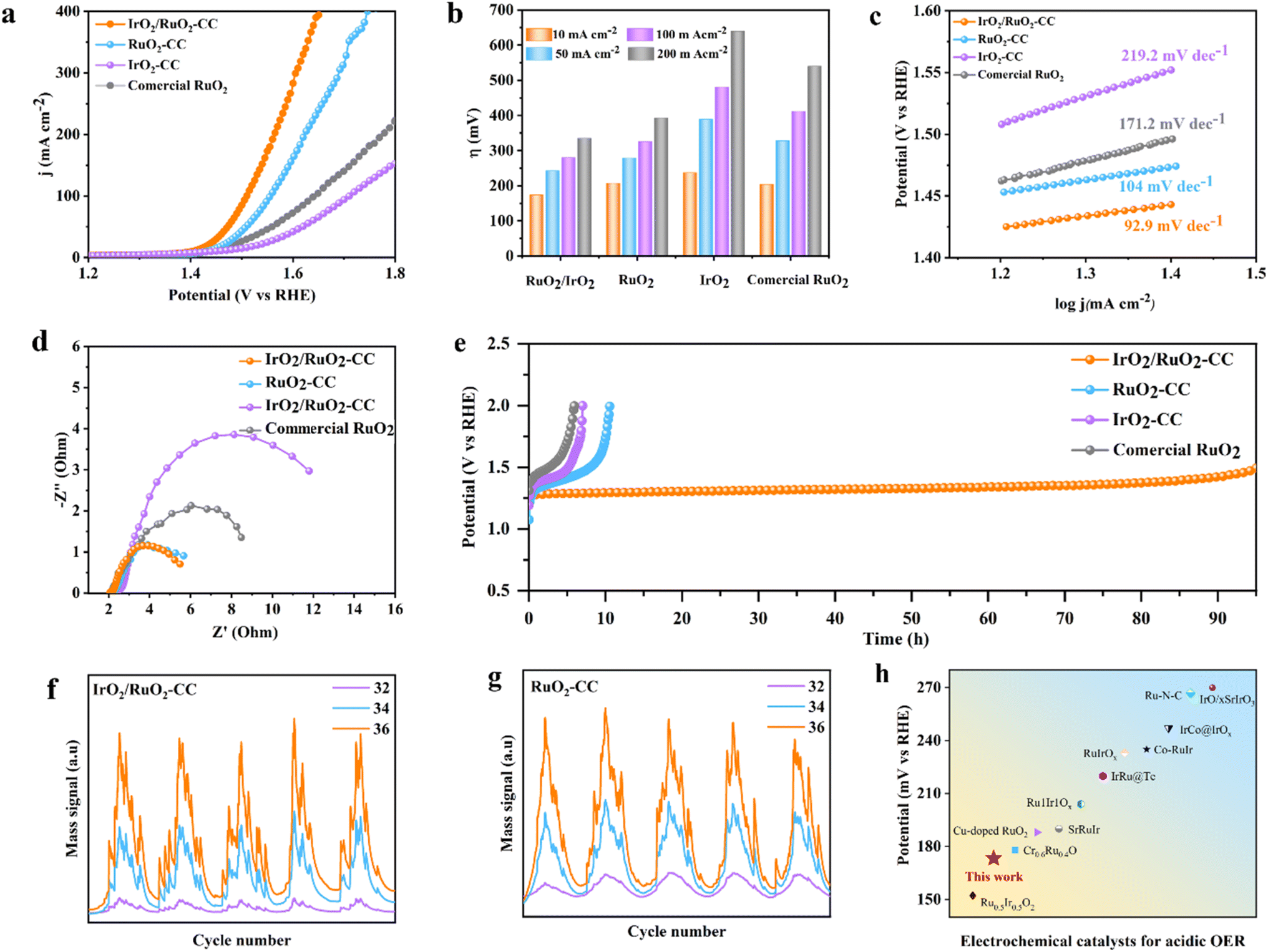 | ||
| Fig. 3 Electrocatalytic OER performances of the catalysts in 0.5 M H2SO4 electrolyte. (a) Polarization curves with iR compensation; (b) the comparison of overpotentials needed to reach 10, 50, 100 and 200 mA cm−2; (c) corresponding Tafel slopes derived from the LSV curves of IrO2/RuO2-CC, RuO2-CC, IrO2-CC, and commercial RuO2; (d) EIS spectra; (e) stability curves of IrO2/RuO2-CC, RuO2-CC, and IrO2/RuO2-CC at 10 mA cm−2. Operando DEMS signals of 36O2, 34O2, and 32O2 for (f) IrO2/RuO2-CC and (g) RuO2-CC in 0.5M H218O aqueous H2SO4 electrolyte; and (h) comparison of the acidic OER activity of IrO2/RuO2-CC with the reported catalysts, sourced from Table S1.† | ||
 | ||
| Fig. 4 Density functional theory calculations: (a) the differential charge density structure of amorphous IrO2/RuO2-CC and (b) crystalline IrO2/RuO2-CC; the model colors are Ru (teal), Ir (green), and O (red); (c) the free energy diagram; and (d) the projected DOS of amorphous IrO2/RuO2-CC and crystalline IrO2/RuO2-CC. | ||
2. Experimental section
2.1 Chemicals
Cobalt nitrate hexahydrate [Co(NO3)2)·6H2O] (MACKLIN, 99.99%), 2-methylimidazole (MACKLIN, 98%), ruthenium(III) chloride [RuCl3] (MERYER, 98%), ruthenium(IV) oxide (MACKLIN, 99.99%), iridium chloride hydrate [IrCl3 xH2O] (Aladdin, 52%), Nafion solution (Sigma-Aldrich ∼5%), and perfluorinated resin solution containing Nafion (Sigma-Aldrich ∼5%) were procured and utilized without additional purification, except for the commercial carbon cloth (Saidemu, MA-EN-CU-09011P), which underwent activation in concentrated nitric acid at 100 °C for five hours.2.2 Synthesis of Co-MOF-CC
The commercial carbon cloth (CC) (MA-EN-CU-09011P) was first activated by treating it with concentrated HNO3 for five hours at 100 °C, followed by multiple washes with distilled water after cooling. Subsequently, 1.33 g of 2-methylimidazole and 0.58 g of Co(NO3)2·6H2O were dissolved separately in 40 mL of deionized (DI) water. These solutions were then combined in a beaker, where a piece of CC was vertically suspended. The combined solutions were thoroughly mixed to ensure homogeneity. After five hours, the CC was removed, washed with DI water, and dried at 50 °C for 12 h to synthesize the Co-MOF-coated carbon cloth precursor.2.3 Synthesis of IrO2/RuO2-CC
A 50 mL solution containing RuCl3 and IrCl3 at a concentration of 5 mg mL−1 was prepared. A piece of CC, previously coated with Co-MOF, was immersed vertically in the solution for 45 min. After immersion, the CC was removed, rinsed with DI water, and annealed at 350 °C for 2 h in a muffle furnace, with a heating rate of 2 °C min−1. This process resulted in the formation of amorphous IrO2/RuO2-coated CC. Similarly, RuO2-CC, IrO2-CC and commercial RuO2 were fabricated by submerging the carbon cloth coated with Co-MOF into separate solutions with a concentration of 5 mg mL−1 of RuCl3 and IrCl3, respectively.2.4 Characterization
The morphologies, crystallography, and surface compositions of the developed catalysts were determined by field-emission scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Some high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and energy dispersive spectroscopy (EDS) results were obtained using an FEI Talos F200X TEM (200 kV). The X-ray absorption fine structure (XAFS) spectra were collected using the VESPERS beamline at the CLS (Canadian Light Source). X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were also employed. The elemental ratios of the catalysts were measured using energy-dispersive X-ray spectroscopy (EDS).2.5 Electrochemical characterization
Electrochemical measurements were carried out in a typical three-electrode system controlled by a CHI workstation, in which graphite was used as the counter electrode and Ag/AgCl as the reference electrode and modified CC as the working electrode, with a geometrical area of 1.0 cm2. The polarization curves were recorded at a scan rate of 5 mV s−1 in 0.5 M H2SO4 with pH = 0. All potentials are converted to the RHE scale according to the Nernst equation. Chronopotentiometry was used to investigate the stability of the electrode at 10 mA cm−2 current density. EIS was performed at a potential of 1.33 V vs. RHE from 105 to 0.1 Hz frequency range by applying an amplitude of 5 mV. For evaluating the ECSA, the Cdl of electrocatalysts was measured by using cyclic voltammograms in a non-faradaic potential window (0.8–0.9 V vs. RHE) at different scan rates from 2 to 10 mV s−1. The plots of the current density differences (ΔJ = (Ja – Jc) at 0.85 V vs. RHE) against the different scan rates have a linear relationship, and their slopes are twice their Cdl values.3. Results and discussion
3.1 Synthesis and structural characterization
The schematic diagram distinctly illustrates the synthesis of RuO2/IrO2-CC, as shown in Fig. 1a. Initially, the carbon cloth (CC) was treated with concentrated nitric acid (HNO3) to introduce oxygen-containing groups on its surface, creating adsorption sites. This modified CC was then immersed in a solution of 2-methyl-imidazole and cobalt nitrate, leading to the formation of Co metal–organic framework (MOF) nanosheets on the CC substrate (Fig. S1a–c and ESI†). The Co-MOF-coated CC was subsequently submerged in a mixed solution of RuCl3 and IrCl3, dried overnight, and annealed at 350 °C, resulting in the formation of the IrO2/RuO2-CC composite. During annealing, the interaction between ruthenium and iridium ions caused the dissociation of ligands bound to the MOF, forming nanoparticles of IrO2/RuO2 on the CC substrate.Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) were employed to investigate the intricate structural characteristics of the IrO2/RuO2-CC composite.
The analysis revealed a spherical morphology with an average size of approximately 150 nm and a disordered atomic configuration (Fig. 1b, S1d, ESI†). The morphology of pristine RuO2-CC and IrO2-CC is depicted through SEM in Fig. S1e and f (ESI†). In a detailed analysis, high-resolution TEM images prominently presented the absence of lattice fringes, a definitive indication of the catalysts' amorphous nature (Fig. 1c). The TEM analyses reveal that the disordered atomic structure of the IrO2/RuO2-CC composite, lacking crystalline features, offers high structural flexibility and enhanced catalytic potential. High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM) observations corroborated these findings, demonstrating that the IrO2/RuO2-CC composite exhibits an amorphous structure devoid of crystallinity (Fig. 1d).35 This crucial observation was further substantiated by the selected area electron diffraction (SAED) pattern, which notably lacked distinct diffraction rings, thereby reinforcing the conclusion of the catalyst's amorphous structure (Fig. 1e).35
To further confirm the amorphous nature of the composite material, we conducted X-ray diffraction (XRD) analysis. XRD analysis revealed weak but distinct diffraction peaks, confirming the presence of crystallinity, despite conventional HRTEM and HAADF-STEM images showing no lattice fringes, initially suggesting a fully amorphous material (Fig. S2, ESI†). To resolve this discrepancy, we conducted targeted high-resolution HAADF-STEM imaging, which identified localized crystalline domains in the core regions, surrounded by an amorphous surface matrix (Fig. S3, ESI†). This dual-phase structure reconciles the XRD-detected long-range order with the amorphous signatures observed in HRTEM, conclusively establishing a hybrid architecture: a crystalline core encapsulated by an amorphous outer layer.
The homogeneous dispersion of Ru, Ir, and O elements throughout the entire structure was meticulously confirmed by Scanning Transmission Electron Microscopy-Energy Dispersive X-ray Spectroscopy (STEM-EDX) elemental mapping (Fig. 1f). A similar uniform distribution was observed for the RuO2-CC and IrO2-CC composites (Fig. S4-S5, ESI†). The STEM-EDX results revealed an approximate atomic ratio of Ru to Ir of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S6, ESI†). This careful control over elemental composition is vital for optimizing the catalytic activity and stability of the composite material.
:1 (Fig. S6, ESI†). This careful control over elemental composition is vital for optimizing the catalytic activity and stability of the composite material.
To explore the chemical state of the amorphous IrO2/RuO2-CC composite, X-ray photoelectron spectroscopy (XPS) was employed. As shown in (Fig. S7, ESI†), the XPS survey for IrO2/RuO2-CC confirmed the coexistence of Ru, Ir, and O elements in the prepared electrocatalyst, which aligns well with the results from EDS. In the amorphous IrO2/RuO2-CC composite, a negative shift was observed in the Ru 3d 5/2 and Ru 3d 3/2 peaks located at 284.7–287.5 eV, in contrast to the peak range of 284.93 to 287.85 eV observed in RuO2-CC (Fig. 2a). This alteration implied a reduced oxidation state of Ru within the composite relative to the Ru(IV) state present in RuO2-CC.36 In the Ir 4f XPS analysis of IrO2/RuO2-CC, a slight positive shift was observed in the peaks located at 61.89 and 64.64 eV, corresponding to Ir 4f7/2 and 4f5/2 of Ir(IV), respectively (Fig. 2b). This shift contrasts with the peak range of 61.58 to 64.53 eV observed in IrO2-CC. The discrepancy indicates that Ir exhibits a higher valence state in the IrO2/RuO2-CC composite compared to IrO2-CC.27 Furthermore, the amorphous IrO2/RuO2-CC structure exhibits no peaks in the Co 2p region, as illustrated in Fig. S8 (ESI†). This observation confirms that the Co element is absent in the structure.
The white line of X-ray absorption near-edge structure (XANES) spectra was employed to elucidate the electron transition behavior and electronic structure of IrO2/RuO2-CC, thereby providing clearer insights into the valence electronic state through intensity analysis. As depicted in Fig. 2c, the white-line region of the Ru K-edge for amorphous IrO2/RuO2-CC displayed an absorption energy that was slightly lower than that of RuO2-CC. This indicates that the valence state of Ru in amorphous IrO2/RuO2-CC spans from 0 to +4.27,37Fig. 2d demonstrates that the white line peak intensity of amorphous IrO2/RuO2-CC is markedly higher than that of IrO2-CC and Ir foil, indicating that the valence states of Ir are in the order: amorphous IrO2/RuO2-CC > IrO2-CC > Ir foil. These observations concerning the valence states of Ru and Ir are consistent with the XPS results previously discussed. The Fourier transforms of the extended X-ray absorption fine structure (EXAFS) spectra (Fig. 2e and f) at the Ru K-edge and Ir L3-edge were conducted to probe the local chemical environment of IrO2/RuO2-CC. The FT-EXAFS spectra indicate that the bond length of Ru–O remains almost the same in the composite IrO2/RuO2-CC compared to pristine RuO2-CC. However, the bond length of Ir–O is slightly reduced in the composite compared to pristine IrO2-CC. This reduction can be attributed to differences in the crystal structure, as metal–oxygen bonds in the 1T phase are inherently shorter than those in the rutile phase.38 These observations suggest significant interactions between Ru, O, and Ir atoms within the composite. The peaks in the R-space at approximately 1.55 and 1.65 Å correspond to the Ru–O and Ir–O coordination shells, respectively, indicating that IrO2/RuO2-CC retains the characteristic RuO6 and IrO6 coordination octahedra. Additionally, the wavelet transforms of EXAFS (WT-EXAFS) analysis revealed Ru–Ir and Ir–Ru scattering signals in the Ru K-edge (Fig. 2g–i) and Ir L3-edge (Fig. 2j–l) WT-EXAFS, respectively, which indicates a strong interaction between Ru and Ir in amorphous IrO2/RuO2-CC.39,40 These robust interactions within the Ru–O–Ir local structures may inhibit the formation of more soluble high-valent Ru/Ir complexes, thereby significantly enhancing the stability of the electrocatalysts.41
3.2 Electrochemical performances
The oxygen evolution reaction (OER) activity of the amorphous IrO2/RuO2-CC composite was assessed in a 0.5 M H2SO4 electrolyte using an electrode system and subsequently compared with pristine RuO2-CC, IrO2-CC, and commercial RuO2 electrocatalysts, respectively. The electrocatalytic activity was typically assessed using the linear sweep voltammetry (LSV) method. The amorphous IrO2/RuO2-CC exhibited the lowest overpotential (η10 = 174 mV) at 10 mA cm−2 compared to RuO2-CC (η10 = 207), IrO2-CC (η10 = 237), and commercial RuO2 (η10 = 227) (Fig. 3a). The OER activity of the electrocatalysts (IrO2/RuO2-CC, RuO2-CC, IrO2-CC and commercial RuO2) without iR correction is given in Fig. S9†. The amorphous IrO2/RuO2-CC composite demonstrates enhanced OER performance, with RuO2-CC acting as the primary active component, while IrO2-CC improves their dispersion and electrical conductivity. This synergistic effect leads to a notable increase in overall OER activity compared to the individual components.Furthermore, Fig. 3b presents the overpotential data for all electrocatalysts at current densities of 10, 50, 100 and 200 mA cm−2, providing a comprehensive comparison of their performances. Similarly, Tafel slopes were calculated to assess the reaction kinetics of the electrocatalysts. The analysis revealed that the amorphous IrO2/RuO2-CC composite exhibited the smallest slope (92.9 mV dec−1) compared to RuO2-CC (104 mV dec−1), IrO2-CC (219.2 mV dec−1), and commercial RuO2 (171.2 mV dec−1), indicating that the amorphous IrO2/RuO2-CC composite facilitates more efficient OER kinetics. These findings suggest that the amorphous IrO2/RuO2-CC composite enables faster electron transfer and reaction kinetics relative to the other catalysts tested.
To determine the optimal concentration, we varied the proportions of IrO2 and RuO2, conducting systematic experiments and STEM analysis at different ratios. Our investigation revealed that a 1:1 ratio of RuO2 to IrO2 yielded the highest catalytic performance. At this specific ratio, the catalyst was completely amorphous, which was crucial for its effectiveness. In contrast, other concentrations resulted in inferior catalytic performance, and the catalyst was not fully amorphous, except for the pristine RuO2-CC (Fig. S10, ESI†). These findings underscore the importance of precise composition in catalyst design. Achieving the right balance between RuO2 and IrO2 is essential for maximizing catalytic activity, highlighting the critical role of the amorphous structure in enhancing performance.
Additionally, as per the acquired Electrochemical Impedance Spectroscopy (EIS) data, the amorphous IrO2/RuO2-CC configuration exhibits the most diminutive semicircular arc at a potential of 300 mV vs. RHE, denoting diminished charge transfer resistance and enhanced reaction kinetics in comparison to all other synthesized specimens (Fig. 3d). To gain deeper insights into the enhanced activity of amorphous IrO2/RuO2-CC in oxygen evolution reaction (OER) performances, electrochemical double-layer capacitance (Cdl) was investigated, and the electrochemical active surface area (ECSA) was determined through Cdl measurements (Fig. S11a–c†). The synthesized amorphous IrO2/RuO2-CC exhibited notably higher Cdl and ECSA activities compared to pristine RuO2-CC and IrO2-CC (Fig. S11d, ESI†), indicating that the amorphous nature of the composite significantly improves and enhances the density of active sites.
Besides activity, durability and stability are also pivotal factors in assessing the oxygen evolution reaction (OER) potential of any catalyst. Therefore, employing chronopotentiometric measurement, the stability of the amorphous IrO2/RuO2-CC electrocatalyst was analyzed at a current density of 10 mA cm−2, revealing that the primary catalyst retained a stable overpotential for 95 h (Fig. 3e). For instance, IrO2-CC remained stable for only 10 h, while RuO2-CC became inactivated after 5 h at a constant current density of 10 mA cm−2. Following durability analysis, a slight decrease in Ru ion concentration relative to Ir ions was observed, which may indicate a potential difference in stability between Ru and Ir under acidic conditions. However, underscoring their favorable stability characteristics is further affirmed by operando Differential Electrochemical Mass Spectrometry (DEMS). In this study, Differential Electrochemical Mass Spectrometry (DEMS) was employed to investigate the oxygen evolution reaction (OER) in both the amorphous IrO2/RuO2-CC composite and pristine RuO2 and IrO2 materials (Fig. 3f, g and S12, ESI†). DEMS, which enables real-time tracking of gaseous products during electrochemical reactions, monitored the isotopic oxygen species 32O2, 34O2, and 36O2, corresponding to molecular oxygen composed of 16O, 17O, and 18O isotopes, respectively. These isotopes are crucial for identifying specific reaction pathways and determining the involvement of lattice oxygen in the OER. The significantly lower intensity of the 32O2 signal in the amorphous IrO2/RuO2 composite indicates a reduced release of lattice oxygen, suggesting lower oxygen degradation compared to the pristine materials. This reduction in lattice oxygen involvement is directly linked to the enhanced catalytic stability observed in the composite. Meanwhile, the signals for 34O2 and 36O2 exhibited no significant changes, further confirming that the composite effectively minimizes the participation of lattice oxygen. By limiting the release of lattice oxygen, the composite not only reduces degradation but also improves its overall stability and performance during the OER.
A comprehensive evaluation of the OER performance of amorphous IrO2/RuO2-CC was conducted by comparing its activity to a range of recently reported OER electrocatalysts. As illustrated in Fig. 3h and detailed in Table S1 (ESI),† the analysis indicates that amorphous IrO2/RuO2-CC exhibits superior catalytic activity. This outstanding performance is evident when compared to numerous Ru/Ir-based OER electrocatalysts reported in the current literature. The data reveal that IrO2/RuO2-CC not only matches but, in many cases, exceeds the activity levels of these contemporary catalysts. Overall, these findings underscore the competitive advantage of amorphous IrO2/RuO2-CC in the field of OER catalysis. The IrO2/RuO2-CC catalyst demonstrates a mass activity of 776 mA mg−1 at 1.65 V vs. RHE in 0.5 M H2SO4, representing 1.6-fold, 6.2-fold, and 3.7-fold enhancements over RuO2-CC (496 mA mg−1), IrO2-CC (126 mA mg−1), and commercial RuO2 (211 mA mg−1), respectively. This performance advantage is further demonstrated through comparison with state-of-the-art Ru- and Ir-based catalysts in Table S2,† where our catalyst ranks among the highest reported activities.
To evaluate the linear sweep voltammetry (LSV) performance differences between amorphous and crystalline forms of RuO2/IrO2, the amorphous form was transformed into its crystalline counterpart. This evidence was provided by HRTEM images, which displayed lattice fringes indicative of the presence of crystalline parts (Fig. S13a, ESI†). This crucial observation was further substantiated by the selected area electron diffraction (SAED) pattern, which prominently exhibited distinct diffraction rings (Fig. S13b, ESI†). Further transformation was confirmed by the appearance of sharp peaks in the X-ray diffraction (XRD) patterns (Fig. S13c, ESI†), which are characteristic of crystalline structures. Our investigation revealed that the crystalline IrO2/RuO2-CC composite displayed significantly lower catalytic activity compared to its amorphous counterpart (Fig. S13d, ESI†). While crystallinity is often associated with improved material properties, our findings suggest that in this particular case, the amorphous structure offered distinct advantages in electrocatalysis. These insights, specific to our study, underscore the importance of considering amorphous structures when designing and optimizing advanced catalysts.
After subjecting the amorphous IrO2/RuO2-CC sample to acidic OER stability testing, morphological analyses were conducted again. The results from microscopy techniques revealed no significant alterations. The morphology of the material remained unchanged under these harsh conditions. HR-TEM/HAADF-STEM images indicated negligible changes in the structure of amorphous IrO2/RuO2-CC, confirming its robust stability (Fig. S14a and b, ESI†). Additionally, the selected area electron diffraction (SAED) pattern, which remains free of distinct diffraction rings even after stability testing, conclusively demonstrates that the structure retains its amorphous nature without any signs of crystallization, closely resembling its original state (Fig. S14c, ESI†). Moreover, STEM-EDX analysis exhibited a modest decrease in the concentrations of ruthenium, iridium, and oxygen following the acidic OER test, suggesting some dissolution of these elements over prolonged stability testing. (Fig. S14d, ESI†). These findings are mutually supportive, reinforcing the conclusion that amorphous IrO2/RuO2-CC retains its structural integrity, stability, and morphology even under harsh conditions, highlighting its potential for durable catalytic applications.
3.3 Theoretical insights into amorphous/crystalline IrO2/RuO2 composites
DFT calculations performed at zero potential provide crucial insights into the superior catalytic activity of the amorphous IrO2/RuO2 composite compared to its crystalline counterparts. Fig. 4a and b illustrates a more pronounced charge transfer at the active interface of the amorphous IrO2/RuO2 composite compared to its crystalline counterpart, with yellow and cyan electron clouds indicating gained and donated electrons, respectively. This enhanced electron transfer in the amorphous IrO2/RuO2 composite leads to more effective interactions at the interface. As a result, the amorphous structure significantly accelerates the OER, improving catalytic activity. It is well-established that the adsorption energies of the intermediates, *OH, *O, and *OOH, play a crucial role in determining the overpotential.42 The free energy diagrams reveal that the *OOH formation from *O, which is the rate-limiting step, exhibits a smaller free energy gap in the amorphous composite (1.67 eV) compared to its crystalline counterpart (1.97 eV).This suggests that the amorphous structure lowers the reaction energy barrier more effectively than the crystalline form, leading to enhanced catalytic performance. The optimized atomic configurations and lower ΔG values for the amorphous composite further confirm its superior electrocatalytic activity.42 Structural models of the clean surface and various OER intermediates (*OH, *O, and *OOH) adsorbed on the surfaces of amorphous IrO2/RuO2-CC and crystalline IrO2/RuO2-CC are shown in Fig. S15, ESI.†
The projected density of states (DOS) analysis reveals that the amorphous IrO2/RuO2-CC composite exhibits a DOS closer to the Fermi level compared to the crystalline form, with values of −1.87 and −1.93, respectively. This closer proximity results in an elevated d-band center energy level, thereby enhancing electron interactions and facilitating more efficient electron transfer. As a result, the amorphous composite demonstrates superior adsorption of reaction intermediates, leading to enhanced catalytic activity, particularly in the oxygen evolution reaction (OER).
Although the peak intensities between the two phases are comparable, the subtle shift in DOS positioning highlights the intrinsic advantages of the amorphous structure in optimizing catalytic performance.34 These findings highlight the significant impact of the amorphous structure in reducing energy barriers and improving the overall efficiency of the OER process, offering valuable insights for the design of advanced electrocatalysts.
4. Conclusions
In summary, theoretical calculations have provided a clear understanding of the intrinsic high electroactivity of the amorphous RuO2/IrO2-CC composite, effectively guiding its experimental synthesis as an efficient OER electrocatalyst. This study presents an amorphous RuO2/IrO2-CC composite that effectively addresses the challenge of creating stable and efficient electrocatalysts for acidic oxygen evolution reaction (OER). Unlike crystalline materials, amorphous structures can undergo significant reorganization during reactions, increasing their adaptability and effectiveness. DFT calculations highlight that the amorphous RuO2/IrO2-CC composite enhances catalytic activity by reducing the energy barrier for reactions and enabling more efficient electron transfer compared to its crystalline counterpart. These findings and strategies in amorphous materials design mark a significant step in advancing electrocatalysis for sustainable energy. By offering a blueprint for high-performance catalysts and deepening our understanding of their structural and electronic properties, this work paves the way for developing next-generation electrocatalysts, setting a solid foundation for future research and broadening the horizon of energy applications.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Muhammad Bilal Hussain: formal analysis, original draft writing and editing. Munir Ahmad: primary investigation, conceptualization and formal analysis. Shabab Hussain: reviewing. Rida Javed: reviewing. Zulakha Zafar: reviewing. Dinghua Zhou: reviewing. Arunpandiyan Surulinathan: reviewing. Renfei Feng: reviewing. Xian-Zhu Fu: supervision and funding acquisition. Shao-Qing Liu: supervision, funding acquisition and reviewing. Jing-Li Luo: funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22309116), the Shenzhen Science and Technology Program (No. ZDSYS20220527171401003, RCBS20231211090519029, and KQTD20190929173914967). The authors thank the VESPERS beamline of the Canadian Light Source for the synchrotron characterization. We thank the Instrumental Analysis Center of Shenzhen University for the assistance with the Electron Microscope.References
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma and Z. Feng, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS PubMed.
- L. Zong, P. Li, F. Lu, C. Wang, K. Fan, Z. Li and L. Wang, Adv. Funct. Mater., 2023, 33, 2301013 CrossRef CAS.
- L. Chen, W. Zhao, J. Zhang, M. Liu, Y. Jia, R. Wang and M. Chai, Small, 2024, 20, 2403845 CrossRef CAS PubMed.
- Y. Lin, Y. Dong, X. Wang and L. Chen, Adv. Mater., 2023, 35, 2210565 CrossRef CAS PubMed.
- Q. Qin, T. Wang, Z. Li, G. Zhang, H. Jang, L. Hou, Y. Wang, M. G. Kim, S. Liu and X. J. Liu, J. Energy Chem., 2024, 88, 94–102 CrossRef CAS.
- X. Long, B. Zhao, Q. Zhao, X. Wu, M.-N. Zhu, R. Feng, M. Shakouri, Y. Zhang, X. Xiao and J. Zhang, Appl. Catal., B, 2024, 343, 123559 CrossRef CAS.
- K. Sardar, E. Petrucco, C. I. Hiley, J. D. Sharman, P. P. Wells, A. E. Russell, R. J. Kashtiban, J. Sloan and R. Walton, Angew. Chem., Int. Ed., 2014, 126, 11140–11144 CrossRef.
- X. Wu, Q. Yan, H. Wang, D. Wu, H. Zhou, H. Li, S. Yang, T. Ma and H. J. Zhang, Adv. Funct. Mater., 2024, 34, 2404535 CrossRef CAS.
- Y. Zhang, X. Zhu, G. Zhang, P. Shi and A.-L. Wang, J. Mater. Chem. A, 2021, 9, 5890–5914 RSC.
- M. Zlatar, D. Escalera-López, M. G. Rodríguez, T. s. Hrbek, C. Götz, R. Mary Joy, A. Savan, H. P. Tran, H. N. Nong and P. Pobedinskas, ACS Catal., 2023, 13, 15375–15392 CrossRef CAS.
- X. Wu, J. Wu, Y. Hu, L. Zhu, B. Cao, K. M. Reddy, Z. Wang and H. J. Qiu, Small, 2024, 20, 2404019 CrossRef CAS PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- W. Li, Y. Bu, X. Ge, F. Li, G. F. Han and J. B. J. C. Baek, ChemSusChem, 2024, 17, e202400295 CrossRef CAS PubMed.
- M. N. Lakhan, A. Hanan, A. H. Shar, I. Ali, Y. Wang, M. Ahmed, U. Aftab, H. Sun and H. Arandiyan, Chem. Commun., 2024, 60, 5104–5135 RSC.
- Y. Jiang, H. Liu, Y. Jiang, Y. Mao, W. Shen, M. Li and R. He, Appl. Catal., B, 2023, 324, 122294 CrossRef CAS.
- M. E. C. Pascuzzi, A. Goryachev, J. P. Hofmann and E. J. Hensen, Appl. Catal., B, 2020, 261, 118225 CrossRef.
- Z. Li, H. Sheng, Y. Lin, H. Hu, H. Sun, Y. Dong, X. Chen, L. Wei, Z. Tian and Q. Chen, Adv. Funct. Mater., 2024, 34, 2409714 CrossRef CAS.
- J. Su, R. Ge, K. Jiang, Y. Dong, F. Hao, Z. Tian, G. Chen and L. Chen, Adv. Mater., 2018, 30, 1801351 CrossRef PubMed.
- S. Park, Y. Shao, J. Liu, Y. J. E. Wang and E. Science, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- X. Cui, P. Ren, C. Ma, J. Zhao, R. Chen, S. Chen, N. P. Rajan, H. Li, L. Yu and Z. Tian, Adv. Mater., 2020, 32, 1908126 CrossRef CAS PubMed.
- D. Chen, T. Liu, P. Wang, J. Zhao, C. Zhang, R. Cheng, W. Li, P. Ji, Z. Pu and S. L. Mu, ACS Energy Lett., 2020, 5, 2909–2915 CrossRef CAS.
- J. Chen, Y. Ma, T. Huang, T. Jiang, S. Park, J. Xu, X. Wang, Q. Peng, S. Liu and G. Wang, Adv. Mater., 2024, 36, 2312369 CrossRef CAS PubMed.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z. S. Wu, Angew. Chem., Int. Ed., 2024, 136, e202316903 CrossRef.
- W. Gou, S. Zhang, Y. Wang, X. Tan, L. Liao, Z. Qi, M. Xie, Y. Ma, Y. Su and Y. Qu, Energy Environ. Sci., 2024, 17, 6755–6765 RSC.
- W. Zhu, X. Song, F. Liao, H. Huang, Q. Shao, K. Feng, Y. Zhou, M. Ma, J. Wu and H. Yang, Nat. Commun., 2023, 14, 5365 CrossRef CAS PubMed.
- T. G. Yun, Y. Sim, Y. Lim, D. Kim, J.-S. An, H. Lee, Y. Du and S.-Y. Chung, Mater. Today, 2022, 58, 221–237 CrossRef CAS.
- G. Chen, R. Lu, C. Ma, X. Zhang, Z. Wang, Y. Xiong and Y. Han, Angew. Chem., Int. Ed., 2024, 63, e20241160 Search PubMed.
- N. Yao, H. Jia, J. Zhu, Z. Shi, H. Cong, J. Ge and W. J. C. Luo, Chem, 2023, 9, 1882–1896 CAS.
- J. Sun, D. Yang, S. Lowe, L. Zhang, Y. Wang, S. Zhao, P. Liu, Y. Wang, Z. Tang and H. Zhao, Adv. Energy Mater., 2018, 8, 1801495 CrossRef.
- X. Yu, X. Gong, H. Qiao, X. Liu, C. Ma, R. Xiao, R. Li and T. Zhang, Small Methods, 2024, 8, 2400793 CrossRef CAS PubMed.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- M. Wang, X. Feng, S. Li, Y. Ma, Y. Peng, S. Yang, Y. Liu, H. Lei, J. Dang and W. Zhang, Adv. Funct. Mater., 2024, 34, 2410439 CrossRef CAS.
- S. Li, T. Liu, W. Zhang, M. Wang, H. Zhang, C. Qin, L. Zhang, Y. Chen, S. Jiang and D. Liu, Nat. Commun., 2024, 15, 3416 CrossRef CAS PubMed.
- J. He, X. Zhou, P. Xu and J. J. Sun, Adv. Funct. Mater., 2021, 11, 2102883 CAS.
- K. Wang, Y. Wang, B. Yang, Z. Li, X. Qin, Q. Zhang, L. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS PubMed.
- Z. Xia, H. Zhang, K. Shen, Y. Qu and Z. Jiang, Phys. B Condens. Matter, 2018, 542, 12–19 CrossRef CAS.
- Y. Wen, P. Chen, L. Wang, S. Li, Z. Wang, J. Abed, X. Mao, Y. Min, C. T. Dinh and P. D. J. Luna, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers and L. Fruchter, Nat. Commun., 2018, 1, 508–515 CAS.
- X. Bai, X. Zhang, Y. Sun, M. Huang, J. Fan, S. Xu and H. Li, Angew. Chem., Int. Ed., 2023, 62, e202308704 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01910a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |