
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced direct air capture by uranyl superoxide: a comprehensive study of the carbonation reaction of uranyl peroxide complexes†
Sarah K.
Scherrer
 and
Tori Z.
Forbes
*
and
Tori Z.
Forbes
*
Department of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: tori-forbes@uiowa.edu
First published on 30th April 2025
Abstract
Reactive oxygen species, such as superoxide, are known to engage in direct air carbon capture within strong base systems. Previous work has also suggested that U(VI) peroxide/superoxide solids may also engage in this process, although the phase behavior and kinetics of this system have not been explored. Herein, we report optimal conditions which promote carbonation of two potassium uranyl triperoxide phases and their analogous diperoxo superoxide forms. Raman spectroscopy and elemental analysis were used to monitor the carbonation reaction, and the data was subjected to Principal Component Analysis (PCA) to gain insight on the complex reactivity of these phases. These systems exhibited full carbon dioxide (CO2) uptake at 22 °C when relative humidity is at least 45%. Further, carbonation of the uranyl peroxide complexes occurred under atmospherically relevant CO2 concentrations, and we utilized a custom-built reaction chamber to evaluate the kinetics of this process. Our results identified conditions favoring the carbonation of uranyl peroxide complexes and provide insight on the mechanism of carbonate formation.
Introduction
Carbon negative methods are employed to remove atmospheric carbon dioxide (CO2) for permanent storage or transformation into chemical species, such as carbonate (CO32−).1–3 The most promising carbon negative technique is direct air capture (DAC), which utilizes a chemical sorbent to selectively bind CO2 in air.4–6 Solid or liquid amines (e.g., monoethanolamine (MEA))7–11 and aqueous strong base (e.g., potassium hydroxide (KOH) or sodium hydroxide (NaOH))6,12–16 are the most established DAC sorbents; however, these methods are currently energy intensive and economically challenging.4,5,17–19 Specifically, these sorbents are limited by slow kinetics due to ultra-dilute CO2 concentration in ambient air (approximately 420 ppm (ref. 20)). Thus, there is a critical need for novel and cost-effective DAC materials and methods to achieve gigaton scale removal.Previous work has demonstrated full mineralization or conversion of CO2 from metal (i.e., Na+, K+) stabilized reactive oxygen species (ROS) such as peroxide (O22−) and superoxide (O2−˙), but fundamental challenges within ROS-based carbon capture persist.21–31 Most significantly, ROS are known to rapidly decompose in the presence of water, and large volumes of highly concentrated (9.25 M) hydrogen peroxide (H2O2) are necessary to remain competitive with established methods. Stoin et al.21 have reported both superior efficiency of CO2 removal and improved kinetics via mixed solutions of alkali hydroxides and H2O2 with the in situ generation of O2−˙ inducing enhanced reactivity. However, this work identified a crucial limitation in that H2O2 spontaneously decomposes in alkaline conditions, thus hindering its overall efficiency without the addition of a chemical stabilizer such as sodium pyrophosphate. More recently, Ribó et al.23 described the reaction between vanadium peroxides (with small amounts of O2−˙ detected) and atmospheric CO2; however, major challenges remain for these materials in that they can undergo auto-decomposition under air instead of reacting with CO2. Additionally, they exhibit slow kinetics for the carbonation reaction with full conversion requiring several months in ambient air. Kravchuk et al.32 previously reported that U(VI) also stabilizes O2−˙ within a potassium uranyl peroxide/superoxide complex (KUPS-1) K4−x[UO2(O2)3−x(O2−˙)x](H2O2)4(H2O)4 (0 < x ≤ 1) which exhibited reactivity with atmospheric CO2 forming uranyl carbonate phases, K4[(UO2)(O2)(CO3)2]·xH2O (x = 1, 2.5) and K4[(UO2)(CO3)3] (Fig. 1). This work also suggested that use of radical initiators such as benzaldehyde may reduce necessary H2O2 input by 90%.32,33 More recently, Scherrer et al.34 demonstrated that the material initially forms as potassium uranyl triperoxide (KUT-1) K4[UO2(O2)3](H2O2)4(H2O)4 with oxidation of O22− to O2−˙ occurring in the solid-state material within 3 weeks under inert conditions. In aqueous media, this process occurred within 48 hours and enhanced water stability of O2−˙ coordinated to U(VI) was observed. These studies also found that the KUT-1 phase is a precursor to the potassium uranyl triperoxide (KUT, K4[UO2(O2)3]·4H2O) reported by Dembowski et al.35 KUT-1 and KUT phases are differentiated by their respective lattices where KUT-1 contains a potassium network with coordinated H2O2 and aqua ligands, while KUT contains only potassium ions and lattice waters (Fig. 1). Both KUT and KUT-1 can undergo O22− oxidation ultimately forming a uranyl diperoxo superoxide monomer in KUPS (K4−x[UO2(O2)3−x(O2−˙)x]·4H2O, 0 < x ≤ 1) and KUPS-1, respectively, with interconversion of these materials illustrated in Fig. 1. While the KUPS-1 compound has previously shown spontaneous carbonation in the presence of air, the behavior of the other related compounds is unknown.
 | ||
| Fig. 1 Conversion of (A) KUT-1 to (B) KUPS-1 following O22− oxidation or to (C) KUT following decomposition of H2O2 within the K+ network. (D) KUPS is formed after H2O2 decomposition of (B) or O22− oxidation of (C). Upon exposure to atmospheric CO2, the superoxide containing materials undergo direct air carbon capture to form uranyl carbonate phases ((E) uranyl peroxo-carbonate and (F) uranyl tricarbonate). | ||
Herein, we investigated optimal conditions promoting the carbonation of KUT-1, KUT, KUPS-1, and KUPS within a custom-built flow-through apparatus using both high purity CO2 and compressed air to mimic both point sources and direct air capture processes. Previous work noted that humid conditions influence the stability and efficiency of carbon capture sorbents, and ROS exhibit water sensitivity, which may lead to sorbent degradation and hinder carbonation.36–38 Hence, we explored carbonation in low, mid, and high relative humidity (RH) conditions. Elemental analysis, powder X-ray diffraction (PXRD), Raman spectroscopy, and electron paramagnetic resonance (EPR) spectroscopy were used to analyze the pure and reacted materials. Finally, the carbonation reaction kinetics for solid KUT-1, KUPS-1, KUT and KUPS were explored using a custom-built chamber equipped with a high sensitivity CO2 sensor.
Experimental methods
Materials synthesis
All aqueous solutions were prepared using Millipore water (18.2 MΩ) and chemicals purchased were used directly without further purification. Caution: UO2(NO3)2·6H2O contains radioactive238U, which is an α emitter, and like all radioactive materials must be handled with care. These experiments were conducted by trained personnel in a licensed research facility with special precautions taken toward the handling, monitoring, and disposal of radioactive materials. KUT-1 was synthesized and isolated as previously reported.34 To synthesize large quantities of material, batches of at least 40 synthetic 20 mL vials were prepared with each containing 0.2 mL 0.05 M uranyl nitrate hexahydrate (International Bio-Analytical Industries Inc., 99.99%) in methanol, 0.15 mL pure methanol (Fischer Chemical, ACS 99.9%) and 2.5 mL benzyl alcohol (Alfa Aesar, 99%). A separate solution containing 0.2 mL 0.9 M aqueous potassium hydroxide (Sigma-Aldrich, 90%), 0.3 mL 0.05 M aqueous N-(phosphonomethyl)iminodiacetic acid hydrate (Sigma-Aldrich, 95%) with potassium hydroxide for solubility, and 0.2 mL 30% aqueous H2O2 (Fisher Chemical, ACS 99.9%) was mixed via vortex and carefully layered on top of the organic layer for each scintillation vial. The vials were capped and stored in the dark for overnight crystallization.To isolate solid KUT-1, the contents of each scintillation vial were transferred to a 15 mL plastic conical tube and centrifuged at 5000 rpm for 5 minutes. Each conical tube was decanted followed by the addition of 5 mL methylene chloride (Fisher Chemical, >99.5%). Conical tubes were centrifuged for an additional 5 minutes at 5000 rpm ensuring any residual water was forced into the top aqueous layer. The aqueous layer was decanted and the KUT-1 solid was transferred to a watch glass for vacuum desiccator drying. Upon drying for a minimum of 20 minutes, the solid material was immediately utilized in the studies described herein. For the studies involving KUPS-1, the material was aged under argon gas. After about 3 weeks, the material was periodically checked for the presence of O2−˙ via EPR spectroscopy. Upon O2−˙ detection, materials were utilized or held under argon or vacuum. KUT-1 samples were characterized for purity using powder X-ray diffraction and Raman spectroscopy.
KUT was synthesized similarly to its previously reported protocol.35 In a 20 mL scintillation vial, 2 mL 0.5 M uranyl nitrate hexahydrate, 2 mL 30% H2O2 and 2 mL 8 M KOH were mixed. Approximately 1 mL of pure methanol was added slowly. Scintillation vials were capped, and a yellow precipitant formed rapidly, typically within five minutes but could take several hours. The resulting solid was isolated via vacuum filtration, rinsed with methanol and dried. Any material that was not used immediately was stored under argon. Before using any of the additional material, samples were checked for O2−˙ via EPR spectroscopy, and the purity was determined using X-ray diffraction and Raman spectroscopy. Studies involving KUPS utilized the material after detection of O2−˙ by EPR spectroscopy.
The uranyl peroxo-carbonate phase K4[UO2(O2)(CO3)2]·2.5H2O was also synthesized to serve as a model carbonated compound using a method adapted from Zehnder et al.39 and Goff et al.40 For the modified synthesis, UO2 powder was dissolved in 2 M K2CO3 and 30% H2O2 at room temperature. The solution was stirred for one hour and additional 30% H2O2 was added to the solution. Four mL aliquots of the solution were transferred to 20 mL scintillation vials and 6 mL methanol was layered onto the solution. The vials were capped and left to crystallize at room temperature in the dark overnight. Dark orange/red precipitate formed on the bottom of the vials and then the solid was vacuum filtered and dried. The resulting materials were characterized for purity using powder X-ray diffraction and Raman spectroscopy.
Details of reaction apparatus
Characterization of the synthesized and reacted materials
Principal Component Analysis (PCA) was implemented for further interpretation of the Raman spectral data in Origin 9.60 (OriginLab, Northampton, MA) 64-bit software PCA app.41 Normalized and background subtracted data was utilized with each wavenumber (2000–200 cm−1) representing the observations and the spectral intensities as the input data. The covariance matrix setting was used, and basic statistics, loadings, and scores were computed for each analysis. The number of significant principal components (PCs) was selected based on the number of components explaining a minimum of 95% total variance. Resulting calculations were plotted in either 2D or 3D, corresponding to the number of significant principal components.
Results and discussion
Confirmation of purity for the starting materials
Prior to flow-through experiments, materials characterization was performed on the as-synthesized materials (KUT-1, KUPS-1, KUT, and KUPS) and K4[UO2(O2)(CO3)2]·2.5H2O to serve as references. PXRD patterns of the synthesized materials matched well with patterns simulated from single-crystal X-ray diffraction data (KUT-1/KUPS-1 ICSD 135984, KUT/KUPS ICSD 243360, K4[UO2(O2)(CO3)2]·2.5H2O ICSD 173289 and K4[UO2(O2)(CO3)2]·H2O ICSD 59925) indicating purity of the bulk phase (ESI, Fig. S3–S7†).EPR spectroscopy was collected for the synthesized materials to confirm the absence/presence of O2−˙ (ESI, Fig. S8–S11†). Predicted features for O2−˙ in KUPS-1 are observed at g‖ = 2.057 and g⊥ = 2.017 (gx = 2.016, gy = 2.017, gz = 2.057).34 The predicted EPR spectrum of KUPS has yet to be reported, though the principal g-factors are presumably the same given the structural similarities with KUPS-1 specifically regarding the uranyl monomeric unit. Experimentally observed values are consistent with the predicted features for both KUPS-1 (g‖ = 2.056 and g⊥ = 2.014) and KUPS (g‖ = 2.057 and g⊥ = 2.014). The additional feature at g = 2.037 observed in the KUPS EPR spectrum could correspond to the g‖ of a second O2−˙ species, hence the apparent broadening of the g⊥ = 2.014 signature, however efforts to understand these interactions between paramagnetic species and U(VI) are ongoing. Lack of these features in the KUT-1 and KUT materials implies the presence of diamagnetic (EPR silent) O22− within the triperoxide phases. Additional features observed in all materials are consistent with trace Cr(V) impurities that are known to exist within these materials.34 The signature at g = 1.982 is consistent with the Cr(V)–peroxo complex [Cr(V)(O)(O2)(OH2)x]+ and hyperfine splitting at g = 1.994 and g = 1.953.42 The isotropic feature at g = 1.972 also matches a Cr(V)–peroxo complex in the form of [Cr(V)(O2)4]3−.42
Raman spectral features for all starting compounds were also collected and the features were analyzed to confirm vibrational modes (ESI, Fig. S12–S15†). Fitted spectrum of pure KUT-1 shows six bands in the spectroscopic window of interest and is well-aligned with previous reports of these phases.32 The highest intensity band at 727 cm−1 corresponds to ν1 symmetric stretch of UO22+.32 Bands at 812, 825, 849 and 880 cm−1 correspond to ν1–3 motions of O22− bound to UO22+.35 The band appearing at 860 cm−1 corresponds to νO–O stretch of H2O2 coordinated to potassium within the potassium network.43 Upon conversion to KUPS-1, the Raman features remain in the same range as in KUT-1 but do exhibit shifts related to the O22− features (from 825 to 821 cm−1) and ν1 symmetric stretch of UO22+ (from 727 to 722 cm−1). The fitted spectrum of KUT is quite similar with vibrational modes observed at 841, 822, and 811 cm−1 for the ν1–3 motions of O22− bound to UO22+ and 712 cm−1 corresponding to the ν1 UO22+ symmetric stretch. These features are in good agreement with those previously reported by Dembowski et al.35 We note additional small features at about 1060 and 756 cm−1 corresponding to surface CO32− species.32,44 Efforts were taken to minimize the presence of any carbonate species in the starting materials, however given the highly reactive nature of these phases, they could not always be prevented from forming during sample transfer processes. When KUT transforms to the superoxide-containing phase KUPS, the Raman features only vary by 2–3 cm−1, which is within error of the instrument.
The uranyl peroxo-carbonate K4[UO2(O2)(CO3)2]·2.5H2O (ESI, Fig. S16†) model compound shows several distinct features in its vibrational spectrum. Most notably are the appearance of three bands at 1065, 1053 and 1030 cm−1 indicative of CO32− species, in addition to the most intense signal at 767 cm−1 corresponding to ν1 UO22+. Lower intensity bands at 693 and 716 cm−1 (CO32−) and 806 cm−1 (UO22+) are also identified. The remaining features at 840 and 850 cm−1 correspond to O22− bound to UO22+. The Raman spectrum of K4[UO2(O2)(CO3)2]·2.5H2O is well aligned with the previously published spectrum.40 Spectral assignments are summarized in Table 1.
| KUT-1 | KUPS-1 | KUT | KUPS | UPDC | Assignment |
|---|---|---|---|---|---|
| 1065 | ν C–O for CO32- | ||||
| 1052 | ν C–O for CO32- | ||||
| 1029 | ν C–O for CO32- | ||||
| 880 | 880 | ν O–O for H2O2 within lattice | |||
| 860 | 868 | ν O–O stretch for H2O2 within lattice | |||
| 849 | 848 | 850 | ν 1–3 for O22− bound to UO22+ | ||
| 841 | 844 | 840 | ν 1–3 for O22− bound to UO22+ | ||
| 825 | 821 | 822 | 826 | ν 1–3 for O22− bound to UO22+ | |
| 812 | 800 | 811 | 814 | 806 | ν 1–3 for O22− bound to UO22+ |
| 727 | 722 | 712 | 715 | 767 | ν 1 for UO22+ |
| 716 | ν C–O for CO32- | ||||
| 692 | ν C–O for CO32- |
To quantitatively assess the carbonation of KUT-1, KUPS-1, KUT and KUPS, we also preformed CHN elemental analysis on the as synthesized samples to determine the amount of CO2 captured by the material. On average, the pristine materials contained 0.045–0.095% carbon, or 0.027–0.057 mol C per mol U, confirming maximum purity. Average carbon contents for the KUT-1, KUPS-1, KUT and KUPS flow-through studies can be found in the ESI section (ESI, Table S1†).
Following flow-through experiments of KUT-1 with pure CO2, amounts of carbon detected were highly dependent on the relative humidity throughout gas exposure (Fig. 2a). At 15% RH, the material contained an average of 0.207% carbon (0.125 mol C per mol U). Given the small change in the carbon content, we hypothesized that the detected carbon is primarily adsorbed CO2. When RH is increased to 45% and 70% throughout CO2 exposure, the average carbon contents of the materials were 4.800% (2.896 mol C per mol U) and 4.290% (2.588 mol C per mol U) respectively, suggesting conversion of KUT-1 to K4[UO2(O2)(CO3)2]·H2O (theoretical 4.025% C) or K4[UO2(O2)(CO3)2]·2.5H2O (theoretical 3.850% C), and likely small amounts of K4[UO2(CO3)3] (theoretical 5.936% C) or other carbonate rich phases (i.e., K2CO3, theoretical 8.683% C).
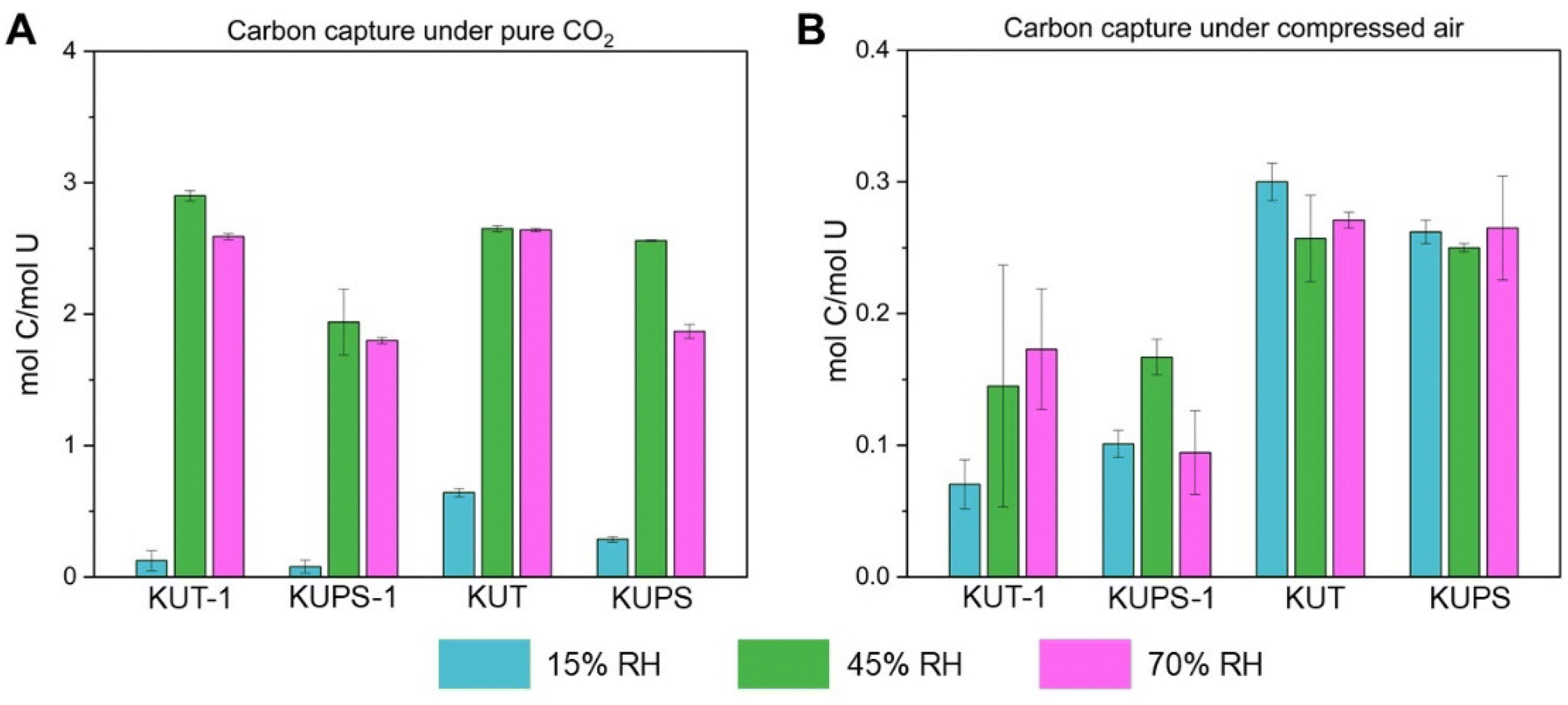 | ||
| Fig. 2 Elemental analysis of KUT-1, KUPS-1, KUT and KUPS following flow-through studies at 15%, 45%, and 70% relative humidity under pure CO2 (A) and compressed air (B). | ||
Flow-through experiments of KUT-1 with compressed air similarly suggest dependence on relative humidity, while additionally revealing the significance of CO2 concentration (Fig. 2b). Samples at 15%, 45% and 70% RH were found to contain 0.117% (0.07 mol C per mol U), 0.240% (0.145 mol C per mol U) and 0.287% carbon (0.173 mol C per mol U), respectively. Therefore, negligible carbonation of KUT-1 occurs under ambient CO2 concentrations in the two hours of air exposure.
Similar trends were observed in flow-through experiments with KUPS-1, KUT and KUPS, such that optimal carbonation occurs at higher relative humidity and higher CO2 content. Both KUT and KUPS exposed to pure CO2 showed improved carbon capture at low relative humidity compared to KUT-1 and KUPS-1, though still significantly less than at higher RH. Carbon contents for KUPS-1 and KUPS exposed to ambient air in the flow-through system, regardless of RH, were comparable to that after pure CO2 15% RH exposure. Following ambient air exposure across all RH values, KUT exhibited consistent carbon contents, which was over a 57% decrease in the carbon captured compared to low RH, pure CO2 exposure. This implies that the presence of O2−˙ enhances the carbon capture reaction under ambient [CO2]. It is highly likely that O2−˙ forms within the triperoxide phases as an intermediate step in the reaction mechanism, thus more CO2 uptake is observed when the starting material already contains O2−˙.
Both KUT and KUPS exhibit increased carbon capture under air at all relative humidity conditions compared to KUT-1 and KUPS-1. As outlined previously, the major difference between these phases is the presence of H2O2 in the crystalline lattice for KUT-1/KUPS-1, which is absent for KUT/KUPS. Therefore, the K+/H2O2 network in the KUT-1 and KUPS-1 compounds must impact the CO2 absorption or reactivity, thus slowing down the process of CO2 reacting with the uranyl peroxide monomer. However, the reasoning behind this change in reactivity was not clear, so we turned to vibrational analysis to understand changes in speciation throughout the process.
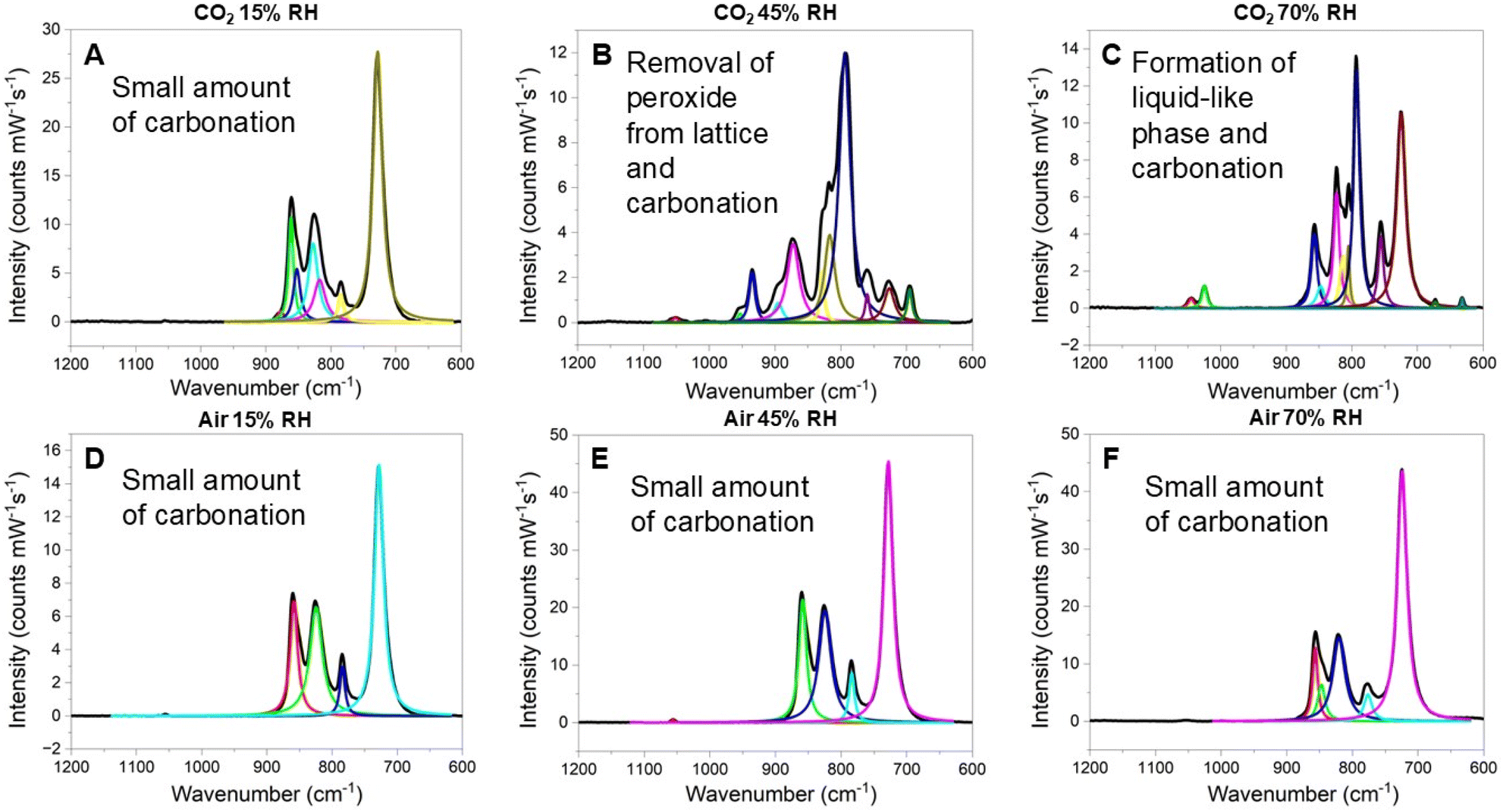
Influence of relative humidity. Following 2 hours of CO2 exposure at 15, 45 and 70% RH, Raman spectra were obtained, and the features were fit to further evaluate the overall changes. Flow-through studies at 15% RH yielded no substantial changes in the vibrational spectra or in the PXRD of the reacted material, indicating no carbonate formation (Fig. 3a and ESI Fig. S17†). This confirms the results from the elemental analysis suggesting that the detected carbon is primarily a small amount of adsorbed CO2. When RH is increased to 45 and 70%, the Raman spectra suggests that the peroxide within the lattice has reacted and overall, the U(VI) peroxo complex begins to convert to a uranyl peroxo-carbonate material (Fig. 3b and c).40 Features associated with peroxide in the lattice (i.e., 849, 860, and 880 cm−1) are absent, indicating that the peroxide within the lattice has reacted under these higher relative humidity conditions. Ingrowth of vibrational bands at 1048 and 1031 cm−1 correspond to ν1 CO32− stretches that are bound to U(VI) as those associated with K2CO3 typically occur at 1067 cm−1.45 The highest intensity band at 800 cm−1 also aligns well with ν1 UO22+ within uranyl carbonate complexes. Additional weaker bands at 677 and 636 cm−1 were also observed and are not typically observed within solid-state carbonate materials. However, these features can be observed for ultra concentrated carbonate and bicarbonate solutions (3.5–8.0 M) and have been assigned to carbonate ion-water restricted rotations.46 As the KUT-1 material absorbs a significant amount of water in the high RH environment, the presence of carbonate-ion water pairing is reasonable. In addition, a small feature at 1004 cm−1 is observed in the 70% RH sample. It could correspond to HCO3− as typical literature examples of HCO3− vibrational modes identify a ν5 C–OH stretch at 1017 cm−1.44,45 The slight redshift would imply secondary interactions within the coordinating environment; however, this assessment has not been fully confirmed. In general, the Raman spectra support that higher RH induces carbon capture with conversion of KUT-1 to a uranyl peroxo-carbonate phase. Lastly, bands at 828, 817, and 727 cm−1 remain and could correspond to the original material, indicating that not all the original KUT-1 has converted at this point.32 Bands at 817 and 727 cm−1 have also been correlated to ν1 UO22+ bound to three CO32− ligands and ν1 CO32− bound to U, respectively, which could indicate some degree of conversion to K4[UO2(CO3)3].32,47 Given the viscous nature of the sample initially after the reaction at 45 and 70% RH, no PXRD data could be collected for these materials.
 | ||
| Fig. 3 Vibrational spectra following flow-through studies with KUT-1 exposed to pure CO2 at (A) 15%, (B) 45% and (C) 70% RH. Vibrational spectra of KUT-1 following exposure to compressed air at 15%, 45% and 70% RH are shown as (D), (E), and (F), respectfully. | ||
Additional transformation of the 45 and 70% RH samples were noted after being aged under ambient conditions for several days. Visually, the orange viscous material transformed to a dry red/orange powder over the course of three days in a sealed glass vial. The Raman spectrum revealed significant changes in the vibrational features, with bands at 1054, 1050, 850, 840, 790, 768, 714, and 692 cm−1, which are consistent with K4[UO2(O2)(CO3)2]·2.5H2O (ESI, Fig. S18†).40 Additional phase confirmation was provided by the PXRD of the dried powder, which confirmed the formation of crystalline K4[UO2(O2)(CO3)2]·2.5H2O and K4[UO2(CO3)3] (ESI, Fig. S19†). This further supports that the vibrational spectra of the samples immediately following flow-through experiments represent an intermediate phase in the carbonation reaction.
Influence of CO2 concentration. After analysis of materials upon exposure to pure CO2, flow-through experiments were repeated utilizing compressed air at 15, 45 and 70% RH. Raman spectra of all samples are largely consistent with the starting KUT-1 material, suggesting that negligible carbonation occurred within the 2 hour period under ultra-dilute CO2 concentrations (Fig. 3d–f). PXRD of the material after the 2 h reaction time also showed no changes from the initial material (ESI, Fig. S20†). Quantification of CO2 captured (vide supra) suggests influence of RH even at low CO2 concentrations, though much less pronounced. This implies that, while CO2 concentration is the primary factor influencing the carbonation of KUT-1, reactivity is enhanced under higher RH. Additionally, we note that the EPR spectrum of KUT-1 following 2 hours of air exposure now reveals a small feature consistent with O2−˙ in KUPS-1 (ESI, Fig. S21†). This indicates rapid formation of O2−˙ under ambient conditions as previously noted and may contribute to the carbonation reaction mechanism.
Transformation of the samples exposed to compressed air was also observed after aging under ambient conditions for three days. The visual change was more subtle, with the yellow powders converting to a yellowish orange color, but numerous spectroscopic variations were noted for the vibrational features. Raman spectrum of the material three days after the exposure to compressed air displayed vibrational modes at 844 (ν1 O22−), 825 (ν3 O22−), 814 (ν2 O22−), and 716 (ν1 UO22+) cm−1 consistent with that of KUT (ESI, Fig. S22†).35 PXRD further confirms that the material converted to the KUT/KUPS phase (ESI, Fig. S23†) suggesting decomposition of the K+/H2O2 network surrounding the uranyl monomer. Further transformation of the material was observed after three additional days sealed under ambient conditions with the vibrational spectrum and diffraction pattern suggesting conversion to K4[UO2(O2)(CO3)2]·2.5H2O (ESI, Fig. S24 and S25†).40 Presence of carbonation after the formation of O2−˙ within material suggests that the ROS species is a component of the reaction mechanism in the direct air carbon capture.
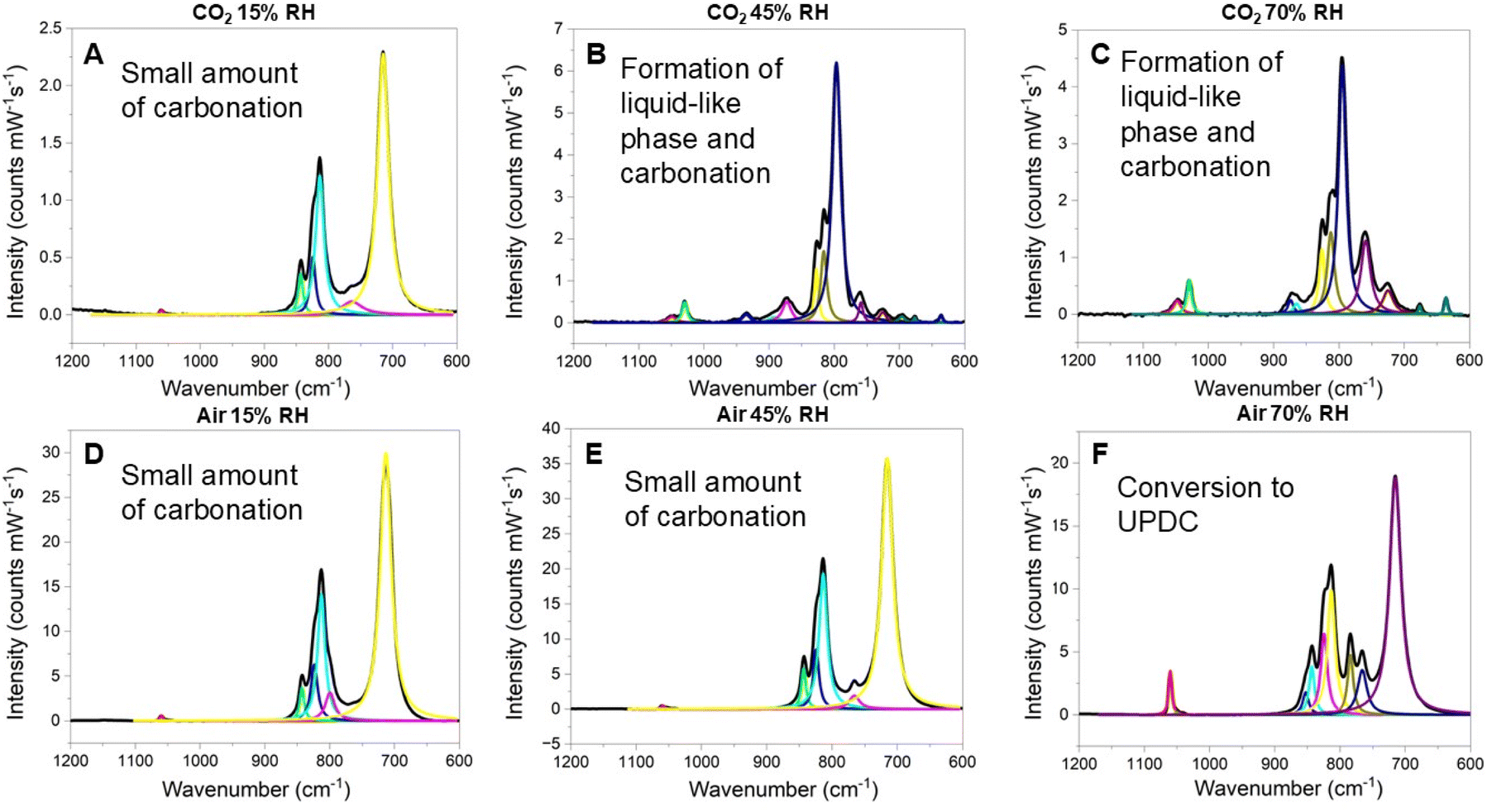
Influence of superoxide and secondary coordination sphere. Flow-through studies varying the relative humidity, and the specific gas (CO2 or compressed air) were repeated with KUPS-1 to gain additional insight on the influence of superoxide for the carbonation reaction. The vibrational spectra (Fig. 4) are largely consistent with those for KUT-1, although a feature, albeit very weak, at 1056 cm−1 is observed in all three of the samples exposed to compressed air. This supports the earlier statement from the elemental analysis data in that KUPS-1 exhibits enhanced carbon capture under dilute CO2 concentrations. PXRD of the KUPS-1 samples exposed to air and CO2 at 15% RH show features consistent with the starting material, in addition to new lower intensity features associated with UO2CO3 (ref. 48 and 49) (ESI, Fig. S26 and S27†). The KUPS-1 45% and 70% RH CO2 spectra differ from those of KUT-1 in that many additional features are observed in the window of interest. KUPS-1 exposed to pure CO2 at 45% RH for two hours includes a band at 1052 cm−1 that corresponds to a carbonate feature. In addition, there are bands at 953, 935 and 895 cm−1 that are not observed in any other KUT-1 or KUPS-1 reacted samples. These bands cannot be definitively assigned, but features in that region were also noted by Mestl et al.50 in the decomposition of BaO while heating and were attributed to non-crystalline peroxide species. Bands at 873, 828, 817, and 727 cm−1 could correspond to residual KUPS-1 in the samples or to carbonate bound to U vibrations similar to those observed in reacted KUT-1.32 Given the spectral overlap, it is difficult to conclusively identify these features. When the samples are exposed to 70% RH, there are more residual uranyl triperoxide spectral signatures remaining in the sample, but we observe additional evidence of carbonation (bands at 1045 and 1025 cm−1), including the formation of the uranyl tricarbonate complex (806 cm−1 consistent with the reported U
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational mode).51 Additionally, there is evidence that the hygroscopic sample becomes a concentrated liquid-like form, with features at 673 and 632 cm−1. It is also important to point out the absence of features that would signify the formation of the uranyl peroxo-carbonate phase, particularly the peroxo stretches at 850 and 840 cm−1 and the uranyl stretch at 767 cm−1.
O vibrational mode).51 Additionally, there is evidence that the hygroscopic sample becomes a concentrated liquid-like form, with features at 673 and 632 cm−1. It is also important to point out the absence of features that would signify the formation of the uranyl peroxo-carbonate phase, particularly the peroxo stretches at 850 and 840 cm−1 and the uranyl stretch at 767 cm−1.
 | ||
| Fig. 4 Vibrational spectra following flow-through studies with KUPS-1 exposed to pure CO2 at (A) 15%, (B) 45% and (C) 70% RH. Vibrational spectra of KUPS-1 following exposure to compressed air at 15%, 45% and 70% RH are shown as (D), (E), and (F), respectfully. | ||
These studies were also repeated with KUT and KUPS to gain insight on the influence of changes in the secondary coordination sphere as both samples do not contain H2O2 coordinated to the K+ cations in the lattice (Fig. 5 and 6). The KUT sample contained a small amount of surface carbonate in the initial material so there was no significant reaction of the compound when exposed to compressed air. PXRD of the KUT materials following exposure to air indicate the remaining KUT phase, as well as small features aligned with UO2CO3 (ESI, Fig. S28 and S29†). Under pure CO2 at 15% RH, there was a second feature at 1055 cm−1, as well as a feature at 766 cm−1, suggesting that the formation of a small amount of the uranyl peroxo-carbonate.40 In addition, there are features at 884 and 783 cm−1 that match well with the formation of UO2CO3.48,49 The KUT vibrational features from the pristine material also blue shifted to those observed for the KUPS material, suggesting that the peroxide is beginning to oxidize. PXRD of the KUT compound exposed to CO2 at 15% RH indicates that some KUT/KUPS remains crystalline, but there are also features that confirm a small amount of UO2CO3 in the sample (ESI, Fig. S30†). As the RH increases, the sample again absorbs a significant amount of water and the reactivity continues with carbonation (1047, 1029, and 635 cm−1), reactive peroxide species (935 cm−1), and free peroxide (875 cm−1). This behavior continues as the RH increases to 70%. Reactivity is also observed for KUPS with a small amount of carbonation of the sample in air confirmed by PXRD (ESI, Fig. S31†) with the appearance of weak diffraction peaks associated with UO2CO3, which matches well with the elemental analysis. Similarly, some level of carbonation is observed for all KUPS samples that are exposed to CO2, with additional uranyl carbonate features appearing with increasing relative humidity.
 | ||
| Fig. 5 Vibrational spectra following flow-through studies with KUT exposed to pure CO2 at (A) 15%, (B) 45% and (C) 70% RH. Vibrational spectra of KUT following exposure to compressed air at 15%, 45% and 70% RH are shown as (D), (E), and (F), respectfully. | ||
 | ||
| Fig. 6 Vibrational spectra following flow-through studies with KUPS exposed to pure CO2 at (A) 15%, (B) 45% and (C) 70% RH. Vibrational spectra of KUPS following exposure to compressed air at 15%, 45% and 70% RH are shown as (D)–(F), respectfully. | ||
PCA-based spectral analysis. Due to complexity of the system, we employed Principal Component Analysis (PCA) on the Raman data to identify precise features unique to phases throughout the progression of the carbonation reaction and determine trends. The first PCA iteration included samples from flow-through studies with pure CO2 and compressed air, aged samples, and K4[UO2(O2)(CO3)2]·2.5H2O associated with the KUT-1 materials. The loadings plot reveals three distinct phases representing various stages in the carbonation reaction including the starting material (PC1), intermediate phase (PC2) and fully converted (PC3) (Fig. 7a). Then analyzing the computed biplot, we can reference the loadings with specific experimental data (e.g., wavenumber). The biplot reveals which Raman features fall within the 95% confidence ellipsoid, meaning those wavenumbers are observed in all samples and not unique to a specific phase in the reaction. Points falling outside of the ellipsoid are thus significant to the spectra of corresponding loadings. These are identified as 728, 800, and 767 cm−1 for PCs 1, 2 and 3 respectively. We can then note that the band at 800 cm−1 occurs most often when there are other features associated with carbonate brine formation, whereas the feature at 728 cm−1 is unreacted KUT-1 (specifically the U
O Raman active mode),32 and 767 cm−1 is the formation of the K4[UO2(O2)(CO3)2]·2.5H2O phase (UO vibrational mode).40 Thus, for KUT-1 and KUPS-1, the material is minimally reactive under air or CO2 at low relative humidities. Under higher relative humidities, the material forms a brine-like substance that then converts to the uranyl peroxo-carbonate forms after drying.
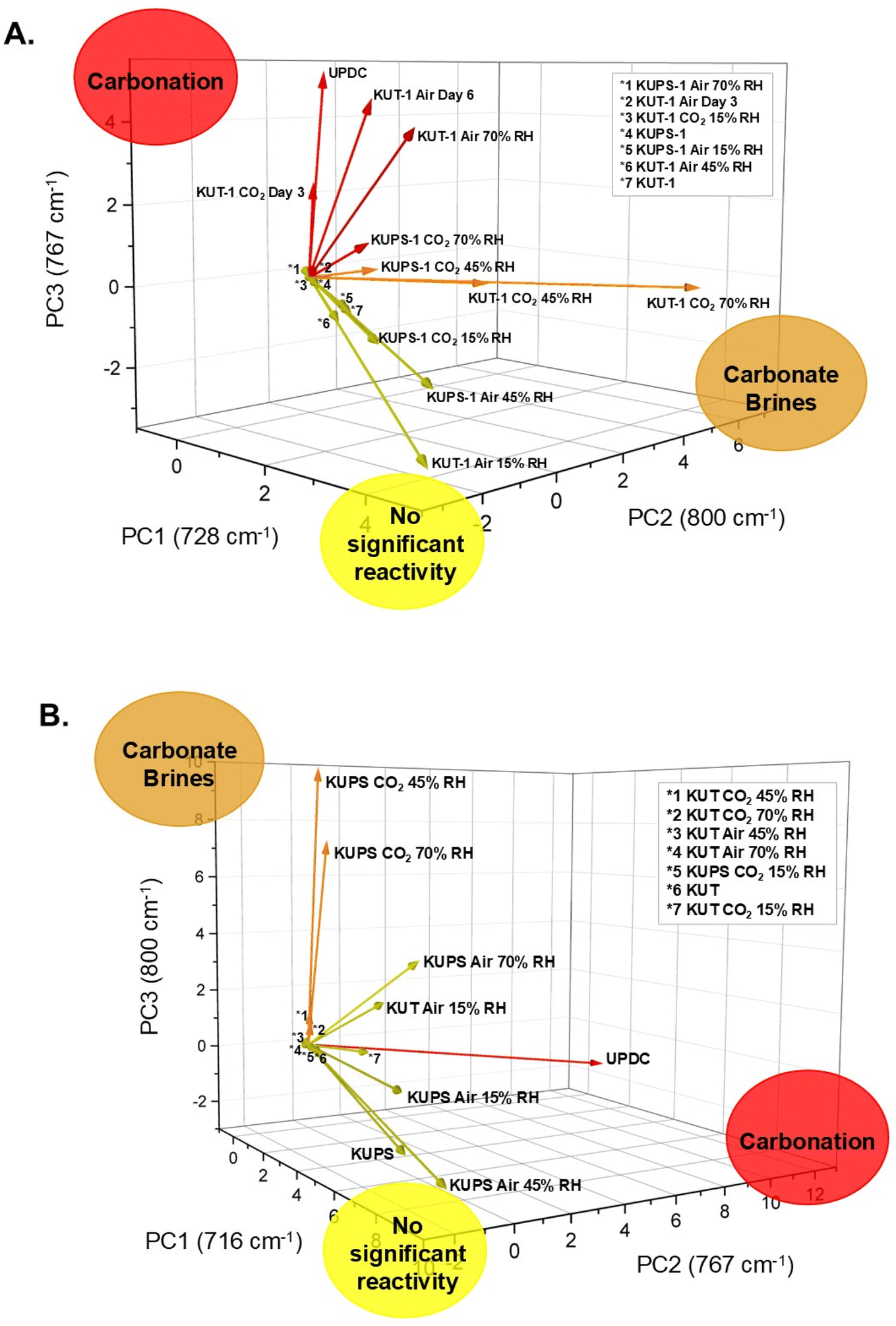 | ||
| Fig. 7 Principal component analysis (PCA) of the Raman data for (A) KUT-1 and KUPS-1, and (B) KUT and KUPS. PC analysis reveals trends in reactivity of these phases under varying relative humidity and [CO2] conditions and indicate no significant reactivity in low RH and/or low [CO2] environments, transformation to carbonate brines under high RH conditions, and transformation to uranyl peroxo-carbonate (UPDC) under mid-high RH environments. The three distinct phases are correlated to the presence of specific Raman features at 728(A)/716(B) cm−1 (no significant reactivity), 800 cm−1 (carbonate brines), and 767 cm−1 (carbonation). | ||
The second PCA iteration included samples from CO2 and compressed air flow-through experiments with KUT and KUPS and K4[UO2(O2)(CO3)2]·2.5H2O. The resulting loadings plot (Fig. 7b) and biplot are largely consistent with the first PCA iteration in that three distinct phases of the carbonation reaction are highlighted. PC1 is again identified as the unreacted materials but corresponds to 716 cm−1 due to the shift in the uranyl symmetric stretch within the pristine materials Raman spectra.35 PC2 (767 cm−1) is identified as the carbonated materials and PC3 (800 cm−1) corresponds to carbonate brines. While the specific identifications of PC2 and PC3 are reversed from the first PCA iteration, this does not affect the overall interpretation of the Raman data and is likely due to the lower intensities of the associated samples. The same conclusions are still drawn in that KUT and KUPS are minimally reactive under air or CO2 at low relative humidities and form liquid-like carbonate brines under high relative humidity.
Influence of gas flow rate and exposure time. Since the transformation of KUT-1 to the potassium uranyl peroxo-carbonate is slower at atmospheric CO2 concentrations, additional factors may be more pronounced under such conditions. As such, we first qualitatively assessed the influence of gas flow rate and exposure time on the reactivity of KUT-1 exposed to either the ambient conditions or within the flow through system. The Raman spectrum of KUT-1 under ambient conditions suggested some degree of carbonation in as little as 24 hours with a band appearing at 1052 cm−1 (ESI, Fig. S32†). Additional bands were observed at 852, 844, 835, 784, 768, 717 and 691 cm−1 and are well aligned with features observed in the earlier flow-through studies. Powder diffraction of the dried materials further revealed that the material is a mixed phase of KUT-1 and K4[UO2(O2)(CO3)2]·2.5H2O (ESI, Fig. S33†). Vibrational spectrum of KUT-1 exposed to constant air flow for approximately 24 hours reveals bands at 844, 825, 814, and a strong band at 716 cm−1 associated with ν1 UO22+ for KUT (ESI, Fig. S34†).35 The Raman spectrum and diffraction pattern (ESI, Fig. S35†) are both consistent with KUT meaning the higher flow rate led to decomposition of the K+/H2O2 network in KUT-1 yielding the KUT phase. Therefore, the H2O2 in the crystalline lattice is reactive to the air but does not directly correlate with carbonation.
Previous studies have identified that the carbonation reaction of both potassium uranyl triperoxide and potassium uranyl diperoxo superoxide are thermodynamically favorable,32,52 though details of the reaction kinetics have not been reported. As such, carbonation kinetics were determined for the reaction of the solid-state materials (KUT-1, KUPS-1, KUT and KUPS) with atmospheric CO2. The kinetic data were modeled to four adsorption models and referenced to the carbonation kinetics for potassium superoxide (KO2) pellets reported by Chegeni et al.53 (Table 2 and ESI Fig. S36–S55†). Based upon the R2 values, carbonation for each of the four systems best follows second-order kinetics. The second order model assumes adsorption rate is dependent on chemisorption meaning the chemical reaction between the solid material and CO2 drives the rate of carbonation.54 The experimental data also fits well to the first-order model, which could indicate that the carbonation reaction for all systems follows pseudo-first-order kinetics. The Elovich55,56 and Richie second-order57 models consistently resulted in the lowest R2 values. The former is used to describe adsorption onto heterogenous surfaces, while the latter describes the adsorption of gases at vacant surface sites on a solid. Behind the first and second-order models, the data fits decently well with the rate controlling model which indicates intraparticle diffusion of the adsorbate through the pores of the adsorbent.56,58,59 Bulk diffusion likely has some effect on the carbonation reaction, however chemisorption appears to be the driving force behind the reaction kinetics.
| Kinetic model | Nonlinear form | Parameter | KUT-1 | KUPS-1 | KUT | KUPS | KO2 |
|---|---|---|---|---|---|---|---|
| First-order | q t = qe(1 − e−tk1) | k 1 | 0.094 | 0.455 | 1.221 | 1.839 | 0.0324 |
| R 2 | 0.978 | 0.959 | 0.998 | 0.992 | 0.9348 | ||
| Second-order |

|
k 2 | 2.195 × 10−6 | 8.441 × 10−6 | 1.107 × 10−4 | 1.310 × 10−4 | 3.3 × 10−4 |
| R 2 | 0.993 | 0.992 | 0.986 | 0.998 | 0.9633 | ||
| Elovich |
q
t
= β × log(α × β) + β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(t) log(t) |
α | 2.173 × 107 | 1.572 × 108 | 4.067 × 105 | 6.627 × 108 | 0.0279 |
| R 2 | 0.777 | 0.851 | 0.937 | 0.883 | 0.9763 | ||
| Ritchie second-order |
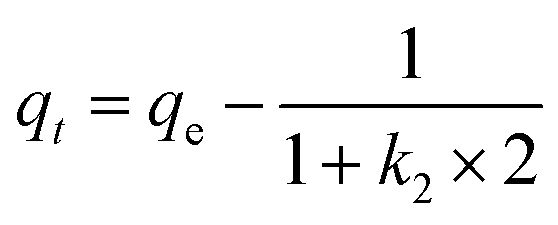
|
k 2 | 0.249 | 3.796 | 7.116 | 13.902 | 348.63 |
| R 2 | 0.912 | 0.782 | 0.921 | 0.928 | 0.5656 | ||
| Rate controlling | q t = kid × t0.5 | k id | 43.245 | 60.282 | 128.171 | 97.610 | 2.1639 |
| R 2 | 0.956 | 0.991 | 0.985 | 0.957 | 0.9849 |
Rate constants from the first- and second-order models reveal multiple trends regarding the carbonation reaction of the four systems. First, KUT-1 and KUPS-1 exhibit considerably slower kinetics compared to KUT and KUPS. This offers further support that the H2O2 in the crystalline lattice of KUT-1 and KUPS-1 is not directly correlated to the carbonation reaction. Additionally, the phases containing O2−˙ at the onset of air exposure (KUPS-1 and KUPS) were kinetically faster than their triperoxide analogues (KUT-1 and KUT). Only trace amounts of peroxide oxidation occur in the bulk solid meaning the O2−˙-containing uranyl monomers exist as defects within the material. Despite forming in such small quantities, the presence of O2−˙ directly influences reactivity of the bulk. This explains why the second-order rate constant for KO2 pellets is slightly larger than the constants reported in this study, as the O2−˙ concentration within KO2 is higher than in the uranyl diperoxo superoxide materials; though, the slightly higher relative humidity utilized for those studies could have also contributed to faster kinetics.53 The reaction kinetics demonstrate enhanced carbonation by uranyl diperoxo superoxide relative to uranyl triperoxide and are consistent with literature reports of enhanced carbon capture with O2−˙ containing materials.21,24,53
Conclusion
Herein, we have reported optimal conditions promoting the carbonation of potassium uranyl triperoxide and potassium uranyl diperoxo superoxide phases within a flow-through system at 22 °C. Carbonate formation was monitored via Raman spectroscopy and elemental analysis. Flow-through studies under pure CO2 revealed conversion to the uranyl peroxo-carbonate phase when relative humidity is at least 45%. Subsequent flow-through studies under compressed air reveal additional factors influencing the carbonation reaction, including CO2 concentration, gas flow rate, and the presence of O2−˙. Further, flow-through studies in which the triperoxide starting materials were exposed to compressed air revealed the formation of O2−˙ but no initial carbonation, suggesting that oxidation of O22− to O2−˙ is an early step in the reaction mechanism. These findings were reflected in the reaction kinetics of solid-state KUT-1, KUPS-1, KUT and KUPS with atmospheric CO2, which follow pseudo first-order kinetics. Notably, these studies found that the carbonation of U(VI) diperoxo superoxide phases are more kinetically favorable than their analogous U(VI) triperoxide phases.Overall, this study demonstrates conditions, namely relative humidity and presence of O2−˙ within the adsorbent, which promote the transformation of uranyl peroxide materials to uranyl carbonate phases via CO2 removal from ambient air. Our findings present a more efficient sorbent than recently reported transition metal (e.g., vanadium) peroxides in terms of uptake capacity and reaction kinetics, provided for by the ability of U(VI) to stabilize ROS long enough to allow for reaction with CO2. Further, our findings provide insight into the carbonation reaction mechanism, but additional efforts are ongoing to identify specific mechanistic steps for this process. While the current study shows enhanced reactivity for the solid-state potassium uranyl diperoxo superoxide materials, we acknowledge that their reactivity in solution is unknown. Additional studies are therefore necessary to evaluate the carbonation reaction of these materials in solution to determine the possible catalytic nature of these materials within strong base DAC processes.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy Basic Energy Sciences (DE-SC0023479). We acknowledge the University of Iowa Materials Analysis, Testing and Fabrication Facility, Dr Daniel Unruh for support associated with the X-ray diffractometer and Raman spectrometer, and Dr Michael Sinnwell for support associated with the elemental analyzer.References
- A. Allangawi, E. F. H. Alzaimoor, H. H. Shanaah, H. A. Mohammed, H. Saqer, A. A. El-Fattah and A. H. Kamel,
C
, 2023, 9, 17–46 CAS
.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed
- M. Ozkan, S. P. Nayak, A. D. Ruiz and W. Jiang, iScience, 2022, 25, 103990 CrossRef CAS
- K. Lackner, H.-J. Ziock and P. Grimes, Report mumber: LA-UR-99-583, United States, 1999.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed
- E. Unveren, B. Monkul, Ş. Sarıoğlan, N. Karademir and E. Alper, Petroleum, 2016, 3, 37–50 CrossRef
- M. Mahmoudkhani and D. W. Keith, Int. J. Greenhouse Gas Control, 2009, 3, 376–384 CrossRef CAS
- A. A. H. I. Mourad, A. F. Mohammad, M. Altarawneh, A. H. Al-Marzouqi, M. H. El-Naas and M. H. Al-Marzouqi, Int. J. Energy Res., 2021, 45, 13952–13964 CrossRef CAS
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed
- S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 16873–16876 CrossRef CAS PubMed
- M. Saeidi, A. Ghaemi, K. Tahvildari and P. Derakhshi, J. Chin. Chem. Soc., 2018, 65, 1465–1475 CrossRef CAS
- P. Smith, S. Davis, F. Creutzig, S. Fuss, J. Minx, B. Gabrielle, E. Kato, R. Jackson, A. Cowie, E. Kriegler, D. Vuuren, J. Rogelj, P. Ciais, J. Milne, J. Canadell, D. McCollum, G. Peters, R. Andrew, V. Krey and C. Yongsung, Nat. Clim. Change, 2015, 6, 42–50 CrossRef
- G. Leonzio, P. S. Fennell and N. Shah, Appl. Sci., 2022, 12, 2618 CrossRef CAS
- X. Zhang, Z. Song, R. Gani and T. Zhou, Ind. Eng. Chem. Res., 2020, 59, 2005–2012 CrossRef CAS
- X. Lan and R. Keeling, https://gml.noaa.gov/ccgg/trends/data.html, accessed April 2025.
- U. Stoin, Z. Barnea and Y. Sasson, RSC Adv., 2014, 4, 36544–36552 RSC
- J. S. Hirschi, M. Nyman and T. J. Zuehlsdorff, J. Phys. Chem. A, 2024, 128, 7785–7794 CrossRef CAS
- E. G. Ribó, Z. Mao, J. S. Hirschi, T. Linsday, K. Bach, E. D. Walter, C. R. Simons, T. J. Zuehlsdorff and M. Nyman, Chem. Sci., 2024, 15, 1700–1713 RSC
- E. Shirman and Y. Sasson, Greenhouse Gases:Sci. Technol., 2024, 14, 1037–1048 CrossRef CAS
-
H. T. Lin, Y. C. Chao, H. W. Hsu, C. A. Chen, G. B. Chen and F. H. Wu, presented in part at the ASPACC 2019, Fukuoka, Japan, 2019 Search PubMed
- B. Yu, B. Zou and C.-W. Hu, J. CO2 Util., 2018, 26, 314–322 CrossRef CAS
- V. Kumar, N. Kumar Lamba, A. Baig, A. Kumar Sonker, N. Sharma, J. Kaushik, K. Malika Tripathi, Sonal and S. Kumar Sonkar, Chem. Eng. J., 2024, 500, 156786 CrossRef CAS
- Z. Mao, M. Rashwan, E. Garrido Ribó, M. Nord, L. N. Zakharov, T. W. Surta, A. Uysal and M. Nyman, J. Am. Chem. Soc., 2024, 146, 19489–19498 CrossRef CAS PubMed
- K. Bach, E. Garrido Ribó, J. S. Hirschi, Z. Mao, M. T. Nord, L. N. Zakharov, K. A. Goulas, T. J. Zuehlsdorff and M. Nyman, Chem. Mater., 2025, 37, 48–61 CrossRef CAS
- M. Fairley, G. E. Sigmon and J. A. LaVerne, Inorg. Chem., 2023, 62, 19780–19785 CrossRef CAS PubMed
- Z. C. Emory, J. A. LaVerne and P. C. Burns, Dalton Trans., 2024, 53, 17169–17178 RSC
- D. V. Kravchuk, N. N. Dahlen, S. J. Kruse, C. D. Malliakas, P. M. Shand and T. Z. Forbes, Angew. Chem., Int. Ed., 2021, 60, 15041–15048 CrossRef CAS PubMed
- D. V. Kravchuk and T. Z. Forbes, Angew. Chem., Int. Ed., 2019, 58, 18429–18433 Search PubMed
- S. K. Scherrer, H. Rajapaksha, D. V. Kravchuk, S. E. Mason and T. Z. Forbes, Chem. Commun., 2024, 60, 10584–10587 RSC
- M. Dembowski, V. Bernales, J. Qiu, S. Hickam, G. Gaspar, L. Gagliardi and P. C. Burns, Inorg. Chem., 2017, 56, 1574–1580 CrossRef CAS PubMed
- K. An, A. Farooqui and S. T. McCoy, Appl. Energy, 2022, 325, 119895 CrossRef CAS
- Y. Gao, Y. Tao, G. Li, P. Shen, R. J. M. Pellenq and C. S. Poon, Proc. Natl. Acad. Sci. U. S. A., 2025, 122, e2418239121 CrossRef CAS PubMed
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed
- R. A. Zehnder, S. M. Peper, B. L. Scott and W. H. Runde, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2005, 61, i3–i5 Search PubMed
- G. Goff, L. Brodnax, M. Cisneros, S. Peper, S. Field, B. Scott and W. Runde, Inorg. Chem., 2008, 47, 1984–1990 CrossRef CAS PubMed
- H. Abdi and L. J. Williams, WIREs Computational Statistics, 2010, 2, 433–459 CrossRef
- L. Zhang and P. A. Lay, Inorg. Chem., 1998, 37, 1729–1733 CrossRef CAS
- H. H. Eysel and S. Thym, Z. Anorg. Allg. Chem., 1975, 411, 97–102 CrossRef CAS
- A. R. Davis and B. G. Oliver, J. Solution Chem., 1972, 1, 329–339 CrossRef CAS
- J. D. Frantz, Chem. Geol., 1998, 152, 211–225 CrossRef CAS
- B. G. Oliver and A. R. Davis, Can. J. Chem., 1973, 51, 698–702 CrossRef CAS
- A. Anderson, C. Chieh, D. E. Irish and J. P. K. Tong, Can. J. Chem., 1980, 58, 1651–1658 CrossRef CAS
- R. L. Frost and J. Čejka, J. Raman Spectrosc., 2007, 38, 1488–1493 CrossRef CAS
- R. L. Frost and J. Čejka, J. Raman Spectrosc., 2009, 40, 1096–1103 CrossRef CAS
- G. Mestl, M. P. Rosynek and J. H. Lunsford, J. Phys. Chem. B, 1998, 102, 154–161 CrossRef CAS
- G. Lu, A. J. Haes and T. Z. Forbes, Coord. Chem. Rev., 2018, 374, 314–344 CrossRef CAS PubMed
- A. Arteaga, T. Arino, G. C. Moore, J. L. Bustos, M. K. Horton, K. A. Persson, J. Li, W. F. Stickle, T. A. Kohlgruber, R. G. Surbella Iii and M. Nyman, Chem.–Eur. J., 2024, 30, e202301687 CrossRef CAS PubMed
- A. Chegeni, V. Babaeipour, M. Fathollahi and S. G. Hosseini, J. Cluster Sci., 2022, 33, 2167–2178 CrossRef CAS
- S. M. Lalji, M. Ayubi, S. I. Ali, S. u. Rahman and M. Mustafa, Discover Chemistry, 2024, 1, 71 CrossRef
- A. N. Ebelegi, N. Ayawei and D. Wankasi, Open J. Phys. Chem., 2020, 10, 166–182 CrossRef CAS
- A.-B. Al-Odayni, F. S. Alsubaie, N. A. Y. Abdu, H. M. Al-Kahtani and W. S. Saeed, Polymers, 2023, 15, 1983–2001 CrossRef CAS PubMed
- A. G. Ritchie, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1650–1653 RSC
- E. Asuquo, A. Martin, P. Nzerem, F. Siperstein and X. Fan, J. Environ. Chem. Eng., 2017, 5, 679–698 CrossRef CAS
- E. Guibal, C. Milot and J. M. Tobin, Ind. Eng. Chem. Res., 1998, 37, 1454–1463 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01836f |
| This journal is © The Royal Society of Chemistry 2025 |