
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic Ag–Au nanoparticles from nanoconfinement: adjusting properties by electrochemical synthesis†
Thais
Schroeder Rossi
 a,
Athanasios A.
Papaderakis
a,
Maximilian
Jaugstetter
a,
Zaher
Jlailati
a,
Miriam
Knoke
a,
Pouya
Hosseini
b,
Paolo
Cignoni
b,
Fengli
Yang
c,
Maximilian
Gerwin
a,
Oliver
Trost
a,
Marius
Spallek
a,
Eduardo
Ortega
c,
Beatriz
Roldan Cuenya
c,
Debbie C.
Crans
d,
Nancy E.
Levinger
de and
Kristina
Tschulik
*ab
a,
Athanasios A.
Papaderakis
a,
Maximilian
Jaugstetter
a,
Zaher
Jlailati
a,
Miriam
Knoke
a,
Pouya
Hosseini
b,
Paolo
Cignoni
b,
Fengli
Yang
c,
Maximilian
Gerwin
a,
Oliver
Trost
a,
Marius
Spallek
a,
Eduardo
Ortega
c,
Beatriz
Roldan Cuenya
c,
Debbie C.
Crans
d,
Nancy E.
Levinger
de and
Kristina
Tschulik
*ab
aChair of Analytical Chemistry II, Faculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: kristina.tschulik@rub.de
bMax Planck Institute for Sustainable Materials GmbH, Max-Planck-Straße 1, 40237 Düsseldorf, Germany
cDepartment of Interface Science, Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
dDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA
eDepartment of Electrical and Computer Engineering, Colorado State University, Fort Collins, CO 80523-1373, USA
First published on 30th June 2025
Abstract
Developing synthetic pathways that exhibit well-controlled yet versatile characteristics to prepare nanoparticles (NPs) with properties tailored to the desired application is a topic of continuous interest. Herein, we introduce an innovative approach to form bimetallic alloy and core–shell-like Ag–Au NPs, employing reverse micelles as nanoreactors at the polarized electrolyte/electrode interface. Encapsulation of the metal precursors in the nanocavities of polystyrene-b-poly(2-vinylpyridine) (PS–P2VP) reverse micelles provides a route to control the NP size without the need for additional chemicals. By investigating the relations between the electrochemical driving force of the process and the complex interplay among the precursor species and the electrolyte medium, bimetallic Ag–Au NPs with sizes ranging from below 10 nm to 140 nm were synthesized with adjustable element configuration (core–shell vs. alloy) and composition. Notably, the resultant NPs were either Ag-rich alloys or Au-rich alloys or had a core–shell-like configuration with adjustable core and shell compositions based on the applied electrode potential and electrolyte medium. Finally, the prepared NPs were evaluated for their catalytic activity based on their physical properties against the hydrogen evolution reaction, where the core–shell-like NPs showed the most promising performance.
Introduction
The distinct properties of particles at the nanoscale and their high surface to volume ratio render nanoparticles (NPs) particularly promising candidates for a range of applications, such as biomedicine,1 energy storage,2,3 optoelectronics,4,5 sensing,6,7 and (electro)catalysis.8,9 The use of NPs, in particular noble metal-based NPs, can result in comparable or even higher overall performance than their bulk counterparts, while also reducing material usage and lowering costs.10–12 Therefore, the generation and fundamental characterization of NPs is important because it could impart better control over future design of systems.Despite the variety of applications1–9 and reports demonstrating the flexibility of bimetallic NP properties13–15 by modifying their size, composition, and element configuration (such as core–shell, inverted core–shell, or alloy), comparably few examples of the exploitation of this potential have been reported. This arises from the challenge of easily adjusting core–shell, inverted core–shell, and alloy nanoparticles beyond their chemical “nobility”, i.e., their position in the electrochemical series. In classic wet-chemical methods involving more than one metallic precursor species in the solution, the more noble element, i.e., the one with a higher reduction potential, typically is enriched in the core, while the less noble one tends to concentrate in the shell, even in nominal alloy systems. For silver–gold nanoparticles (Ag–Au NPs), for example, the commonly reported core–shell structure following the single step wet-chemical synthetic route is Au@Ag (core@shell), comprising the more noble element, i.e., Au, in the particle core, as Au is preferentially reduced.16–20
Decorating the more noble component onto the outermost layers of the shell is also possible, for instance, by techniques such as galvanic replacement21,22 or by appropriately controlling the reaction environment (e.g., different gas atmospheres or vacuum to induce surface segregation23,24). The configuration with the more noble metal in the shell can be achieved by wet-chemical routes too, when synthesized in two steps, that is, first the core composed of the less noble metal is formed, and in the second step the more noble metal is deposited onto these seeds.25–29
Due to their low cost and versatility when compared with other methods, wet-chemical methods are the most widely used approaches to synthesize metallic NPs, starting at the molecular level and growing to the nanoscale. These methods require various chemical reagents that serve as reducing agents (e.g., NaBH4 and N2H4) and stabilizing agents (e.g., citrate), which can significantly alter the NP properties. This variability makes it difficult to study the structure–property relation of nanoparticles, as altering the NPs' configuration (core–shell, inverted core–shell, or alloy) while keeping all other conditions identical has not been possible using these wet-chemical approaches. Furthermore, the NP growth process is complex,30 and their functionalization on an electrode surface via additional steps, such as drop casting, spin coating or dip coating, can lead to agglomeration upon solvent removal, which is usually performed via evaporation.31
Electrosynthesis is a promising alternative to circumvent the drawbacks introduced in wet-chemical synthesis, as the addition of reducing and/or stabilizing agents is not always necessary. At the same time, the metal precursor(s) can be reduced and functionalized directly on the electrode substrate by the electrochemically induced reduction reaction. Appropriate tuning of the experimental parameters, such as applied potential bias and the precursor concentration, can lead to the desired composition and morphology of the NPs. While electrosynthesis offers significant advantages, potential challenges include achieving uniform surface coverage, which can complicate precise nanoparticle localization.
An emerging method alternative to traditional (electro)synthetic routes involves encapsulation of the precursor in the nanocavities of droplets, emulsions, or reverse micelles, rendering them nanoreactors.16,31–36 Besides the control of the NP size,37,38 the introduction of nanoreactors in the electrodeposition process can be used for adjusting the otherwise difficult-to-alter reactivity as it can influence additional physicochemical processes, such as the interplay between the precursor interactions with the electrode and the electrolyte in the vicinity of the electrode, its mass transport, and the interfacial structure. Such effects induced by the nanoreactor species can be classified as electrochemical confinement effects.31
In this work, we demonstrate how the composition and structure of Ag–Au NPs can be tailored by adjusting the electrosynthesis parameters, independently of the position of the individual element precursor species in the electrochemical series. This is achieved by selectively controlling the reactivity and stabilization of each component in a reverse micellar environment. Additionally, we provide insights into the role of the electrolyte medium in the electrosynthesis process from nanoconfinement. Scheme 1 provides a concise summary of the findings presented in this paper.
 | ||
| Scheme 1 Overview of the synthesis outcomes of Ag–Au NPs under varying conditions. Schematic representation not to scale. | ||
We synthesized mixed Ag–Au NPs via a one-step electrochemical reduction of encapsulated metal precursors in diblock copolymer polystyrene-b-poly(2-vinylpyridine) (PS–P2VP) reverse micelles. The reverse micelles are dispersed either in an organic electrolyte or an ionic liquid. The influence of the electrolyte medium and applied potential on the morphological characteristics, size, composition, and element configuration of the NPs formed is explored. The derived results are rationalized based on the complex interplay between the charged precursors, electrolyte, solvent, and substrate in the interfacial region.
By tuning the electrosynthesis parameters, we demonstrate the single-step formation of Ag–Au core–shell-like NPs with a Ag-rich core. Starting with both precursors in solution and synthesizing such a type of nanoparticle in a one-step approach is not yet reported and is accomplished in this work by adjusting the electrolyte composition and the applied potential, with the precursors encapsulated within the PS–P2VP reverse micelles. Single entity electrochemistry measurements were employed to further explore the mechanism of electrodeposition at the nanoscale.
Finally, the mixed Ag–Au NPs were evaluated as electrocatalysts towards the hydrogen evolution reaction (HER). The performance was analyzed to link synthesis conditions to electrochemical activity in NPs with altered element configurations but identical capping agents. Our work conceptualizes a strategy for controlled synthesis of NPs with adjustable properties for diverse applications. By varying reduction potentials to control reduction rates, rather than changing reducing or capping agents, we maintain identical chemical components. This enables more systematic adjustment and comparison of nanomaterials than existing approaches.
Results and discussion
Nanoparticle synthesis and characterization
Ag–Au bimetallic NPs were prepared from PS–P2VP reverse micelles by electrochemical reduction on highly oriented pyrolytic graphite (HOPG) electrodes using (i) the organic electrolyte (OE) 33.5 mM tetra n-butylammonium hexafluorophosphate (TBAPF6) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio mixture of dichloroethane (DCE) and toluene at applied potentials of −0.7, −1.0 and −1.3 V vs. a Pt quasi-reference electrode (QRE) or (ii) the ionic liquid (IL) EMIMNTf2 at applied potentials of −0.7 or −1.0 V vs. Pt QRE. We selected these electrolytes to shift the reducibility of silver and gold precursors with respect to each other, aiming to influence the configuration of the bimetallic NPs (see Fig. 2 and S5 and the detailed discussion in Section 1.3 in the ESI†). The choice of the applied biases relies on preliminary cyclic voltammetry (CV) experiments used to investigate the potential range within which the reduction of the metal precursors occurs (Fig. S1†). By varying the applied potential for the electrodeposition process, we change the electrochemical driving force (i.e., the applied overpotential) for the precursor reduction in each medium, enabling us to investigate their effect on the physical properties (i.e., composition, morphology and configuration) of the NPs deposited on the electrode surface.
:1 volume ratio mixture of dichloroethane (DCE) and toluene at applied potentials of −0.7, −1.0 and −1.3 V vs. a Pt quasi-reference electrode (QRE) or (ii) the ionic liquid (IL) EMIMNTf2 at applied potentials of −0.7 or −1.0 V vs. Pt QRE. We selected these electrolytes to shift the reducibility of silver and gold precursors with respect to each other, aiming to influence the configuration of the bimetallic NPs (see Fig. 2 and S5 and the detailed discussion in Section 1.3 in the ESI†). The choice of the applied biases relies on preliminary cyclic voltammetry (CV) experiments used to investigate the potential range within which the reduction of the metal precursors occurs (Fig. S1†). By varying the applied potential for the electrodeposition process, we change the electrochemical driving force (i.e., the applied overpotential) for the precursor reduction in each medium, enabling us to investigate their effect on the physical properties (i.e., composition, morphology and configuration) of the NPs deposited on the electrode surface.
Scanning electron microscopy (SEM) images (Fig. 1A–C) show the morphological characteristics of a series of mixed Ag–Au NPs deposited on HOPG from the organic electrolyte (see the Experimental section for the detailed protocol). At the most negative potential used, the resulting NPs, on average, appear much smaller than those created at less negative potentials (Fig. 1A–C). Size distribution plots (Fig. 1D–F) obtained by image analysis of at least 200 nanoparticles for each sample demonstrate that NPs in Fig. 1A are ∼15 times smaller than those shown in Fig. 1B and C. These small NPs formed at the most negative applied potential also display a higher degree of substrate coverage, with well dispersed NPs in the nanometer range as opposed to large, isolated agglomerates at less negative biases. Similar results were obtained for the electrodeposition in IL, as shown in Fig. S2.†39 Additional SEM images of samples prepared in both organic electrolyte and IL are shown in Fig. S4.†
 | ||
| Fig. 1 (A–C) SEM images and (D–F) size distribution plots with mean and standard deviation values of Ag–Au NPs synthesized from confined precursors in PS–P2VP reverse micelles in organic electrolyte (TBAPF6 in DCE + toluene) at −1.3 V, −1.0 V, and −0.7 V. Note: mean and standard deviation values were determined from the lognormal fitting parameters μ and σ, as described in the ESI, Section 1.2.† | ||
To elucidate the underlying physicochemical factors resulting in the observed morphological characteristics and identify the corresponding electrochemical processes for the bimetallic system, we investigated the electrodeposition of each individual encapsulated metal precursor in the electrolytes used for the preparation of the bimetallic NPs (Fig. 2 and S5†). Fig. 2 shows voltammograms recorded in organic electrolyte for bare HOPG and for PS–P2VP reverse micelles loaded with AgNO3, HAuCl4 and mixed AgNO3 + HAuCl4 (for a detailed discussion see Section 1.3 in the ESI†40–52). These voltammograms show that Au electrodeposition requires a more negative applied potential compared to Ag in the electrolytic medium used, which is the opposite of what is observed in aqueous electrolytes53 and is further supported by the unsuccessful attempt to deposit Au–Ag NPs at less negative potentials, i.e., at −0.5 V and −0.35 V (data not shown).
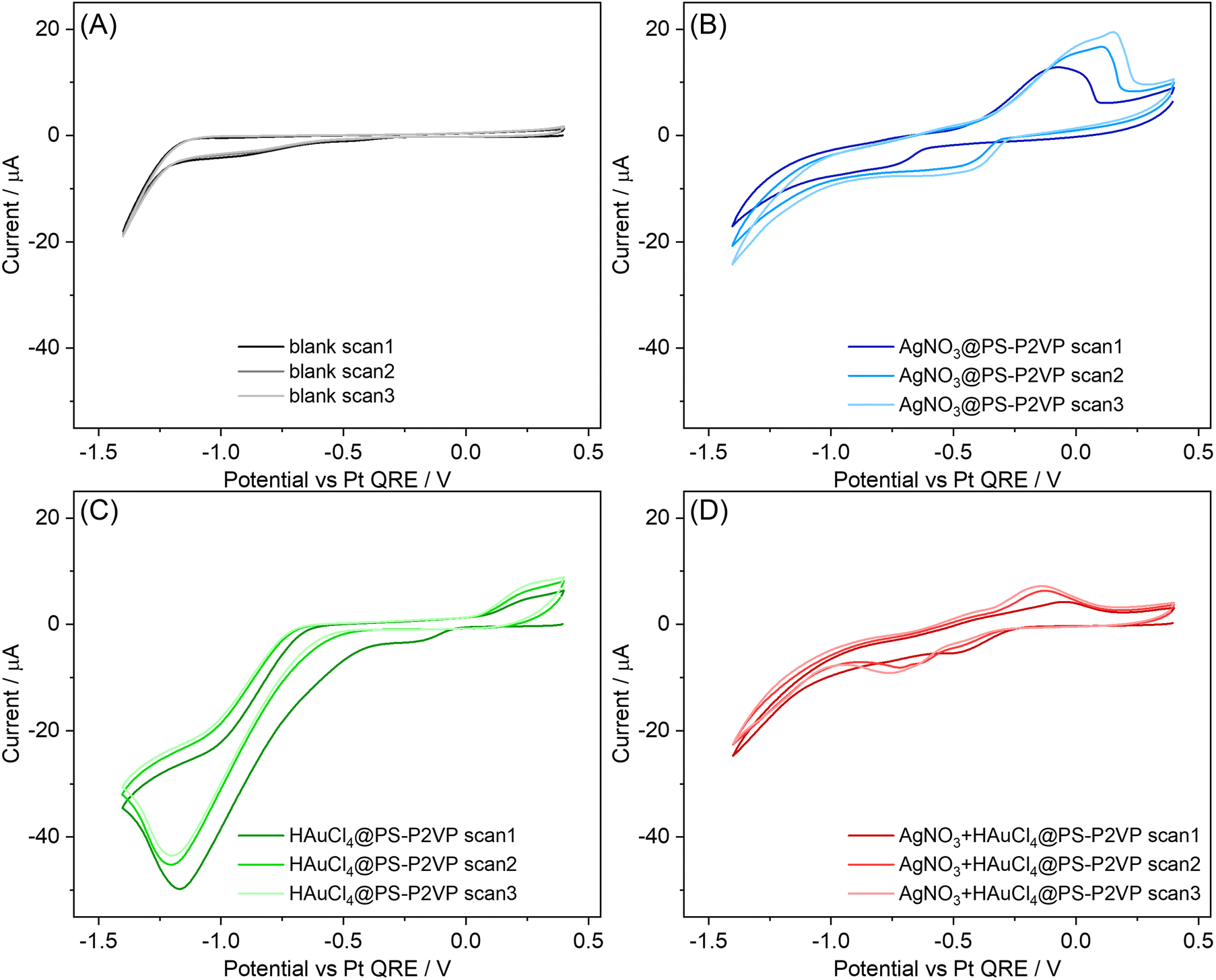 | ||
| Fig. 2 Consecutive cyclic voltammograms of (A) bare substrate, (B) AgNO3@PS–P2VP, (C) HAuCl4@PS–P2VP and (D) AgNO3 + HAuCl4@PS–P2VP reverse micelles in organic electrolyte (TBAPF6 in DCE + toluene). CV plotting convention: IUPAC. WE: HOPG (ZYB grade, 10 × 10 mm, exposed area = 0.5 cm2). QRE: Pt wire. CE: Pt gauze. The WE compartment is not separated from the CE. Applied voltage is not IR corrected. Starting point: 0.4 V vs. Pt QRE. Direction of the scan: cathodic. Scan rate: 50 mV s−1. | ||
To interpret this finding, we consider the interplay between the precursors–solvent–electrolyte specific interactions. AuCl4− reduction to metallic Au involves the release of Cl− ions. In general, the solubility of small anions is determined by the electrophilicity of the solvent, which can be assessed using its acceptor number.54 DCE has a low acceptor number of 16.7 kJ mol−1 and toluene exhibits no electrophilic properties. This predicts that the solvation degree of Cl− in the organic electrolyte is much weaker than it would be in water, rendering the anion release thermodynamically less likely.46 This shifts the reaction reduction potential to more negative values46,54 compared to the reduction of Ag+, where no such step is involved. This difference in the electrochemical behavior of the two metal precursors with the electrolyte used influences both the composition and morphological characteristics of the prepared binary NPs.
When using IL as the electrolyte, the thermodynamic barrier for Cl− release is expected to be less pronounced due to the solvent-free nature of IL, which also leads to an enhanced interaction between the precursor species and the medium. The role of electrolyte in NP morphology is, however, less straightforward when using the IL, as the voltammograms display no clear features of Ag deposition (see Fig. S5†). Nonetheless, in addition to tracking the individual precursors, the EMIM+ cations are known to adsorb strongly to the surface of both HOPG and various metals,47–51 with the extent of adsorption increasing with more negative biases with respect to the potential of zero charge, Epzc. The strong interaction between EMIM+ and Au can lead to a dynamic state of disordered crystallographic-dependent adsorption that results in surface etching due to the formation of soluble complexes, e.g., [EMIM]+[AuCl2]− (similar complexes, e.g., [EMIM]+[AgCl2]−, can also be formed with Ag),52 with accompanied surface reconstruction. Various in situ surface sensitive microscopic and spectroscopic techniques have reported this phenomenon,47,48,50 which becomes more pronounced as the applied potential shifts to more negative values, thus strongly influencing the surface morphology and possibly composition, e.g., for alloys or mixtures of the substrate. On this basis, we attribute the decreased size of the NPs prepared at −1.0 V to the combined effect of increased overpotential for the deposition process and more intense etching. We also emphasize that the determination of Epzc at HOPG in ionic liquids, such as EMIMNTf2, via common electrochemical techniques, e.g., electrochemical impedance spectroscopy, is very challenging due to the complex nature of the underlying processes, e.g., the space-charge-like capacitance arising from HOPG and the electrode-ion interactions.55 Nonetheless, the potential values applied in our study during the electrodeposition experiments correspond to significantly more negative values than those lying in the potential region where Epzc is expected in similar systems55 and hence the adsorption of EMIM+ is a reasonable assumption. An additional phenomenon that needs to be accounted for here is the increased density of Cl− ions inside the electrochemical double layer (EDL) region, emerging from the release of Cl− during the complete reduction of AuCl4−. On this basis, the excess negative charge introduced in the electrolyte side of the EDL is screened by attracting more EMIM+ near the electrode. Considering that the lower the applied potential the higher the rate of the deposition reaction (hence the release of Cl−), it is expected that the density of EMIM+ near the surface of the electrode would also increase.
Having clarified the electrochemical reduction of the individual encapsulated precursors, we return to the reverse micelle assisted electrodeposition of the mixed precursors and the NPs presented in Fig. 1. If the reverse micelle (size: 28 ± 4 nm, Fig. S6†) acts as a nanoreactor that can limit the size of the formed NP, we interpret the formation of larger NPs to result from the reduction of precursors encapsulated inside more than one reverse micelle. In agreement with established nucleation theories,56–60 this suggests that at more negative potentials a higher number of NPs is formed by instantaneous reduction and nucleation of precursors from single reverse micelles, due to the higher electrochemical driving force for precursor reduction. Hence, small NPs are formed. Conversely, at less negative potentials, with a smaller driving force for precursor reduction, NPs are formed more progressively. Fewer NPs nucleate progressively at a lower rate and growth of these NPs results from successive reduction of precursors from other micelles leading to a higher polydispersity.59,60 The driving forces resulting from varying applied potentials can be conceptualized as different reducing strengths and reduction rates. For example, in a typical wet-chemical synthetic procedure, the use of different reducing agents, e.g., NaBH4 instead of citrate, can result in varying sizes and degrees of polydispersity.61,62 The electrochemical route we present here is highly advantageous, as it does not require the introduction of additional chemical compounds and subsequent removal steps. Furthermore, the electrochemical approach can be tuned continuously via the applied potential (down to a few mV steps) and is not restricted by the reduction potential of typical chemical reducing agents.
We used scanning transmission electron microscopy (STEM), secondary electron imaging and energy dispersive X-ray spectroscopy (EDX) to explore the NP morphology. Both the STEM images in Fig. 3A–C and the secondary electron images in Fig. 3D–F show that NPs formed at more negative potentials have a smoother, more spherical surface than the irregular NPs formed at less negative potentials and further support the differences in nucleation and growth behaviour discussed above. We interpret the irregular morphology as originating from anisotropic particle growth resulting from reduction of precursors released from additional reverse micelles on top of the initially nucleated NPs. This is in contrast to the formation of NPs from single reverse micelles, which we discuss in more detail, in section “Single entity electrochemistry”.
 | ||
| Fig. 3 (A–C) Dark-field STEM image, (D–F) corresponding secondary electron image and (G–I) EDX mapping of Ag–Au NPs synthesized from confined precursors in PS–P2VP reverse micelles in organic electrolyte (TBAPF6 in DCE + toluene) at −1.3 V, −1.0 V, and −0.7 V. Note that NPs seen in STEM images may look larger or more aggregated than in SEM images due to the scratching step in sample preparation for STEM measurements (details in the electron microscopy experimental section). | ||
EDX mapping (Fig. 3G–I) suggests different element configurations for the NPs formed at different potentials in the organic electrolyte. At −1.0 and −0.7 V, EDX measurements reveal that NPs with different bimetallic compositions between the inner (“core”) and outer (“shell”) parts are formed, resulting in a “core–shell-like” NP. At a more negative potential, −1.3 V, alloy-like NPs are produced. The NPs' overall composition is Ag-rich, corroborating the favored Ag deposition in this organic electrolyte compared to Au. According to EDX analysis, the alloy-like NPs formed at −1.3 V have a composition of 79 ± 6% Ag and 21 ± 6% Au, which matches with the Ag:Au ratio for the core of the NPs formed at −1.0 V (80 ± 8% Ag and 20 ± 8% Au). The shell in the NPs prepared at −1.0 V comprises 58 ± 10% Ag and 42 ± 10% Au. For the NPs synthesized at −0.7 V, the core and shell compositions are Ag:Au 88 ± 2:12 ± 2 and 68 ± 1:32 ± 1, respectively (see Fig. S7†). The overall composition of NPs was determined from at least 100 nanoparticles for each sample. Therefore, these latter NPs have a core–shell-like configuration with a Ag-richer core and a Ag-poorer shell. The Ag content in both the core and shell increases when a less negative potential is applied during synthesis, as anticipated when considering the deposition processes of the individual encapsulated precursors (Fig. 2). Starting with both precursors in solution and forming core–shell-like NPs with a Ag-rich core in a single step have not been reported previously; we achieve this novel synthesis by tuning the applied potential parameter when the precursors are encapsulated inside PS–P2VP reverse micelles. Therefore, the synthetic approach reported might be an early-stage proof of concept study demonstrating the one-step preparation of core–shell-like NPs using reverse micelles as morphology regulators, while simultaneously tailoring their physicochemical characteristics by establishing structure–composition relations through the elaborate control of the electrolyte–precursor interactions and electrochemical driving force.
We observe similarities and differences for Ag–Au NPs synthesized in IL compared to the organic electrolyte. The morphology observed in STEM and secondary electron images of Ag–Au NPs prepared from confined precursors in PS–P2VP reverse micelles in EMIMNTf2 (Fig. 4) is similar to that observed in the organic electrolyte. NPs prepared at more negative potential are smaller with a smoother surface than the bigger irregular-surfaced NPs formed at less negative potential. In IL, alloy NPs are also produced. However, contrary to the NPs synthesized in the organic electrolyte, EDX mappings of the NPs formed in IL (Fig. 4) do not exhibit core–shell-like configuration. Instead, these NPs have a uniform composition throughout their structure and are Au-rich, rather than the Ag-rich alloy, at both applied biases used (Ag:Au 10 ± 6:90 ± 6 at −1.0 V and 8 ± 1:92 ± 1 at −0.7 V). This indicates the role of electrolyte in NP formation and morphology.
 | ||
| Fig. 4 Dark-field STEM image, corresponding SEI image and EDX mapping of Ag–Au NPs synthesized from confined precursors in PS–P2VP reverse micelles in ionic liquid (EMIMNTf2) at (A–C) −1.0 V, (D–F) −0.7 V. Note that NPs seen in STEM images may look larger or more aggregated than in SEM images due to the scratching step in sample preparation for STEM measurements (details in the electron microscopy experimental section). Apparent high background Ag content may be misleading due to different intensity color scales for Ag and Au (see Fig. S8† for individual elements EDX mappings). | ||
Anodic stripping voltammograms of the Ag–Au NPs synthesized from precursors-filled PS–P2VP reverse micelles provide additional insight into the NPs formed in organic and IL electrolytes. For samples prepared in the organic electrolyte (Fig. S9†53,63–65), we observe progressive dealloying for Ag–Au NPs formed at all three investigated potentials. Because the core–shell-like NPs have an alloy shell, the electrolyte can easily reach the Ag-rich core before oxidizing all Au that is in the shell. The voltammetric curves of samples prepared in IL (Fig. S10†) show primarily anodic peaks that we assign to Au species based on the relatively high potential. This observation agrees with the high Au content (∼90%) in the NPs formed in ionic liquid.
To explore the role that the reverse micelles play in the NP formation, composition and morphology, we also investigated the deposition from precursors not encapsulated inside the reverse micelle but free in the same electrolyte (organic electrolyte or IL), resulting in completely different coverages. In the organic electrolyte (Fig. S11†), STEM and secondary electron images show aggregated islands of around 1 μm or more on the electrode surface. The EDX mapping of these deposits indicates that neither alloy nor core–shell-like Ag–Au NPs form in the bulk electrolyte. Instead, Ag NPs and Au NPs are separately synthesized. The overall composition percentage of the islands is Ag 80 ± 6%, Cl 16 ± 6%, and Au 4 ± 4%, indicating that the NPs are mostly formed by Ag and that AgCl is also significantly formed. This differs substantially from what is observed for Ag–Au NPs formed from precursors confined in reverse micelles, where the overall Cl content is less than 1%. Deposition from unconfined precursors in IL (Fig. S12†) also forms Au-rich alloy particles; however, the particles are around 400 nm, significantly larger than the NPs obtained under confined conditions. The results from unconfined precursors synthesis further confirm the importance of reverse micelles as nanoreactors to successfully form alloy or core–shell-like Ag–Au NPs.
By selecting synthesis parameters, i.e., applied potentials and electrolyte type, we demonstrate Ag–Au NP formation with adjustable size, composition and element configuration from precursors confined in PS–P2VP reverse micelles. Table 1 provides a summary of NPs formed under each synthesis condition, including how the single-step formation of a Ag-rich core in a Ag–Au core–shell-like NP was possible. This is in contrast to the vast majority of Ag–Au core–shell-like NPs reported, that include a Au-rich core.16–20 Hence, our work showcases manipulation of core and shell Ag and Au compositions independent from the element position in the electrochemical series. For both organic and IL electrolytes, the synthesis at a more negative potential leads to significantly smaller NPs. The different sizes and surface morphologies corroborate the hypothesis of NP formation from nucleation of precursors entrapped inside single reverse micelles at more negative potentials and from nucleation and further growth from precursors encapsulated in more reverse micelles at less negative potentials.
| Medium | Applied potential/V | Size/nm | Configuration | Composition | Ag/% | Au/% |
|---|---|---|---|---|---|---|
| OE | −1.3 | 8 ± 5 | Alloy-like | Ag-rich | 79 ± 6 | 21 ± 6 |
| −1.0 | 134 ± 44 | Core–shell-like | Ag-rich | 80 ± 8 (core) | 20 ± 8 (core) | |
| 58 ± 10 (shell) | 42 ± 10 (shell) | |||||
| −0.7 | 111 ± 39 | Core–shell-like | Ag-rich | 88 ± 2 (core) | 12 ± 2 (core) | |
| 68 ± 1 (shell) | 32 ± 1 (shell) | |||||
| IL | −1.0 | 8 ± 6 | Alloy-like | Au-rich | 10 ± 6 | 90 ± 6 |
| −0.7 | 75 ± 36 | Alloy-like | Au-rich | 8 ± 1 | 92 ± 1 |
Single entity electrochemistry
To reveal if the single-digit-nanometer-sized NPs formed at more negative potentials in both the organic electrolyte and the IL arise from single reverse micelles, nanoimpact measurements were performed. In these measurements, when single reverse micelles collide with a polarized electrode, the precursors loaded in the reverse micelles are reduced forming a Ag–Au NP, which is observed as a spike in the current-time plot.10 We only observed these spikes in the presence of precursor-loaded reverse micelles, not in their absence (Fig. S13†).We determine the charge associated with the amount of Ag–Au deposited by integrating the area of the electrochemical spike. Eqn (1) correlates the charge to the diameter of the NP through the volume of a sphere and Faraday's law.10
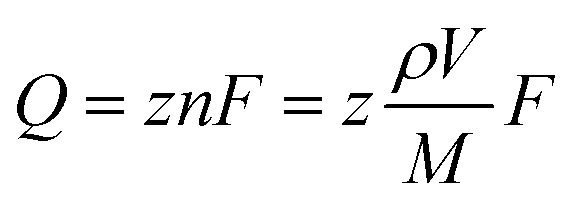 | (1) |
Assuming a spherical geometry for the NPs formed (Fig. S2† and 4), eqn (1) can be rearranged into eqn (2) to yield the particle diameter, d.
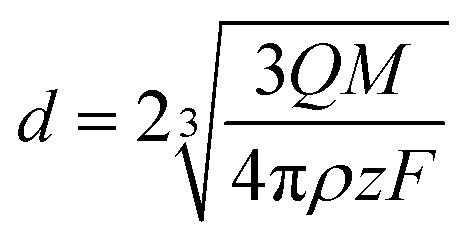 | (2) |
Based on the oxidation state of the metallic precursors (AgCl and HAuCl4), the number of transferred electrons for each Ag and Au atom is 1 and 3, respectively.10,66,67 Assuming a direct electron transfer path:
| Ag+ + 1e− → Ag0 | (3) |
| AuCl4− + 3e− → Au0 + 4Cl− | (4) |
Because the NP formed is a bimetallic mixture of Ag and Au, the total molar mass, total density, and total number of transferred electrons are related to the final NP composition. For NPs formed in the ionic liquid at −1.0 V applied potential, the composition is 10% Ag and 90% Au, as demonstrated by EDX and X-ray photoelectron spectroscopy (XPS) (Section 3 in the ESI†68). We determined the size of the Ag–Au NPs formed from single reverse micelles to be 9 ± 5 nm from a total of 593 individual impacts (size distribution in Fig. 5). This agrees with the sizes of NPs formed by electrodeposition at the most negative potential described in the previous section.
 | ||
| Fig. 5 (A) Size distribution plot of Ag–Au NPs synthesized from confined precursors in PS–P2VP reverse micelles in ionic liquid (EMIMNTf2) from 593 individual collision events during single entity electrochemical measurements. Inset: a cartoon of a reverse micelle colliding with a polarized electrode, generating a nanoparticle via precursors reduction. (B) Transient current during one measurement and enlarged impact features indicated with colored asterisks, with the corresponding peak height in pA, duration in ms, area in fC and calculated NP diameter in nm. The last enlarged region (dark yellow asterisk) shows the background current, i.e., in the absence of impacts. | ||
The single entity electrochemical measurements indicate that the NPs formed at the most negative potentials occur instantaneously from single micelles, corroborating the previous explanation that at more negative potentials, instantaneous nucleation of NPs from individual reverse micelles dominates, while at less negative potentials nucleation and further growth from precursors contained in multiple reverse micelles occurs.
Hydrogen evolution reaction electrocatalysis
The electrocatalytic activities of Ag–Au NPs synthesized from precursors-loaded PS–P2VP reverse micelles were investigated using the hydrogen evolution reaction as a model reaction (Fig. 6) to showcase how the overall activity of the NPs can be influenced by adjusting the synthetic route parameters. As NPs formed in organic electrolyte at −0.7 and −1.0 V showed very similar properties in the previous characterization techniques, only the NPs synthesized at −0.7 V are shown in Fig. 6. To provide insights into the influence of the deposition process on the electrocatalytic activity of Ag–Au NPs towards the HER, we need to exclude surface area effects. To achieve this, the electrochemically active surface area (ECSA) of the prepared NPs should be determined. ECSA was estimated, following an electrochemical stripping procedure, as described in Section 4 in the ESI.† The resulting ECSA for each sample was then used to normalize the respective raw current values per ECSA. In this regard, the results in Fig. 6 provide an estimation of the intrinsic catalytic activity of the prepared NPs based on their composition and element configuration.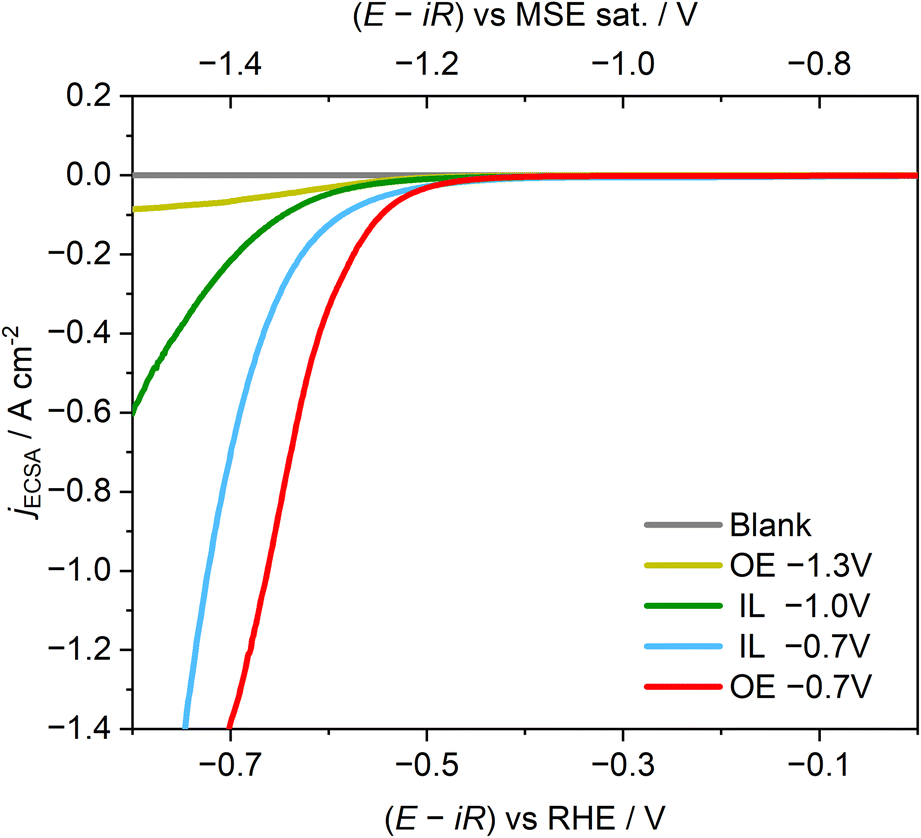 | ||
| Fig. 6 Averaged hydrogen evolution reaction linear sweep voltammetry (LSV) curves from at least three independent experiments of Ag–Au NPs synthesized from confined precursors in PS–P2VP reverse micelles in organic electrolyte (OE, TBAPF6 in DCE + toluene) or in ionic liquid (IL, EMIMNTf2). Electrolyte: Ar-saturated 0.1 M H2SO4. WE: HOPG (ZYB grade, 10 × 10 mm, exposed area = 0.5 cm2). RE: MSE sat. CE: carbon rod (∅ = 6 mm). The WE compartment is not separated from the CE. Applied voltage is IR corrected. Current is normalized by the electrochemical surface area. Scan rate: 10 mV s−1. | ||
The Ag-rich alloy-like NPs formed in organic electrolyte at −1.3 V exhibit the lowest catalytic activity, which is consistent with the fact that Ag is less catalytically active than Au for the hydrogen evolution reaction.69–72 As a result, it is reasonable to expect that Ag-rich NPs would be less active than the Au-rich alloy-like NPs, which were formed in IL. However, the specific differences in activity between NPs generated in IL at different potentials remain unclear. Among the samples, the NPs formed in organic electrolyte at −0.7 V are the most active, demonstrating the remarkable properties that can be achieved using the core–shell-like configuration produced in this work. Even being Ag-rich, i.e., its composition is mostly the less catalytically active element, the final electrocatalytic activity surpasses that of the alloy-like NPs that are around 90% formed by Au. This result introduces an alternative strategy for the use of less expensive materials to achieve similar or better performance in electrocatalysis, which can be realized by synthesizing NPs from confined precursors. The latter further highlights the advantages of using reverse micelles as nanoreactors in electrodeposition. Future directions should be focused on the optimization of the control over the size and composition of the NPs and aim towards the establishment of size–composition–activity relations for various electrocatalytic reactions of interest. At this point, we need to emphasize that developing a new high-performance catalyst for the HER is not the primary objective of this work. Instead, HER activity was employed as a model reaction to further highlight the distinct properties achieved through our tailored synthetic approach. This functional assessment supports our central aim of demonstrating how different nanoreactor environments and deposition conditions influence the resulting characteristics of the NPs. Finally, an additional noteworthy point in the data reported in this section is the evident activity of the prepared NPs despite the use of reverse micelles as encapsulating reagents. Polymer-based stabilizing reagents are reported to introduce impurities on the surface of NPs that often require rather intense purification methods (e.g., plasma treatment37,38) for their removal. NPs not being purified show negligible or particularly low activities due to the blocking of their active sites. However, the evident activity of Au NPs previously reported10 and of the Ag–Au NPs prepared following the approach described in this study implies that the presence of the stabilizing ligands on the surface of the NPs is rather minimal.
Experimental
Materials
Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, Alfa Aesar, 99.99% metal basis), silver nitrate (AgNO3, Alfa Aesar, >99.9%), tetra n-butylammonium hexafluorophosphate (TBAPF6, Alfa Aesar, 98%), 1,2-dichloroethane (DCE, Carl Roth, ≥99%), and ultrapure water purified using a Barnstead GenPure xCAD Plus (Thermo Fisher Scientific, conductivity 0.055 μS cm−1 at 25 °C) were used. Toluene (VWR Chemicals, 99.99%), isopropanol (VWR Chemicals, ACS) and acetone (Sigma Aldrich, ACS, >99.5%) were filtered with a 200 nm pore size PTFE filter prior to use. Ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMIMNTf2, IoLiTec, 99%) was washed with ultrapure water and dried overnight under vacuum prior to use. Aqueous electrolyte solutions were prepared by diluting 95% H2SO4 (Fisher Scientific, analytical reagent grade) and 37% HCl (Sigma Aldrich, reagent grade).AgNO3 + HAuCl4@PS–P2VP reverse micelle synthesis
PS–P2VP reverse micelles were prepared in toluene as previously reported10,73 and filled with silver and gold precursors in equimolar amounts (AgNO3 + HAuCl4@PS–P2VP) using two main solutions. The detailed synthesis procedure is outlined below.Solution 1: 50 mg of poly(styrene-b-2-vinylpyridine) [P1330–S2VP, Mn: PS(48500)–P2VP(70000), sourced from Polymer Source] were dissolved in 5 mL of toluene. This formed spherical micelles with polar poly-2-vinylpyridine (P2VP) heads as the core and nonpolar polystyrene (PS) tails extending outward.
Solution 2: 33.23 mg of HAuCl4·3H2O and 14.35 mg of AgNO3 were dissolved in 5 mL of toluene. 1 mL of tetrahydrofuran (THF, Sigma Aldrich, >99.9%) was added to fully dissolve the Au and Ag precursors.
Both solutions were vigorously stirred for 24 hours at room temperature. Subsequently, solution 2 was added to solution 1, and the mixture was stirred for an additional 48 hours at room temperature. The presence of THF in the final reverse micelles formed in toluene was confirmed by nuclear magnetic resonance spectroscopy (Section 5 in the ESI†74). The effective encapsulation of AgNO3 and HAuCl4 in the reverse micelles suspended in toluene was confirmed by inductively coupled plasma mass spectrometry (ICP-MS). ICP-MS was also performed in organic electrolyte and ionic liquid to determine the amount of Ag and Au that leaches from the reverse micelles into the different electrolyte solutions (Section 6 in the ESI†).
Electrochemical measurements
The electrochemical ensemble synthesis and electrochemical characterization were performed in a three-electrode setup using an Autolab PGSTAT128N (Metrohm) potentiostat. Ensemble electrodeposition was performed using highly ordered pyrolytic graphite (HOPG, TipsNano, ZYB grade, 10 × 10 mm, exposed area = 0.5 cm2) as the working electrode (WE), a platinum wire as the quasi-reference electrode (QRE), and a platinum gauze as the counter electrode (CE, 1024 mesh cm−2). The WE surface was renewed by exfoliating the top layers.Cyclic voltammetry (CV) and electrodeposition were performed in two media: (i) organic electrolyte (10 μL of the micellar suspension described in the previous section was added to 670 μL of 50 mM TBAPF6 in DCE + 320 μL of toluene) or (ii) ionic liquid (3 μL of micellar suspension was transferred to 300 μL of EMIMNTf2 and toluene was removed by applying vacuum overnight). For CV and electrodeposition using unconfined precursors, 1 mM of AgNO3 and 1 mM of HAuCl4 were used in the same media described above (for the organic electrolyte case, we substituted 10 μL of micellar suspension with toluene).
For CV measurements, the potential window was from −1.4 to 0.4 V in organic electrolyte and from −2.0 to 1.0 V in ionic liquid, starting with the cathodic scan, with a scan rate of 50 mV s−1. For electrodeposition, a potential of −0.7, −1.0, −1.3 or −1.8 V vs. Pt QRE was applied for 30 min in chronoamperometric measurements. After deposition, the WE was washed with toluene, acetone, isopropanol (three times each) and ultrapure water (four times) and dried under Ar flow.
Linear sweep voltammetry (LSV) was performed in a three-electrode setup using HOPG as the WE, mercury–mercurous sulphate in saturated K2SO4 (MSE sat., SI analytics GmbH, 0.64 V vs. the standard hydrogen electrode, SHE75) as the reference electrode (RE) and a carbon rod (∅ = 6 mm) as the CE. The linear sweep voltammetry curve was recorded in an Ar-saturated 0.1 M H2SO4 solution, from −0.2 to −1.6 V vs. MSE sat. (from 0.5 to 0.9 V vs. the reversible hydrogen electrode, RHE), at a scan rate of 10 mV s−1.
Anodic stripping voltammetry was performed in a three-electrode setup using HOPG as the WE, homemade silver–silver chloride (Ag/AgCl/KCl 3 M, 0.21 V vs. SHE75) as the RE and a carbon rod (∅ = 6 mm) as the CE. To prevent silver contamination, the RE was equipped with a double junction containing 0.25 M KNO3 solution. The scans were recorded in a 0.1 M HCl solution, from −0.3 to 1.3 V vs. Ag/AgCl (from −0.03 to 1.57 V vs. RHE), at a scan rate of 10 mV s−1.
Single entity experiments were performed using an analogue potentiostat (VA-10X, npi electronic GmbH) using a three-electrode setup with a homemade carbon fiber (C, ∅ = 7 μm) as the WE and a platinum wire as the quasi-reference counter electrode (QRCE). Nano impacts were recorded in ionic liquid at an IL:micelles of 20:1 ratio, i.e., 15 μL of micellar suspension was transferred to 300 μL of EMIMNTf2 and toluene was removed by applying vacuum overnight, 20 μL of the micellar suspension in EMIMNTf2 was transferred to the electrochemical cell and different potentials from −0.8 V to −1.65 V vs. Pt QRE were applied. By using the software SignalCounter, the signals were evaluated and the size of the formed NPs was determined.
Electron microscopy
Samples were measured both with scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM). SEM images were recorded directly on the substrate (HOPG) using a JEOL JSM-7200F electron microscope operating at a 20 kV acceleration voltage. For STEM imaging, an ultrathin C film on a Lacey carbon support film, 400 mesh Cu grid (Ted Pella) was wet in ultrapure water and scratched on the substrate surface. STEM images were recorded using a JEOL JEM-2800 electron microscope operating at 200 kV acceleration voltage. Energy dispersive X-ray spectroscopy (EDX) mappings were acquired with the equipped double silicon drift detectors, with a solid angle of 0.98 steradians, a detection area of 100 mm2, and 133 eV spectral resolution, until at least 20000 counts of Ag or Au were acquired. Nanoparticle size distributions were determined by analyzing SEM and STEM images using ImageJ software.
Conclusions
Different Ag–Au NPs were successfully electrochemically synthesized using precursors-loaded PS–P2VP reverse micelles as nanoreactors. The resulting nanoparticles show how size, composition, element configuration and electrocatalytic properties can be adjusted using electrosynthesis conditions, such as electrolyte and applied potential. For each investigated medium (organic electrolyte and ionic liquid), electrosynthesis from precursors encapsulated inside individual reverse micelles yields smaller alloy Ag–Au NPs with a better-defined geometry when more negative potentials were applied. At less negative potentials, morphologically irregular, larger Ag-rich-core alloy-shell (in organic electrolyte) or Au-rich alloy NPs (in ionic liquid) resulted from precursors encapsulated inside more than one reverse micelle nanoreactor. The formation of such core–shell-like NPs in a single step shows how the NP composition and element configuration can be adjusted despite the position of elements in the electrochemical series when the precursors are encapsulated inside reverse micelles. The unique properties of these core–shell-like NPs are also illustrated by their superior catalytic activity towards the hydrogen evolution reaction among all samples described herein. Despite having a high silver content, the Ag–Au core–shell-like NPs outperformed the predominantly gold alloy-like NPs as catalysts for the hydrogen evolution reaction. This finding suggests that more cost-effective materials may deliver comparable or even better electrocatalytic performance, with a synthesis method utilizing confined precursors in reverse micelles playing a key role in this advancement.This work offers an outline for choosing electrosynthesis parameters to produce a variety of bimetallic Ag–Au NPs from confined conditions and the possibility to customize the core and shell composition as needed. We expect that this approach can be extended to non-precious elements in the future and might be used to synthesize NPs from different precursor salts due to the versatility of PS–P2VP reverse micelle loading.
Data availability
The data supporting this article have been included as part of the ESI† or will be made available upon request.Author contributions
TSR: conceptualization, methodology, investigation, validation, data analysis, writing – original draft, review and editing. AAP: methodology, data analysis, validation, writing – review and editing. MJ: conceptualization, methodology, investigation, validation, data analysis. ZJ: methodology, investigation, validation. MK: methodology, investigation, data analysis. PH: XPS data analysis, validation, writing – review and editing. PC: ICP-MS sample preparation, data analysis, writing – review and editing. FY: synthesis of precursors-loaded PS–P2VP reverse micelles. MG: electron microscopy characterization; data analysis. OT: XPS investigation, data analysis. MS: investigation, data analysis. EO: electron microscopy characterization; data analysis. BRC: supervision and validation of micelle fabrication. DCC: supervision and validation of NMR measurements. NEL: supervision and validation of NMR measurements, writing – review and editing. KT: conceptualization, supervision, project administration, funding acquisition, writing – review and editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Germany's Excellence Strategy – EXC-2033 – 390677874 – RESOLV, the Research Training Group “Confinement-Controlled Chemistry” – grant GRK2376/331085229, SFB 1316 – project number 327886311 and SFB 1625 – project number 506711657 for the funding. TSR acknowledges the Deutscher Akademischer Austauschdienst (DAAD, German Academic Exchange Service) – funding program number 57572629 for the scholarship fundings. TSR and KT thank SFB 1316 – project number 327886311. AAP and KT thank the European Union's Horizon 2020 Research and Innovation Programme (ERC project MITICAT, grant 949724). ZJ and KT acknowledge SFB 1625 – project number 506711657. PH, PC, and KT are grateful for the support of the Max Planck Society through the Max Planck Fellowship Program. MS acknowledges the German Chemical Industry Fund (FCI) for a Kekulé Fellowship. The authors acknowledge the Zentrum für Grenzflächendominierte Höchstleistungswerkstoffe (ZGH, Center for Interface-Dominated High Performance Materials) for the use of the JEOL JSM-7200F SEM, the Center for Solvation Science ZEMOS (Zentrum für molekulare Spektroskopie und Simulation solvensgesteuerter Prozesse) for the use of the JEOL JEM-2800 TEM and access to further infrastructure and laboratories, and the Analytical Resources Core (ARC) of Colorado State University for the use of the Agilent 400 MR NMR spectrometer. The authors are grateful to Linda Stefen and the Analytiklabor of the Fraunhofer-Institut für Umwelt-, Sicherheits- und Energietechnik UMSICHT for the execution of the ICP-MS measurements. Dr Aleksander Kostka, Dr Yenting Chen, Dr Michele Mailhot and Dr Lennart Sobota are acknowledged for the SEM, STEM/EDX, NMR and Schlenk line training, respectively. The authors thank Arian Saghri for creating the graphical abstract and Scheme 1.References
- X. Han, K. Xu, O. Taratula and K. Farsad, Applications of nanoparticles in biomedical imaging, Nanoscale, 2019, 11, 799–819 RSC.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Energy storage: the future enabled by nanomaterials, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- H. Jin, L. Zhou, C. L. Mak, H. Huang, W. M. Tang and H. L. Wa Chan, Improved performance of asymmetric fiber-based micro-supercapacitors using carbon nanoparticles for flexible energy storage, J. Mater. Chem. A, 2015, 3, 15633–15641 RSC.
- K. M. Noone and D. S. Ginger, Doping for speed: colloidal nanoparticles for thin-film optoelectronics, ACS Nano, 2009, 3, 261–265 CrossRef CAS PubMed.
- L. de Sio, T. Placido, R. Comparelli, M. Lucia Curri, M. Striccoli, N. Tabiryan and T. J. Bunning, Next-generation thermo-plasmonic technologies and plasmonic nanoparticles in optoelectronics, Prog. Quant. Electron., 2015, 41, 23–70 CrossRef.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Advances in the Application of Magnetic Nanoparticles for Sensing, Adv. Mater., 2019, 31, e1904385 CrossRef PubMed.
- A. M. Parambil, E. Priyadarshini, S. Paul, A. Bakandritsos, V. K. Sharma and R. Zbořil, Emerging nanomaterials for the detection of per- and poly-fluorinated substances, J. Mater. Chem. A, 2025, 13, 8246–8281 RSC.
- B. R. Cuenya, Synthesis and catalytic properties of metal nanoparticles: size, shape, support, composition, and oxidation state effects, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS.
- J. Li and S. Sun, Intermetallic Nanoparticles: Synthetic Control and Their Enhanced Electrocatalysis, Acc. Chem. Res., 2019, 52, 2015–2025 CrossRef CAS PubMed.
- M. V. Evers, M. Bernal, B. Roldan Cuenya and K. Tschulik, Piece by Piece - Electrochemical Synthesis of Individual Nanoparticles and their Performance in ORR Electrocatalysis, Angew. Chem., Int. Ed. Engl., 2019, 58, 8221–8225 CrossRef CAS PubMed.
- Q. Zheng, H. Kang, J. Yun, J. Lee, J. H. Park and S. Baik, Hierarchical construction of self-standing anodized titania nanotube arrays and nanoparticles for efficient and cost-effective front-illuminated dye-sensitized solar cells, ACS Nano, 2011, 5, 5088–5093 CrossRef CAS PubMed.
- N. Deogratias, M. Ji, Y. Zhang, J. Liu, J. Zhang and H. Zhu, Core@shell sub-ten-nanometer noble metal nanoparticles with a controllable thin Pt shell and their catalytic activity towards oxygen reduction, Nano Res., 2015, 8, 271–280 CrossRef CAS.
- L. Wang, M. H. Kafshgari and M. Meunier, Optical Properties and Applications of Plasmonic-Metal Nanoparticles, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- J. F. Gomes, A. C. Garcia, C. Pires, E. B. Ferreira, R. Q. Albuquerque, G. Tremiliosi-Filho and L. H. S. Gasparotto, Impact of the AuAg NPs Composition on their Structure and Properties: A Theoretical and Experimental Investigation, J. Phys. Chem. C, 2014, 118, 28868–28875 CrossRef CAS.
- F. Fiévet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J.-Y. Piquemal, L. Sicard and G. Viau, The polyol process: a unique method for easy access to metal nanoparticles with tailored sizes, shapes and compositions, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
- S. Mahmud, S. S. Satter, A. K. Singh, M. M. Rahman, M. Y. A. Mollah and M. A. B. H. Susan, Tailored Engineering of Bimetallic Plasmonic Au@Ag Core@Shell Nanoparticles, ACS Omega, 2019, 4, 18061–18075 CrossRef CAS PubMed.
- A. M. Mostafa, E. A. Mwafy, N. S. Awwad and H. A. Ibrahium, Au@Ag core/shell nanoparticles prepared by laser-assisted method for optical limiting applications, J. Mater. Sci.: Mater. Electron., 2021, 32, 14728–14739 CrossRef CAS.
- P. Rani, R. S. Varma, K. Singh, R. Acevedo and J. Singh, Catalytic and antimicrobial potential of green synthesized Au and Au@Ag core-shell nanoparticles, Chemosphere, 2023, 317, 137841 CrossRef CAS PubMed.
- M. Perez-Lloret, A. Fraix, S. Petralia, S. Conoci, V. Tafani, G. Cutrone, A. Vargas-Berenguel, R. Gref and S. Sortino, One-Step Photochemical Green Synthesis of Water-Dispersible Ag, Au, and Au@Ag Core-Shell Nanoparticles, Chemistry, 2019, 25, 14638–14643 CrossRef CAS PubMed.
- C. Hamon and D. Constantin, Growth Kinetics of Core–Shell Au/Ag Nanoparticles, J. Phys. Chem. C, 2020, 124, 21717–21721 CrossRef CAS.
- A. Papaderakis, I. Mintsouli, J. Georgieva and S. Sotiropoulos, Electrocatalysts Prepared by Galvanic Replacement, Catalysts, 2017, 7, 80 CrossRef.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Synthesis, Properties, and Applications of Hollow Micro-/Nanostructures, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- S. Zafeiratos, S. Piccinin and D. Teschner, Alloys in catalysis: phase separation and surface segregation phenomena in response to the reactive environment, Catal. Sci. Technol., 2012, 2, 1787 RSC.
- N. Gilis, L. Jacobs, C. Barroo and T. Visart de Bocarmé, Surface Segregation in Au–Ag Alloys Investigated by Atom Probe Tomography, Top. Catal., 2018, 61, 1437–1448 CrossRef CAS.
- H. Lee, S. Hong, J. Lee, Y. D. Suh, J. Kwon, H. Moon, H. Kim, J. Yeo and S. H. Ko, Highly Stretchable and Transparent Supercapacitor by Ag-Au Core-Shell Nanowire Network with High Electrochemical Stability, ACS Appl. Mater. Interfaces, 2016, 8, 15449–15458 CrossRef CAS PubMed.
- Y. Zhu, S. Kim, X. Ma, P. Byrley, N. Yu, Q. Liu, X. Sun, D. Xu, S. Peng, M. C. Hartel, S. Zhang, V. Jucaud, M. R. Dokmeci, A. Khademhosseini and R. Yan, Ultrathin-shell epitaxial Ag@Au core-shell nanowires for high-performance and chemically-stable electronic, optical, and mechanical devices, Nano Res., 2021, 14, 4294–4303 CrossRef CAS.
- E. Csapó, A. Oszkó, E. Varga, Á. Juhász, N. Buzás, L. Kőrösi, A. Majzik and I. Dékány, Synthesis and characterization of Ag/Au alloy and core(Ag)–shell(Au) nanoparticles, Colloids Surf., A, 2012, 415, 281–287 CrossRef.
- I. Shmarakov, I. Mukha, N. Vityuk, V. Borschovetska, N. Zhyshchynska, G. Grodzyuk and A. Eremenko, Antitumor Activity of Alloy and Core-Shell-Type Bimetallic AgAu Nanoparticles, Nanoscale Res. Lett., 2017, 12, 333 CrossRef PubMed.
- A. Sakthisabarimoorthi, S. A. M. B. Dhas and M. Jose, Preparation of composite Ag@Au core–shell nanoparticles and their linear and nonlinear optical properties, J. Mater. Sci.: Mater. Electron., 2019, 30, 1677–1685 CrossRef CAS.
- M. Wuithschick, B. Paul, R. Bienert, A. Sarfraz, U. Vainio, M. Sztucki, R. Kraehnert, P. Strasser, K. Rademann, F. Emmerling and J. Polte, Size-Controlled Synthesis of Colloidal Silver Nanoparticles Based on Mechanistic Understanding, Chem. Mater., 2013, 25, 4679–4689 CrossRef CAS.
- M. Jaugstetter, N. Blanc, M. Kratz and K. Tschulik, Electrochemistry under confinement, Chem. Soc. Rev., 2022, 51, 2491–2543 RSC.
- J. Reyes-Morales and J. E. Dick, Electrochemical-Shock Synthesis of Nanoparticles from Sub-femtoliter Nanodroplets, Acc. Chem. Res., 2023, 56, 1178–1189 CrossRef CAS PubMed.
- N. Anton, J.-P. Benoit and P. Saulnier, Design and production of nanoparticles formulated from nano-emulsion templates-a review, J. Contr. Release, 2008, 128, 185–199 CrossRef CAS PubMed.
- A. Guiet, T. Reier, N. Heidary, D. Felkel, B. Johnson, U. Vainio, H. Schlaad, Y. Aksu, M. Driess, P. Strasser, A. Thomas, J. Polte and A. Fischer, A One-Pot Approach to Mesoporous Metal Oxide Ultrathin Film Electrodes Bearing One Metal Nanoparticle per Pore with Enhanced Electrocatalytic Properties, Chem. Mater., 2013, 25, 4645–4652 CrossRef CAS.
- M. W. Glasscott, A. D. Pendergast and J. E. Dick, A Universal Platform for the Electrodeposition of Ligand-Free Metal Nanoparticles from a Water-in-Oil Emulsion System, ACS Appl. Nano Mater., 2018, 1, 5702–5711 CrossRef CAS.
- A. D. Pendergast, M. W. Glasscott, C. Renault and J. E. Dick, One-step electrodeposition of ligand-free PdPt alloy nanoparticles from water droplets: controlling size, coverage, and elemental stoichiometry, Electrochem. Commun., 2019, 98, 1–5 CrossRef CAS.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- H. Mistry, R. Reske, P. Strasser and B. Roldan Cuenya, Size-dependent reactivity of gold-copper bimetallic nanoparticles during CO2 electroreduction, Catal. Today, 2017, 288, 30–36 CrossRef CAS.
- D. Alwast, J. Schnaidt, K. Hancock, G. Yetis and R. J. Behm, Effect of Li+ and Mg2+ on the Electrochemical Decomposition of the Ionic Liquid 1-Butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide and Related Electrolytes, ChemElectroChem, 2019, 6, 3009–3019 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Molecular mechanism of the photochemical generation of gold nanoparticles in ethylene glycol: support for the disproportionation mechanism, J. Phys. Chem. B, 2006, 110, 14014–14019 CrossRef CAS PubMed.
- M. Deen, C. Shamshoom, A. Gamble, D. Bejan and N. J. Bunce, Electrodeposition of metal cations from the wet ionic liquid [EMIM][TFSI], Can. J. Chem., 2016, 94, 170–175 CrossRef CAS.
- L. Aldous, D. S. Silvester, C. Villagrán, W. R. Pitner, R. G. Compton, M. Cristina Lagunas and C. Hardacre, Electrochemical studies of gold and chloride in ionic liquids, New J. Chem., 2006, 30, 1576–1583 RSC.
- C. Villagrán, C. E. Banks, C. Hardacre and R. G. Compton, Electroanalytical determination of trace chloride in room-temperature ionic liquids, Anal. Chem., 2004, 76, 1998–2003 CrossRef PubMed.
- D. J. Lomax and R. A. Dryfe, Electrodeposition of Au on basal plane graphite and graphene, J. Electroanal. Chem., 2018, 819, 374–383 CrossRef CAS.
- D. Pltcher and F. C. Walsh, Metal finishing, in Industrial Electrochemistry, Springer, Dordrecht, 1993 Search PubMed.
- L. M. Monzon, F. Byrne and J. Coey, Gold electrodeposition in organic media, J. Electroanal. Chem., 2011, 657, 54–60 CrossRef CAS.
- Y.-Z. Su, Y.-C. Fu, J.-W. Yan, Z.-B. Chen and B.-W. Mao, Double layer of Au(100)/ionic liquid interface and its stability in imidazolium-based ionic liquids, Angew. Chem., Int. Ed. Engl., 2009, 48, 5148–5151 CrossRef CAS PubMed.
- Y.-Z. Su, J.-W. Yan, M.-G. Li, Z.-X. Xie, B.-W. Mao and Z.-Q. Tian, Adsorption of Solvent Cations on Au(111) and Au(100) in Alkylimidazolium-Based Ionic Liquids – Worm-Like versus Micelle-Like Structures, Z. Phys. Chem., 2012, 226, 979–994 CrossRef CAS.
- R. Atkin, S. Z. El Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, AFM and STM Studies on the Surface Interaction of [BMP]TFSA and [EMIm]TFSA Ionic Liquids with Au(111), J. Phys. Chem. C, 2009, 113, 13266–13272 CrossRef CAS.
- J. Yang, F. Hilpert, Y. Qiu, E. Franz, V. Briega-Martos, S. Cherevko, K. Mayrhofer, O. Brummel and J. Libuda, Interactions of the Ionic Liquid [C 2 C 1 Im][DCA] with Au(111) Electrodes: Interplay between Ion Adsorption, Electrode Structure, and Stability, J. Phys. Chem. C, 2024, 128, 2834–2843 CrossRef CAS.
- M. Jitvisate and J. R. T. Seddon, Direct Measurement of the Differential Capacitance of Solvent-Free and Dilute Ionic Liquids, J. Phys. Chem. Lett., 2018, 9, 126–131 CrossRef CAS PubMed.
- A. Pearson, A. P. O'Mullane, V. Bansal and S. K. Bhargava, Galvanic replacement mediated transformation of Ag nanospheres into dendritic Au-Ag nanostructures in the ionic liquid BMIMBF4, Chem. Commun., 2010, 46, 731–733 RSC.
- V. Grasmik, C. Rurainsky, K. Loza, M. V. Evers, O. Prymak, M. Heggen, K. Tschulik and M. Epple, Deciphering the Surface Composition and the Internal Structure of Alloyed Silver-Gold Nanoparticles, Chemistry, 2018, 24, 9051–9060 CrossRef CAS PubMed.
- U. Mayer, V. Gutmann and W. Gerger, The acceptor number - a quantitative empirical parameter for the electrophilic properties of solvents, Monatsh. Chem., 1975, 1235–1257 CrossRef CAS.
- J. Yang, A. A. Papaderakis, J. S. Roh, A. Keerthi, R. W. Adams, M. A. Bissett, B. Radha and R. A. W. Dryfe, Measuring the Capacitance of Carbon in Ionic Liquids: From Graphite to Graphene, J. Phys. Chem. C, 2024, 128, 3674–3684 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Mechanisms of nucleation and growth of nanoparticles in solution, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- M. W. Glasscott and J. E. Dick, Fine-Tuning Porosity and Time-Resolved Observation of the Nucleation and Growth of Single Platinum Nanoparticles, ACS Nano, 2019, 13, 4572–4581 CrossRef CAS PubMed.
- Y.-S. Jun, Y. Zhu, Y. Wang, D. Ghim, X. Wu, D. Kim and H. Jung, Classical and Nonclassical Nucleation and Growth Mechanisms for Nanoparticle Formation, Annu. Rev. Phys. Chem., 2022, 73, 453–477 CrossRef CAS PubMed.
- J. Polte, Fundamental growth principles of colloidal metal nanoparticles – a new perspective, CrystEngComm, 2015, 17, 6809–6830 RSC.
- B. Scharifker and G. Hills, Theoretical and experimental studies of multiple nucleation, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- B. Pal, S. Rana and R. Kaur, Influence of Different Reducing Agents on the Ag Nanostructures and Their Electrokinetic and Catalytic Properties, J. Nanosci. Nanotechnol., 2015, 15, 2753–2760 CrossRef CAS PubMed.
- S. Karimi, A. Moshaii and M. Nikkhah, Controlled synthesis of colloidal monodisperse gold nanoparticles in a wide range of sizes; investigating the effect of reducing agent, Mater. Res. Express, 2019, 6, 1150f2 CrossRef.
- B. J. Plowman, B. Sidhureddy, S. V. Sokolov, N. P. Young, A. Chen and R. G. Compton, Electrochemical Behavior of Gold–Silver Alloy Nanoparticles, ChemElectroChem, 2016, 3, 1039–1043 CrossRef CAS.
- E. N. Saw, V. Grasmik, C. Rurainsky, M. Epple and K. Tschulik, Electrochemistry at single bimetallic nanoparticles - using nano impacts for sizing and compositional analysis of individual AgAu alloy nanoparticles, Faraday Discuss., 2016, 193, 327–338 RSC.
- P. Cignoni, P. Hosseini, C. Kaiser, O. Trost, D.-R. Nettler, L. Trzebiatowski and K. Tschulik, Validating Electrochemical Active Surface Area Determination of Nanostructured Electrodes: Surface Oxide Reduction on AuPd Nanoparticles, J. Electrochem. Soc., 2023, 170, 116505 CrossRef CAS.
- S. K. Panda, S. Sen, S. Roy and A. Moyez, Synthesis of Colloidal Silver Nanoparticles by Reducing Aqueous AgNO3 Using Green Reducing Agents, Mater. Today: Proc., 2018, 5, 10054–10061 CAS.
- S. Ghosh, R. Hawtof, P. Rumbach, D. B. Go, R. Akolkar and R. M. Sankaran, Quantitative Study of Electrochemical Reduction of Ag + to Ag Nanoparticles in Aqueous Solutions by a Plasma Cathode, J. Electrochem. Soc., 2017, 164, D818–D824 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- C. Rurainsky, A. G. Manjón, F. Hiege, Y.-T. Chen, C. Scheu and K. Tschulik, Electrochemical dealloying as a tool to tune the porosity, composition and catalytic activity of nanomaterials, J. Mater. Chem. A, 2020, 8, 19405–19413 RSC.
- A. Londono-Calderon, C. A. Campos-Roldan, R. G. González-Huerta, M. L. Hernandez-Pichardo, P. Del Angel and M. J. Yacaman, Influence of the architecture of Au Ag Pt nanoparticles on the electrocatalytic activity for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2017, 42, 30208–30215 CrossRef CAS.
- J. T. Bender, A. S. Petersen, F. C. Østergaard, M. A. Wood, S. M. J. Heffernan, D. J. Milliron, J. Rossmeisl and J. Resasco, Understanding Cation Effects on the Hydrogen Evolution Reaction, ACS Energy Lett., 2023, 8, 657–665 CrossRef CAS.
- G. K. Gebremariam, A. Z. Jovanović, A. S. Dobrota, N. V. Skorodumova and I. A. Pašti, Hydrogen Evolution Volcano(es)—From Acidic to Neutral and Alkaline Solutions, Catalysts, 2022, 12, 1541 CrossRef CAS.
- F. Behafarid, J. Matos, S. Hong, L. Zhang, T. S. Rahman and B. R. Cuenya, Structural and Electronic Properties of Micellar Au Nanoparticles: Size and Ligand Effects, ACS Nano, 2014, 8, 6671–6681 CrossRef CAS PubMed.
- N. M. Correa, J. J. Silber, R. E. Riter and N. E. Levinger, Nonaqueous polar solvents in reverse micelle systems, Chem. Rev., 2012, 112, 4569–4602 CrossRef CAS PubMed.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier, Amsterdam, Boston, 1st edn, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01833a |
| This journal is © The Royal Society of Chemistry 2025 |