DOI:
10.1039/D5TA01405K
(Paper)
J. Mater. Chem. A, 2025,
13, 18948-18958
A data-driven framework to accelerate the discovery of hybrid cathode materials for metal-based batteries†
Received
20th February 2025
, Accepted 26th March 2025
First published on 29th March 2025
Abstract
Selecting materials for hybrid cathodes (HCs) for batteries, which involve a combination of intercalation and conversion materials, has gained interest due to their synergistic and averaged properties, enabling enhanced energy density and stability. Herein, we present a data-driven, chemistry-agnostic inverse material design framework for discovering HCs for metal-based batteries. This framework systematically explores the potential materials space for any given working ion, evaluates the candidate's stability, and identifies the growth modes/adsorption of the components to identify stable HCs. To demonstrate the application of the framework, we performed a case study to discover HCs with an average gravimetric energy density surpassing that of the widely used high energy density NMC333 cathode material. The framework identified LiCr4GaS8–Li2S as a promising HC with an average energy density of 1424 Wh kg−1 (on a lithiated cathode basis) that exceeds NMC333's maximum theoretical energy density of 1028 Wh kg−1. The identified HC has several additional desirable features: (1) its lithiated and delithiated phases are thermodynamically stable; (2) it undergoes minimal volumetric change upon (de)lithiation that mitigates the high-volumetric change of the conversion material; (3) it has a high energy density that ameliorates the low energy density of the intercalation material; (4) its intercalation component serves as both a conductive additive and support for sulfur species, immobilizing S while simultaneously contributing to the total cathode energy density; and finally, (5) it is expected to enhance durability and capacity retention over conventional Li–S batteries.
1 Introduction
Li-ion batteries (LIBs) have become a key commercial rechargeable (secondary) battery technology for a plethora of applications due to their superior performance metrics, including high energy and power densities, high cycling stability, and low maintenance. This unique combination enables practical electric vehicles, portable/mobile/wearable electronic devices, and is rapidly scaling to provide grid energy storage to buffer intermittent renewable energy harvesting technologies.1 However, the energy density, cost, and safety of LIBs fall short of meeting the increasingly ambitious targets in sectors such as electric vehicles and grid storage, which has driven extensive investigation into cathode materials over the past three decades.2 Generally, cathode materials are classified based on their structural changes induced by their lithiation (discharging) and delithiation (charging) processes. This classification results in two main cathode categories; intercalation materials and conversion materials. Intercalation materials undergo a topotactical phase transition upon (de)lithiation, which involves a two-phase transformation that preserves the host crystal structure upon Li extraction and insertion. In contrast, conversion materials undergo a reconstructive phase transition, resulting in a two-phase transformation that results in a final phase with little to no crystallographic relationship to the initial phase.3–6
Lithium intercalation materials represent the archetypical cathode materials for LIBs and are more mature than conversion materials in that they have nearly attained their maximum theoretical energy densities. However, they often rely on relatively scarce and expensive elements (e.g., Co and Ni) and suffer potential safety issues arising from oxygen inclusion. On the other hand, conversion materials are relatively new and possess high energy densities with low materials costs, especially in the case of redox-anion chemistries. Nonetheless, they face considerable obstacles to their commercialization, primarily stemming from their reconstructive phase transition upon (de)lithiation. This phase transition causes large volumetric changes, sluggish kinetics, and thus slow charging/discharging, and poor reversibility (rechargeability). Additionally, they also tend to exhibit poor electronic and ionic conductivities.7–9 These contrasting strengths and limitations suggest a hybrid approach could yield cathode materials with a balance of high energy density and stability.
Several efforts have aimed to design hybrid (heterostructure) cathodes (HCs) that combine both intercalation and conversion material building blocks to at least achieve averaged cathode properties that are unattainable by their monolithic counterparts. Heterostructure electrodes have several advantages:10 first, heterostructure electrodes integrate the advantages of various components to mitigate the inherent shortcomings of the individual component materials; second, heterostructures are an effective approach for enhancing the electronic conductivity via the modification of the band structures to narrow the band gap; third, an interfacial electric field between the component materials can lower the ion diffusion barrier and accelerate ion diffusion; fourth, the chemical bonds and van der Waals and electrostatic interactions between the conversion and intercalation materials can stabilize the structure to improve the cycling stability; finally, the charge redistribution within the component materials of electrode heterostructures introduces additional ion binding sites that increase energy density. Xu et al. studied combining VS2 as an intercalation material with sulfur chemistry as the conversion component in conjunction with a solid electrolyte. They reported that VS2 exhibited excellent Li+ and electronic conductivities, contributed to the total capacity, and concluded that VS2 is an ideal platform to unlock the high energy density of sulfur chemistry.11
Several recent studies have demonstrated the potential of hybrid cathode structures by combining intercalation and conversion components to achieve balanced properties. Xue et al. investigated a hybrid cathode of Mo6S8 Chevrel-phase as the intercalation material and sulfur as the conversion material.12 The Chevrel-phase enhanced capacity, reduced reliance on conductive additives due to its high electronic conductivity, and maintained structural integrity with minimal volumetric changes during (de)lithiation. Additionally, strong bonding between the Chevrel-phase and sulfur immobilizes sulfur species and thus mitigates polysulfide shuttling and capacity fading. The authors concluded that Chevrel-phase materials provide a robust substrate to support the conversion of sulfur species. Huang et al. developed a simple model to describe the two-stage discharging–charging behavior of the intercalation and conversion components of HCs.13
Although no systematic approach currently exists for exploring HC materials, previous research has identified promising designs despite the challenges posed by the vast combinatorial space of potential materials. A data-driven framework would provide an efficient pathway for navigating this space and accelerating the discovery of high-performance HCs. Fortunately, the pursuit of high-performance HCs inherently lends itself to the approaches of data-driven materials discovery.
Herein, we describe a data-driven, chemistry-agnostic inverse material design framework to accelerate the discovery of HCs for metal-based batteries. The framework systematically explores the material space of potential HCs for any given working ion, evaluates the stability of HCs, and determines the growth modes/adsorption of conversion material components to identify stable HCs. We conducted a case study aimed at discovering novel HCs with an average gravimetric energy density greater than that of the widely used high energy density NMC333 cathode to demonstrate the application and potential outcomes of the framework for designing HCs.14 The framework identified LiCr4GaS8–Li2S as a promising HC that achieves the design objective of having an average energy density (1424 Wh kg−1 on a lithiated cathode basis) that exceeds the maximum theoretical energy density of NMC333 (1028 Wh kg−1). Additionally, it demonstrates the thermodynamic stability of both the lithiated and delithiated phases. In comparison to conventional Li–S batteries, this HC heterostructure is predicted to have high electronic conductivities and lower volumetric changes. Further, the durability, self-discharge, mechanical integrity, and capacity fading of the identified HC are all anticipated to be superior to those of conventional Li–S batteries. Overall, the framework proved effective in achieving pre-defined battery material design objectives by evaluating potential HCs.
2 Methodology
The developed framework comprises a sequence of six steps, as shown in Fig. 1a. The data, methods, algorithms, analytical tools, and software packages exploited in this framework have been previously established and are readily accessible. Thereby, the unique contribution of this framework lies in the integration and application of these resources to accelerate the discovery of HCs. This framework can be used for any working ion; however, herein we only considered Li+. A schematic of the framework is depicted in Fig. S1.†
 |
| | Fig. 1 (a) The data-driven framework scheme developed in this work for the discovery of HCs for metal-based batteries; (b) data acquisition and pairing of the lithiated and delithiated phases, where the datasets of lithiated and delithiated/decomposed phases were retrieved from the Materials Project repository. Convex hull composition phase diagrams were constructed using SLSQP optimization to identify stable delithiated phases by minimizing the decomposition energy. Only pairs with single delithiated phases were selected for further analysis due to their better anticipated reversibility; (c) determination of the lithiation reaction type via structural comparison between the lithiated-delithiated pair, where crystallographic similarity between lithiated and delithiated structures was assessed with Pymatgen's StructureMatcher to classify the (de)lithiation-induced structural changes as intercalation or conversion reactions. | |
2.1 Data acquisition
The Materials Project (MP),15 including the MP repository and Pymatgen,16 provides core capabilities for the framework. In defining this framework, we assumed that a given lithiated phase (LinAB) represents the fully discharged state and the corresponding completely delithiated phase (AB) is the stable charged state. As shown in Fig. 1b, two distinct materials datasets of lithiated and delithiated/decomposed phases with energies up to 100 meV per atom of the convex hull were retrieved from the MP repository to accommodate all the possible material classes including nitrides, which have high metastability.17 Each materials dataset encompasses various materials entries, with key features including chemical formula, formation energy per atom, structure, energy above the hull, bandgap energy, and density.
2.2 Pairing of the lithiated and delithiated phases
To thermodynamically pair the Li phases to the delithiated/decomposed phases, the Sequential Least SQuares Programming (SLSQP) optimizer from the SciPy python library18 was exploited to construct a convex hull composition phase diagram to perform a thermodynamic stability analysis for each composition space available in the materials dataset. SLSQP was applied to the formation energies of the possible delithiated/decomposed phases to minimize the decomposition energy of a given lithiated phase, consequently determining the corresponding delithiated/decomposed phase(s), as shown in Fig. 1b. The pairs with single delithiated phases were adopted for the subsequent steps, while pairs with multiple delithiated (decomposed) phases were excluded from further consideration as components of any potential HC due to their anticipated poor reversibility upon (de)lithiation.
2.3 Determination of lithiation reaction type
To determine the (de)lithiation reaction type for pairs with single delithiated phases, the crystallographic similarity between the lithiated and delithiated structures was evaluated for a given pair. This crystallographic comparison determines whether the (de)lithiation reaction is a topotactic phase transition (intercalation) or a reconstructive phase transition (conversion), as shown in Fig. 1c. The StructureMatcher module from the Pymatgen library was utilized for structural comparison. The flowchart of the StructureMatcher module is shown in Fig. S2.† In some cases, a single lithiated phase (LinAB) has several possible single delithiated polymorphs that share the same chemical formula (AB), yet that have different structures. Accordingly, crystallographic comparisons for a given lithiated phase can identify multiple possible competing lithiation reactions. Thus, two cases arise; either all possible reactions are: (1) of the same lithiation reaction type, or (2) a mix of the two lithiation reaction types. In the former case, the lithiated phase was paired to the delithiated phase that exhibits the lowest formation energy, thereby allowing direct identification of the lithiation reaction type. In the latter case, the lithiated phase was paired to the delithiated phase with the lowest formation energy within the intercalation lithiation type, as the formation energies of all potential delithiated polymorphs are closely matched and mostly fall within the range of DFT error. Moreover, the thermodynamic feasibility of a lithiation reaction (Li+ intercalation) was substantiated by the comparatively lower kinetic barriers associated with topotactic intercalation reactions, which induce marginal crystallographic changes. In contrast, reconstructive conversion reactions induce significant crystallographic changes.
2.4 Calculation of reaction voltage, theoretical capacity, and volumetric change
For each lithiated–delithiated (Li–deLi) pair, key cathode metrics are computed using the materials' features obtained from the MP to guide subsequent analyses and procedures. The reaction voltage, gravimetric energy density, and volumetric change (Δvol) are evaluated using eqn (1)–(4), respectively.17,18| | | ΔGr ≈ EDFT (LinAB) − EDFT (Lin−xAB) − xEDFT (Li) | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
where ΔGr is the Gibbs free energy of reaction, EDFT represents the energies of LinAB, Lin−xAB, and Li that are calculated from DFT-computed total energies, x is the number of Li+ ions involved in the lithiation reaction, V is the reaction voltage, z is the charge of the working-ion, which is +1 in case of Li+, F is Faraday's constant, εm is the gravimetric energy density, and volphase represents the volumes of the lithiated and delithiated phases.
2.5 Chemical and electrochemical stability evaluation
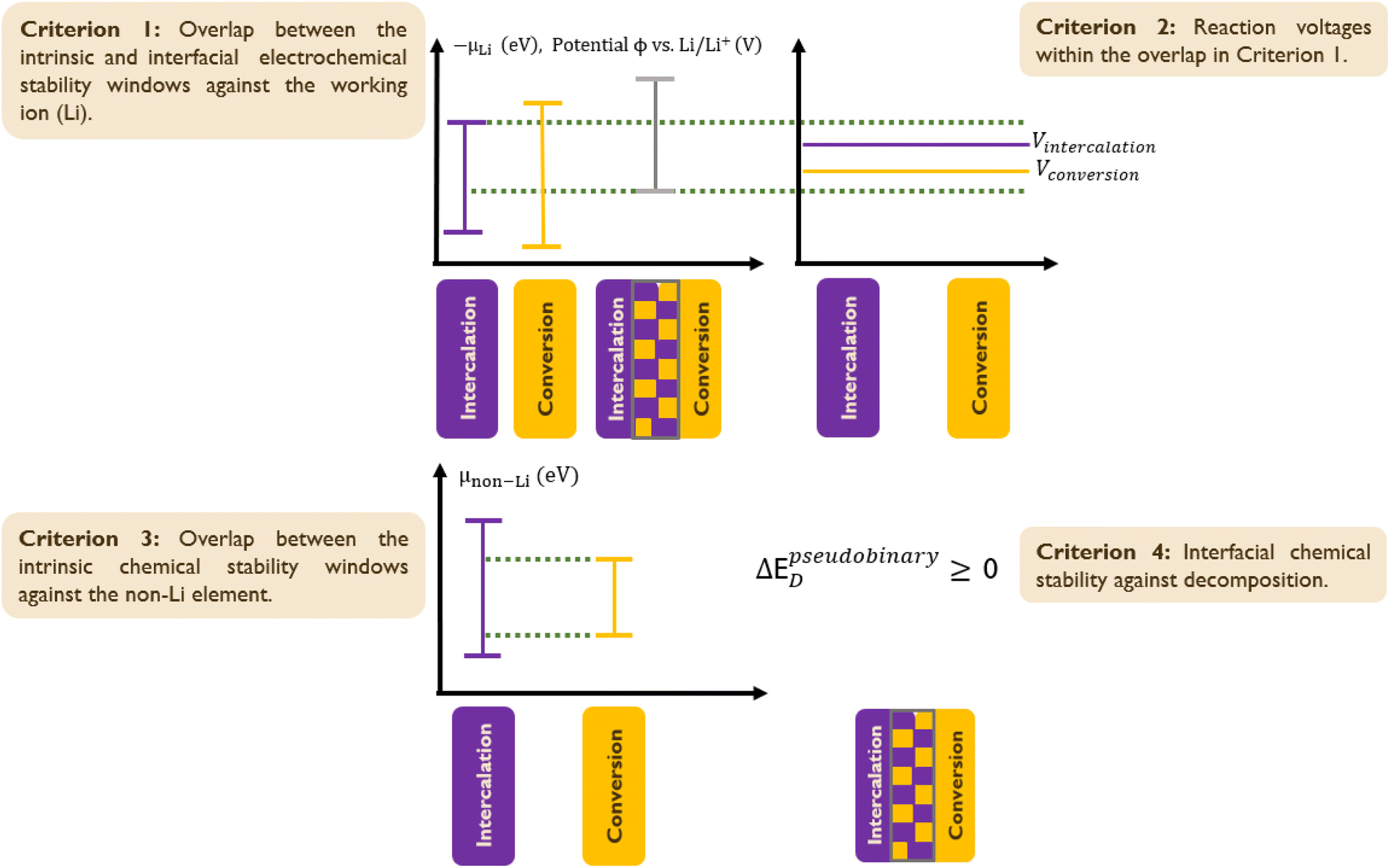
Herein, the computational scheme to evaluate the chemical and electrochemical stabilities of heterogeneous solid interfaces was adopted from Zhu et al.19 However, we introduced Criterion 2, as shown in Fig. 2, to account for the energy density contribution requirement for both components of the HC. For HC candidates that successfully passed previous down selection steps, the intercalation (intcl) and conversion (conv) component materials must have compatible chemical and electrochemical windows, as illustrated in Fig. 2. There are four criteria for the chemical and electrochemical stabilities of the two cathode building blocks.
 |
| | Fig. 2 Chemical and electrochemical stability evaluation scheme. The stability of hybrid cathode candidates is assessed based on four criteria: Criterion 1 ensures overlapping electrochemical stability windows of intercalation and conversion materials to prevent decomposition (eqn (5)–(8)); Criterion 2 requires reaction voltages within the overlap of Criterion 1 (eqn (2) and (5)–(8)); Criterion 3 mandates overlapping chemical stability windows for non-Li elements to avoid decomposition/cross-diffusion (eqn (9)). Criterion 4 ensures interface stability in the absence of an applied voltage (eqn (5), (6) and (10)). | |
2.5.1 Criterion 1.
The intrinsic and interfacial electrochemical stability windows of the component materials against the working ion (Li+) must overlap to ensure that both building blocks along with their interface coexist over the same voltage range (ΔΦ) without decomposition. This criterion is evaluated using eqn (5)–(8).
2.5.2 Criterion 2.
The reaction voltages of the intercalation and conversion component materials must lie within the overlap determined by Criterion 1 to ensure that both component materials can be charged/discharged without decomposing. This criterion is evaluated using eqn (2) and (5)–(8).
2.5.3 Criterion 3.
For each non-Li element within the combined compositional space of component materials, the intrinsic chemical stability windows of the building blocks against the non-Li element must overlap to ensure that a common chemical potential value (μnon-Li) can be achieved across the interface to avoid decomposition/cross-diffusion. This criterion is evaluated using eqn (9).
2.5.4 Criterion 4.
The interface must be chemically stable against decomposition to ensure the chemical stability of the interface under conditions of no applied voltage. This criterion also ensures material stability during cell preparation and is evaluated using eqn (5), (6) and (10).
For the interfacial Criteria 1 and 4, Cinterface, Cintcl, Cconv, Einterface, Eintcl, and Econv represent the compositions and energies of the pseudobinary interface, intercalation (intcl) and conversion (conv) materials, as shown in eqn (5) and (6).
| | | Cinterface (Cintcl, Cconv, x) = xCintcl + (1 − x)Cconv | (5) |
| | | Einterface (Eintcl, Econv, x) = xEintcl + (1 − x)Econv | (6) |
where
x is assumed to be 0.5 because it fairly provides the minimum mutual reaction energies, as the results reported by Zhu
et al. suggest.
19
The grand potential phase diagrams at T = 300 K (ref. 20) were constructed using the Pymatgen library and the MP database to identify the competing phase equilibria (eq) and to calculate the decomposition energies and stability windows of the intercalation and conversion phases using the following equations:
| | | ΔEopenD (phase, μLi) = Eeq[Ceq(C, μLi)] − E(phase) − ΔnLiμLi | (7) |
| | | ΔEopenD(intcl, conv, μLi) = Eeq[Ceq(Cinterface, μLi)] − Einterface (intcl, conv) − ΔnLiμLi | (8) |
| | | ΔEopenD (phase, μnon-Li) = Eeq[Ceq(C, μnon-Li)] − E(phase) − Δnnon-Liμnon-Li | (9) |
| | | ΔED (intcl, conv) = Eeq(Cinterface) − Einterface(intcl, conv) | (10) |
where Δ
EopenD is the decomposition reaction energy at a given chemical potential
μ,
Ceq and
Eeq are the composition and the total formation energy of the competing phase equilibria at a given chemical potential
μ,
E(phase) is the formation energy of the phase of interest, and Δ
n is the change in the number atoms of a given element at chemical potential
μ.
In this study, the stable chemical and electrochemical windows were defined as the potential ranges over which the phase of interest exhibits decomposition energies ≥ −50 meV per atom. This threshold was selected to account for the DFT margin of error that introduces errors into the calculated thermodynamic formation energies.
Relying solely on thermodynamic criteria to screen materials suitable for various applications can be overly stringent and often prohibitively difficult to satisfy. This is especially true for grand canonical composition phase diagrams that include numerous elemental compositions. To illustrate this, the intrinsic electrochemical stability windows against Li+ (working ion) are calculated based on the stability of the decomposition products. Accordingly, these calculations usually underestimate the experimental electrochemical stability windows due to their inability to account for the kinetics that arise from the activation energies to form the decomposition products. Consequently, they fail to identify materials that are sufficiently metastable because they are kinetically trapped from forming decomposition products at the conditions of interest. To remedy this, the electrochemical stability windows can be computed based on indirect decomposition via (de)lithiation of the cathode material, as explained in Schwietert et al. and Zhu et al.21,22 Unfortunately, this approach requires extensive DFT calculations, which renders it impractical for high-throughput data-driven materials screening studies. Alternatively, reasonable tolerances can be considered to widen the electrochemical stability windows for Criteria 1 and 2 to account for the metastability provided by the kinetics of decomposition into competing phases. For Criterion 3, if there are reasonably small gaps between the chemical windows, transition-state theory (TST) calculations can be used to predict whether the material is kinetically trapped by estimating the diffusivity of the thermodynamically unstable species. This allows one to assess the probability that these species diffuse out of the cathode material to cause decomposition to occur at a given temperature. These species are usually less mobile than Li, increasing the likelihood that these materials are sufficiently metastable to persist sufficiently for a given application.
2.6 Evaluation of hybrid cathode candidates via quantum mechanical modeling
For stable hybrid cathode candidates that pass previous screening steps, quantum mechanical modeling was exploited to study the nature of the lithiated and delithiated hybrid cathode interfaces. For the case of two crystalline solid cathode materials, the dominant interfacial growth mode can be identified to offer insights in cases where interface modifications are needed, e.g., the application of surfactants. For the case of a crystalline cathode and a molecular species (e.g., S8), the thermodynamic favorability of adsorption is computed to assess the possible immobilization of the molecular species to the cathode surface, for example to evaluate cathode stability against polysulfide shuttling.
2.6.1 Interfacial growth mode.
For a given hybrid cathode, low-index facets of the intercalation and conversion components that minimize the interfacial von Mises strain, with a maximum limit of 5%23 were chosen. DFT calculations were then performed to compute the surface (γ), adhesion (Wad) and interfacial (σinterface) energies using the following equations:24–26| |  | (11) |
| |  | (12) |
| |  | (13) |
where A is the surface area of the surface/interface, E(slab) is the total energy of a given facet, E(bulk) is the bulk total energy per atom of the phase of interest, N is the number of atoms in the slab model of the surface, and Esubstrate, Efilm, and Einterface are the DFT energies of the substrate, film, and interface, respectively.
The relation between γfilm, γsubstrate, and σinterface defines the dominant interfacial growth mode. Thus, if γfilm ≤ γsubstrate − σinterface, the dominant growth mode will be layer-by-layer (Frank–van der Merwe) growth. Otherwise, the dominant interfacial growth mode is island (Volmer–Weber) growth.27
2.6.2 Adsorption study.
An adsorption study was conducted for hybrid cathodes when one of its components is a molecular species. Surface energies at neutral charge (unbiased) conditions for the low-index facets were evaluated using DFT to identify the facet with the lowest surface energy, and thus the most abundant facet at equilibrium, for the adsorbent. Binding energies were then computed for adsorption at different sites on the most stable facet. To make binding energies representative of working conditions, all DFT calculations were evaluated using the grand canonical ensemble thermodynamic potential, where the electron reservoir potential was sampled over the operating potential window of the cathode.
3 Results and discussion
The primary objectives for applying the framework in this work are twofold: (1) to provide an illustrative case study that demonstrates its application and potential outcomes and (2) to achieve the design goal of discovering novel HCs with an average gravimetric energy density that surpasses that of NMC333. NMC333, a widely-used intercalation cathode material, has a maximum theoretical gravimetric energy density of 1028 Wh kg−1, based on the mass of the lithiated cathode.14 The detailed flowchart for designing high-energy density HCs is shown in Fig. S3.†
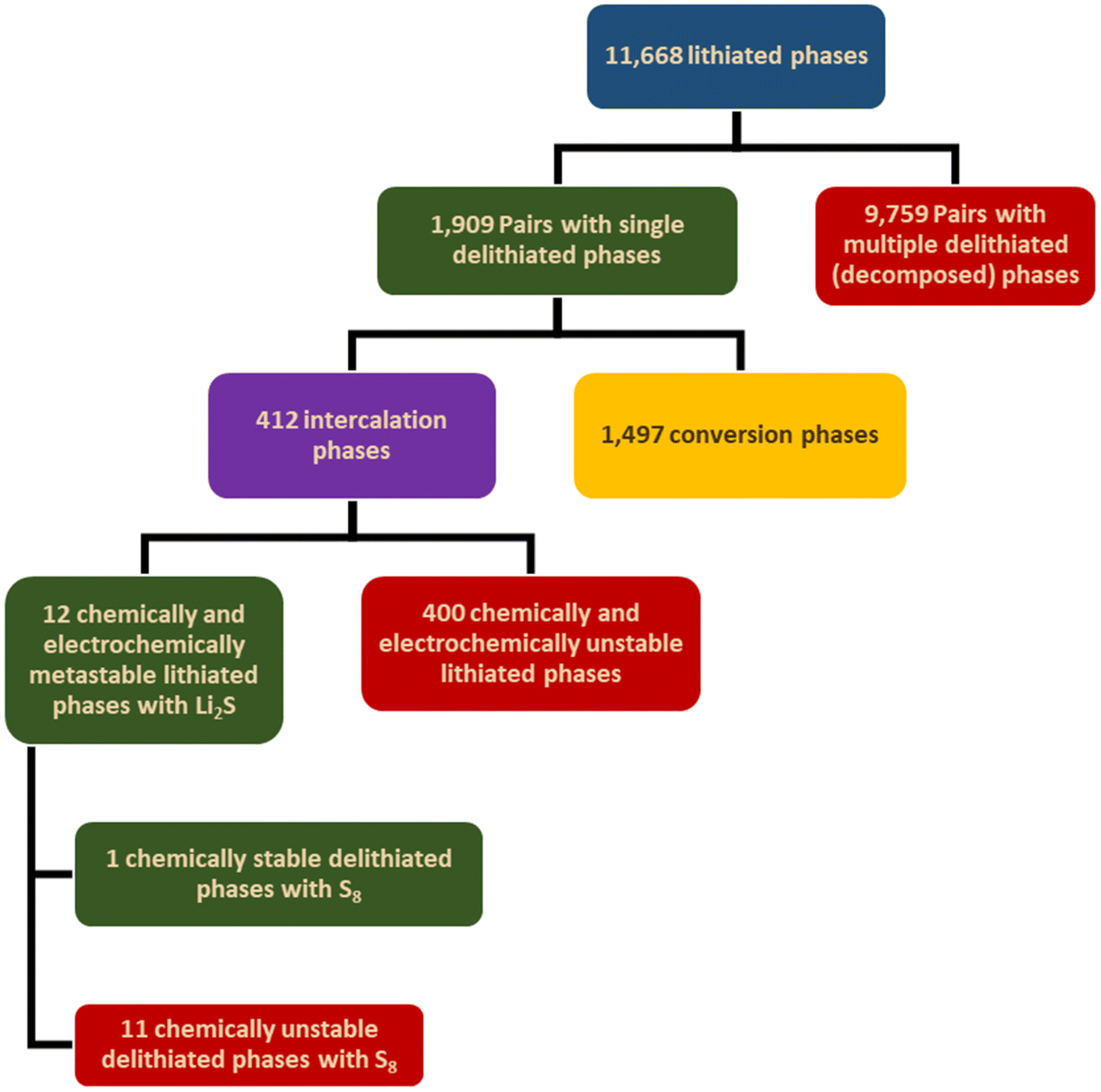
Two distinct datasets containing lithiated and delithiated/decomposed phases were retrieved from the MP repository. These datasets were filtered to include only phases with energies within 100 meV per atom of the convex hull, excluding phases containing elements that are scarce, toxic, or radioactive, as detailed in Table S1.† Phases containing fluorine or hydroxyl groups were also excluded due to their limited reversibility and propensity to trigger parasitic reactions, respectively. The lithiated phases dataset contained 11688 phases. Pairing these lithiated phases with their corresponding delithiated phases resulted in 1909 delithiated single phase pairs and 9759 delithiated multi-phase pairs. Using the StructureMatcher module from the Pymatgen library yielded 412 intercalation phases and 1497 conversion phases. Our evaluation of the calculated theoretical capacities indicated that surpassing NMC333's gravimetric energy density of 1028 Wh kg−1 is only feasible by incorporating sulfur as the conversion component.
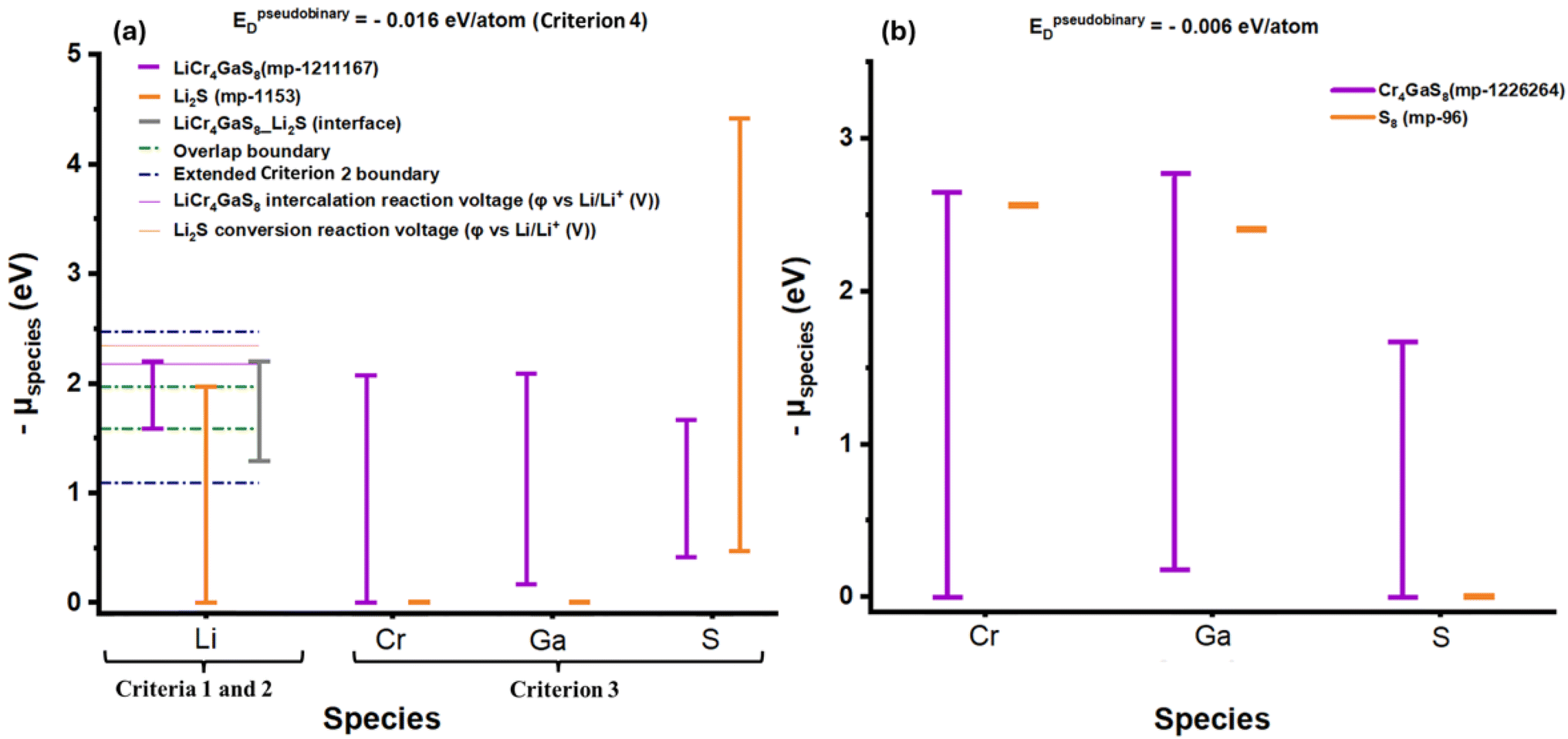
The chemical and electrochemical stability evaluation scheme was then exclusively applied to the 412 HCs, each composed of one of the 412 intercalation components and Li2S as the conversion component. Applying the complete thermodynamic stability criteria of the chemical and electrochemical stability evaluation scheme resulted in all 412 HCs failing. This is primarily due to most candidate HCs failing Criterion 2 because of the multiple oxidation states, high polarization and reactivity of sulfur. Nonetheless, Schwietert et al. showed that the estimation of the thermodynamic formation energy of the decomposed products frequently underestimates the actual electrochemical stability range.22 Hence, a less conservative version of Criterion 2 was adopted to account for the overpotentials of the (de)lithiation reaction, which is an inner-sphere electron transfer redox reaction involving bond breaking/formation that typically requires activation energies > 0.4 eV.28 Thus, a margin of 0.5 eV was added to both limits of the overlap window of Criterion 2. This modified criterion led to 12 lithiated intercalation-Li2S pairs passing as metastable HCs. The chemical stability evaluation scheme based on Criteria 3 and 4, as Criteria 1 and 2 are applicable only to the lithiated phases, were then applied to the delithiated phases of the 12 HCs. This resulted in only 1 stable candidate (LiCr4GaS8–Li2S), as depicted in Fig. 3a and b and tabulated in Table S2.†Fig. 4 summarizes the results of the framework through step 5.
 |
| | Fig. 3 (a) Chemical and electrochemical stability evaluation scheme for each species in Li2S and LiCr4GaS8 (lithiated phases), where the hybrid material passed Criterion 1 by having overlapping electrochemical stability windows of intercalation and conversion components, passed the modified version of Criterion 2 by straddling reaction voltages within the extended Criterion 2 boundaries, passed Criterion 3 by having overlapping chemical stability windows for non-Li elements, and passed Criterion 4 by having EDpseudobinary ≥ −50 meV per atom. (b) Chemical stability evaluation scheme (Criteria 3 and 4) for each species in S8 and Cr4GaS8 (delithiated hybrid components), where Criteria 1 and 2 are irrelevant here because the intercalation and conversion components are Li+-free (the working ion). The delithiated hybrid components passed Criterion 3 by having overlapping chemical stability windows for non-Li elements, and passed Criterion 4 by having EDpseudobinary ≥ −50 meV per atom. | |
 |
| | Fig. 4 Summary of the computational framework case study for designing high-energy density HCs. From a dataset of 11688 lithiated phases retrieved from the MP repository, 1909 single-phase and 9759 multi-phase pairs were identified. Classification categorized 412 materials as intercalation phases and 1497 materials as conversion phases. Surpassing NMC333's energy density requires sulfur as the conversion component; thus, each intercalation phase candidate was paired with Li2S as the conversion phase. Stability evaluation of these 412 HC pairs (intercalation phase + Li2S) led to all candidates failing Criterion 2. By modifying Criterion 2 by adding a 0.5 V margin, 12 metastable HCs were identified. Further chemical stability evaluation of the delithiated HCs narrowed the candidates to one viable HC pair: LiCr4GaS8–Li2S. | |
3.1 Examination of the identified hybrid cathode candidate by DFT
As the lone surviving HC candidate, LiCr4GaS8–Li2S (mp-1211167–mp-1153) was then evaluated for its metastability by computing the barriers to Ga diffusion using DFT to determine whether Ga is kinetically stabilized in LiCr4GaS8, given the slight gap (0.168 eV) between the μGa windows of LiCr4GaS8 and Li2S, as depicted in Fig. 3a. Additionally, this study identified the dominant interfacial growth mode of Li2S on LiCr4GaS8 and the adsorption energetics of S8 molecules to Cr4GaS8 to evaluate the stabilization effect of Cr4GaS8 for S8.
3.1.1 Ga migration barrier calculation.
To determine the stability of LiCr4GaS8 against decomposition, we computed the energy barrier to Ga diffusion using the climbing-image solid-state nudged elastic band (CI SS-NEB) method at the PBE DFT level of theory.29,30 The details of the calculations are provided in the Computational details section. Ga resides on one unique site within LiCr4GaS8, shown in Fig. 5a, and the minimum energy pathway (MEP) for Ga migration between adjacent Ga sites was computed. The computed MEP results in a predicted Ga migration energy barrier in LiCr4GaS8 of 3.32 eV. Along the MEP, Ga reaches a transition state when it passes through an S3 triangular plane, as shown in Fig. 5a. This is because the sulfur triangular plane has an area of only 0.57 Å2, which is smaller than Ga3+’s cross-sectional area of 0.69 Å2, and is the bottleneck along the Ga MEP. The predicted barrier results in a Boltzmann factor for the rate of diffusion of Ga in LiCr4GaS8 at 298 K ∼50 orders of magnitude smaller than that of Li+ diffusion in some ceramic electrolytes,31 indicating that Ga is kinetically-stabilized in LiCr4GaS8.
 |
| | Fig. 5 (a) DFT-computed approximate minimum energy pathway (MEP) for Ga (red) migration in LiCr4GaS8. Green circles represent the energies of the nudged elastic band (NEB) images, and the green curve illustrates the interpolated MEP derived from these points. The green, yellow, blue, red and hollow red spheres represent Li, S, Cr, Ga and Ga-symmetric sites, respectively. The area available within the sulfur triangular plane (0.57 Å2) constitutes the narrowest point along the Ga migration pathway, which is smaller than the Ga3+ cross-sectional area of 0.69 Å2. The yellow plane is defined by the 3 sulfur atoms (yellow) and indicates the position of the diffusion transition state for Ga migration. NEB at the PBE level predicts the diffusion barrier to be 3.32 eV. (b) The interface formed between LiCr4GaS8 (111) and Li2S (110) has the lowest von Mises strain of 1.1% and was thus adopted for the interfacial growth mode study. | |
3.1.2 The interfacial growth mode of Li2S on LiCr4GaS8.
The interfacial growth mode of Li2S on LiCr4GaS8 was evaluated as described in Section 2.6.1 and in the Computational details. The interface between LiCr4GaS8 (111) and Li2S (110) achieved the lowest von Mises strain of 1.1% and was adopted for the interfacial growth mode analysis, as shown in Fig. 5b. The interfacial growth mode results show that the dominant mode of Li2S growth on LiCr4GaS8 is Volmer–Weber island growth because γLi2S > γLiCr4GaS8 − σinterface.
3.1.3 The adsorption of S8 on Cr4GaS8.
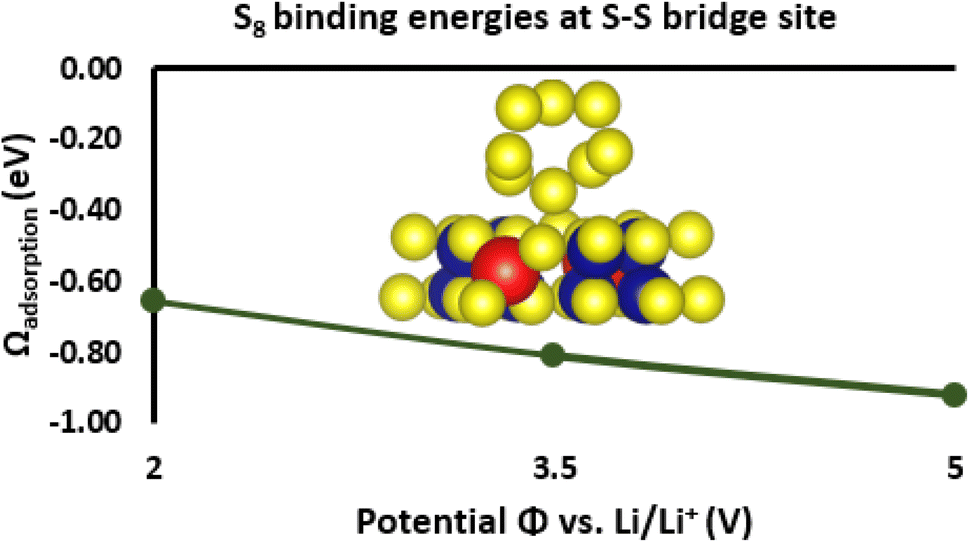
Solid crystals of S are considered weak molecular solids held together by relatively weak van der Waals forces. Therefore, to understand the adsorption of S8 on Cr4GaS8 we first examined the interfacial growth of sulfur on Cr4GaS8. To this end, we evaluated the adsorption of S8 on Cr4GaS8 as described in Section 2.6.2 and the Computational details section. Surface energies for the (100), (110), and (111) facets of Cr4GaS8 were compared at neutral charge, as shown in Fig. S4.† The (100) facet has the lowest surface energy and was adopted for the adsorption study, as shown in Fig. 6. Binding energies for sulphur were computed within the grand canonical ensemble with the electron reservoir potential sampled at +2.00 V, +3.50 V, and +5.00 V (Li/Li+), which spans the range over which S8 exists (above the oxidation onset potential of Li2S). Sulphur binding was investigated by direct adsorption of S8. Regardless of the binding site, S8 prefers binding at oxidative potentials. This likely indicates a positive correlation between S8 binding energy to positive surface charge. Given the relatively high potential of zero charge (PZC) of the (100) surface facet (3.61 V (Li/Li+)), other surfaces that may exist with lower surface areas would likely bind S8 stronger at equivalent biases. At +2.00 V, +3.50 V, and +5.00 V (Li/Li+), S8 binds via an S–S bridge site. At +2.00 V, S8 prefers binding to a Cr atop site by a margin of 0.2 eV. The S binding grand energy to the (100) facet has a bias dependence of −0.7 eV V−1 for the Cr atop site, and −0.1 eV V−1 for S–S bridge site. The (100) facet is shown in Fig. S6.† Cr-atop binding occurs at all Cr sites. Favorable S–S bridge binding occurs between adjacent S atoms bound to Cr and S atoms bound to Ga, as shown in Fig. S5.† Binding sites of S–S bridge and Cr-atop sites are indicated by red boxes in Fig. S6.† The results reveal favorable adsorption energies of S8 on the S–S bridge site of Cr4GaS8 between −0.65 and −0.85 eV, suggesting that Cr4GaS8 stabilizes and immobilizes S8 within the studied potential region (Fig. 6).
 |
| | Fig. 6 The adsorbed S8 species on the (100) facet of Cr4GaS8. The (100) facet has the lowest surface energy and was adopted for the adsorption study. The yellow, blue, and red spheres represent S, Cr, and Ga atoms, respectively. S8 binding energies at the S–S bridge site of Cr4GaS8, where the electron reservoir potential was sampled over the charging (delithiation) operating window. The results reveal that adsorption to Cr4GaS8 stabilizes and immobilizes S8. | |
3.2 Features of the discovered HC
The LiCr4GaS8–Li2S HC, which we identified using our framework, has a DFT-predicted average energy density of 1424 Wh kg−1 on a lithiated cathode basis, and is thus predicted to achieve the design objective of exceeding the maximum theoretical energy density of NMC333 of 1028 Wh kg−1 on a lithiated cathode basis. Furthermore, LiCr4GaS8–Li2S provides several advantages. First, according to MP, both the lithiated and delithiated intercalation and conversion phases of LiCr4GaS8–Li2S lie on the hull, meaning that they are thermodynamically stable with respect to decomposition. Second, according to MP, both the lithiated and delithiated phases of LiCr4GaS8 have 0 eV bandgaps, which compensates for the low electronic conductivities of the lithiated and delithiated Li2S conversion building blocks with bandgaps of 3.54 eV and 2.51 eV, respectively. Third, the volumetric change of LiCr4GaS8 upon (de)lithiation is only 7%, which mitigates the large volumetric change of (de)lithiation of Li2S (80%). Fourth, Li2S possesses a high energy density of 2739 Wh kg−1, which compensates for LiCr4GaS8's low energy density of only 108 Wh kg−1. Fifth, LiCr4GaS8 acts as both a conductive additive and immobilizer of S to suppress shuttling of sulfur species while actively contributing to the energy density of the cathode. Sixth, LiCr4GaS8 serves as an ideal substrate for supporting the molecular S species. Finally, we expect that the life span, self-discharge, mechanical integrity, and capacity fading of batteries with this HC will be superior to conventional Li–S batteries.
4 Conclusions
This work establishes a data-driven framework for the systematic design of hybrid cathodes for metal-based batteries that can be customized to specific design objectives. Our framework demonstrates the potential of combining data-driven approaches with inverse design principles to identify hybrid cathode structures that meet or exceed high-energy benchmarks, as illustrated in our case study. Here, LiCr4GaS8–Li2S (mp-1211167–mp-1153) emerged as a promising HC that achieves an average energy density of 1424 Wh kg−1, surpassing the maximum theoretical energy density of the conventional NMC333 cathode (1028 Wh kg−1). The comprehensiveness and fidelity of this case study is limited by the available phases and the level of theory used to predict the properties of the materials in the Materials Project repository.
The framework highlights both the promise and challenges inherent in the field. Specifically, the intricate thermodynamic criteria involving electrochemical and chemical stability evaluations surfaced as key bottlenecks, especially for composition phase diagrams with the extensive elemental compositions of the HCs and under the grand canonical ensemble conditions relevant to battery environments. Finding stable interfaces is a challenging task for materials spaces including sulfur because it possesses multiple oxidation states, high polarization and reactivity, causing these interfaces to fail thermodynamic and electrochemical stability criteria. These insights underscore the need for robust computational schemes and an expansion of thermodynamic data to improve framework fidelity in complex battery environments.
We suggest that experimental validation of the identified HC, LiCr4GaS8–Li2S, is needed, particularly with respect to its overall ionic conductivity and interfacial evolution, which remain challenging to determine computationally. Moreover, a multi-objective optimization study is crucial to identifying the critical ratio of the intercalation to conversion component materials that maximizes both the rate capability and the energy density of the HC. Additionally, the properties of the HC can be further refined through compositional modifications, such as substituting Ga with B or Al and exploiting defect engineering techniques. Further avenues of exploration could involve investigating and maximizing the HC lithiation capacity. The framework described here can be systematically exploited to explore the potential HC space for any given working ion and any chemistry of interest with pre-defined battery material design objectives to enable targeted performance-driven exploration of the vast materials space to guide the discovery of next-generation battery materials with enhanced energy storage and operational efficiency.
5 Computational details
DFT calculations to compute the Ga migration barrier and interfacial growth mode were performed using the Vienna ab initio simulation package (VASP)32 with the Perdew, Burke and Ernzerhof (PBE) generalized gradient approximation (GGA)33 exchange-correlation (XC) functional and the projector augmented-wave (PAW) method.34A plane wave cutoff energy of 520 eV was applied.
5.1 Ga migration barrier calculation
To compute the kinetic stability of Ga in Cr4GaS48, a Γ-point centered 2 × 2 × 2 k-point mesh was used for Brillouin zone integration. The relaxation of electronic degrees of freedom was terminated when the change in total energies between two consecutive electronic steps was < 10−5 eV. The structural relaxations were completed when the residual force on each atom was < 0.01 eV Å−1. The bulk structure of Cr4GaS48 (mp-1226264) was pulled from the MP repository. Ga migration in Cr4GaS4 occurs via a vacancy hopping mechanism, which was modeled using a 2 × 2 × 2 supercell model of bulk Cr4GaS4 with a Ga vacancy defect. Prediction of the MEP for Ga diffusion used the climbing image solid-state nudged elastic band (CI SS-NEB) method29,30,35 where the migration path was divided into a number of equidistant configurations (images). We report the 13-image MEP, although because the MEP is symmetric, only seven images representing steps along the MEP from the initial state to the midpoint were computed.
5.2 The interfacial growth mode of Li2S on LiCr4GaS8
To optimize heterostructures, the relaxation of electronic degrees of freedom stops when the change in total energies between two consecutive electronic steps was < 10−5 eV. The structural relaxations were completed when the change in total energies between two consecutive ionic steps was < 10−4 eV. Pymatgen was employed to automatically create a Γ-point centered k-point mesh for Brillouin zone integration with a grid density of 1200 k-points per the total number of atoms of a given structure.16 The bulk structures of LiCr4GaS8 (mp-1211167) and Li2S (mp-1153) were pulled from the MP repository. Pymatgen was used to cut the surface facets, calculate the von Mises strains, and construct the interfaces. An interface between LiCr4GaS8 (111) and Li2S (110) achieved the lowest von Mises strain and was adopted for the interfacial growth mode study. The interface formed between LiCr4GaS8 (111) and Li2S (110) was constructed using one layer of each component with 1.5 Å of interfacial spacing and a vacuum slab thickness of 25 Å to avoid spurious effects from interactions with periodic images. Constrained optimization was used to freeze all the atoms except for the atoms at the interface.
5.3 The adsorption of S8 on Cr4GaS8
All calculations to predict adsorption energies were performed using the JDFTx program36 with the PBE functional with dispersion interactions corrected with the Grimme semi-empirical D3 dispersion correction.37 Core electrons were represented using the GBRV pseudopotential set.38 The starting geometry for the bulk material was taken from the MP. The electronic energy at a planewave cutoff energy of 544 eV was compared to a convergence analysis over a planewave cutoff range of 490–600 eV and was shown to be near convergence.
For optimization of the bulk phase, a 5 × 5 × 5 k-point mesh was chosen as a balance between computational efficiency and accuracy based on a convergence test. For surface calculations, surfaces were cut using Pymatgen, and a standard k-point density of 24 k-points per atom was used for choosing the k-point mesh. Solvation of surfaces was modeled implicitly using the GLSSA13 linear PCM model,39 where glyme was modeled using 0.5 M Na+ and 0.5 M F− as the electrolyte. Surface energies for the (100), (110), and (111) surface facets were compared at neutral charge (unbiased) conditions. Alternative terminations to these surface facets were constructed to explore possible lower energy configurations. As shown in Fig. S4,† the (100) surface facet was computed to be the most stable surface, and thus was chosen for investigating sulphur binding energies.
Data availability
The data that supports the findings of this study are available in the supplemental material of this article.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We gratefully acknowledge the support from the National Science Foundation, Award No. NSF CBET-2323065. The views expressed in this article do not necessarily represent the views of the National Science Foundation nor the U.S. Government. We would like to express our sincere gratitude to Dr Nicholas R. Singstock for his invaluable contributions to the conceptualization and design of this study.
References
-
M. Dixit, N. Muralidharan, A. Parejiya, R. Amin, R. Essehli and I. Belharouak, in Management and Applications of Energy Storage Devices, ed. K. E. Okedu, IntechOpen, 2022 Search PubMed.
- J. Ma, Y. Li, N. S. Grundish, J. B. Goodenough, Y. Chen, L. Guo, Z. Peng, X. Qi, F. Yang, L. Qie, C.-A. Wang, B. Huang, Z. Huang, L. Chen, D. Su, G. Wang, X. Peng, Z. Chen, J. Yang, S. He, X. Zhang, H. Yu, C. Fu, M. Jiang, W. Deng, C.-F. Sun, Q. Pan, Y. Tang, X. Li, X. Ji, F. Wan, Z. Niu, F. Lian, C. Wang, G. G. Wallace, M. Fan, Q. Meng, S. Xin, Y.-G. Guo and L.-J. Wan, J. Phys. D: Appl. Phys., 2021, 54, 183001 CrossRef CAS.
- D. Andre, H. Hain, P. Lamp, F. Maglia and B. Stiaszny, J. Mater. Chem. A, 2017, 5, 17174 RSC.
-
R. A. Huggins, Advanced Batteries: Materials Science Aspects, Springer US, Boston, MA, 2009 Search PubMed.
- J. Wang, Z. Wang, J. Ni and L. Li, Electrochem. Energy Rev., 2022, 5, 211 CrossRef CAS.
- A. Van der Ven, Z. Deng, S. Banerjee and S. P. Ong, Chem. Rev., 2020, 120, 6977 CrossRef CAS PubMed.
- S.-H. Yu, X. Feng, N. Zhang, J. Seok and H. D. Abruña, Acc. Chem. Res., 2018, 51, 273 CrossRef CAS PubMed.
- H. D. Yoo, E. Markevich, G. Salitra, D. Sharon and D. Aurbach, Mater. Today, 2014, 17, 110 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252 CrossRef CAS.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Adv. Mater., 2021, 33, 2100855 CrossRef CAS PubMed.
- S. Xu, C. Y. Kwok, L. Zhou, Z. Zhang, I. Kochetkov and L. F. Nazar, Adv. Funct. Mater., 2021, 31, 2004239 CrossRef CAS.
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374 CrossRef CAS.
- J. Huang, Y. Gao and Z. Peng, Battery Energy, 2022, 1, 20210016 CrossRef CAS.
- A. Eftekhari, ACS Sustainable Chem. Eng., 2019, 7, 3684 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314 CrossRef CAS.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. Van Der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. Van Mulbregt, A. Vijaykumar, A. P. Bardelli, A. Rothberg, A. Hilboll, A. Kloeckner, A. Scopatz, A. Lee, A. Rokem, C. N. Woods, C. Fulton, C. Masson, C. Häggström, C. Fitzgerald, D. A. Nicholson, D. R. Hagen, D. V. Pasechnik, E. Olivetti, E. Martin, E. Wieser, F. Silva, F. Lenders, F. Wilhelm, G. Young, G. A. Price, G.-L. Ingold, G. E. Allen, G. R. Lee, H. Audren, I. Probst, J. P. Dietrich, J. Silterra, J. T. Webber, J. Slavič, J. Nothman, J. Buchner, J. Kulick, J. L. Schönberger, J. V. De Miranda Cardoso, J. Reimer, J. Harrington, J. L. C. Rodríguez, J. Nunez-Iglesias, J. Kuczynski, K. Tritz, M. Thoma, M. Newville, M. Kümmerer, M. Bolingbroke, M. Tartre, M. Pak, N. J. Smith, N. Nowaczyk, N. Shebanov, O. Pavlyk, P. A. Brodtkorb, P. Lee, R. T. McGibbon, R. Feldbauer, S. Lewis, S. Tygier, S. Sievert, S. Vigna, S. Peterson, S. More, T. Pudlik, T. Oshima, T. J. Pingel, T. P. Robitaille, T. Spura, T. R. Jones, T. Cera, T. Leslie, T. Zito, T. Krauss, U. Upadhyay, Y. O. Halchenko and Y. Vázquez-Baeza, Nat. Methods, 2020, 17, 261 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253 RSC.
- C. J. Bartel, S. L. Millican, A. M. Deml, J. R. Rumptz, W. Tumas, A. W. Weimer, S. Lany, V. Stevanović, C. B. Musgrave and A. M. Holder, Nat. Commun., 2018, 9, 4168 CrossRef PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685 CrossRef CAS PubMed.
- T. K. Schwietert, A. Vasileiadis and M. Wagemaker, JACS Au, 2021, 1, 1488 CrossRef CAS PubMed.
-
A. A. Demkov, in Handbook of Materials Modeling: Applications: Current and Emerging Materials, 2020, pp. 1119–1149 Search PubMed.
-
I.-H. Chu, in Handbook of Materials Modeling: Applications: Current and Emerging Materials, 2020, pp. 1231–1254 Search PubMed.
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 2, 264 CrossRef CAS.
- D. Dardzinski, M. Yu, S. Moayedpour and N. Marom, J. Phys.: Condens. Matter, 2022, 34, 233002 CrossRef CAS PubMed.
-
K. W. Kolasinski, Surface Science: Foundations of Catalysis and Nanoscience, John Willey & Sons, Ltd, 2008 Search PubMed.
-
W. Schmickler and E. Santos, Interfacial Electrochemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- R. Sundararaman, K. Letchworth-Weaver, K. A. Schwarz, D. Gunceler, Y. Ozhabes and T. A. Arias, SoftwareX, 2017, 6, 278 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446 CrossRef CAS.
- D. Gunceler, K. Letchworth-Weaver, R. Sundararaman, K. A. Schwarz and T. A. Arias, Modell. Simul. Mater. Sci. Eng., 2013, 21, 074005 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Benjamin S.
Rich
b and
Charles B.
Musgrave
a,
Benjamin S.
Rich
b and
Charles B.
Musgrave