
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polythieno[3,4-b]pyrazine: pathways to metallic charge transport†
Dominik
Farka‡
 *abc,
Elisabeth
Leeb
b,
Olvido Irrazabal
Moreda
d,
Theresia
Greunz
e,
Christoph
Ulbricht
b,
Jiří
Duchoslav
d,
Jaroslav
Vacek
a,
Kristian
Kříž
af,
Jindřich
Fanfrlík
a,
Cigdem
Yumusak
b,
Jakub
Drnec
d,
Jozef
Kajčovič
g,
David
Stifter
e and
Niyazi Serdar
Sariciftci
b
*abc,
Elisabeth
Leeb
b,
Olvido Irrazabal
Moreda
d,
Theresia
Greunz
e,
Christoph
Ulbricht
b,
Jiří
Duchoslav
d,
Jaroslav
Vacek
a,
Kristian
Kříž
af,
Jindřich
Fanfrlík
a,
Cigdem
Yumusak
b,
Jakub
Drnec
d,
Jozef
Kajčovič
g,
David
Stifter
e and
Niyazi Serdar
Sariciftci
b
aInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences (CAS), Flemingovo Náměstí 542/2, 160 00 Prague, Czech Republic. E-mail: dominik.farka@uochb.cas.cz; farka@jcu.cz
bLinz Institute for Organic Solar Cells (LIOS) Physical Chemistry, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
cUniversity of South Bohemia in České Budějovice (JU), Branišovská 1645/31a, 370 05 České Budějovice, Czechia
dEuropean Synchrotron Radiation Facility (ESRF), 71 Av. des Martyrs, 38000 Grenoble, France
eCenter for Surface and Nanoanalytics (ZONA), Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
fDepartment of Cell and Molecular Biology, Computational Biology and Bioinformatics, Uppsala University, Husargatan 3, 751 24 Uppsala, Sweden
gFaculty of Chemistry, Brno University of Technology (BUT), Purkyňova 118, CZ-612 00 Brno, Czech Republic
First published on 26th June 2025
Abstract
For the first time, an alkyl-chain free variant of polythieno[3,4-b]pyrazine (PTP) was created in a conductive state via oxidative chemical vapour deposition (oCVD). The charge transport properties of this new PTP are superior to those of the state-of-the-art ones by ten-fold with a conductivity of 0.5 S cm−1 and the material is found in the metallic regime of the metal–insulator-transition as the conductivity is vastly independent on temperature. Herein, we investigate the chemical and physical composition of the material in order to explain this improvement and discuss the results in light of the polythiophenes produced using the same method. Using GIWAXS, we discover large crystalline features combined with smaller, highly organized assemblies and gain insight into the chain length. The chemical composition of the material is determined via XPS and FTIR and further supplemented by computational methods. We employ DFT calculations (ωB97X-V/def2-QZVP) to understand the differences between unsubstituted and alkylated PTP. The former shows stronger proclivity to form planar structures, and the conjugation is broken upon distortion. Alternatively, in the latter case, such disorder does not considerably affect the conjugation. Furthermore, we simulate the effects of doping based on XPS results and reveal the possibility of protonation, despite the low pKa of the materials. Furthermore, this protonation allows the materials to achieve a cis conformer despite the typical expectation suggesting the prevalence of the trans form. Molecular dynamics simulations (NPT MD) of 24 dodecamers confirm the prevalence of a trans conformer. However, at multiple occasions, trimers of the cis motif form. The simulations predict stronger charge transport in the trans conformer. However, the cis isomer exhibits relevant levels and an extremely low HOMO–LUMO difference (<0.1 eV). This indicates the possibility of high conductivities and explains the hallmarks of metallic charge transport we observe experimentally.
Introduction
For thirty years, polythieno[3,4-b]pyrazine (PTP; Fig. 1a–d) has been the preferred material with regard to low band-gap polymers.1,2 Typically, this polymer is considered a structural analogue of polyisothianaphtalene (PITN) as their monomer subunits are rigid and conjugated.1 PTP combines the electron-rich properties of thiophene with the electron-poor pyrazine, which results in a small band-gap and is the reason for the distinction of the materials. Therein, the two ring systems of the monomer subunit fuse into a large conjugated structure.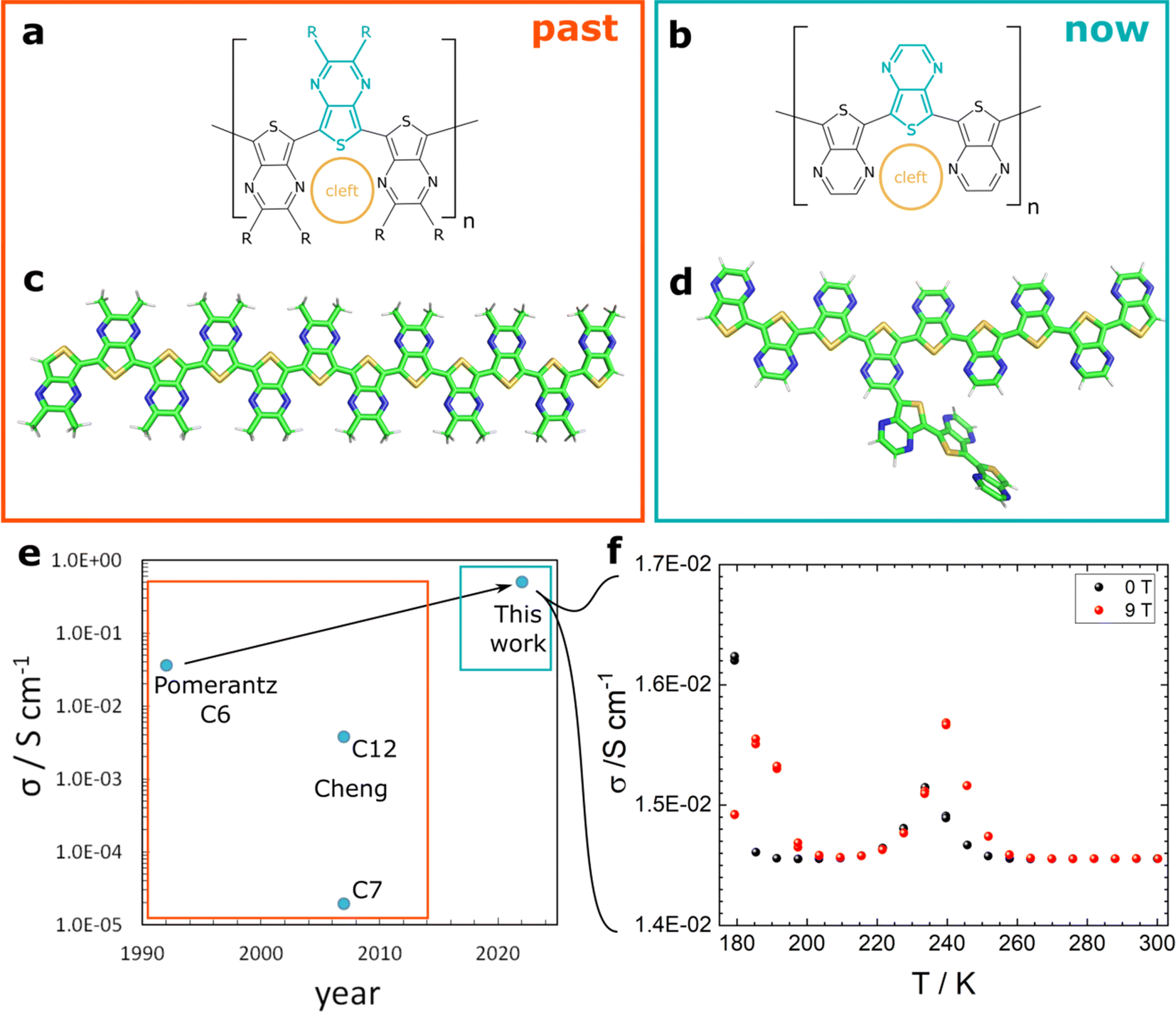 | ||
| Fig. 1 The history of PTP development with the achievements of past studies (orange rectangle) and our work (turquoise). Trimer of (a) poly-(2,3-dialkyl thieno[3,4-b]thienopyrazine) and (b) poly-(thieno[3,4-b]thienopyrazine). The respective monomer is highlighted in turquoise, and the substituents “R” stand for the respective alkyl chain as a representative for the previously studied materials. The iron-binding site “cleft” is shown in gold.9 (c) The 3D structure of methyl-PTP as a representative alkylated species optimized via DFT (ωB97X-V/QZVP). (d) The 3D structure of PTP with representative branching from the proposed side reaction.14 (e) Conductivity of PTP species reported over the years. Pomerantz et al.7 reported a dihexyl-substituted (C6) material, Cheng et al. (2007) reported on a thiophene-thienopyrazine co-polymer (heptyl-C7- and dodecyl-C12-substituted).19 (f) Temperature-dependent conductivity measurements of PTP reveal a peak upon cooling, typical for a disordered metal with metallic behaviour.3 Below a temperature of 180 K, contact issues were encountered and negative voltages were measured. The behaviour below 200 K was reproducible in a second measurement and enforces the metallic hallmarks observed at higher temperature. | ||
Alternatively, PTP can be considered a structural analogue of PEDOT.3,4 Here, the focus lies with the heteroatoms and the possible intra-molecular bonds that they may form.5 The monomer subunit of PEDOT and PTP contains sulphur—a large polarisable atom that can form a σ-hole.4,6 During the extension of the thiophene rings' C–S σ-bond, an absence of electrons results in a partial positive charge on the otherwise negatively charged sulphur (a “σ-hole donor”). Thus, it attracts the free electron pair of the σ-hole acceptor of the neighbouring monomer subunit, i.e. the free electron pair of oxygen and nitrogen for PEDOT and PTP, respectively. Consequently, these weaker attractions stabilize the planar conformation, which typically improves conjugation. This presents a new approach to viewing this polymer and may therefore lead to new unexpected insights.4
Where PEDOT is robust and easy to work with, PTP must be handled with care. The first synthetic approaches for PTP achieved a HOMO–LUMO gap of 0.95 eV.7,8 A decade later, a predicted band gap of 0.66 eV was achieved.9 This achievement became possible through the avoidance of iron ions and over-oxidation by the use of an alternative doping agent. Iron was computationally predicted to be readily complexed in the cavity formed by two orthogonal trimer subunits, causing the polymer to loop upon itself and decrease conjugation (termed “backbiting”).10,11
The thieno[3,4-b]pyrazine (subsequently thienopyrazine) building block found various applications, such as in co-polymers,12–15 or as a building block for stable hole transport materials.16 However, in terms of general charge transport, this material is virtually unknown.7,17 Furthermore, many of its physical properties (such as the dielectric constant, its chemical identity and chain-length, long-range order, and its work function) have never been measured.
Virtually18 all work on PTP relies on materials with alkylated pyrazine rings (Fig. 1a and c). However, since the focus was not on the conductivity, no correlation with the bulkiness of the substituents was established (Fig. 1e).7,19 The omnipresent alkyl-substituents address the issue of solubility during synthesis and arguably limit disorder.9,13 As we reveal in this work, they also act as a protecting group for the less reactive positions on the pyrazine ring. Over this potential branching site, the spatial presence of the PTP HOMO orbital is significant.14,16 Therefore, we consider the branched PTP in our calculations.
Instead of the prevalent electrochemical approach,9–11 we employed oxidative chemical vapour deposition (oCVD) with a mild, in situ generated oxidizing agent.20,21 This method was previously developed for PEDOT. It has been shown to be applicable for poly(3,4-ethylenedithiathiophene) (PEDTT)22 and even non-thiophene biopolymers, such as polydopamine23,24 and polyguanidine.25
The design of the synthetic apparatus includes a gas chromatography (GC) feature (Fig. 2a), and thus cleans the emergent material in situ before it is deposited. Non-volatile impurities in the starting materials (i.e., from stainless steel industrial reactors or any catalyst) are excluded from the product, as they do not participate in the reaction. Volatile impurities either go to the exhaust or can be avoided by careful placement of the substrate in the relevant part of the deposition zone. In the same manner, different crystalline properties of the sample can be achieved, as discussed in this work. The air-borne oxidant emerges from a mixture of sulphuric acid and its sodium salt,26 which in turn polymerizes and dopes the monomer to yield the final insoluble polymer.
 | ||
| Fig. 2 (a) Schematic representation of the synthetic apparatus. Brighter red frame represents the insulation part, where high-conductivity PTP:sulf is deposited. (b) Deposition zone of PTP (greenish material). (c) Optical microscopy of PTP:sulf with the highlighted AFM measurement area (red circle). (d) GIWAXS measurement of the “upstream” deposition zone. (e) GIWAXS measurement of PTP:sulf in the deposition zone of interest. (f) AFM image of PTP:sulf on the Au (111) (deposition zone of interest). (g and h) Inner region of the GIWAXS results shown in (d) and (e). (i) Interpretation of the GIWAXS results. Two types of assembly are found in the deposition zone of interest: the edge-on (7 polymers chains) and perpendicular forms. A stacking distance of 3.2 Å was determined. | ||
Since we rely on volatility rather than solubility (electrochemistry,9 wet-chemistry18), the alkyl side chains were omitted (Fig. 1b and d). This might affect conjugation, and different modes of charge transport may become dominant. This omission of alkyl chains was previously only attempted once and an insoluble, black material was achieved,18 as opposed to the yellow-green colour reported for alkylated PTP.7 Our strategy was rewarded with a turquoise material, which sets the new record room temperature conductivity of 0.5 S cm−1 (Fig. 1e). This corresponds to an improvement by an order of magnitude, and presents a first step towards the records set by other polythiophenes made in the same manner.20,22 We characterize the material in-depth with a strong theoretical elucidation of the involved effects to form a basis for future improvement, and to understand the fundamental PTP-design.
The chemical composition is characterized via optical methods and X-ray photoelectron spectroscopy (XPS). We employ grazing-incidence wide-angle X-ray spectroscopy (GIWAXS) to gain insights into the material structure and chain length, and analyze two parts of the deposition zone to gain insight into the differences in the structural properties. This allows us to understand the existing challenges to reach the performance of similar polythiophenes.
These results are supplemented by DFT calculations via the ωB97X-V functional (def2-QZVP basis set), which was a particularly good match for polythiophenes.27 We add calculations using the COSMO28 approach to support the XPS results of the chemical doping, and investigate the possible protonation of the rather acidic amines of the pyrazine ring.11 The calculations indicate dual doping via protonation and a counter ion similar to emeraldine polyguanidine.25 The distribution of the HOMO and LUMO suggest excellent conjugation. The HOMO is also delocalized over the pyrazine ring. Hence, there is a non-zero probability of branching off. This is further enhanced when protonated. Thus, in acidic environments such as ours, the probability rises. Protonation also increases the stability of the Z-, or cis-rotational isomer. The bond length alternation along the axis of conjugation revealed the material to intrinsically be in a quinoid form, i.e., the monomer subunits are connected via double bonds. It could therefore be argued to use the E-/Z-terminology over the typical trans-/cis-terms.
Molecular dynamics simulations (NPT-MD, 300 K, see ESI Methods†) indicate the formation of the cis-conformer. Subsequent charge transport simulations indicate an extremely small band-gap (>0.1 eV), which may in part explain the observed conductive properties.
This combined experimental and computational effort illustrates the strengths and limitations of the previous and current approaches towards PTP, and gives a perspective on future improvement and the material application for charge- and proton-transport, or the much needed artificial cellular signalling.29,30
Methods
The methods are found in the ESI† of this work.Results
Experimental results
In the scope of previous results,20,22 we were initially most concerned with the conductivity of this novel material. Three optimized films were measured; the highest conductivity of 0.5 S cm −1 was achieved at room temperature with electrodes in van-der Pauw geometry (Fig. 1e). This presents an improvement of about an order of magnitude compared to previous results by Pomerantz et al.7 Temperature-dependent measurements (0 and 9 T) were possible down to 173 K. Below that temperature, only negative voltages oscillating around −0.125 V were measured in both cases, possibly the consequence of a phase-transition.For the samples in 4-line geometry, significantly lower conductivities were observed (Fig. 1f), and yet temperature-dependent measurements were possible. These revealed the rise-and-fall behaviour typical for metallic conductors31 even at strong magnetic fields of 9 T, which should typically have a negative impact (Fig. 1f).3 Conductivity maxima were observed at 230 and 240 K for ambient and high-field, respectively. Below 200 K, another set of peaks was observed, further confirming the metallic behaviour in our material. Save for these regions, the conductivity was virtually temperature-independent until 173 K, where the supposed phase transition occurred.
The lower absolute value of conductivities of these samples is possibly caused by larger electrode distances and the polymers inhomogeneity. Optical microscopy on sapphire and gold surfaces revealed the presence of large, possibly crystalline structures surrounded by differently organized material (ESI Fig. S1e and f†). This explains the dramatic change in transport behaviour with increased electrode distances: there are fewer conductive channels that effectively span the distance between the electrodes at larger distances to allow for efficient charge transport (percolation).
Measurements on sapphire further support this hypothesis. The macroscopically transparent, turquoise film consists of bright-green elliptical features – an archipelago-like structure, as previously obtained for PEDOT20,32 and PEDTT22 synthesized by tube-furnace oCVD (schematic representation of the apparatus is given in Fig. 2a). The “greenish-yellow” colour indicates the presence of highly-doped PTP.7 The turquoise colour observed by bare-eye (Fig. 2b) results from the mixing of two colours: green (doped) material and “dark black-blue” (undoped) material, or it may indicate the protonated, lower band-gap species predicted from computational results.
Mechanically, the obtained films were extremely robust and exhibited strong adhesion to both glass and sapphire. Even on thicker films, the exertion of extreme force such as attempted removal of the films by a metal spatula only resulted in small scratches. This is quite different from the similarly synthesized PEDOT and PEDTT, where adhesion to the surfaces was much weaker. This strong adhesion gives hope for industrial applications, where mechanical robustness is desirable.
The signature feature of PTP is its low band gap. Therefore, we considered estimating our material's band gap via the Tauc plot (Fig. S1c and d†).33 For this, we investigated the materials optical properties by means of Fourier-transform infra-red (FTIR, Fig. S1a†) and ultra-violet visible spectra (UV-vis, Fig. S1b†). Based on the former approach, the hallmarks of a highly doped material (absorption peak in the near-IR region) caused by the presence of polaronic and bipolaronic states are found (analogous to PEDOT).34,35 Based on the latter approach and the assumption of a direct band-gap transition, we arrive at a band-gap of 1.38 eV, which by far exceeds any expectation.5,7,11 We must point out that the absorption peak is blue-shifted by 200 nm as opposed to the alkylated PTP-variety reported by Pomerantz,7 which we will explain in the theoretical part of this work.
The UV-vis spectra (Fig. S1b†) show a minimum in absorption at ∼2.3 eV (540 nm), which corresponds to the observed colour of the material. The line shape resembles the previously reported spectra of PTP, although the minimum is less pronounced and blue-shifted.7 Furthermore, the observed absorption increases with increasing photon energy and multiple shoulders are observable, which matches the previously reported behaviour. Since this was observed even in the alkylated variety, this is possibly due to the disorder-induced breakage of planarity and various polymer chain lengths rather than chemical diversity (i.e., side reactions, branching).
The chemical identity of the material can be deduced from FTIR and XPS data. The FTIR spectra (Fig. S1a†) show strong IRAV bands in the region up to 0.1 eV, followed by a peak at 0.14 eV, which is typical for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (aromatic) signal and is observed in the form of two distinct peaks.25 Next, at around 0.2 eV, we observe a strong, broad absorption that may indicate a SO2 species interacting with water-complexes36 or species of H2SO4.37 The presence of water is confirmed by the broad absorption at around 0.4 eV. However, the identity of the dopant cannot be concluded from these measurements alone.
N (aromatic) signal and is observed in the form of two distinct peaks.25 Next, at around 0.2 eV, we observe a strong, broad absorption that may indicate a SO2 species interacting with water-complexes36 or species of H2SO4.37 The presence of water is confirmed by the broad absorption at around 0.4 eV. However, the identity of the dopant cannot be concluded from these measurements alone.
Additionally, we electrochemically determine the band gap of samples deposited on carbon felt (CF, Fig. S2†), carbon paper (CP, Fig. S3†), and an electropolymerized material on platinum (Fig. S4†), which resulted in band gaps of 1.18, 1.90, and 1.6 eV, respectively. These results are in good agreement with the optically determined band gap (ESI, Fig. S1d†). A full discussion of these results can be found in the ESI.†
A new batch of monomer was used for electrochemistry (and the measurements described below). The purity was confirmed via NMR (Fig. S5 and S6†). Due to the tanned colour of the starting material and in the absence of measurable impurities, we conclude that the onset of oligomer formation has occurred in the second batch. Polymers obtained from oCVD of both batches were not distinguishable in terms of their physical properties. In electropolymerized PTP, these oligomers likely caused larger band-gaps. The material grown on CF showed the cleanest deposition pattern and the lowest band gap. Since the tube furnace-based oCVD brings intrinsic gas chromatographic properties, we believe that the heavier oligomers were precipitated prior to reaching our deposition zone of interest. Thus, a band gap of 1.18 eV is realistic.
We conducted measurements via atomic force microscopy (AFM) on the material deposited on atomically flat gold (111) on mica (muscovite). A zoomed-out image of the general area can be seen in the optical microscopy image (Fig. 2c) with the measurement area in the red circle (AFM image given in Fig. 2f). The optical microscopy image reveals a very rough, inhomogeneous film as the colours result from interference (i.e., they indicate different thicknesses). Similar changes in thickness were not observed in samples deposited on bare glass – for those samples, the bright-green features possibly indicate highly doped material (Fig. S1e†).
Even in the more homogeneous region measured in AFM, we can see height differences of about 30 nm (Fig. 2f). Given the overall film thickness of 56 nm, this indicates a very rough material that is possibly formed via the Stranski–Krastanow growth mode.38 Elliptical features are observed, which are organized into two assembly-types: disorganized assemblies or linear shapes. These are hundreds of nanometres long, growing roughly in the same direction, which is consistent with the lines observed in the optical images (top-left to bottom-right, Fig. 2c and f). Given the observed non-uniform directionality, we believe that this is not directly caused by epitaxial effects – which we later observe by GIWAXS. These directional features are below the diffraction limit of light and were not observed on bare glass (Fig. S7†), where only the spherical type of assembly was observed. Hence, it is highly possible that like the larger features, their origin is related to the gold substrate, but not due to epitaxial effects.
To prove the long-range order hypothesis, we investigate our material via GIWAXS. We used two samples: one from the deposition zone of interest (Fig. 2d and g) and another one from the hotter, “upstream” part of the deposition zone (Fig. 2e and h).
Since PTP contains nitrogen where oxygen is present in PEDOT, we expect similarities in the structures observed and use the established nomenclature thereof.39
Both samples appear to be rather rough, as can be seen from the signal-broadening and uneven intensity distribution (spottiness) of the outer rings (1.5–2.0 Å−1). The signal around 2.0 Å−1 in the “upstream” sample indicates the presence of an edge-on oriented material, given the increased intensity of the ring along the vertical scattering direction (Q∥ = 0),39 with a π-stacking distance of approximately 3.2 Å – this is significantly denser than what is found for PEDOT and reflects PTP's flat monomer subunit, similar to poly(isothinaphatelene).40
In both deposition regions, a crystalline material of hundreds of nanometers in size was formed. The spotted pattern indicates the presence of crystals larger than 200 nm. Their orientation is non-uniform in nature since we observe a ring pattern. Hence, we can rule out conventional epitaxial effects (1.5–2.0 Å−1). These are possibly the linear patterns observed in AFM and optical microscopy (Fig. 2c and f).
In the deposition zone of interest, we observed an additional phase that was not seen in the “upstream” sample. We observe a face-on oriented material (inner circles, Fig. 2h). These rings correspond to crystalline planes with interplanar distances of 12, 24, and 35 Å. The first two are different order diffractions of the same family of planes (24 Å) and suggest a variety of less homogeneously organized edge-on oriented material, i.e., π-stacks of approximately seven polymers that form a unit cell. The signal of the innermost ring (35 Å) corresponds to the length expected of a polymer chain of 10 repetitive units – consequently, we interpret this signal as upright standing chains, as reported previously for PEDOT.32 This indicates an average chain length of 10 monomer subunits, which is in good agreement with the experimental and theoretical values for polythiophenes.4,20,34
We employed the Scherrer formula21 in order to calculate the exact size of these crystallites, and came up with 24 and 34 nm for the 35 and 24 Å signals, respectively. This is similar to values found for PEDTT synthesized by the same method.22
The mix of various types of organization found by the GIWAXS measurements indicates that despite the local order, the transport across differently oriented crystallites may cause significant disruptions and decreased measurable conductivities. This can also manifest as increased scattering at grain boundaries or temperature-activated phenomena (see the conductivity maximum at 220–240 K, Fig. 1f). Other sources of disorder can further contribute to this in the disordered regions of the material. These regions, including trans- and cis-isomers and protonation, are investigated in the theoretical part of this work and are another source for the observed behaviour of temperature-dependent charge transport.
In comparison to the similarly obtained PEDOT (4050 S cm−1)20 and PEDTT (1050 S cm−1),22 only a fraction of the possible conductivity was achieved (0.5 S cm−1). To improve these results, we suggest prolonging the deposition zone by insulation of the deposition zone, so that a more resolved material can be achieved.41 Alternatively, the focus may be shifted to more “upstream” deposited materials, where a more face-on oriented material is expected.39 Alternatively, we suggest the use of functionalized surfaces to alter the materials organization on the molecular level.32,42
Next, we consider the XPS spectra (Fig. 3). The XPS survey spectrum (Fig. 3a) reveals carbon, oxygen, sulfur and nitrogen as the major surface elemental constituents. In addition, very small amounts of sodium and silicon can be detected, which are likely impurities introduced when rinsing the samples after synthesis.
 | ||
| Fig. 3 XPS spectra of the PTP-sample on glass, including the (a) survey, (b) carbon, (c) nitrogen, (d) oxygen, and (e) sulphur high-resolution spectra. | ||
The elemental composition of the film was determined from the survey spectra as follows: C (37.1 at%), N (10.0 at%), O (35.5 at%), and S (14.4 at%), and remaining elements with 3 at%.
A detailed analysis of the XPS spectra is given in the ESI† in the corresponding chapter.
The C 1s spectrum (Fig. 3b) indicates the presence of two main carbon species, which correspond to the thiophene (285.0 eV; 16.3 at%) and pyrazine (286.3 eV; 17.4 at%) ring systems, i.e., carbon bound to sulphur and nitrogen, respectively. Two minor peaks correspond to the positively charged carbon (287.8 eV; 2.2 at%) and π–π* satellites (1.2 at%).
The N 1s spectrum (Fig. 3c) reveals three distinct peaks. The first peak corresponds to pristine nitrogen atoms of the pyrazine ring (400.0 eV; 6.0 at%), while the second (401.9 eV; 3.5 at%) corresponds to their protonated form. Traces of over-oxidation9 were found in the form of the last peak (0.5 at%).
From these results, it became apparent that about 40% of all amines in the topmost layers were protonated. This presents a valid contribution towards doping, similar to what is observed in amine-containing conductive polymers such as polyguanidine.25 The second conclusion is that we have intrinsic disorder in the material, i.e., contamination by oximes, which will have a detrimental effect on the conductivity.
The oxygen spectrum (Fig. 3d) hints at the identity of the counter-ion present as a consequence of doping. The ratio of 2.7 between the lower energy peak (SO/S–O−) and higher energy peak (S–OH) is lower than expected for pure hydrogen sulphate (a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which was reported for previous syntheses based on this method. Consequently, small amounts of sulphur dioxide or sulphuric acid are possibly present in the material.
:1), which was reported for previous syntheses based on this method. Consequently, small amounts of sulphur dioxide or sulphuric acid are possibly present in the material.
The S 2p spectrum (Fig. 3e) indicates the presence of two sulphur species – sulphur found in thiophene (lower energy, 4.9 at%) and sulphur found in hydrogen sulphate (9.5 at%). However, it must be noted that any sulphur dioxide signal would overlap with the signal of thiophene. Given the structure of PTP, we conclude that 4.4 at% are found in the polymer, while 0.5 at% come from SO2.
The FTIR and XPS data therefore confirmed the doping mechanism to be complex in nature and of two kinds: doping by hydrogen sulphate and by protonation. This is surprising as a very low pKa11 is expected in pyrazine, and high acidity must be present in the material. This in turn indicates that PTP has a strong tendency to retain protons, and is possibly capable of their conduction and storage. In lieu of a large presence of experimental data on this polymer, meaningful insights had to be obtained from computational methods. As such, we resorted to DFT to gain insights at the molecular level.
Computational results
Initial calculations were performed on various PTP-trimers. Dimethylated PTP (MePTP) served as a representative of the alkylated species used in prior studies. Additionally, equivalent selenophenes (PSP and MePSP) were studied to test the role of σ-holes, with the larger polarizability amplifying any effects caused by the emerging chalcogen bonds.43 The structures of these trimers are found in the ESI, Fig. S8.† Optimizations confirmed the above findings on structural order in PTP:MePTP showed a second, local energetic minimum in its non-planar, disordered form (ESI, Fig. S9 and Table S1†). Thus, conjugation in methylated PTP variants may be essentially broken (dihedral angles of 89.5° and 120.1° for MePTP and MePSP, respectively). These minima are both more than 15 kcal mol−1 less stable than the planar minimum. However, impurities, mechanical disorder, and/or heat (kinetic control) may stabilize this product.To rule out hetero-atom interactions causing this behaviour, the distances between the nitrogen and sulphur and the nitrogen-atoms in adjacent monomer subunits across the “cleft” were measured (ESI, Table S2†).4,9
In all observed trimers, the N–N distance was significantly larger than the vdW radii (>163%). Hence, their Pauli exclusion repulsion was not the determining factor. However, they can still repel electrostatically, resulting in the formation of a non-planar minimum to minimize this repulsion. In all global minima, the S–N distance was 10–14% below the vdW radii; thus, chalcogen bond formation is likely. Since the distances were smaller in the unsubstituted species than the methylated ones, the omission of alkyl groups should have a positive effect on the attraction and conjugation. Hence, this will be reflected in an improved conductivity. To quantify this effect, we performed angular scans, followed by the characterization of the electrostatic potential (ESP) molecular surfaces.
For reasons of symmetry, only half the rotation was calculated (0–180°). Like in the PEDOT analogues,4 in terms of energies, a sinusoidal behaviour was detected with a peak at 90°, where conjugation is expected to be the worst. The maximum repulsion was found in the cis-conformer, where nitrogen–nitrogen repulsion would become relevant (Fig. S10a†). Hence, the local minimum at 120° is the result of this repulsion and stabilization due to improved conjugation (Fig. S10b†). Most relevant to us, however, is the initial increase of energy below 90°.
The observed energetic barriers are significantly larger than those observed in PEDOT (Fig. S10a,† 4 kcal mol−1).4 At lower angles (0–15°), however, all species require only a small amount of energy to diverge from planarity. In the selenophenes, this effect is less pronounced and larger energetic barriers are observed (3–4 kcal mol−1), which support our hypothesis that chalcogen bonds do play a significant role in planarization. The difference between methylated and non-methylated structures is much smaller (≤1 kcal mol−1), yet it is above the accuracy of the method employed.44,45 However, it does not explain the larger S–N distance in MePTP. The attraction between heteroatoms in the non-methylated species consequently removes the kinetic barrier observed in MePTP, and thus prevents the formation of defects (Fig. S10 and Table S1†).7,19 Therefore, the larger energetic barriers observed indicate the significant energy gains achieved by the electronic connection of the large conjugated monomer subunits.46,47 This is reflected in the differences of the band gaps, where alkylation of the respective trimer leads to an increased difference between the HOMO and LUMO (Fig. S10b†). To confirm the role of the chalcogen bonds, we calculated electrostatic potential (ESP) molecular surfaces in our trimers and expected a less pronounced σ-hole in the methylated species, an effect enhanced in the more polarisable polyselenophenes (PSP and MePSP).
 | ||
| Fig. 4 ESP molecular surfaces of the trimers (and branched PTP-tetramer). | ||
The conjugation within each monomer subunit7,46 distributes any substitution effect to all heteroatoms, thus profoundly affecting the ESP. Hence, the σ-hole becomes less electropositive and almost neutral, weakening the resulting chalcogen bond. Furthermore, the monomer subunit ESP appears to be more heterogeneous, possibly affecting the charge transport and intermolecular interactions. This electron excess will also lead to more repulsion between nitrogens across the “cleft” and foster the formation of a local, non-planar minimum in MePTP (Fig. 4).
Upon protonation of the amines (H2-PTP), the situation is very similar as in MePTP. Only the region of the protonated nitrogen becomes electron-positive (Fig. 4), which is reflected in the relaxation studies, as the trimer readily relaxes into the planar form. This is relevant to us as the XPS results indicate a large portion of our material to be protonated.
While branching is not very likely to occur and has only a small effect on the chalcogen bond akin to methylation, its effects might be rather disruptive. On top of spatial effects, the ESP surface of the branches is significantly changed, which is particularly true for the hydrogen atoms. This may cause the formation of traps within the system. Consequently, it explains why our synthetic route achieves significantly improved conductivities, which are intrinsically limited by disorder, and traps in the disordered regions in-between crystallites. Additionally, chalcogen bond formation between the main chain and its branch is possible. This may result in the formation of alternative conduction pathways, and can lead to electron-scattering. This should be reflected in the distribution of HOMO and LUMO orbitals, which we calculated for dodecamers, where this should be most pronounced. Conversely, the branch may distort the matrix in a way to allow the formation of pores within the material necessary for proton transport.
Since XPS revealed large amounts of SOx species and the amines of the monomer subunits are known to be easily deprotonated, all prerequisites for proton transport are present – we therefore investigated the possibility in a later chapter of this work.
Dodecamers
While the trimers served us well as models for investigating the relationship of the chalcogen bonds and substitution, delocalization effects over the polymer chain will only become visible fully at longer chain lengths.4 Herein, we focus entirely on the effects of the methyl functionalities and protonation, and optimize three dodecamers from disordered states: PTP with a central trimer branch, MePTP, and the aforementioned branched, diprotonated PTP (H2-PTP) at the branching point and in the peripheral “cleft” (Fig. S11†).Whether protonated or not, both non-methylated PTP variants are optimized into a single, global minimum, highlighting the limited effects of protonation. Conversely, MePTP exhibited two minima again, just as it was the case in the trimer.
Dihedral angle and bond length alternation (BLA) analysis revealed similar results for the planar conformer of PTP and MePTP, and was slightly in favour of the methylated variety (ESI, Table S3†). This is despite the disruptive influence of the branch on the conformation. In fact, parts of the branched PTP separated from it are more in the quinoid form than MePTP.5
The energetic difference between the two MePTP conformers was 38.6 kcal mol−1 (Table S3†), and semi-planar islands were formed this time. Still, two significant distortions (−53.8 and −58.5°) were found separating the material from planarity and discontinuing planarity. In a real material, this would significantly impact charge transport.
We now understand one reason why it was possible to overcome prior transport issues.7 However, we need to understand the limitation of our own synthetic method. Therefore, we repeated angular scans for dodecamers of PTP and MePTP (Fig. S12a and c†) and protonated H2-PTP (Fig. S12b and d†). We focused on the benzoid, peripheral part (D7) and the central, quinoid (D3; in the case of PTP, branched labelled as “subst.“) part, as revealed by BLA (ESI, Table S3†). In the latter case, the bond was expected to be closer to sp2 hybridization and a steeper increase in energy was expected upon rotation. The expected sinusoid arrangements occurred in angular scans of the unprotonated species and the energy penalties for the quinoid part were always larger (Fig. S12a†).
Again, at low angles (0–15°), the investigated bond of all structures behave quite similarly, and 2 kcal mol−1 is needed to disrupt its planarity. Then, the behaviour diverges significantly. For PTP, 50% more energy is needed for further rotation than for MePTP. However, they are almost equal at 60°. This illustrates how alkylation gives the polymer more leeway in terms of structural movement. However, it must be noted that these events occur in energy ranges far exceeding those of PEDOT.4
In protonated H2-PTP, we were surprised to find that the minima obtained from optimization were not the global minima (Fig. S12b†). As opposed to the trans conformer, the cis-variety was the more stable one by 2 and 4 kcal mol−1 for the central and peripheral parts, respectively. Since the two amines were previously shown to be too close in the cis-conformer, the proton consequently has to be shared and allows for this surprising structure.
The central region of both PTP and MePTP is highly sensitive to disorder in terms of the band-gap. Upon rotation, a large increase is observed (Fig. S12c†). This is much less so in the peripheral region of PTP. Furthermore, it is utterly absent in the MePTPs HOMO–LUMO gap, where no change occurs, which is a testament to the poor conjugation in the latter.
To relate these results to experimental values, we conducted a qualitative comparison of the HOMO–LUMO gap ratios for the unsubstituted and alkylated PTP. Computationally, a ratio of 0.73 is observed, i.e., 0.11 and 0.15 eV for PTP MePTP and PTP, respectively (ESI Fig. S12c†). The ratio of the literature value of the bandgap of alkylated PTP7 and our experimental values is 0.68 and 0.81 for the optically and electrochemically determined band gap (Fig. S1d and S2†), respectively. This indicates that a large blue-shift in the optical absorption is certainly conceivable.
In H2-PTP, the band gap was insensitive to rotation (=disruption) in the peripheral region, which was expected from a benzoid structure, as indicated by BLA (Fig. S12d†).This indicates either poor conjugation or independence of the planarity and conjugation. Conversely, the central part close to the substitution site was highly sensitive to disorder, but in an inverse manner as the band-gap decreased upon rotation. This is puzzling, as it suggests improved conjugation. For both unusual cases, we visualized the HOMO and LUMO orbitals in the planar and disordered states for all three structures (ESI, Fig. S13–15†).
For PTP (Fig. S13†), disruption of the central part leads to substantial changes in the HOMO and LUMO distribution, effectively dissecting the conjugated system in half. When the peripheral part is changed, however, the branch of the polymer becomes significantly involved in the LUMO, while the HOMO remains relatively unaffected.
For MePTP (Fig. S14†), disruption in the central part only plays a minor role, and does not entirely break the conjugation in either the HOMO or the LUMO orbitals. The reason for the observed changes lies in the increased contribution of the pyrazine ring to the frontier orbitals, effectively raising the band-gap. Rotation in the peripheral part plays an even smaller role. This reflects why optimization was crucial to achieve the predicted low bandgaps.1,7,9
For H2-PTP (Fig. S14†), the angular scans revealed the lowest HOMO LUMO gap when the central, quinoid bond is rotated by 90°. This is possibly caused by the fact that when the central bond is rotated, the HOMO and LUMO orbitals sit on the same atoms with a strong involvement of the branch. No such effect is observed in the peripheral region.
These distribution patterns reveal that PTP is apparently much more sensitive to disorder in terms of p-type conduction than its alkylated versions. Hence, although the energetic cost of bond rotation is prohibitively expensive even at low angles, charge transport in PTP is significantly decreased once the polymer becomes disordered. If the polymerization reaction occurs in the cis-conformation, the large energetic barrier would essentially lock the polymer in this position – which is possibly unsurpassable even by post-processing methods.3,31,48,49
Future attempts at the synthesis of PTP should therefore avoid iron and overoxidation.9 Furthermore, the avoidance of a protic environment during synthesis should also be considered if the trans-conformer is to be achieved, or protic environments when the cis-variant is the preferred outcome should be considered. An alternative might be found by the use of cleavable substituents,50 or a substituent which improves the strength of the materials chalcogen bonds.
Doping of branched hexamers
Since we now understand the intramolecular and noncovalent interactions of PTP and their impact on the optical properties of the materials, it is time to focus on the doping process. XPS suggests a combination of hydrogen sulphate and sulphur dioxide, yet the presence of the latter is surprising. We therefore set up simulations in order to investigate the composition of the material using the COSMO method. We created twelve systems of various doping agent combinations surrounding a single, branched PTP-hexamer. All of them were constitutional isomers to each other to make the results comparable. We present the three most stable geometries in Fig. 5, and include two of the least stable variants in the ESI (Table S4).† | ||
| Fig. 5 Comparison of the constitutional isomers of the branched, doped hexamers of PTP:sulf. Comparison of the most stable compositions (x-axis in the plot) and their relative energies calculated via COSMO and a dielectric function of 10 (orange triangles) and 30 (black squares, corresponds to the high concentration of acid). The three most stable chemistries obtained indicate that the sulphur species are most probably a combination of HSO4− and SO2, HSO4− and HSO3− (+0.5 kcal mol−1) or HSO4− and SO2 in combination with protonated PTP (+2.5 kcal mol−1). At lower dielectric constants, PTP would immediately deprotonate and result in an apparently higher stability. | ||
The COSMO method was involved in order to make these calculations meaningful, yet it requires knowledge of the dielectric constant – a property which was never reported for PTP but was beyond our reach. We therefore estimated it based off of XPS results. Since high concentrations of sulphuric acid/hydrogen sulphate were determined, we took the dielectric constant of (aqueous) sulphuric acid (Table S5†). The resulting dielectric constant was around 30, much higher than that in typical conductive polymers.51 Therefore, a second calculation for a more reasonable value of 10 was conducted.
At the lower dielectric constant, protonation was not possible. At the higher dielectric constant, protonation proved possible when the sulphur species were HSO4− and SO2. This indicates that our initial guess was reasonable. Still, multiple protonations were impossible to achieve under our conditions and were unfavourable by more than 200 kcal mol−1. For comparison, a similar value was achieved for the extremely unlikely coexistence of sulphuric acid and sulphate (ESI, Table S4†).
The most stable chemistry under our conditions was unprotonated PTP with the sulphur species HSO4− and SO2. Protonation of PTP under these conditions was unfavourable by 2.5 kcal mol−1. Only 0.5 kcal mol−1 above the most stable chemistry was HSO4− and HSO3−, which is close to the limit of the method employed.44,45 Consequently, theoretical and experimental results agree that HSO4− and SO2 are the sulphur species found in the synthesized PTP.
Interestingly, even in the doped state, all of the polymers had a strong tendency to form a planar structure (ESI, Table S4 and Fig. S11†). The only exception is constituted by the tri-protonated variant, where the attraction between the heteroatoms would naturally be the weakest.
Interestingly, the side branch remained rather planar with respect to the main chain. This indicates that the branch gains stability through interaction with the main chain. The counter ions and water are also of interest (Fig. 5). The visualization of noncovalent contacts revealed many interactions between the oligomers and the small molecules/ions in all observed cases. This indicates a similar situation as in PEDTT,22 where the polymer is thought to form a planar structure due to a strong interaction with the counter ion.4 Consequently, even in PTP, the sulphur species and water play an active role in improving the charge transport, and do not merely serve the part of passive counter ions.
Molecular dynamics
Based on the obtained energetically most stable geometry of hexamers (HSO4− and SO2), we constructed a matrix of dodecamers (the branches were removed for simplicity) and doping agents, including water. These presented the starting point of our molecular dynamics (MD) simulations.As DFT calculations showed that PTP had a stronger tendency towards planarity than PEDOT4 (Fig. S11 and S12†), we decided to start with the most adverse conditions, i.e., where disorder is possibly the strongest and hardest to overcome due to the disruptive presence of water and dopants. The results can be seen in ESI Fig. S16.†
This revealed PTP's strong proclivity to overcome disorder and to form a network of nearly planar structures connected at hubs where multiple chains formed (staggered) π-stacks (ESI, Fig. S16a†). These are the optimal conditions for delocalization (based on Fig. S13 and S15†) and inter-chain transport (Fig. S16a†).52,53
The majority of the chains were found in the expected trans-conformation, but multiple occasions of the cis-conformer were found (Fig. S16b–d†). Since DFT calculations indicate a substantial energetic barrier to adapt this conformation (Fig. S12†), there needs to be an alternative energetically feasible pathway in order to lead to multiple instances of this conformer in our simulations. Therefore, we attempted to find their origin in external influences. Although these structures were typically in the vicinity of “pores” within the matrix where the disruptive influence of water and SOx was most likely to occur (Fig. S16c and d†), we were unable to find evidence of a clear connection.
Since these motifs were rather infrequent and always embedded within the polymer matrix of trans-conformers, GIWAXS would be unable to detect their presence. Given the high degree of doping and the subsequently large amount of protons, it is highly likely that these motifs are found in our material too. This would suggest the intrinsic presence of disorder on which backscattering may occur due to a mismatch in the wavefunctions.
Transport of pentamer-based geometries
To link these structural anomalies to charge transport, we picked three pentamers of the most prevalent motifs found in our MD simulations and calculated their electron-transmission spectra and pathways: the (almost) planar trans-conformer (M1), the disordered trans-conformer (M2), and the partial cis-conformer (M3). The results can be found in Fig. 6. The full depiction of the geometries of the simulated devices is found in the ESI, Fig. S17.†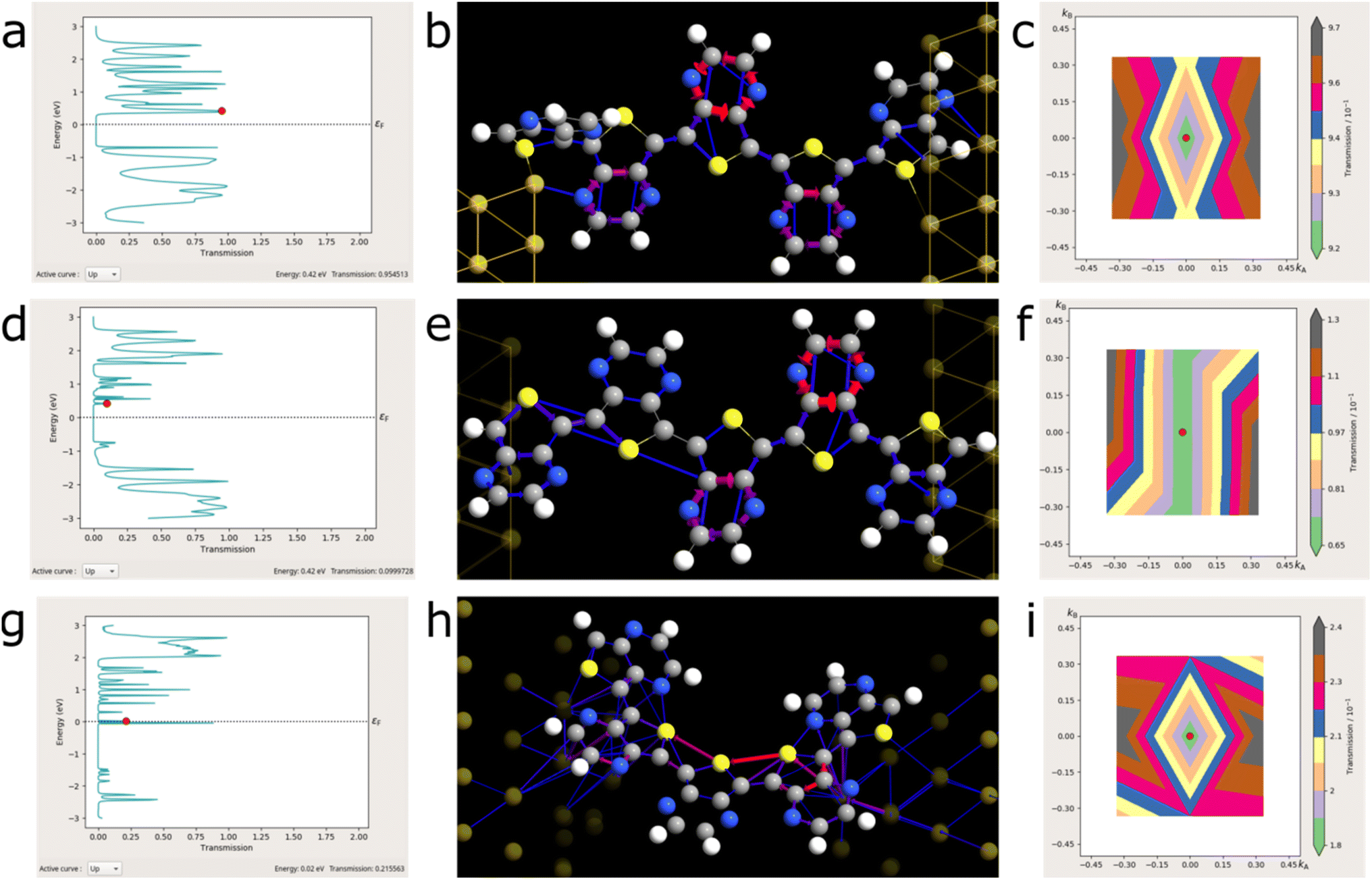 | ||
| Fig. 6 Transport calculations of the M1–M3 LUMO (π*, highlighted by the red dot in the transmission spectra). Transmission spectra with the LUMO level indicated by the red dot (a, d, and g) and the corresponding LUMO transmission pathways (b, e and h). The contribution of the pathway towards the transmission is weighted from blue to red for the least to greatest charge-transport, respectively. (c, f, and i) The respective transmission coefficients (M1–M3) plotted against the reciprocal space vectors ka and kb. | ||
The transmission spectra showed only minor differences between the two trans-conformers (Fig. 6a and d) with respect to their π and π* (or HOMO–LUMO) levels. Both molecules showed a π* at 0.42 eV, yet their transmission differed by an order of magnitude at 0.955 and 0.099 for M1 and M2, respectively.
In both molecules, the pyrazine rings were disproportionately involved in transmission, which indicates that a form of charge-hopping phenomenon is responsible for transport (Fig. 6b and e).54 In M2, additional tunnelling phenomena were observed between the most disordered monomer subunits. This was reflected in the transmission coefficient plots (plotted versus the reciprocal space vectors, ka and kb). Whereas in M1, a symmetrical, diamond-shaped distribution transmission in two dimensions is indicative of a proclivity to low-dimensional charge transport, M2 showed symmetry along the kb vector.
Since the LUMO was significantly less conductive in M2, we investigated its HOMO (ESI, Fig. S18a–c†). This gave a transmission of 0.160 or twice the value found for the LUMO (0.099). This was reflected in the inversion of the spatial distribution of transmission coefficients (from short to long range).
The charge transport properties of M1 are an intrinsic property of the molecular geometry (Fig. S18d–f†), and remained even in an alternative electrode model. Even the transmission pathway and coefficient remained unchanged (Fig. S18e and f†).
The switch from trans- to cis-conformation (M1/M2 to M3) resulted in a significant change with regards to the obtained transmission spectrum (Fig. 6g).
What immediately stood out was the presence of states around the Fermi energy, EF. If confirmed experimentally, the cis-conformer presents a true synthetic metal.3 This may be counterintuitive, yet of little surprise once we consider the remote similarity to polysulphurnitride.55,56 Furthermore, the transmission pathway truly indicates a strong role of the sulphur-atoms in the charge transmission (Fig. 6h, red transmission arrows). The contributions of these states to the transmission were 0.883 and 0.216 for the HOMO (−0.04 eV relative to EF) and the LUMO (+0.02 eV relative to EF), respectively. This is comparable to the M1 conformer, yet at a miniscule HOMO–LUMO gap of <0.1 eV: an energy difference which would foster charge transport even at room temperature without the necessity of doping.
Contrary to expectation, the calculated transmission pathways in M3-LUMO (Fig. 6h) resembled ballistic transport and occurred through sulphur, instead of delocalization through the π-orbitals.57 Upon closer inspection, this resembled parts of M2-LUMO (Fig. 6e) and was in stark contrast to M1-LUMO (Fig. 6b), where delocalization through π-orbitals dominated. This might indicate a different mode of charge transport in the non-planar parts of PTP. Transport in M2-LUMO and M3-LUMO is thus reminiscent of the situation found in polysulphur-nitrile.55
Because of the involvement of the sulphur atoms in both M2 and M3, we were interested in their similarities with regards to charge transport, i.e., if the transmission values would be stronger in the HOMO, and if an inverse behaviour of the transmission coefficients in relation to k-space would be revealed (Fig. 6i and S19†). This was confirmed for both HOMO and LUMO+1 (Fig. S19d and f†). The transmission value of the M3-HOMO (0.88) was a multiple of the LUMO and LUMO+1 values (0.22 and 0.18, respectively). Thus, transport through the HOMO orbitals is preferred in the non-planar and cis-oriented material over LUMO-based transport.
Since M1 and M3 differ substantially in the atoms involved in charge transport, observable conductance may suffer greatly in a hybrid material where both conformers coexist. One conformer will effectively act as an impurity for the other, and introduce traps to the material. This will require thermal activation (hopping)- or worse-it will present deep traps that do not contribute to charge conduction.58,59 As the overlap of their wavefunctions is diminished by their different nature, scattering phenomena will become more likely.59,60
This is particularly visible when we focus on the transmission pathways of M1-LUMO and M3-LUMO (Fig. 6b and h). Where the latter red and purple arrows indicate strong conductions between sulphur atoms (ballistic transport), the transmission pathway occurs along the polymer backbone (blue, thin arrows) with prolonged stays at the pyrazine rings (red arrows, akin to weak localization).60,61 The wavefunctions of two such different molecules will vary greatly and will require additional energy to delocalize.
In the case of our material, we observe temperature-independent charge transport down to 173 K(Fig. 1f), and band-like transport is expected. Thus, if the cis-conformed material is present, its states are beyond the conduction edge and do not contribute to charge transport, as they effectively present traps. The geometric factors observed via GIWAXS (Fig. 2c–i) indicate the presence of trans-isomers in the crystalline part, but high amounts of acidic dopants are observed via XPS (Fig. 3). Thus, we conclude that we have a hybrid material. This is further supported by the fact that we have low conductivities in coexistence with hallmarks of metallic transport-even under the influence of a strong magnetic field (Fig. 1f).3,31 Thus, we likely underestimate our real conductivities as the macroscopic, spatial dimensions of the measured thin-film possibly include large swathes of disordered material which do not participate in charge transport. This does not discard the potential of the cis-conformer, but rather suggests that we must focus on the separation of both conformers to access the full potential of both. The proclivity to form the cis-conformer could be further enhanced by the use of photochemical methods50 or self-assembled monolayers.32,42 In that way, the extremely low HOMO–LUMO band gap (<0.1 eV vs. ∼1 eV in trans) may result in a synthetic (semi-)metal.
Conclusions
The conductivity of polythieno[3,4-b]pyrazine (PTP) has been greatly improved and a new conductivity record has been established. However, we fell short of the achievements in PEDOT and had to search for the reason why this occurred.We set out with the hypothesis that PEDOT and PTP share similarities in terms of their heteroatoms, and confirmed that this is a valid assumption. The low band-gap of PTP gives us hope that conductivities superior to that of PEDOT are achievable. However, virtually all results indicate that PEDOT's unique robustness is missing. To achieve great conductivities, the removal of alkyl substituents is key to success. However, this also makes the conjugation of the materials extremely dependent on planarity. Still, at 230 K, 240 K, and below 200 K, the conductivity peaks reveal metallic properties within our semi-crystalline PTP material. At 173 K, a phase transition occurs, which requires more attention-including the improvement of the synthetic method to produce either of the PTP-conformers. Proton-free environments should be used for the trans-conformer. Meanwhile, for the cis-conformer, our method may be adapted, i.e., via photochemistry50 or SAMs.42 Acidic environments may suffer from the increased prevalence of branched PTP. However, its presence cannot be confirmed nor discarded experimentally.
As such, this work suggests that the quest for high-conductivity PTP is far from over, and this work lays the foundation for future progress and applied research. The complex character, and the electron- and (possible) proton transport make it an interesting target to test in the scope of the electrochemical CO2 reduction reaction (CO2RR24,25), or even cellular signalling.29
Data availability
Datasets generated and/or analysed during the current study are not publicly available online at the time the manuscript is submitted to the journal for technical reasons. Before the necessary repositories are established, all data will be provided by the corresponding author where possible upon request. Crystallographic data, XPS and GIWAXS measurement-data as well as computational inputs will be obtained from collaborators; provision of data can therefore be delayed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the institutional project RVO 61388963. We thank Prof. P. Hobza and Dr Ivo Starý for his support and for making this work possible. The authors thank ESRF and ID31 beamline for beamtime provision under the experiment identifier IHCH1777.62 D. F. acknowledges the support of the Institute of Organic Chemistry and Biochemistry (IOCB) of the Czech Academy of Sciences (CAS) through the “IOCB fellowship”-scheme. D. F. would also like to express his thanks to Prof. Jean-Pierre Roncali for the inspiration to investigate this material and to Vojtěch Košťál, Jiří Rybáček, and Andrei Ionut Mardare for scientific discussions. J. D. and D. S. acknowledge the government of Upper Austria for financial support (project ASAES). J.K. thanks the AKTION project No. 95p3.References
- K. Nayak and D. S. Marynick, The interplay between geometric and electronic structures in polyisothianaphthene, polyisonaphthothiophene, polythieno(3,4-b)pyrazine, and polythieno(3,4-b)quinoxaline, Macromolecules, 1990, 23, 2237–2245 Search PubMed.
- P. Otto and J. Ladik, Theoretical search for low-gap polymers based on polythiophen, Synth. Met., 1990, 36, 327–335 CrossRef CAS.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Progress in understanding structure and transport properties of PEDOT-based materials: A critical review, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- D. Farka, K. Kříž and J. Fanfrlík, Strategies for the Design of PEDOT Analogues Unraveled: the Use of Chalcogen Bonds and σ-Holes, J. Phys. Chem. A, 2023, 127, 3779–3787 Search PubMed.
- J. Roncali, Molecular Engineering of the Band Gap of π-Conjugated Systems: Facing Technological Applications, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- N. Biot and D. Bonifazi, Chalcogen-bond driven molecular recognition at work, Coord. Chem. Rev., 2020, 413, 213243 Search PubMed.
- M. Pomerantz, B. Chaloner-Gill, L. O. Harding, J. J. Tseng and W. J. Pomerantz, Poly(2,3-dihexylthieno[3,4-b]pyrazine). A new processable low band-gap polyheterocycle, J. Chem. Soc., Chem. Commun., 1992, 1672–1673 RSC.
- J. Kastner, et al., Raman spectra and ground state of the new low bandgap polymer poly(thienopyrazine), Synth. Met., 1995, 69, 593–594 Search PubMed.
- D. D. Kenning and S. C. Rasmussen, A Second Look at Polythieno[3,4-b]pyrazines: Chemical vs. Electrochemical Polymerization and Its Effect on Band Gap, Macromolecules, 2003, 36, 6298–6299 CrossRef CAS.
- D. D. Kenning, M. R. Funfar and S. C. Rasmussen, Polymerization Studies of Thieno[3,4-b]Pyrazines, Polym. Prepr., 2001, 42, 506–507 CAS.
- D. D. Kenning, et al., Thieno[3,4- b ]pyrazines: Synthesis, Structure, and Reactivity, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS.
- W.-Y. Lee, et al., High hole mobility from thiophene-thienopyrazine copolymer based thin film transistors, J. Polym. Res., 2009, 16, 239–244 CrossRef CAS.
- R. Mondal, S. Ko and Z. Bao, Fused aromatic thienopyrazines: structure, properties and function, J. Mater. Chem., 2010, 20, 10568 RSC.
- E. W. Culver, T. E. Anderson, J. T. López Navarrete, M. C. Ruiz Delgado and S. C. Rasmussen, Poly(thieno[3,4- b ]pyrazine- alt -2,1,3-benzothiadiazole)s: A New Design Paradigm in Low Band Gap Polymers, ACS Macro Lett., 2018, 7, 1215–1219 CrossRef CAS.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Thieno[3,4-b]pyrazines and their applications to low band gap organic materials, Chem. Commun., 2011, 47, 11394 RSC.
- S. N. Afraj, et al., 2,3-Diphenylthieno[3,4- b ]pyrazines as Hole-Transporting Materials for Stable, High-Performance Perovskite Solar Cells, ACS Energy Lett., 2022, 7, 2118–2127 CrossRef CAS.
- C. Wang, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, Poly(3,4-ethylenedithiathiophene). A New Soluble Conductive Polythiophene Derivative, Chem. Mater., 1995, 7, 58–68 CrossRef CAS.
- A. J. Hagan, S. C. Moratti and I. C. Sage, Synthesis of low band gap polymers: Studies in polyisothianaphthene, Synth. Met., 2001, 119, 147–148 CrossRef CAS.
- K.-F. Cheng, C.-L. Liu and W.-C. Chen, Small band gap conjugated polymers based on thiophene-thienopyrazine copolymers, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5872–5883 CrossRef CAS.
- D. Farka, et al., Anderson-Localization and the Mott-Ioffe-Regel Limit in Glassy-Metallic PEDOT, Adv. Electron. Mater., 2017, 3, 1700050 CrossRef.
- D. Farka, A. O. F. Jones, R. Menon, N. S. Sariciftci and P. Stadler, Metallic conductivity beyond the Mott minimum in PEDOT: Sulphate at low temperatures, Synth. Met., 2018, 240, 59–66 Search PubMed.
- D. Farka, et al., Overcoming intra-molecular repulsions in PEDTT by sulphate counter-ion, Sci. Technol. Adv. Mater., 2021, 22, 985–997 CrossRef CAS PubMed.
- H. Coskun, et al., Biofunctionalized conductive polymers enable efficient CO 2 electroreduction, Sci. Adv., 2017, 3, e1700686 CrossRef PubMed.
- H. Coskun, et al., Chemical vapor deposition - based synthesis of conductive polydopamine thin-films, Thin Solid Films, 2018, 645, 320–325 CrossRef CAS.
- H. Coskun, et al., Cofunction of Protons as Dopant and Reactant Activate the Electrocatalytic Hydrogen Evolution in Emeraldine-Polyguanine, Adv. Mater. Interfaces, 2020, 7, 1901364 CrossRef CAS.
- D. Schwartz, R. Gadiou, J.-F. Brilhac, G. Prado and G. Martinez, A Kinetic Study of the Decomposition of Spent Sulfuric Acids at High Temperature, Ind. Eng. Chem. Res., 2000, 39, 2183–2189 Search PubMed.
- N. Mardirossian and M. Head-Gordon, ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904 Search PubMed.
- A. Klamt and G. Schüürmann, COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Organic Bioelectronics: Bridging the Signaling Gap between Biology and Technology, Chem. Rev., 2016, 116, 13009–13041 Search PubMed.
- E. Stavrinidou, et al., Electronic plants, Sci. Adv., 2015, 1, e1501136 CrossRef PubMed.
- M. N. Gueye, et al., Structure and Dopant Engineering in PEDOT Thin Films: Practical Tools for a Dramatic Conductivity Enhancement, Chem. Mater., 2016, 28, 3462–3468 Search PubMed.
- D. H. Apaydin, et al., Nanometer-Thick Thiophene Monolayers as Templates for the Gas-Phase Epitaxy of Poly(3,4-Ethylenedioxythiophene) Films on Gold: Implications for Organic Electronics, ACS Appl. Nano Mater., 2022, 5, 3194–3200 Search PubMed.
- J. Tauc, Optical properties and electronic structure of amorphous ge and si, Mater. Res. Bull., 1968, 3, 37–46 Search PubMed.
- I. Zozoulenko, et al., Polarons, Bipolarons, And Absorption Spectroscopy of PEDOT, ACS Appl. Polym. Mater., 2019, 1, 83–94 Search PubMed.
- C. Cobet, et al., Influence of molecular designs on polaronic and vibrational transitions in a conjugated push-pull copolymer, Sci. Rep., 2016, 6, 35096 CrossRef PubMed.
- S. Hirabayashi, F. Ito and K. M. T. Yamada, Infrared spectra of the (H2O)n–SO2 complexes in argon matrices, J. Chem. Phys., 2006, 125, 034508 Search PubMed.
- J. P. Misiewicz, K. B. Moore and P. R. Franke, Sulfurous and sulfonic acids: Predicting the infrared spectrum and setting the surface straight, J. Chem. Phys., 2020, 152, 024302 Search PubMed.
- I. N. Stranski and L. Krastanow, Zur Theorie der orientierten Ausscheidung von Ionenkristallen aufeinander, Monatsh. Chem., 1937, 71, 351–364 Search PubMed.
- M. Heydari Gharahcheshmeh, M. T. Robinson, E. F. Gleason and K. K. Gleason, Optimizing the Optoelectronic Properties of Face-On Oriented Poly(3,4-Ethylenedioxythiophene) via Water-Assisted Oxidative Chemical Vapor Deposition, Adv. Funct. Mater., 2021, 31, 2008712 CrossRef CAS.
- F. Wudl, M. Kobayashi and A. J. Heeger, Poly(isothianaphthene), J. Org. Chem., 1984, 49, 3382–3384 Search PubMed.
- M. Heydari Gharahcheshmeh, et al., Tuning, optimization, and perovskite solar cell device integration of ultrathin poly(3,4-ethylene dioxythiophene) films via a single-step all-dry process, Sci. Adv., 2019, 5, eaay0414 CrossRef PubMed.
- D. Farka, M. Ciganek, D. Veselý, L. Kalina and J. Krajčovič, Epitaxial Guidance of Adamantyl-Substituted Polythiophenes by Self-Assembled Monolayers, ACS Omega, 2024, 9, 38733–38742 Search PubMed.
- K. Srivastava, T. Chakraborty, H. B. Singh and R. J. Butcher, Intramolecularly coordinated azobenzene selenium derivatives: Effect of strength of the Se⋯N intramolecular interaction on luminescence, Dalton Trans., 2011, 40, 4489 RSC.
- K. Kříž and J. Řezáč, Non-covalent interactions atlas benchmark data sets 4: σ-hole interactions, Phys. Chem. Chem. Phys., 2022, 24, 14794–14804 Search PubMed.
- K. Kříž, M. Nováček and J. Řezáč, Non-Covalent Interactions Atlas Benchmark Data Sets 3: Repulsive Contacts, J. Chem. Theory Comput., 2021, 17, 1548–1561 Search PubMed.
- P. D. Homyak, J. Tinkham, P. M. Lahti and E. B. Coughlin, Thieno[3,4-b]thiophene Acceptors with Alkyl, Aryl, Perfluoroalkyl, and Perfluorophenyl Pendants for Donor–Acceptor Low Bandgap Polymers, Macromolecules, 2013, 46, 8873–8881 Search PubMed.
- J.-L. Bredas, Mind the gap!, Mater. Horiz., 2014, 1, 17–19 RSC.
- N. Massonnet, A. Carella, A. de Geyer, J. Faure-Vincent and J.-P. Simonato, Metallic behaviour of acid doped highly conductive polymers, Chem. Sci., 2015, 6, 412–417 RSC.
- X. Wang, et al., High electrical conductivity and carrier mobility in oCVD PEDOT thin films by engineered crystallization and acid treatment, Sci. Adv., 2018, 4, eaat5780 Search PubMed.
- D. Farka, M. Scharber, E. D. Głowacki and N. S. Sariciftci, Reversible Photochemical Isomerization of N, N′-Di( t -butoxycarbonyl)indigos, J. Phys. Chem. A, 2015, 119, 3563–3568 CrossRef CAS PubMed.
- A. E. Mansour, et al., Conductive Polymer Work Function Changes due to Residual Water: Impact of Temperature-Dependent Dielectric Constant, Adv. Electron. Mater., 2020, 6, 2000408 Search PubMed.
- R. Noriega, et al., A general relationship between disorder, aggregation and charge transport in conjugated polymers, Nat. Mater., 2013, 12, 1038–1044 Search PubMed.
- J. F. Coker, et al., Perpendicular crossing chains enable high mobility in a noncrystalline conjugated polymer, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2403879121 Search PubMed.
- A. Olejnik, B. Dec, W. A. Goddard and R. Bogdanowicz, Hopping or Tunneling? Tailoring the Electron Transport Mechanisms through Hydrogen Bonding Geometry in the Boron-Doped Diamond Molecular Junctions, J. Phys. Chem. Lett., 2022, 13, 7972–7979 CrossRef CAS PubMed.
- V. V. Walatka, M. M. Labes and J. H. Perlstein, Polysulfur Nitride—a One-Dimensional Chain with a Metallic Ground State, Phys. Rev. Lett., 1973, 31, 1139–1142 Search PubMed.
- M. M. Labes, P. Love and L. F. Nichols, Polysulfur nitride - a metallic, superconducting polymer, Chem. Rev., 1979, 79, 1–15 CrossRef CAS.
- N. Velasquez, et al., X-ray induced ultrafast charge transfer in thiophene-based conjugated polymers controlled by core-hole clock spectroscopy, Phys. Chem. Chem. Phys., 2024, 26, 1234–1244 Search PubMed.
- V. Coropceanu, et al., Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 Search PubMed.
- S. D. Baranovskii, Theoretical description of charge transport in disordered organic semiconductors: Charge transport in disordered organic semiconductors, Phys. Status Solidi B, 2014, 251, 487–525 CrossRef CAS.
- G. Bergmann, Physical interpretation of weak localization: A time-of-flight experiment with conduction electrons, Phys. Rev. B: Condens. Matter Mater. Phys, 1983, 28, 2914–2920 Search PubMed.
- G. Bergmann, Localization in thin films — a time-of-flight-experiment with conduction electrons, Physica B+C, 1984, 126, 229–234 CrossRef CAS.
- J. Drnec, O. Irrazabal Moreda, M. Mirolo, M. Ronovsky and P. Sallés Perramon, Ex situ samples, European Synchrotron Radiation Facility, 2027, DOI:10.15151/ESRF-ES-1525926441.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01145k |
| ‡ D. F. would like to dedicate this work to his grandfather, polymer chemist Herman Ernst Schenk, who left this world while this manuscript was being prepared. |
| This journal is © The Royal Society of Chemistry 2025 |