
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed simultaneous iron and cerium redox reactions for durable chemical looping dry reforming of methane†
Minjung
Kim
a,
Michael
Tomechko
b and
Shang
Zhai
 *ac
*ac
aDepartment of Mechanical and Aerospace Engineering, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: zhai.218@osu.edu
bWilliam G. Lowrie Department of Chemical and Biomolecular Engineering, The Ohio State University, Columbus, OH 43210, USA
cSchool of Earth Sciences, The Ohio State University, Columbus, Ohio 43210, USA
First published on 2nd June 2025
Abstract
This study investigates the synergistic interaction among elements in nickel–(iron and cerium oxide) for chemical looping dry reforming of methane (CLDRM) at 700 °C to 900 °C. Nickel catalyzes methane conversion to enable simultaneous reduction of iron and cerium. We quantified methane conversion by two co-existing mechanisms: partial oxidation to syngas and pyrolysis to solid carbon and H2. In the CO2 step, the bulk interaction between Fe and Ce forms cerium orthoferrite (CeFeO3) that enhances methane conversion, despite the anomalously reduced Ce3+ in the oxidation product, CeFeO3. An optimal nickel loading enhances methane conversion and CeFeO3 formation while limiting solid carbon accumulation, and it decreases with increasing CLDRM temperature, because high temperature also facilitates the reactions. The optimal nickel loading in Ni0.34–(Fe0.67Ce0.50Ox)-900 maintained 76% conversion rates for methane and CO2 over 100 CLDRM cycles at 900 °C, with only 0.26% of the carbon in methane accumulated as solid carbon across the cycles. Highly adjustable syngas ratio is achieved, because methane step generates mostly H2 while CO is mostly from CO2 step. Our findings illustrate the mechanisms of the nickel–(iron and cerium oxide) materials for efficient and durable CLDRM, offering valuable insights about mixed catalyst and oxygen carrier material design.
1. Introduction
Anthropogenic carbon emission level was estimated at 11.1 ± 0.8 Gt carbon per year in 2023, emphasizing the critical need for effective CO2 conversion techniques as part of the global transition towards carbon neutrality.1,2 Carbon conversion techniques turn CO2 into value-added products. One such promising product is syngas, a mixture of carbon monoxide (CO) and hydrogen (H2), a feedstock to produce fuels, chemicals, and energy carriers such as methanol, ammonia, and liquid hydrocarbons. Various techniques exist for syngas production, including steam reforming of methane (SMR), partial oxidation of methane (POM), coal gasification, and biomass gasification. Of these methods, SMR is currently the most prominent technology in the industry using steam as a partial oxidizing agent for methane.3–5Dry reforming of methane (DRM) emerged as a more sustainable alternative by converting two major greenhouse gases, methane and carbon dioxide, into value-added syngas (eqn (1)). However, DRM faces significant challenges that hinder its broad industrial application: (1) carbon deposition from methane pyrolysis (eqn (2))6–10 and the Boudouard reaction (eqn (3)),11–13 which deactivates catalyst at high-temperature (typically well above 600 °C); (2) the need for high operating temperature to activate and break the C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in methane and carbon dioxide;14–16 (3) some of the hydrogen product can be consumed by unreacted carbon dioxide via the reverse water gas shift reaction (eqn (4)).
O bonds in methane and carbon dioxide;14–16 (3) some of the hydrogen product can be consumed by unreacted carbon dioxide via the reverse water gas shift reaction (eqn (4)).
| CH4 + CO2 → 2H2 + 2CO | (1) |
| CH4 → C + 2H2 | (2) |
| 2CO → C + CO2 | (3) |
| CO2 + H2 → CO + H2O | (4) |
Many metals have been explored as catalysts to mitigate these challenges. Noble metal catalysts, such as Pt, Rh, and Ru often demonstrate lower carbon deposition compared to non-noble metal catalysts like Ni and Co but are less viable for industrial use due to their high cost.17,18 Ni based catalysts have been intensively studied due to their low cost and high reactivity with CH4.19 Hou et al.20 utilized rhodium over Ni supported on mesoporous alumina. The addition of a small amount of rhodium led to higher catalytic performance without coke formation. Ni-based catalysts for methane reforming suffer from severe carbon deposition, while rhodium, though effective, is costly and exhibits poor stability. Consequently, the long-term stability of the material remains a significant concern for practical application. Another Ni catalyst composed of 5% Ni–3% CeO2 supported on mesoporous nanocrystalline zirconia demonstrated effective catalytic performance.21 The CH4 conversion was initially about 70% but it decayed to 40% after 1500 hours of continuous operation. The deactivation issue was possibly due to the coke formation and material sintering.
Chemical looping uses a chemical intermediate to selectively transfer mass between each step of a global reaction. Certain metal oxides are often used as the chemical intermediate due to their ability to exchange oxygen with the gas phase depending on oxygen partial pressure and temperature. This approach has been extensively studied for DRM (eqn (5) and (6)).
| δCH4 + MOx → δCO + 2δH2 + MOx−δ | (5) |
| δCO2 + MOx−δ → δCO + MOx | (6) |
Chemical looping dry reforming of methane (CLDRM) offers the advantage of oxidizing deposited carbon by CO2, thereby preventing long-term catalyst deactivation.22,23 Also, the separation of methane and carbon dioxide feeds prevents hydrogen product loss in the presence of CO2 through the reverse water gas shift reaction. Furthermore, a preferred syngas H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio can be obtained by controlled mixing of the two product streams. Thus, CLDRM presents a promising strategy for optimizing syngas production while addressing the limitations of traditional DRM. Kim et al.24 investigated a Ni–Fe–Al oxide oxygen carrier, which enabled stable CLDRM with 99% methane conversion, overcoming deactivation issues by incorporating Ni into Fe–Al oxide through spinel phase formation, enhanced CH4 activation, and CO2-assisted carbon gasification. Shao et al.25 demonstrated the in situ formation of a halite-structured (MgCoNiMnFe)Ox high entropy oxide catalyst for CLDRM, achieving exceptional syngas selectivity and cyclic stability over 100 redox cycles at 800 °C. Rao et al.26 found that a nickel-modified iron oxide oxygen carrier supported on La0.8Sr0.2FeO3 significantly enhances methane conversion, CO selectivity, and stability over 160 cycles in CLDRM.
:CO ratio can be obtained by controlled mixing of the two product streams. Thus, CLDRM presents a promising strategy for optimizing syngas production while addressing the limitations of traditional DRM. Kim et al.24 investigated a Ni–Fe–Al oxide oxygen carrier, which enabled stable CLDRM with 99% methane conversion, overcoming deactivation issues by incorporating Ni into Fe–Al oxide through spinel phase formation, enhanced CH4 activation, and CO2-assisted carbon gasification. Shao et al.25 demonstrated the in situ formation of a halite-structured (MgCoNiMnFe)Ox high entropy oxide catalyst for CLDRM, achieving exceptional syngas selectivity and cyclic stability over 100 redox cycles at 800 °C. Rao et al.26 found that a nickel-modified iron oxide oxygen carrier supported on La0.8Sr0.2FeO3 significantly enhances methane conversion, CO selectivity, and stability over 160 cycles in CLDRM.
Metallic Ni has been shown to activate C–H bond to enhance the kinetics for methane conversion.27,28 Hill et al. found that Ni supported on ceria significantly enhances syngas production through CLDRM compared to non-catalyzed ceria because of low activation energy (<50 kJ mol−1).29 Cao et al.30 investigated that Ni/CeZrO2 catalysts selectively oxidize methane and subsequently split CO2 and H2O via oxygen-vacancy-rich surfaces, demonstrating stable redox performance at 650 °C. Han et al.31 reported a ultralow Ni loading on ceria achieved nearly 100% methane conversion and CO selectivity in CLDRM without carbon deposition over 50 cycles due to strong metal–support interaction that stabilized Ni nanoparticles. Löfberg et al. also used Ni supported on CeO2.27 Ni has two important roles: (1) activation of the reactants and (2) enhancing the rate of oxygen supply from the solid.
However, Ni cannot be oxidized by carbon dioxide at typical CLDRM operating temperatures, limiting its role in oxygen exchange.32 More et al.33 demonstrated that the metallic Ni catalyzes methane cracking and acts as carbon carrier, while remaining unoxidized throughout the cycle. Therefore, other redox active metals or support is needed for oxygen exchange. Iron undergoes a wide range of phases from Fe to Fe3O4 by methane reduction and carbon dioxide reoxidation, providing a large oxygen exchange capacity. Iron has also been shown to dissociate CO bonds, aiding in CO2 decomposition kinetics.34 Zhang et al.35 reported iron-containing perovskite nanocomposites enabled efficient thermochemical CO2 splitting and methane partial oxidation in a cyclic redox scheme, achieving about 96% syngas selectivity and high CO2 conversion at 980 °C. Ceria (CeO2) is an abundant support material with high oxygen mobility and redox reactivity, increasing resistance to carbon deposition and oxygen exchange capacity.36–38
Galvita et al.39 proposed the addition of Ni to CeO2–Fe2O3 led to a deeper reduction of the metal oxide in a chemical looping complete oxidation of methane. By adding Ni, the CO yield became 10 times higher than without during the CO2 step. Carbon accumulation on the surface was successfully removed by CO2 re-oxidation step by lattice oxygen from CeO2–Fe3O4 providing a higher CO yield. Bhavsar et al.40 utilized mixed Ni–Fe oxide supported on ceria for chemical looping partial oxidation of methane to produce syngas, while using air to re-oxidize the solid oxygen carrier.
Mixed Fe–Ce oxides without Ni have also been explored for CLDRM where CeFeO3 phase was identified. Zhang et al.41 proposed using Fe2O3/CeO2 which enhances oxygen release from both components in CLDRM. During the re-oxidation step, the reduced Fe disperses into ceria forming CeFeO3 that provides enhanced methane conversion (81.3%) and CO selectivity (96.6%). García-García et al.42 reported Fe and Ce mixed oxide demonstrated a synergetic interaction and fine dispersion of Fe oxide particles on the Ce support forms CeFeO3.
To our knowledge, the material reaction mechanisms of Ni mixed with Fe and Ce oxides have not been fully understood in CLDRM cycles with CO2 as the oxidizer to produce syngas. Additionally, the role of CeFeO3 and its cyclability over long cycles have not been demonstrated and studied. In this paper, we investigate the distinct role of each metal and their synergistic effect in Ni–(Fe and Ce oxide) for CLDRM. X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS) complemented each other to clearly identify the solid-state phase transformation and redox activity of each element. Our experiments showed three principal findings: (1) nickel is not redox active across CLDRM cycles due to thermodynamic limitation, and it acts as a metallic catalyst for methane pyrolysis, (2) ceria is a bulk oxygen carrier rather than a support, as it undergoes phase transformation among different cerium oxides and mixed Fe–Ce oxide when activated by sufficient amount of Ni, and (3) ceria is anomalously reduced to +3 oxidation state during CO2 step to form CeFeO3, giving an overall large redox capacity via oxidation of Fe from metallic to +3 oxidation state. These results demonstrate that the synergistic roles of Ni–(Fe and Ce oxide) (shown in Fig. 1A) enhance syngas production efficiency from CLDRM.
 | ||
| Fig. 1 (A) General reaction mechanisms we discovered for chemical looping dry reforming of methane (CLDRM) with Ni–(Fe and Ce oxide). CH4 reduces the oxide to Fe–Ni alloy and oxygen-deficient ceria to produce syngas (H2 + CO), while CH4 can also decompose into solid carbon and H2. In the CO2 step, CO2 is converted to CO by reacting with the solid carbon and oxidizing Fe and reduced cerium oxide to cerium orthoferrites (CeFeO3). (B) Influence of Fe/(Fe + Ni) ratio and temperature on average syngas production rate (over the 3rd to the 10th cycle, solid lines, left axis) and accumulated carbon deposition percentages (carbon accumulation divided by the total carbon in supplied CH4 over the 10 cycles, bars, right axis). All samples contain 60 wt% ceria. | ||
2. Materials and methods
2.1 Ni–(Fe and Ce oxide) synthesis
All samples were prepared via sol–gel synthesis. The masses of iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, Sigma-Aldrich, ≥98%) and nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, Sigma-Aldrich, 99.999%) were determined such that the total mass of iron and nickel elements in each sample was 2 g, with Fe and Ni molar ratio controlled. Masses of citric acid (C6H8O7, Spectrium Chemical Mfg Corp., 99.5%) and cerium(IV) oxide (CeO2, Sigma-Aldrich, 99.9%) were used to reach a molar ratio of 3 moles citric acid to 1 mol of iron plus nickel elements, and a mass ratio of 3 g CeO2 to 2 g of iron and nickel (Fe + Ni) for samples we refer to as “60 wt% CeO2”. For example, preparing Ni0.50–(Fe0.50Ce0.50Ox)-800 and Ni0.50–(Fe0.50Ce0.50Ox)-900 used 7.0534 g Fe(NO3)3·9H2O, 5.0779 g Ni(NO3)2·6H2O, 20.1278 g citric acid, and 3 g CeO2. We write Ni separately at the front of material formula because we found that Ni always remains metallic in the looping. The number following the material name indicates the temperature (in °C) at which it was synthesized and tested. For example, Ni0.50–(Fe0.50Ce0.50Ox)-800 refers to Ni0.50–(Fe0.50Ce0.50Ox) material was synthesized and tested at 800 °C. A detailed naming protocol for all samples is explained in the ESI.†To isolate the effects of Fe and Ni, Fe–Ce oxides (Fe0.33Ce0.50Ox, Fe0.50Ce0.50Ox, and Fe0.67Ce0.50Ox)-800 and Ni supported on cerium oxide (Ni0.34–Ce0.50Ox, Ni0.50–Ce0.50Ox, and Ni0.66–Ce0.50Ox)-800 were prepared. We chose the amount of Fe–Ce oxides or Ni–Ce oxides for CLDRM reaction measurement such that the amount of Fe or Ni and Ce are the same as those in the corresponding Ni–(Fe and Ce oxides)-800. For instance, 0.3464 g of Fe0.33Ce0.50Ox-800 contains the same amount of Fe and Ce as 0.5 g of Ni0.66–(Fe0.33Ce0.50Ox)-800, and 0.4207 g of Ni0.66–Ce0.50Ox-800 contains the same amount of Ni and Ce as 0.5 g of Ni0.66–(Fe0.33Ce0.50Ox)-800, so that we can compare them to understand the roles of Fe and Ni. Detailed explanation is in the ESI.†
To prepare the samples, a solution of citric acid, Fe(NO3)3·9H2O, and Ni(NO3)2·6H2O in 50 mL ethanol (C2H6O, Decon Laboratories, Inc., 95%) was created at room temperature. CeO2 was added, and the suspension was covered and allowed to stir continuously over 6–10 hours. The ethanol was then evaporated at 90 °C for 10–15 hours, creating a thick gel. The gel was dried at 200 °C for 2 hours in a box furnace, forming a solid porous cake. A mortar and pestle were used to grind the solid cake into powder, which was subsequently added to an alumina crucible and calcined in a box furnace in air for 5 hours at 700 °C, 800 °C, or 900 °C after a temperature ramping at 15 °C min−1. The calcination temperature was set the same as the CLDRM reaction temperature. The product was then ground into a fine powder using a mortar and pestle.
2.2 Material characterizations
X-ray diffractometer (XRD) analysis was conducted on an Empyrean Series 3 from Malvern Panalytical X-ray diffractometer operated at 45 kV and 40 mA. The reflection angle was from 20° and 80° at a scan rate of 0.041° per second with a step size of 0.00164°. Phase identification was conducted through PDXL software.43 Crystallographic information files (CIF) were downloaded from ICSD database.44 After the desired CLDRM cycles finished, the sample was cooled down in 150 sccm argon flow to room temperature in U-shaped tubular reactor. Then the tubular reactor was sealed by parafilm and transported into an Ar glovebox. In the glovebox, the cycled sample was spread on a glass slide with vacuum grease. Then the powder was covered with Kapton tape and stored in the glovebox until the time for XRD, to prevent/minimize oxidation of the sample by air. All XRD patterns have a wide peak from 25° to 27° in 2θ due to the Kapton tape.X-ray absorption spectroscopy (XAS) analysis was conducted on easy XAFS 300+ equipment. All cycled samples were prepared in the same way as those for XRD to prevent air exposure. The samples were spread on and wrapped between two Kapton tapes instead of being placed on the glass slide. X-ray Absorption Near Edge Structure (XANES) measurements were performed at the Ni K-edge, Fe K-edge, and Ce LIII-edge. The scan energy range for the Ni K-edge was set from 8200 eV to 9800 eV, divided into four sections: 8200–8300 eV (step size: 2 eV, step duration: 1.5 s), 8300–8600 eV (step size: 0.25 eV, step duration: 3 s), 8600–9000 eV (step size: 2 eV, step duration: 3 s), and 9000–9800 eV (step size: 10 eV, step duration: 3 s). For the Fe K-edge, the energy range of 6900–8100 eV was divided into four sections: 6900–7050 eV (step size: 2 eV, step duration: 1.5 s), 7050–7200 eV (step size: 0.25 eV, step duration: 3 s), 7200–7600 eV (step size: 2 eV, step duration: 3 s), and 7600–8100 eV (step size: 10 eV, step duration: 3 s). For the Ce LIII-edge, the energy range of 5650–6100 eV was measured in three sections: 5650–5700 eV (step size: 2 eV, step duration: 1.5 s), 5700–5900 eV (step size: 0.25 eV, step duration: 3 s), and 5900–6100 eV (step size: 2 eV, step duration: 3 s). Data analysis was performed using Athena software45 to quantify the fractions of different oxidation states of each element in each sample by linear combination fitting (LCF) method.
Reference materials for XANES were: iron(II) oxide (≥99.6%), and iron(II,III) oxide (97%), from Sigma-Aldrich, and iron(III) oxide (99.99%) from BeanTown Chemical. Cerium(IV) oxide (99.9%) is from Sigma-Aldrich. Cerium(IV) oxide was loaded on the U-shape tube reactor and reduced for 20 hours under 25 sccm of H2 at 1100 °C to get Ce2O3 which was confirmed by XRD (Fig. S1A†). CeFeO3 was synthesized by using iron, iron(III) oxide, and cerium(IV) oxide, and confirmed by XRD (Fig. S1B†); the precursor powders were pressed at 3 tons using a circular die of 14.85 mm diameter and calcined at 850 °C for 48 hours under vacuum created by a vacuum pump to avoid air oxidation. NiO was synthesized via sol–gel method by using nickel(II) nitrate hexahydrate and citric acid with ethanol, and confirmed by XRD (Fig. S1C†). The solution was covered and stirred for 6–10 hours, evaporated for 10–15 hours at 90 °C, and calcined for 5 hours at 900 °C. Iron and nickel metals are foils from the XAS lab facility.
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) were conducted with a Thermo Scientific Apreo 2S at 20 kV and 0.8 nA to reveal the material morphology and elemental distribution. Fresh sample was dispersed in deionized water, sonicated, and deposited onto carbon tape before drying at 100 °C for 2 hours. Cycled samples, being highly reactive, were stored and prepared in the glovebox. The samples were loaded on the carbon tape without dispersing in deionized water and transferred to the SEM equipment using a special transfer capsule to avoid air exposure.
2.3 Chemical looping dry reforming procedure
To run a chemical looping dry reforming test, 0.5 g material was measured and placed between thin layers of quartz wool inside a quartz U-shaped packed bed reactor. The packed bed vessel was added to a Micromeritics ChemiSorb TPx furnace unit and it was heated to 700 °C, 800 °C, or 900 °C at a rate of 15 °C min−1 under 150 sccm of Ar. Gas flows were controlled using Aalborg DPC17 units. For each cycle, the sample was reduced under 50 sccm of CH4 mixed with 150 sccm of Ar for 2 minutes, purged with 150 sccm of Ar for 3 minutes, oxidized under 50 sccm of CO2 mixed with 150 sccm of Ar for 2 minutes, and purged with 150 sccm of Ar for 3 minutes.After the desired number of cycles was completed, the samples were oxidized under 80 sccm of 20% O2 balance Ar at the looping temperature before being cooled to room temperature. This fully oxidized all accumulated carbon on the sample to produce CO and CO2, so that carbon accumulation ratio could be quantified by the CO and CO2 gas measurement and expressed as a percentage of carbon in total methane feed. All gaseous products were measured with a Hiden HPR-20 R&D mass spectrometer. To protect the mass spectrometer, H2O in the exhaust was removed using a desiccant; therefore, the H2O product quantity was calculated by hydrogen molar balance among H2, H2O, and CH4.
To characterize the materials after CH4 step and CO2 step, the samples went through 2 complete CLDRM cycles and stopped in the 3rd cycle after CH4 step or CO2 step. For cyclability investigation, Ni0.34–(Fe0.67Ce0.50Ox)-900 samples underwent ninety-nine looping cycles and stopped after CH4 or after the CO2 step in the 100th cycle.
3. Results and discussion
3.1 Ni–(Fe and Ce oxide) performance and two methane conversion mechanisms
The optimal Ni loading for methane conversion varies with temperature. Optimizing Ni loading requires both high syngas production and low carbon accumulation. Seven different Fe-to-Ni ratios mixed with 60 wt% ceria were tested at 700 °C (Fig. S5†), 800 °C (Fig. S6†), and 900° (Fig. S7†). The summarized results are in Fig. 1B and 2S,† where the syngas production rate per gram of material was calculated from the 3rd to the 10th cycle due to the performance instability of the first two cycles. As temperature increased, the syngas production rate improved, and the optimal Fe/(Fe + Ni) ratio for syngas production also increased. This can be explained by a fact that Ni generally catalyzes methane C–H bond cleavage.46–50 As the temperature increased, high Ni loading was no longer necessary to achieve high syngas production rate, as the high temperature also facilitates methane conversion. Additionally, an excess of Ni loading led to increased carbon accumulation. For example, Ni0.85–(Fe0.15Ce0.50Ox)-700 and Ni0.85–(Fe0.15Ce0.50Ox)-900 exhibited high syngas production rates but suffered from significant carbon accumulation over 10 cycles (Fig. S2†).For all samples, mass ratio of Fe plus Ni elements to CeO2 was 2:3, namely CeO2 at 60 wt%, unless otherwise noted. Two representative materials, Ni0.66–(Fe0.33Ce0.50Ox)-800 (iron-poor composition for 800 °C CLDRM) and Ni0.34–(Fe0.67Ce0.50Ox)-800 (iron-rich for 800 °C CLDRM), are discussed here for 10 cycle experimental results. 0.5 g material was packed with quartz wool and tested in a U-shaped tube reactor. 150 sccm Ar was fed into the reactor throughout the experiment and 50 sccm CH4 or CO2 was introduced for 2 minutes for each reaction step. The tube was purged with Ar in between reduction and oxidation steps to separate the two different chemical reactions. The gas flow rate profiles at the reactor exhaust for Ni0.66–(Fe0.33Ce0.50Ox)-800 and Ni0.34–(Fe0.67Ce0.50Ox)-800 are in Fig. 2 and S3,† respectively. For benchmarking, performance of various previously published CLDRM materials and several materials in this paper are summarized in Fig. S4.†
 | ||
| Fig. 2 CLDRM reactor exhaust gas profiles for Ni0.66–(Fe0.33Ce0.50Ox)-800 over (A) 10 cycles and (B) a zoomed-in view of the final three cycles at 800 °C. The black curve is unreacted methane. Hydrogen (red), carbon monoxide (blue), and carbon dioxide (green) are the products during the methane step. In the following CO2 step, CO (blue) is the product, and green curve is unreacted CO2 in the exhaust. At the end of the 10 cycles, 20% O2 in Ar was fed into the reactor to combust and help quantify accumulated carbon deposition by the formed CO2 and CO. | ||
Methane undergoes two chemical reaction mechanisms: (1) taking oxygen from the oxygen carrier to produce syngas, and (2) being pyrolyzed into solid carbon and hydrogen.7 Only the first cycle showed considerable amount of CO2 and H2O produced during the CH4 step (Fig. 2, S3 and S5–S7†), because fresh material was fully oxidized by air during synthesis; as the CLDRM cycles continued, the material could not be fully re-oxidized by CO2.
Ni0.66–(Fe0.33Ce0.50Ox)-800 sample demonstrated rapid production of CO and H2 at the beginning of the methane step, indicating the partial oxidation of methane to syngas occurred immediately; afterwards, CH4 pyrolysis became dominant as the available oxygen from the oxygen carrier was consumed, evidenced by quickly diminishing CO production (Fig. 2). When CO2 step just started, almost all CO2 was reacted. The carbon was almost fully removed by CO2, and the reduced oxide was regenerated by CO2. For all samples, in the end of cycles, 20% O2 balance Ar was introduced to fully oxidize the accumulated solid carbon to CO and CO2 to quantify carbon accumulation shown in Fig. 1B and S2.† For Ni0.66–(Fe0.33Ce0.50Ox)-800, only 0.27% of the total carbon in introduced CH4 ended up as the accumulated solid carbon after 10 cycles.
On the other hand, Ni0.34–(Fe0.67Ce0.50Ox)-800 sample exhibited significantly lower reaction rates than Ni0.66–(Fe0.33Ce0.50Ox)-800, as illustrated in Fig. S3.† The continuous increase of CO and H2 production during methane step suggests that the reaction was mostly methane partial oxidation rather than pyrolysis, which is also supported by Fig. 3A. During CO2 step, CO production occurred rapidly, and the amount of CO generated increased gradually over the cycles (Fig. S3†). This indicates the material became progressively more reduced over cycles, requiring greater conversion of CO2 to regenerate the oxygen carrier, since carbon deposition from methane pyrolysis was minor (Fig. 3A).
 | ||
| Fig. 3 Average methane conversion over the 3rd to the 10th cycle for different Fe/(Fe + Ni) ratios at (A) 800 °C and (B) 900 °C. The methane conversion is divided into partial oxidation and pyrolysis. All samples contain 60 wt% ceria. | ||
Fig. 3A and B further illustrate methane conversions divided into methane partial oxidation and pyrolysis mechanisms by using eqn (7)–(9), for various Fe-to-Ni ratios mixed with the same amount of ceria. At 800 °C, when Fe is more than Ni, methane primarily underwent partial oxidation, while the other compositions exhibited significant pyrolysis with partial oxidation (Fig. 3A). At 900 °C, all materials demonstrated significant portions of both pyrolysis and partial oxidation except pure iron oxide, meaning that even a small amount of Ni was able to significantly promote methane pyrolysis at the elevated temperature of 900 °C (Fig. 3B).
| Solid carbon deposition = (CH4in − CH4out) − CO produced − CO2 produced (all in moles) | (7) |
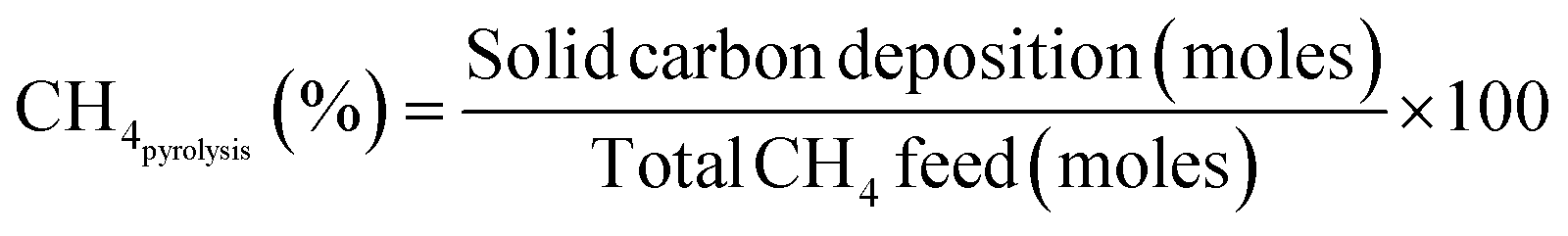 | (8) |
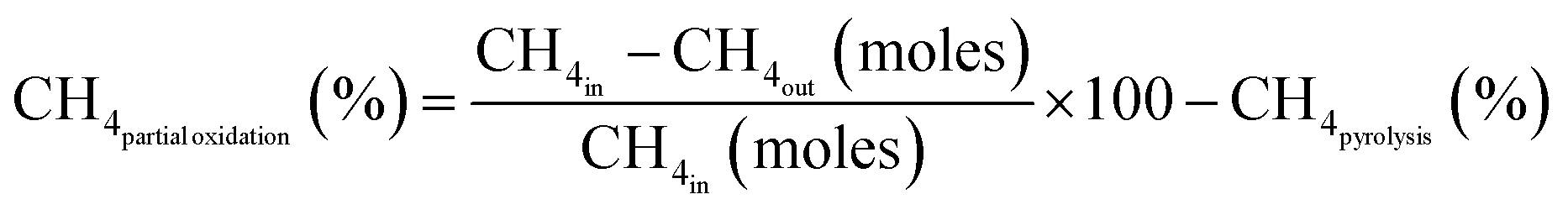 | (9) |
3.2 Nickel as a catalyst and iron as an oxygen carrier
To investigate the roles of Fe and Ni, samples were compared about their CLDRM performance at 800 °C: Niβ–(FeαCe0.50Ox)-800, FeαCe0.50Ox-800, and Niβ–(Ce0.50Ox)-800 where α = 0.33, 0.50, and 0.67 and correspondingly β = 0.66, 0.50, and 0.34. For example, 0.3846 g Fe0.50Ce0.50Ox-800 contains the same amount of Fe and Ce as 0.5 g of Ni0.50–(Fe0.50Ce0.50Ox)-800. Similarly, 0.3808 g Ni0.50–(Ce0.50Ox)-800 has the same amount of Ni and Ce as 0.5 g of Ni0.50–(Fe0.50Ce0.50Ox)-800. Thus, we can compare their performance to understand the roles of Fe and Ni.Although Fe is a well-known catalyst and oxygen carrier for CO2 splitting,51–55 the reactivity between methane and iron oxide mixed with ceria at various loadings was measured to be minimal (Fig. 4A–C), and insignificant Fe and Ce oxide reduction by CH4 led to low CO2 conversion in the subsequent step. Conversely, Ni on ceria samples demonstrated high CH4 and CO2 conversions (Fig. 4D–F), as Ni is a good catalyst for methane conversion.46–50 Hydrogen production significantly exceeded CO production during the methane step, suggesting that with Ni, methane pyrolysis was significantly more favored over syngas production via methane partial oxidation. The excessive carbon formation from methane pyrolysis could not be effectively removed during the CO2 step, giving significant carbon accumulation and performance decay over the cycles (Fig. 4D–F). Finally, Niβ–(FeαCe0.50Ox)-800 demonstrated high and stable reactivity over 10 cycles: syngas production by CH4 partial oxidation occurred along with methane pyrolysis (Figs. S6C–E†). Additionally, carbon accumulation ratios at the end of the 10 cycles of Niβ–(FeαCe0.50Ox)-800 were very small (0.27%, 0.42%, and 1.06%). These results indicate that Fe and Ni create a synergistic effect on both feedstock conversion and carbon elimination.
 | ||
| Fig. 4 Performance of Fe–Ce oxides and Ni–Ce oxides without the synergy between Fe and Ni for CLDRM cycles at 800 °C: (A) Fe0.33Ce0.50Ox-800, (B) Fe0.50Ce0.50Ox-800, (C) Fe0.67Ce0.50Ox-800, (D) Ni0.34–Ce0.50Ox-800, (E) Ni0.50–Ce0.50Ox-800, and (F) Ni0.66–Ce0.50Ox-800. H2, H2O, CO, and CO2 are in red, purple, blue and green bars respectively. CH4 (red square) and CO2 (blue dot) conversions are presented as two curves. | ||
XANES was measured to determine the bulk average oxidation state of each element for Ni0.66–(Fe0.33Ce0.50Ox)-800, Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 and Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900, and Ni0.34–(Fe0.67Ce0.50Ox)-900 samples. The reduced samples were after CH4 step of the 3rd cycle, while the oxidized samples were after CO2 step of the 3rd cycle. All samples were cooled down and protected in Ar until the end of characterization. Ni and Fe XANES spectra are in Fig. S8 and S9.† To quantify the fractions of different oxidation states and get an average oxidation state for each element in each sample, a linear combination fitting (LCF) method was employed, considering co-existing phases and oxidation states of each element. The results are in Tables S1, S2,† and Fig. 5.
 | ||
| Fig. 5 XANES derived average oxidation state of Ni [(A) and (B)] and Fe [(C) and (D)] in CH4-reduced (red square) and CO2-oxidized (blue dot) samples collected from the 3rd cycle, including (A) and (C) Ni0.66–(Fe0.33Ce0.50Ox)-800, Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 at 800 °C and (B) and (D) Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900, and Ni0.34–(Fe0.67Ce0.50Ox)-900 at 900 °C. Ni metal and NiO reference spectra, and Fe metal, Fe3O4, and CeFeO3 reference spectra were used in linear combination fitting to quantify the fractions of different oxidation states for each element in each sample (detailed results in Tables S1 and S2†), so that their average oxidation states in this figure could be calculated. | ||
Ni spectra in all reduced and oxidized samples overlapped well with the metallic Ni reference, so Ni remained a metallic catalyst rather than an oxygen carrier in all cycles (Fig. 5A and B). The thermodynamic limitation of Ni oxidation by CO2 was also demonstrated by its large and positive reaction Gibbs free energy change (Fig. S10†). On the other hand, Fe serves as an oxygen carrier. At 800 °C, Ni0.66–(Fe0.33Ce0.50Ox)-800 sample had minimal Fe redox activity and Fe remained predominantly metallic (Fig. 5C). This suggests there was another redox active element in Ni0.66–(Fe0.33Ce0.50Ox)-800 rather than Fe or Ni to achieve the measured partial oxidation of methane (Fig. 3A). Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 samples exhibited clear Fe redox activity at 800 °C. For the Ni0.50–(Fe0.50Ce0.50Ox)-800 material, Fe was completely reduced to metallic by CH4, while the Fe in Ni0.34–(Fe0.67Ce0.50Ox)-800 sample was not fully reduced. Overall, at 800 °C, Fe redox capacity of Ni0.50–(Fe0.50Ce0.50Ox)-800 material was the highest among the three samples. In comparison, at 900 °C, Fe redox capacity generally increased compared to 800 °C, and Fe redox capacity increased significantly with increasing Fe/(Fe + Ni) ratio (Fig. 5D). Additionally, the Fe spectra of CO2-oxidized Ni0.50–(Fe0.50Ce0.50Ox)-800, Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900, and Ni0.34–(Fe0.67Ce0.50Ox)-900 samples resemble the CeFeO3 reference spectrum (Fig. S9†), implying the formation of a large amount of CeFeO3 phase.
3.3 Ceria as an oxygen carrier with anomalous redox behavior
Ni0.66–(Fe0.33Ox)-800 and Ni0.34–(Fe0.67Ox)-800 with and without ceria, as well as pure ceria, were separately cycled at 800 °C to assess the role of ceria. Pure ceria (Fig. S6H†) and Ni–(Fe oxides)-800 (Fig. 6A and B) exhibited minimal reactivity. However, when ceria was mixed with Ni–(Fe oxides)-800, they achieved significantly enhanced CH4 and CO2 conversions with negligible carbon accumulation (Fig. 6C and D). Also, the conversions were influenced by Fe/(Fe + Ni) ratio and Ni0.66–(Fe0.33Ce0.50Ox)-800 has higher conversions than Ni0.34–(Fe0.67Ce0.50Ox)-800 at 800 °C. | ||
| Fig. 6 Effect of ceria (60 wt%) and Fe-to-Ni ratio for CLDRM at 800 °C. (A) Ni0.66–(Fe0.33Ox)-800, (B) Ni0.34–(Fe0.67Ox)-800, (C) Ni0.66–(Fe0.33Ce0.50Ox)-800, and (D) Ni0.34–(Fe0.67Ce0.50Ox)-800. CO, CO2, H2O and H2 are blue, green, purple, and red in the bars, respectively. The two curves are the conversions of CH4 (red square) and CO2 (blue circle). 0.5 g samples were tested. | ||
XANES was used to reveal Ce oxidation states in 800 °C and 900 °C cycles (Fig. 7 and S11†). At 800 °C, Ni0.66–(Fe0.33Ce0.50Ox)-800 sample showed the largest change in Ce oxidation state (Fig. 7A). In Fig. S11A,† CH4-reduced Ni0.66–(Fe0.33Ce0.50Ox)-800 has Ce spectrum similar to Ce2O3 and CeFeO3 references, indicating Ce mostly at +3 oxidation state. CO2-oxidized Ni0.66–(Fe0.33Ce0.50Ox)-800 has Ce spectrum very similar to CeO2 reference, indicating Ce is almost fully re-oxidized to +4. Therefore, in Ni0.66–(Fe0.33Ce0.50Ox)-800, Ce served as a high-capacity oxygen carrier giving methane partial oxidation (Fig. 3A), while neither Ni nor Fe demonstrated meaningful redox capacity (Fig. 5A and C). In contrast, for Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 samples, the average oxidation state of Ce remained almost unchanged between CH4-reduced and CO2-oxidized states (Fig. 7A, S11B and S11C†).
 | ||
| Fig. 7 (A) and (B) XANES derived average oxidation state of Ce in CH4-reduced (red square) and CO2-oxidized (blue dot) samples collected from the 3rd cycle, including (A) Ni0.66–(Fe0.33Ce0.50Ox)-800, Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 compositions and (B) Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900, and Ni0.34–(Fe0.67Ce0.50Ox)-900 compositions. Reference materials Ce2O3, CeO2, and CeFeO3 were used in linear combination fitting to quantify the fractions of different Ce oxidation states (detailed results in Tables S1 and S2†), so that the average oxidation states in this figure could be calculated. (C) and (D) Ce LIII edge XANES spectra for CH4-reduced and CO2-oxidized Ni0.66–(Fe0.33Ce0.50Ox)-900 and Ni0.34–(Fe0.67Ce0.50Ox)-900, respectively, from the 3rd cycle at 900 °C, along with reference spectra. | ||
At 900 °C, anomalous Ce redox behavior was observed for Ni0.34–(Fe0.67Ce0.50Ox)-900 sample, meaning on average Ce was oxidized during CH4 step, and reduced during CO2 step (Fig. 7B and D). In Fig. 7D, after CH4 step, the Ce LIII edge XANES spectrum is a combination of CeO2 reference plus a small contribution from Ce3+. After CO2 step, the Ce spectrum mostly includes Ce3+ contribution. This anomalous behavior will be further explained by material phase composition shown by X-ray diffraction. The redox behaviors of Ni0.66–(Fe0.33Ce0.50Ox)-900 and Ni0.50–(Fe0.50Ce0.50Ox)-900 samples are similar to their respective 800 °C counterparts (Fig. 7A, B and S11†).
We conducted a sanity check by quantifying oxygen gain by Ni–(Fe and Ce oxides) during CO2 step using two independent measurement methods. We measured CH4-reduced and CO2-oxidized Ni0.66–(Fe0.33Ce0.50Ox)-800, Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800 materials and Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900, and Ni0.34–(Fe0.67Ce0.50Ox)-900 materials in their 3rd CLDRM cycle. From their XANES data, we derived oxygen gain by the reduced material to form the oxidized material. From reactor gas exhaust analysis, we also calculated oxygen gain amount via CO2 step in the 3rd cycle. The detailed result is in Table S3.† The XANES-derived and gas analysis-based results agree with each other qualitatively in trend, though their quantitative results are different likely due to limitations of lab-scale XANES measurement and LCF method.
3.4 X-ray diffraction analysis of material phase transformation
XRD was conducted to investigate material phase transformation between the reduced and oxidized states in the CLDRM cycles. The samples were collected from the 3rd cycle in the same way as for XANES. At 700 °C, almost no phase transformation was shown between reduced and oxidized samples (Fig. S12†). In Ni0.66–(Fe0.33Ce0.50Ox)-700 sample, Fe and Ni remained metal alloy. On the other hand, Ni0.34–(Fe0.67Ce0.50Ox)-700 contained both Fe3O4 and Fe–Ni alloy. Ceria was always at a fully oxidized state as CeO2 among all 700 °C samples.For 800 °C, XRD results of samples with seven different Fe-to-Ni ratios were shown in Fig. 8, and S13.† For Ni0.66–(Fe0.33Ce0.50Ox)-800 sample, Ce is the redox active element involving several cerium oxides (CeO1.67, CeO1.82, CeO1.71, and CeO2), while Fe and Ni remained in a metallic alloy without redox activity. For Ni0.34–(Fe0.67Ce0.50Ox)-800 sample, Fe was the main redox active element partially transitioning among metallic alloy, Fe3O4, and CeFeO3, while ceria redox was absent or too small to be identified by XRD. These behaviors are consistent with their XANES results (Table S1,†Fig. 5A, C and 7A).
 | ||
Fig. 8 XRD phase analysis of CH4-reduced and CO2-oxidized samples with different Fe/(Fe + Ni) ratios collected from the 3rd cycle at 800 °C, including Ni0.66–(Fe0.33Ce0.50Ox)-800, Ni0.50–(Fe0.50Ce0.50Ox)-800, and Ni0.34–(Fe0.67Ce0.50Ox)-800. Red tones are reduced materials and blue tones are oxidized materials. Circular shape symbols represent different types of cerium oxides.  : Fe0.4Ni0.6 (PDF#: 010777972), : Fe0.4Ni0.6 (PDF#: 010777972),  : Fe0.45Ni0.55 (PDF#: 010777968), : Fe0.45Ni0.55 (PDF#: 010777968),  : Fe0.50Ni0.50 (PDF#: 010889593), : Fe0.50Ni0.50 (PDF#: 010889593),  : Fe0.65Ni0.35 (PDF#: 010777970), : Fe0.65Ni0.35 (PDF#: 010777970),  : Fe3O4 (PDF#: 010751372), : Fe3O4 (PDF#: 010751372),  : CeFeO3 (PDF#: 000220166), : CeFeO3 (PDF#: 000220166),  : Ce3O5 (PDF#: 010814450), : Ce3O5 (PDF#: 010814450),  : Ce7O12 (PDF#: 010898432), : Ce7O12 (PDF#: 010898432),  : Ce11O20 (PDF#: 010898435), : Ce11O20 (PDF#: 010898435),  : CeO1.866 (PDF#: 010786854), : CeO1.866 (PDF#: 010786854),  : CeO2 (PDF#: 010758371). : CeO2 (PDF#: 010758371). | ||
The phase transformation of Ni0.50–(Fe0.50Ce0.50Ox)-800 sample can help explain it having the highest redox capacity. After CH4 step, Fe and Ni were fully reduced to metal alloy, while ceria was mostly reduced to CeO1.866 showing its XRD peaks shift towards lower 2θ; after CO2 step, a major portion of Fe was fully oxidized to Fe3+ in CeFeO3, while cerium oxides were converted to a mixture of Ce4+ in CeO2 and Ce3+ in CeFeO3. On average, Fe showed a large redox capacity, while Ce gave a small redox capacity, explaining XANES results in Fig. 5C and 7A. Overall, the complex Ce redox behaviors accompanied by phase transformation facilitate high Fe redox capacity, and an optimal Fe/(Fe + Ni) ratio activates both Fe and Ce redox reactions.
At 900 °C, both Fe and Ce go through redox reactions via phase transformations in Ni0.66–(Fe0.33Ce0.50Ox)-900, Ni0.50–(Fe0.50Ce0.50Ox)-900 and Ni0.34–(Fe0.67Ce0.50Ox)-900 samples (Fig. S14†). For all samples, Ce redox occurred involving various cerium oxides and CeFeO3, while Fe redox occurred involving Fe metal (Ni0.34–(Fe0.67Ce0.50Ox)-900), Fe–Ni alloy, and CeFeO3. The elevated temperature resulted in deep reduction of ceria by CH4 in Ni0.66–(Fe0.33Ce0.50Ox)-900, and Ni0.50–(Fe0.50Ce0.50Ox)-900, where there was no CeO2 remaining, and a remarkably reduced ceria phase showed up with a larger lattice constant than cerium oxides in the XRD database. Uniquely for Ni0.34–(Fe0.67Ce0.50Ox)-900 sample after CH4 step, a separate Fe metal phase formed along with Fe–Ni alloy, and some CeO2 remained. The limited Ce reduction extent gives an average Ce oxidation state close to +4 as XANES showed in Fig. 7B and D. After CO2 step, the Ni0.34–(Fe0.67Ce0.50Ox)-900 sample formed a large portion of CeFeO3 where Ce is at +3, so the sample has an average Ce oxidation state close to +3 by XANES (Fig. 7B and D). The phase transformation explains the anomalous Ce redox behavior in Ni0.34–(Fe0.67Ce0.50Ox)-900.
Overall, the phase transformation analysis reveals that when Ce and Fe co-participate in redox reaction giving more CeFeO3, overall CH4 conversion was significantly improved (Figs. 3, 8, and S14†). Specifically, Ni0.50–(Fe0.50Ce0.50Ox)-800 and Ni0.34–(Fe0.67Ce0.50Ox)-900 have the maximum CeFeO3 amount after CO2 step and the maximum methane conversion. Therefore, the CeFeO3 phase potentially has a catalytic effect for CH4 conversion. The anomalous Ce redox itself does not help with high overall redox capacity, but it enables Fe to reach a large redox range between metallic state and +3 oxidation state in CeFeO3. Additionally, sufficient Ni composition can enable reduction of ceria by CH4.
3.5 Cyclability and durability of Ni0.34–(Fe0.67Ce0.50Ox)-900
100 CLDRM cycles showed stable performance of Ni0.34–(Fe0.67Ce0.50Ox)-900 at 900 °C (Fig. 9A). Approximately 76% conversion was achieved for both CH4 and CO2. Additionally, carbon accumulation was only 0.26% over the 100 cycles, indicating that catalyst deactivation due to solid carbon did not occur. Also, the XRD results of samples after the CH4 and CO2 steps respectively in the 100th cycle were compared to those in the 3rd cycle (Fig. 9B). After the CH4 step of the 100th cycle, reduced cerium oxide phases, Fe–Ni alloy, and metallic Fe phase showed up along with an unknown reduced phase, similar to after the CH4 step of the 3rd cycle. After the CO2 step of the 100th cycle, CeO2, CeFeO3, and Fe–Ni alloy phases existed, and the fraction of CeFeO3 phase was higher compared to after the CO2 step in the 3rd cycle. Two Ce2O3 reference patterns were included to reflect the presence of two distinct reduced cerium oxide phases in the sample (Fig. 9B). One phase, PDF#: 010825921, corresponds to the cubic bixbyite structure, likely derived from CeO2 through the formation of oxygen vacancies. The other phase, PDF#: 000231048, exhibits a trigonal structure, which is attributed to further reduction and subsequent structural rearrangement under the harsh reducing environment. This indicates that as the cerium oxide got reduced further in the later cycles, it was easier for Fe to react with Ce to form CeFeO3 during the CO2 step. Overall, the above results demonstrated good cyclability of reactivity and phase transformation of Ni0.34–(Fe0.67Ce0.50Ox)-900. | ||
Fig. 9 Ni0.34–(Fe0.67Ce0.50Ox)-900 cyclability at 900 °C. (A) 100-cycle CLDRM test, (B) XRD of fresh, CH4-reduced and CO2-oxidized samples in the 3rd vs. the 100th cycles. Circular shape labels in different colors were used to differentiate the different types of cerium oxides. Red tone curves represent CH4-reduced materials and blue tone curves represent CO2-oxidized materials.  : NiFe2O4 (PDF#: 010880380), : NiFe2O4 (PDF#: 010880380),  : FeNi (PDF#: 010718321), : FeNi (PDF#: 010718321),  : Fe (PDF#: 010714409), : Fe (PDF#: 010714409),  : CeFeO3 (PDF#: 000220166), : CeFeO3 (PDF#: 000220166),  : cubic bixbyite Ce2O3 (PDF#: 010825921), : cubic bixbyite Ce2O3 (PDF#: 010825921),  : Ce3O5 (PDF#: 010814450), : Ce3O5 (PDF#: 010814450),  : CeO1.66 (PDF#: 010898434), : CeO1.66 (PDF#: 010898434),  : trigonal Ce2O3 (PDF#: 000231048), : trigonal Ce2O3 (PDF#: 000231048),  : CeO2 (PDF#: 010758371), : CeO2 (PDF#: 010758371),  : unknown reduced phases. XANES derived average oxidation state changes of CH4-reduced and CO2-oxidized samples from 3rd and 100th cycles: (C) XANES Ce LIII edge of CH4-reduced (red square) and CO2-oxidized (blue dot) samples in the 3rd vs. the 100th cycles; (D) XANES Fe K edge of CH4-reduced (red square) and CO2-oxidized (blue dot) samples in the 3rd vs. the 100th cycles. : unknown reduced phases. XANES derived average oxidation state changes of CH4-reduced and CO2-oxidized samples from 3rd and 100th cycles: (C) XANES Ce LIII edge of CH4-reduced (red square) and CO2-oxidized (blue dot) samples in the 3rd vs. the 100th cycles; (D) XANES Fe K edge of CH4-reduced (red square) and CO2-oxidized (blue dot) samples in the 3rd vs. the 100th cycles. | ||
The average oxidation state changes of 3rd and 100th cycled samples for Ce and Fe are presented in Fig. 9C, D, and S15.† The Cycle 100 samples still exhibit the anomalous redox behavior of Ce as in Fig. 9C, and Fe oxidation state change is similar to the Cycle 3 samples as in Fig. 9D. The CO2-oxidized sample in Cycle 100 has more fraction of CeFeO3 phase than that in Cycle 3 (Fig. 9B), which is further confirmed by the fact that CO2-oxidized sample in Cycle 100 has more resemblance to CeFeO3 XANES than that in Cycle 3 (Figs. S15B and S15D†). Overall, the material behaves almost the same between the 3rd and 100th cycles, and both Ce and Fe maintain stable oxygen exchange capacity and phase transformation over 100 cycles (Fig. 9C and D).
The morphology and elemental distribution were investigated with SEM and EDS (Fig. 10 and S16†). Round shaped particles can be observed in fresh sample mostly at 0.1–0.5 μm size (Fig. 10A). After cycling, particles sintered together and their size grew up as expected, and there is no major difference in particle size between the reduced and oxidized samples of the 3rd and 100th cycles (Fig. 10B through Fig. 10E). While such morphological changes can affect gas diffusion by changing transport resistance and gas access to internal active sites, the stable performance shown in Fig. 9A implies that gas diffusion into the particles remains sufficient to sustain the cyclability over 100 cycles. This indicates that, the reaction is not gas diffusion-limited, and the catalytic performance is not significantly influenced by the observed agglomeration. Based on the spatial distribution of each element, EDS shows several major phases (Fig. 10F through Fig. 10I and S16†). The blue dashed circles are regions where Fe and Ni co-exist without presence of O and Ce, indicating Fe–Ni alloy (Fig. 10F and G). The red dashed circles show where Ni does not exist, but O, Fe, and Ce concentrate, and they can be CeO2 and/or CeFeO3, or a combination of both (Fig. 10F, 10H and 10I).
 | ||
| Fig. 10 Particle morphology and elemental distribution of fresh and cycled Ni0.34–(Fe0.67Ce0.50Ox)-900 using SEM. (A) Fresh, (B) CH4-reduced in the 3rd cycle, (C) CO2-oxidized in the 3rd cycle, (D) CH4-reduced in the 100th cycle, and (E) CO2-oxidized in the 100th cycle. EDS elemental distribution of CO2-oxidized in the 100th cycled Ni0.34–(Fe0.67Ce0.50Ox)-900: (F) iron (yellow), (G) nickel (orange), (H) oxygen (blue), and (I) cerium (green). Blue dashed circles represent where only Fe and Ni are present and the red dashed circles indicate where Ce, Fe, O but Ni does not exist. | ||
4. Conclusions
In this paper, Ni–(Fe and Ce oxides) have been demonstrated for durable chemical looping dry reforming of methane (CLDRM). The simultaneous redox reactivity of Fe and Ce accompanied by phase transformations, as well as catalytic activity of metallic Ni, was investigated using XRD and XANES. Nickel is a catalyst in this material system to facilitate methane conversion and promote the reduction of cerium oxide. In the CO2 step, Fe can chemically interact with reduced cerium oxide to form cerium orthoferrites (CeFeO3) as an oxidation product, whose amount correlates with methane conversion. The formation and removal of CeFeO3 in each cycle gives synergy between Fe and Ce, where full redox of Fe between 0 and +3 oxidation states occurred to most of Fe while Ce shows anomalous redox behavior. 100 cycles at 900 °C confirmed the durability of Ni0.34–(Fe0.67Ce0.50Ox)-900, which maintained high conversions of approximately 76% and minimal carbon accumulation of only 0.26%. Despite the slight increase in particle size, the material sustained its performance over 100 cycles with cyclable redox behavior and phase transformation over the 100 cycles. Comparative analysis with previously published materials revealed that Ni–(Fe and Ce oxides) successfully achieve durable high CH4 and CO2 conversions at high weight hourly space velocity. Overall, our results with Ni–(Fe and Ce oxide) material system offer valuable insights into understanding and optimizing mixed catalyst and oxygen carrier materials for thermochemical redox cycles.Data availability
Data for gas profiles during CLDRM experiments, XAS, and XRD characterization measurements are available at Google Drive at https://drive.google.com/drive/folders/1zn1_rBBqyv-0skbP2JSJMhEfS74sDctd. Data for LCF and other data supporting this article has been included in the ESI.†Author contributions
Minjung Kim: data curation, formal analysis, investigation, methodology, visualization, writing – original draft, software, validation. Michael Tomechko: data curation, formal analysis, software, validation, writing – original draft. Shang Zhai: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank Henk Colijn, Denis Pelekhov, and Yehia Khalifa for guidance on XRD, SEM, and XAS analyses conducted at the Center for Electron Microscopy and Analysis (CEMAS), NanoSystems Laboratory (NSL), and Surface Analysis Lab at The Ohio State University. We acknowledge Pr. In-Ho Jung at Seoul National University for guidance on FactSage and Fe–Ce–O material system thermodynamics. Partial funding for shared facilities used in this research was provided by the Center for Emergent Materials: an NSF MRSEC under award number DMR-2011876. Part of the project was supported by President's Research Excellence (PRE) Catalyst Award at The Ohio State University. We also acknowledge Dr Liang-Shih Fan, Sonu Kumar, and Ashin Sunny at The Ohio State University for research discussions.References
- Credible pathways to 1.5 °C – Four pillars for action in the 2020s, https://www.iea.org/reports/credible-pathways-to-150c, accessed August 2024.
- P. Friedlingstein, M. O'Sullivan, M. W. Jones, R. M. Andrew, D. C. E. Bakker, J. Hauck, P. Landschützer, C. Le Quéré, I. T. Luijkx, G. P. Peters, W. Peters, J. Pongratz, C. Schwingshackl, S. Sitch, J. G. Canadell, P. Ciais, R. B. Jackson, S. R. Alin, P. Anthoni, L. Barbero, N. R. Bates, M. Becker, N. Bellouin, B. Decharme, L. Bopp, I. B. M. Brasika, P. Cadule, M. A. Chamberlain, N. Chandra, T.-T.-T. Chau, F. Chevallier, L. P. Chini, M. Cronin, X. Dou, K. Enyo, W. Evans, S. Falk, R. A. Feely, L. Feng, D. J. Ford, T. Gasser, J. Ghattas, T. Gkritzalis, G. Grassi, L. Gregor, N. Gruber, Ö. Gürses, I. Harris, M. Hefner, J. Heinke, R. A. Houghton, G. C. Hurtt, Y. Iida, T. Ilyina, A. R. Jacobson, A. Jain, T. Jarníková, A. Jersild, F. Jiang, Z. Jin, F. Joos, E. Kato, R. F. Keeling, D. Kennedy, K. Klein Goldewijk, J. Knauer, J. I. Korsbakken, A. Körtzinger, X. Lan, N. Lefèvre, H. Li, J. Liu, Z. Liu, L. Ma, G. Marland, N. Mayot, P. C. McGuire, G. A. McKinley, G. Meyer, E. J. Morgan, D. R. Munro, S.-I. Nakaoka, Y. Niwa, K. M. O'Brien, A. Olsen, A. M. Omar, T. Ono, M. Paulsen, D. Pierrot, K. Pocock, B. Poulter, C. M. Powis, G. Rehder, L. Resplandy, E. Robertson, C. Rödenbeck, T. M. Rosan, J. Schwinger, R. Séférian, T. L. Smallman, S. M. Smith, R. Sospedra-Alfonso, Q. Sun, A. J. Sutton, C. Sweeney, S. Takao, P. P. Tans, H. Tian, B. Tilbrook, H. Tsujino, F. Tubiello, G. R. Van Der Werf, E. Van Ooijen, R. Wanninkhof, M. Watanabe, C. Wimart-Rousseau, D. Yang, X. Yang, W. Yuan, X. Yue, S. Zaehle, J. Zeng and B. Zheng, Earth Syst. Sci. Data, 2023, 15, 5301–5369 CrossRef.
- A. Boyano, A. M. Blanco-Marigorta, T. Morosuk and G. Tsatsaronis, Energy, 2011, 36, 2202–2214 CrossRef CAS.
- S. A. Bhat and J. Sadhukhan, AIChE J., 2009, 55, 408–422 CrossRef CAS.
- M. A. Nieva, M. M. Villaverde, A. Monzón, T. F. Garetto and A. J. Marchi, Chem. Eng. J., 2014, 235, 158–166 CrossRef CAS.
- M. Szymańska, A. Malaika, P. Rechnia, A. Miklaszewska and M. Kozłowski, Catal. Today, 2015, 249, 94–102 CrossRef.
- M. Msheik, S. Rodat and S. Abanades, Energies, 2021, 14, 3107 CrossRef CAS.
- J. Boo, E. H. Ko, N.-K. Park, C. Ryu, Y.-H. Kim, J. Park and D. Kang, Energies, 2021, 14, 8182 CrossRef CAS.
- X.-Y. Wu and A. F. Ghoniem, Proc. Combust. Inst., 2019, 37, 5517–5524 CrossRef CAS.
- E. Sun, S. Zhai, D. Kim, M. Gigantino, V. Haribal, O. S. Dewey, S. M. Williams, G. Wan, A. Nelson, S. Marin-Quiros, J. Martis, C. Zhou, J. Oh, R. Randall, M. Kessler, D. Kong, J. Rojas, A. Tong, X. Xu, C. Huff, M. Pasquali, R. Gupta, M. Cargnello and A. Majumdar, Cell Rep. Phys. Sci., 2023, 4, 101338 CrossRef CAS.
- H. Wang, S. Kim and E. Sasmaz, Chem. Eng. J., 2022, 450, 138111 CrossRef CAS.
- C. M. Damaskinos, M. A. Vasiliades and A. M. Efstathiou, Appl. Catal. Gen., 2019, 579, 116–129 CrossRef CAS.
- X. Zhao, S. Sun, Y. Wang, Y. Zhang, Y. Zhu, B. Zong, J. Hu, P. Williams and C. Wu, Chem. Eng. J., 2024, 491, 151668 CrossRef CAS.
- M. Yusuf, M. Beg, M. Ubaidullah, S. F. Shaikh, L. K. Keong, K. Hellgardt and B. Abdullah, Int. J. Hydrogen Energy, 2022, 47, 42150–42159 CrossRef CAS.
- J. Guerrero-Caballero, T. Kane, N. Haidar, L. Jalowiecki-Duhamel and A. Löfberg, Catal. Today, 2019, 333, 251–258 CrossRef CAS.
- K. Mette, S. Kühl, H. Düdder, K. Kähler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2014, 6, 100–104 CrossRef CAS.
- M. Usman, W. M. A. Wan Daud and H. F. Abbas, Renew. Sustain. Energy Rev., 2015, 45, 710–744 CrossRef CAS.
- L. P. Teh, H. D. Setiabudi, S. N. Timmiati, M. A. A. Aziz, N. H. R. Annuar and N. N. Ruslan, Chem. Eng. Sci., 2021, 242, 116606 CrossRef CAS.
- G. R. Schoofs and J. B. Benziger, Langmuir, 1988, 4, 526–532 CrossRef CAS.
- Z. Hou, P. Chen, H. Fang, X. Zheng and T. Yashima, Int. J. Hydrogen Energy, 2006, 31, 555–561 CrossRef CAS.
- M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, Scr. Mater., 2009, 61, 173–176 CrossRef CAS.
- V. Shah, Z. Cheng, D. S. Baser, J. A. Fan and L.-S. Fan, Appl. Energy, 2021, 282, 116111 CrossRef CAS.
- N. L. Galinsky, A. Shafiefarhood, Y. Chen, L. Neal and F. Li, Appl. Catal., B, 2015, 164, 371–379 CrossRef CAS.
- Y. Kim, H. S. Lim, M. Lee and J. W. Lee, Catal. Today, 2021, 368, 86–95 CrossRef CAS.
- Y. Shao, C. Wu, S. Xi, P. Tan, X. Wu, S. Saqline and W. Liu, Appl. Catal. B Environ. Energy, 2024, 355, 124191 CrossRef CAS.
- Q. Rao, J. Zhang, T. Yang, Y. Li, Z. Gai, P. Li, X. Wang, Y. Pan and H. Jin, Chem. Eng. J., 2024, 485, 150033 CrossRef CAS.
- A. Löfberg, J. Guerrero-Caballero, T. Kane, A. Rubbens and L. Jalowiecki-Duhamel, Appl. Catal., B, 2017, 212, 159–174 CrossRef.
- U. P. M. Ashik, W. M. A. Wan Daud and H. F. Abbas, Renew. Sustain. Energy Rev., 2015, 44, 221–256 CrossRef CAS.
- C. Hill, R. Robbins, P. Furler, S. Ackermann and J. Scheffe, Sustain. Energy Fuels, 2023, 7, 574–584 RSC.
- Z. Cao, X. Zhu, K. Li, Y. Wei, F. He and H. Wang, Chem. Eng. J., 2020, 397, 125393 CrossRef CAS.
- Y. Han, M. Tian, C. Wang, Y. Kang, L. Kang, Y. Su, C. Huang, T. Zong, J. Lin, B. Hou, X. Pan and X. Wang, ACS Sustain. Chem. Eng., 2021, 9, 17276–17288 CrossRef CAS.
- Z. Huang, F. He, D. Chen, K. Zhao, G. Wei, A. Zheng, Z. Zhao and H. Li, Energy, 2016, 116, 53–63 CrossRef CAS.
- A. More, C. J. Hansen and G. Veser, Catal. Today, 2017, 298, 21–32 CrossRef CAS.
- A. More, S. Bhavsar and G. Veser, Energy Technol., 2016, 4, 1147–1157 CrossRef CAS.
- J. Zhang, V. Haribal and F. Li, Sci. Adv., 2017, 3, e1701184 CrossRef PubMed.
- A. E. Abasaeed, A. S. Al-Fatesh, M. A. Naeem, A. A. Ibrahim and A. H. Fakeeha, Int. J. Hydrogen Energy, 2015, 40, 6818–6826 CrossRef CAS.
- M. S. Aw, I. G. Osojnik Črnivec, P. Djinović and A. Pintar, Int. J. Hydrogen Energy, 2014, 39, 12636–12647 CrossRef CAS.
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc. A Math. Phys. Eng. Sci., 2010, 368, 3269–3294 CrossRef CAS PubMed.
- V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Appl. Catal., B, 2015, 164, 184–191 CrossRef CAS.
- S. Bhavsar and G. Veser, RSC Adv., 2014, 4, 47254–47267 RSC.
- J. Zhang, T. Yang, Q. Rao, Z. Gai, P. Li, Y. Shen, M. Liu, Y. Pan and H. Jin, Fuel, 2024, 366, 131344 CrossRef CAS.
- F. R. García-García and I. S. Metcalfe, Catal. Commun., 2021, 160, 106356 CrossRef.
- A. Sasaki, A. Himeda, H. Konaka and N. Muroyama, Integrated X-ray powder diffraction software PDXL, Rigaku J., 2010, 26(1), 23–27 Search PubMed.
- FIZ Karlsruhe – Leibniz Institute for Information Infrastructure, Inorganic Crystal Structure Database (ICSD), https://icsd.products.fiz-karlsruhe.de/, accessed February 11, 2025.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541, DOI:10.1107/S0909049505012719.
- J. Ashok, G. Raju, P. S. Reddy, M. Subrahmanyam and A. Venugopal, Int. J. Hydrogen Energy, 2008, 33, 4809–4818 CrossRef CAS.
- A. F. Cunha, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal. Gen., 2008, 348, 103–112 CrossRef CAS.
- J. Ashok, M. Subrahmanyam and A. Venugopal, Catal. Surv. Asia, 2008, 12, 229–237 CrossRef CAS.
- A. Venugopal, S. Naveen Kumar, J. Ashok, D. Hari Prasad, V. Durga Kumari, K. B. S. Prasad and M. Subrahmanyam, Int. J. Hydrogen Energy, 2007, 32, 1782–1788 CrossRef CAS.
- J. Ashok, M. Subrahmanyam and A. Venugopal, Int. J. Hydrogen Energy, 2008, 33, 2704–2713 CrossRef CAS.
- A. Steinfeld, S. Sanders and R. Palumbo, Sol. Energy, 1999, 65, 43–53 CrossRef CAS.
- M. E. Gálvez, P. G. Loutzenhiser, I. Hischier and A. Steinfeld, Energy Fuels, 2008, 22, 3544–3550 CrossRef.
- A. Tofighi and F. Sibieude, Int. J. Hydrogen Energy, 1984, 9, 293–296 CrossRef CAS.
- A. Stamatiou, P. G. Loutzenhiser and A. Steinfeld, Energy Fuels, 2010, 24, 2716–2722 CrossRef CAS.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048–4077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01774b |
| This journal is © The Royal Society of Chemistry 2025 |