
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-performance hydrogen evolution reaction by ReS2/TiO2 hollow microcones created through microwave-hydrothermal consecutive synthesis†
Yoonsun
Choi‡
,
Geonwoo
Kim‡
,
Dongbeom
Kim
,
Kyu-Su
Kim
,
Junhyeok
No
,
In-Ho
Baek
,
Sang-Mun
Jung
 *,
Yong-Tae
Kim
* and
Unyong
Jeong
*
*,
Yong-Tae
Kim
* and
Unyong
Jeong
*
Department of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), Cheongam-ro 77, Namgu, Pohang, 37673, Korea. E-mail: ujeong@postech.ac.kr; yongtae@postech.ac.kr; sangmun0104@postech.ac.kr
First published on 12th March 2025
Abstract
There is a need to develop a high-performance and low-cost hydrogen evolution reaction (HER) catalyst without noble metal. Transition metal dichalcogenides are some of the most promising catalytic materials, and rhenium disulfide (ReS2) emerges as a favorable material due to its unique structural properties. However, ReS2 is still lacking in research for effective catalytic performance; especially, increasing the electrochemical surface area has not been investigated. Herein, we present a 3D-structured ReS2/TiO2 catalyst with a high electrochemically active surface area (ECSA) obtained through a simple two-step process: (i) formation of TiO2 hollow microcones by microwave treatment and ReS2−x coating and (ii) ReS2 nanowall formation by a hydrothermal process. The microconical structure of the catalyst obtained via the two-step process significantly improved the HER activity of ReS2, exhibiting a low overpotential (61 mV), and a large ECSA (3652 cm2) with negligible degradation after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles of potential scanning and for 72 h of chronopotentiometric measurements at a current density of 500 mA cm−2, which is a superior result compared to previously reported ReS2-based HER catalysts.
000 cycles of potential scanning and for 72 h of chronopotentiometric measurements at a current density of 500 mA cm−2, which is a superior result compared to previously reported ReS2-based HER catalysts.
Introduction
Hydrogen is a promising energy source in consideration of energy depletion and global warming due to its high energy efficiency and eco-friendly product after use.1 Green hydrogen evolution reaction (HER) based on water electrolysis has been considered as a clean solution to environmental problems.2 As HER consumes a large amount of electrical energy and uses only the surfaces of the catalysts, synthesis of efficient and noble metal-free catalysts is essential for practical hydrogen production.3–5 Transition metal dichalcogenides (TMDCs) have received much attention as HER catalysts due to the high surface area resulting from the layered structure and their earth-abundance.6–10 Rhenium disulfide (ReS2), as an emerging TMDC member, is a candidate material for HER on account of its unique chemical and electronic properties: weak interlayer coupling,11 stable distorted 1T-phase unlike other TMDCs,12 low ion diffusion barrier,13 and large active site exposure.14 There have been studies on controlling the HER performance of ReS2, by regulating the internal structures through vacancy-engineering,15 formation of grain boundaries,16 and designing superlattices. Zhao et al. compared HER catalytic performances while varying the edge density and the length of pure ReS2 under various synthetic conditions.17 Nonetheless, pure ReS2 still did not reach performance levels comparable to those of noble metals. Recently, doping and decoration of ReS2 with heterogeneous elements have been explored to improve the HER performance. Kim et al. presented a Pt/ReS2 hybrid HER catalyst, in which Pt nanoclusters were impregnated in ReS2 nanoflowers.18 They reported an excellent overpotential (20 mV at 10 mA cm−2) and a high electrochemical surface area (4.07 cm2). Liu et al. doped fluorine in ReS2 through post-fluorination.19 They found that F dopants accelerated the charge transfer and increased the active sites, resulting in a low overpotential (142 mV at 10 mA cm−2) and a large electrochemical active surface area (ECSA) of 400 cm2. ReS2 without containing noble metals deserves more extensive studies to achieve high HER performance.Titanium dioxide (TiO2) has been synthesized in various forms20–24 and is widely used as an energy-related electrode, such as in electrocatalysis (hydrogen evolution, oxygen evolution, and oxygen reduction),25–28 photocatalysis,29 lithium-ion batteries,30 energy conversion,31 and energy storage.32 In water electrolysis, the 3D TiO2 structures have been explored as a supporting body, which expands the surface area to improve the catalytic activity. Nong et al. proposed a mesoporous TiO2 crystal by etching SrTiO3 under hydrothermal conditions, and doped it with well-dispersed Ru for HER catalytic application.21 Chen et al. also prepared a porous TiO2 nanotube array through anodic oxidation and introduced Pt nanoclusters on the array by atomic layer deposition to improve the HER performance.24 Although the composites of 3D-structured TiO2 and noble metal catalysts have made remarkable advances in performance and durability, preparation of 3D-structured TiO2 has limitations in terms of time, cost, and methodological complexity. In addition, there have been few studies that improved the HER performance only with TiO2 and non-noble metals.
In this paper, we report a unique 3D-structured, noble metal-free, high-performance ReS2 HER catalyst. The catalyst is produced on a Ti fiber felt, creating ReS2 nanowalls on TiO2 hollow microcones generated in situ on the Ti felt. We suggest a two-step synthesis: (i) microwave-assisted 3D structuring of TiO2 and ReS2 coating on the surface and (ii) hydrothermal refining of the ReS2 nanowalls to significantly improve the HER performance such as a low overpotential (61 mV at 10 mA cm−2), a high ECSA (3652 cm2), excellent cyclic durability (no degradation after 10000 cycles), and good potential retention (for 72 h at a current density of 500 mA cm−2). The mechanism of microcone formation, the chemistry of the crystal structures, and long-term catalytic performances are thoroughly investigated.
Results and discussion
Formation of the ReS2/TiO2 hollow microcones
The two-step synthesis of the hollow ReS2/TiO2 microcones is depicted in Fig. 1a. Ammonium perrhenate (NH4ReO4) and thiourea (SC(NH)2) were used as a Re precursor and as a S precursor, respectively. The precursors were dissolved in an ionic material (BMIM:BF4), which acted both as a solvent and as a microwave (MW) absorber. Ti fiber felt was selected as a Ti source and a self-supported electrode. When MW was applied to the Ti felt immersed in the precursor solution for 2 min, TiO2 microcones were created on the surface of the Ti felt, and ReS2−x nanoparticles were coated onto the TiO2 microcones. The hydrothermal (HT) process maintained the microcone structure and generated stoichiometric ReS2 nanowalls. Scanning electron microscopy (SEM) images in Fig. 1b–d exhibit the surface morphologies of (b) the bare Ti felt, (c) ReS2 (MW), ReS2−x/TiO2 created by the MW process, and (d) ReS2 (MW-HT), ReS2/TiO2 created by the MW-HT consecutive process. The bare Ti felt did not have any protruding structures on the surface. The average bottom diameter of the microcones of the ReS2 (MW) specimen was about 5 μm. Based on energy-dispersive X-ray spectroscopy (EDS), the average stoichiometry of ReS2 (MW) was determined to be ReS1.62 (Table S1†), whereas it was stoichiometric (ReS1.97) in the ReS2 (MW-HT) specimen. The nanowall structure of ReS2 became prominent after the HT process (Fig. 1d). | ||
| Fig. 1 Morphological evolution of the ReS2/TiO2 microcones. (a) Illustration of the ReS2/TiO2 microcones with ReS2 nanowalls on the surfaces. The structure was synthesized by consecutive processes of microwave (MW) treatment and hydrothermal synthesis. The insets magnify the top of the microcones corresponding to the processes. FE-SEM images of bare Ti felt (b), ReS2−x/TiO2 (c), and ReS2/TiO2 (d). | ||
MW treatment can electromagnetically agitate ionic species. For instance, during the synthesis of ReS2, the Re precursor was ionized to form NH4+ and ReO4−. These ions, together with the ionic material (BMIM:BF4), could absorb MW and cause volumetric heating,33 which is essential for rapid synthesis. Because the solubility and degree of ionization of the precursor depend on ionic molecules, proper combination of the precursors and ionic molecules is crucial for the MW-assisted synthesis. Various materials (ionic molecules, metal precursors) were explored to find out the appropriate combinations. Specifically, BMIM:BF4 actively generated the TiO2 microcones (Fig. S1a†); thus we used it in this study. Presence or absence of SC(NH)2 did not affect the formation of the microcones, whereas the transition metal precursors affected it considerably. The microcones could be formed using various Re and Mo precursors (NH4ReO4, ReCl5, (NH4)2MoO4, MoO2(acac)2, MoCl5, and (NH4)2MoS4) (Fig. S1b†). However, the microcones were not formed by using other transition metal precursors (WCl4, ZrCl4, NbCl5, FeCl2·4H2O, CoCl3, and CrCl3) (Table S2†), and no 3D structures were found on the Ti felt surface, which is because they have a low decomposition degree under MW heating in BMIM:BF4.
To confirm the decomposition degree and reactivity of each precursor, the amount of metal oxides formed from the Re, Mo, and W precursors in the ionic solution by the MW process, in the absence of SC(NH)2, was compared. The W precursor was selected as a representative precursor with a low reactivity. After 2 min of MW reaction in the ionic solution, metal oxide nanoparticles were deposited on the Ti felt, and the remaining amount was dispersed in the reaction solution (Fig. S2†). The dispersed metal oxide nanoparticles in the solution were centrifuged and washed with pure ethanol twice. The collected nanoparticles were dispersed again in fresh ethanol (5 mL). Inductively coupled plasma mass spectroscopy (ICP-MS) was conducted with 5 mL of this dispersion to quantify the amount of metal ions in the dispersion. The amounts of ions of Re (14.1 μmol) and Mo (50.6 μmol) were more than 10 times greater than that of W ions (1.9 μmol) (Table S3†). This result obviously indicates that Re and Mo precursors could be decomposed actively during the MW process, unlike other metal precursors such as the W precursor. In addition, as Re was better deposited on the Ti felt than Mo, the amount of Re remaining in the solution was less than that of Mo. This is why the microcones synthesized using the Re precursor were larger and denser than when Mo precursor was the reactant.
Characterization of the ReS2/TiO2 hollow microcones
Fig. 2a–d characterize the microcones peeled off from the Ti felt of ReS2 (MW-HT). The inside of the microcone was hollow (Fig. 2a). The elemental mapping (Fig. 2b) and the SEM image (Fig. S3†) reveal that the inside surface was coated with ReS2 as well as the outside surface. The transmission electron microscopy (TEM) image in Fig. 2c shows the ReS2 nanowalls grown on TiO2 which were collected by ultrasonication for 1 h. The high-resolution TEM (HR-TEM) image in Fig. 2d shows the 0.62 nm lattice spacings and 0.34 nm spacing which correspond to the (001) plane of ReS2 and the anatase TiO2 (101) plane. | ||
| Fig. 2 Characterization of the ReS2/TiO2 microcones obtained by the MW-HT consecutive process. (a) SEM image and (b) elemental mappings of the ReS2/TiO2 microcone. (c) TEM and (d) HR-TEM images taken from ReS2/TiO2. The spacings of ReS2 (001) and anatase TiO2 (101) are indicated. (e) Comparison of XRD patterns and (f) Raman spectra of the ReS2 (MW) specimen and ReS2 (MW-HT) specimen (powder ReS2 was a reference, and bare Ti was the background). | ||
X-ray diffraction (XRD) patterns of the bare Ti felt, ReS2 (MW) specimen, and ReS2 (MW-HT) specimen are exhibited in Fig. 2e. The peaks corresponding to anatase TiO2 are marked with solid triangles. New peaks were generated at 25.4° and 47.8° after the MW process and they were maintained after the MW-HT process, indicating new generation of anatase TiO2 on the Ti felt surfaces by MW and maintenance of the crystals during the HT process. The peak of the ReS2 nanowalls was not observed due to the relatively small amount compared to Ti and TiO2. Fig. 2f shows the Raman spectra of the bare Ti felt, ReS2 (MW) specimen, and ReS2 (MW-HT) specimen. Raman peaks from pure ReS2 crystalline powder are compared, denoted with hollow triangles. For the pure ReS2, the peaks at 148 cm−1 and 204 cm−1 are assigned to the in-plane (Eg) and out-of-plane (Ag-like) vibration modes of ReS2, respectively. The ReS2 peaks appeared after the MW process, and they became more prominent after the HT process, implying that the crystal structure of ReS2 was refined by the HT process. The anatase TiO2 peaks are marked with solid triangles. The spectrum of bare Ti did not have peaks corresponding to crystalline TiO2. After the MW process, the TiO2 peaks appeared and remained the same after the HT process.
X-ray photoelectron spectroscopy (XPS) also confirmed the formation of ReS2. Fig. 3a and b compare the Re 4f XPS spectra of ReS2 obtained only by HT (Fig. 3a) and by the MW-HT two-step process (Fig. 3b). The ReS2 (HT) specimen contained 32% oxidation states of Re in the Re2O7 phase. In contrast, the ReS2 (MW-HT) specimen showed a negligible fraction of oxidized Re, which indicates that the ReS2 phase created an interface with the TiO2 support layer without the presence of an oxidized Re layer. On the basis of the results, ReS2 is considered as the active site for HER, while TiO2 is a material serving as a support. The HER mechanism for ReS2 has been well investigated already in many theoretical studies.34–36 The activity of the catalyst prepared in this study can be characterized according to the reported theoretical results.
 | ||
| Fig. 3 Difference in the structural evolution when only the HT process was applied and when the MW-HT consecutive process was applied. Re 4f XPS spectra of the ReS2 (HT) (a) and ReS2 (MW-HT) (b) specimens. (c) XANES Re L3-edge spectra of ReS2 (MW-HT) and ReS2 (HT) (ReS2 powder and Re foil were references). Illustration for the competitive oxidation and sulfurization for ReS2 (HT) (d) and ReS2 (MW-HT) (e) specimens. | ||
We analysed X-ray absorption near edge structure (XANES) spectra to confirm the changes in the local electronic structure of the ReS2 (MW-HT) catalyst (Fig. 3c). Typically, the white line intensity or area of XANES spectra for platinum group metal (PGM) correlates with the oxidation state of a metal, where higher intensity or area indicates a higher oxidation state.18,37 The Re L3-edge spectra in Fig. 3c reveal that the white line in the Re L3-edge adsorption edge for ReS2 (MW-HT) catalyst decreased compared to the ReS2 (HT) catalyst, indicating lower oxidation state in the ReS2 (MW-HT) catalyst. The XANES results were consistent with the XPS results.
The ReS2 growth mechanisms are illustrated for the HT process on Ti (Fig. 3d) and for the MW-HT process on the TiO2 microcones (Fig. 3e). In the HT process (12 h) without prior surface coating with ReS2−x, the S elements decomposed from the precursor should compete with oxygen for the reaction with Re; therefore, Re2O7 could exist to a significant degree. Conversely, in the MW-HT consecutive process, as the ReS2−x coating layer on the TiO2 surfaces provided plenty of Re–S bonds, bond breaking by oxygen to form oxides is not relevant during the later HT step. Thus, there were no Re oxides on the TiO2 microcones. Because Re oxides interfere with the catalytic reaction on the surface of ReS2, prevention of Re oxidation is critical for HER performance.38
Formation mechanism of the ReS2/TiO2 hollow microcones
SEM images in Fig. 4a exhibit the temporal evolution of the ReS2−x/TiO2 microcones during the MW process. The evolution of the conical structure is schematically described in Fig. 4b. Two events occurred in parallel during the MW process: (i) formation of a thick TiO2 layer (∼500 nm) by oxidation of Ti and (ii) deposition of Re on the surface of the TiO2 layer. Delamination occurs along the cracks in the TiO2 layer or at the inherent defects on the surface of the commercial Ti fiber felt. The conical structures with a diameter of 1 μm were formed in 45 s of the MW delamination and the ReS2−x layer covered the outer surface of the TiO2 microcones within 60 s, which was confirmed through the SEM study (Fig. S4†). The top of each microcone began to be perforated as the size of the microcone increased. The bottom dimension of the microcone reached 5 μm in 90 s of MW treatment and was saturated. All the microcones were perforated within 120 s of MW treatment. The top opening of the microcones allowed deposition of ReS2−x on their interior surfaces. In addition, the opening enabled formation of new TiO2 on the Ti felt area corresponding to the bottom of the microcones, on which ReS2−x could be further deposited. The microcones began to be peeled off from the Ti felt in 150 s of MW treatment and large fractions were peeled off at 180 s (Fig. S5†). Therefore, we set the time of the MW process at 120 s to prevent loss of catalyst. The same vertical structure was also obtained on a Ti foil by applying the same MW process (Fig. S6†).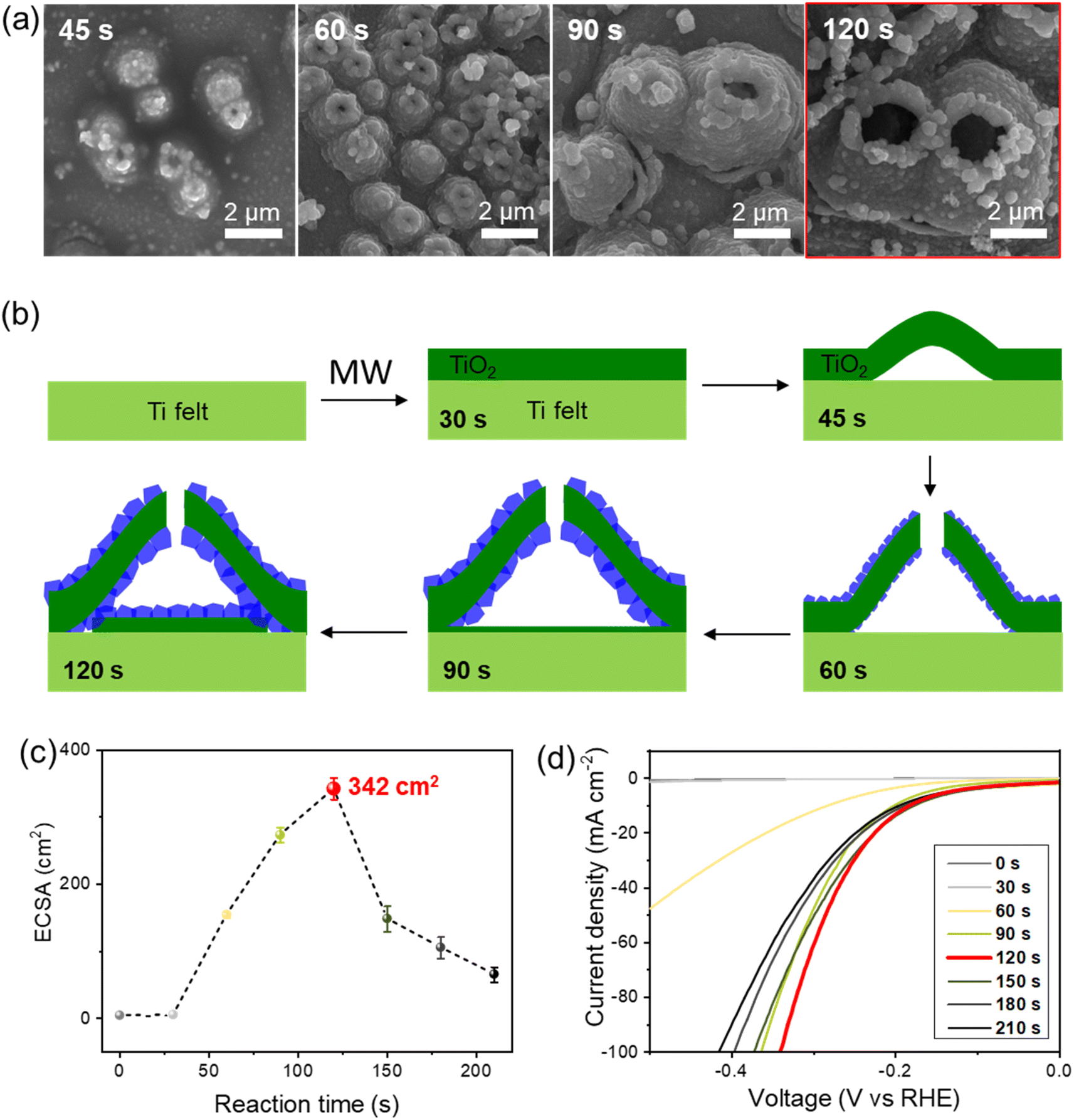 | ||
| Fig. 4 Temporal process of microcone formation and the catalytic performance of ReS2 (MW) depending on the MW reaction time. (a) SEM images at each growth stage. (b) Schematic description for the growth stages of the ReS2 (MW) during the MW process. Changes of the ECSA (c) and the LSV curves (d) depending on the MW reaction time (from 0 s to 120 s). | ||
The unique 3D morphology resulted in gradual enhancement of ECSA according to the MW process time from 3.4 cm2 (0 s) to 4.5 cm2 (30 s), 154 cm2 (60 s), 273 cm2 (90 s), and 342 cm2 (120 s) (Fig. 4c). Because the microcones were delaminated from the Ti felt by further MW process, the ECSA curve showed a decreasing trend with MW time, for instance 148 cm2 in 150 s. In line with the ECSA trend, the HER activity showed the best performance at 120 s of MW treatment, with the lowest overpotential (η = 181 mV) at 10 mA cm−2 current density (Fig. 4d).
Electrocatalytic HER performance of the catalysts
The HER performance of the ReS2 (MW-HT) catalyst was measured using a three-electrode system in H2-saturated 0.1 M HClO4 electrolyte at room temperature. The potential associated with zero current was recorded as the potential of Ag/AgCl (Fig. S7†), and then all electrode potentials were noted against the reversible hydrogen electrode (RHE).39 The HER polarization curves were acquired using linear sweep voltammetry (LSV). The fifth scan was chosen for the LSV curve in each sample, and all the acquired data were calibrated with a RHE. The ReS2 (MW) catalyst showed a relatively poor catalytic performance (η = 181 mV at 10 mA cm−2) (Fig. 5a). The ReS2 (HT) catalyst showed slightly better performance (η = 140 mV at 10 mA cm−2), but still much lower than the performance of a reported Pt/C catalyst (η = 30 mV at 10 mA cm−2).18 The ReS2 (MW-HT) catalyst showed a remarkable improvement of the HER performance, reaching 61 mV at 10 mA cm−2. | ||
| Fig. 5 HER performance of the catalysts. (a) LSV polarization curves, (b) ECSA calculated from the double-layer capacitance, and (c) Nyquist plots. (d) Cyclic voltammetry cycles and chronopotentiometry. (e) HER performance comparison with previously reported Pt-free ReS2-based catalysts in acidic media. | ||
We measured the double-layer capacitance in cyclic voltammograms and compared the calculated ECSA (Fig. 5b). The ECSA of ReS2 (MW-HT) was 3652 cm2, which was in contrast to the values of bare Ti (3.4 cm2), ReS2 (MW) (342 cm2), and ReS2 (HT) (1302 cm2). The electrocatalytic properties of the same specimens were characterized by electrochemical impedance spectroscopy (EIS) analysis (Fig. 5c). The diameter of the semicircles in the Nyquist plot represents the charge-transfer resistance (Rct), which has a significant effect on the reaction kinetics. The value of Rct for ReS2 (MW-HT) was lowest (Rct = 7 Ω) at η = 100 mV, compared to ReS2 (HT) (Rct = 17 Ω), ReS2 (MW) (Rct = 63 Ω), and bare Ti (Rct = 1500 Ω). Quantitative HER kinetics was compared with the Tafel slope (Fig. S8†). The Tafel slope of ReS2 (MW-HT) was 153 mV dec−1, which was lower than that of ReS2 (HT) (160 mV dec−1) and ReS2 (MW) (185 mV dec−1). These results indicate that the superior HER activity of ReS2 (MW-HT) is attributed to the high ECSA, fast charge-transfer rate and reaction kinetics.
Long-term durability is a crucial performance indicator for water electrolysis (Fig. 5d). We conducted an accelerated degradation test (ADT) for the ReS2 (MW-HT) catalyst. After 10000 voltammetry cycles in the range of −0.2 to 0.1 V (vs. RHE), the catalyst showed a negligible negative shift (≤2 mV at 10 mA cm−2). Furthermore, the ReS2 (MW-HT) catalyst showed excellent HER stability for 72 h at a high current density of 500 mA cm−2, with a negligible activity loss. The amount of dissolved Re in the electrolyte, analysed by ICP-MS, was approximately 44 ppb, indicating a negligible level. XPS analysis further confirmed the high HER stability of ReS2 (MW-HT) (Fig. S9†), displaying no changes in the chemical state after the chronopotentiometry. The increased intensity of the H–O peak in the O 1s spectrum was attributed to the water-based nature of the HER process.
The ReS2 (MW-HT) catalyst investigated in this study exhibited superior HER performance compared with that of previously reported ReS2-based Pt-free electrodes in acid media. Fig. 5e and Table S4† compare the performances with literature values.10,17,19,40–42 The performances of the ReS2 (MW-HT) catalyst were comparable with the recent values obtained for ReS2-based HER catalysts containing Pt in acidic media (Table S5†). On the basis of the results, we can conclude that our catalytic electrode had a low overpotential and a high ECSA in addition to excellent stability.
Experimental
Chemicals
All the chemicals were of analytical grade and used as received without further purification. Ammonium perrhenate (NH4ReO4), thiourea (CH4N2S), hydroxylamine hydrochloride (NH2OH·HCl), 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM:BF4), and oxalic acid ((COOH)2·2H2O) were purchased from Sigma-Aldrich. Ethanol was purchased from Daejung Chemicals, Inc. (Korea). Ti fiber felt was purchased from Bekaert.Synthesis of ReS2 powder
The ReS2 powder was synthesized using the microwave-irradiation method.18 16.8 mg of ammonium perrhenate (NH4ReO4) and 23.8 mg of thiourea (CH4N2S) were first dissolved in 2.5 mL of BMIM:BF4 at room temperature. After being stirred for 4 h, the solution was transferred to a 20 mL quartz vial. Subsequently, the solution was heated in a domestic microwave oven at 200 W for 2 min and then allowed to cool to room temperature naturally. The product was washed with water and then ethanol three times using centrifugation. The ReS2 powder was dried at 60 °C for 6 h.Synthesis of ReS2 (MW)
Prior to synthesis, the Ti fiber felt substrate was acid-treated with 5 wt% oxalic acid solution at 80 °C for 30 min to remove surface oxide and contaminants.43 16.8 mg of ammonium perrhenate (NH4ReO4) and 23.8 mg of thiourea (CH4N2S) were first dissolved in 2.5 mL of BMIM:BF4 at room temperature. After being stirred for 4 h, the solution was transferred to a 20 mL quartz vial, and a 1 × 1 cm2 part of 1 × 3 cm2 Ti felt was immersed into the solution. Subsequently, the solution was heated in a domestic microwave oven at 200 W for 2 min and then allowed to cool to room temperature naturally. The felt was washed with ethanol and then water three times each with vigorous shaking. The ReS2−x/TiO2 on the Ti felt was dried at room temperature for 2 h.Synthesis of ReS2 (HT)
134 mg of ammonium perrhenate (NH4ReO4), 190 mg of thiourea (CH4N2S) and 104 mg of hydroxylamine hydrochloride (NH2OH·HCl) were dissolved in 20 mL of deionized water under magnetic stirring at room temperature for 30 min. The aqueous solution was transferred to a 50 mL Teflon-lined autoclave, and a 1 × 1 cm2 part of 1 × 3 cm2 Ti felt was immersed into the solution. The autoclave was sealed and maintained at 220 °C for 12 h. After cooling down to room temperature, the felt was washed with ethanol and then water three times each with vigorous shaking and then dried at room temperature for 2 h.Synthesis of ReS2 (MW-HT)
134 mg of ammonium perrhenate (NH4ReO4), 190 mg of thiourea (CH4N2S) and 104 mg of hydroxylamine hydrochloride (NH2OH·HCl) were dissolved in 20 mL of deionized water under magnetic stirring at room temperature for 30 min. The aqueous solution was transferred to a 50 mL Teflon-lined autoclave, and a 1 × 1 cm2 part of 1 × 3 cm2 ReS2 (MW) was immersed into the solution. The autoclave was sealed and maintained at 220 °C for 12 h. After cooling down to room temperature, the felt was washed with ethanol and then water three times each with vigorous shaking and then dried at room temperature for 2 h.Structural characterization
The morphology of the synthesized catalyst was observed using field-emission SEM (Hitachi S-4800), TEM (JEOL JEM-2100F) at 200 kV, and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM; JEOL JEM-2100F with Cs Corrector) at 200 kV. Powder XRD (Rigaku D/MAX-2500/PC) was performed with Cu Kα radiation (λ = 1.54 Å). The Raman spectrum was obtained using a micro-Raman spectrometer (Witec Alpha 300 RA Confocal Raman) with a wavelength of 532 nm and a laser spot size of 1 μm. The amounts of Re and S were measured using the SEM instrument equipped with an EDS facility (INCA X-sight 7421, Oxford Instruments). The XANES data at the Re k-edge were obtained at the 7D beamline in the PAL with fluorescence mode. The X-ray beam was monochromated by a Si (111) double crystal, where the beam intensity was reduced by 30% to eliminate higher-order harmonics.ICP-MS characterization
The metal ion concentration was measured using ICP-MS (PerkinElmer, NexION 300S). To ensure analysis reliability, calibration curves were obtained by linear fitting of the raw intensities of each element at 1, 5, and 10 ppb before the ion concentration measurements. The electrodes were fully dissolved in aqua regia using an ultrasonic bath, and the ion concentration measurements were conducted by diluting the solution to fit within the calibration curves.Electrochemical measurements
All the electrochemical characterizations were performed using an electrochemical workstation (Metrohm Autolab, PGSTAT204). Electrochemical measurements were conducted using a standard three-electrode electrochemical system in 0.1 M HClO4 (Sigma-Aldrich) solution at room temperature. The prepared samples, a graphite rod, and an Ag/AgCl (sat. KCl) were used as the working, counter, and reference electrodes, respectively. The graphite rod was used as the counter electrode to prevent undesirable back reactions and cross-contamination by trace Pt metal species.44 The calibration of the Ag/AgCl reference electrode was conducted in high-purity (99.999%) H2-saturated 0.1 M HClO4 with a polycrystalline Pt pellet as working electrode.39,45 Linear sweep voltammetry curves were obtained with sweeping the potential between hydrogen evolution (−100 mV) and oxidation (+100 mV) regions with a scan rate of 1 mV s−1, at room temperature. The Tafel plots were replotted to the HER polarization curves as overpotential (η) versus log current density (logj) The polarization curves for HER were obtained using H2-saturated 0.1 M HClO4 at a scan rate of 10 mV s−1 and the potential was iR-corrected with workstation-assisted iR drop compensation (iR = 2 Ω). EIS measurements were performed at −0.02 V vs. RHE, using a sinusoidal wave with a frequency scan range from 105 to 10−1 Hz and an amplitude of 10 mV. The measured EIS data were analyzed with the NOVA software package. The ECSACdl values of the electrodes were calculated by the potential-sweep method in the electrochemical double-layer region.46,47 The capacitance was measured via cyclic voltammetry in a non-faradaic potential range of 0.15 to 0.3 V (vs. RHE), using 5 to 400 mV s−1. An accelerated durability test was conducted with an extended potential cycling test (10000 cycles) in the range of −0.2 V to 0.1 V (vs. RHE) with a scan rate of 50 mV s−1. Chronopotentiometry measurements were also performed with a current density of 500 mA cm−2 for 72 h.
Conclusions
We have synthesized ReS2/TiO2 composites directly on Ti felt with an ultra-high electrochemical surface area by employing two sequential processes: rapid microwave irradiation and a well-established hydrothermal method. Microconical TiO2 structures with nonstoichiometric ReS2−x fully covering the surface were grown during 2 min of microwave irradiation. Subsequently, the deficient sulfur was replenished to convert ReS2−x to ReS2 through sequential 12 h hydrothermal synthesis. We could identify the growth mechanism of the catalytic materials on 3D TiO2 through microwave treatment. The optimized ReS2 (MW-HT) showed HER catalytic activity with a low overpotential of 61 mV, Rct of 7 Ω, robust durability (showing negligible activity loss after 10000 cycles of cyclic voltammetry and over 72 h at 500 mA cm−2), and an ultra-high electrochemical surface area of 3652 cm2.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
U. J. and Y.-T. K. conceived the original idea, supervised the work. U. J., Y.-T. K., and S.-M. J. edited the final manuscript. S.-M. J. and I.-H. B. conducted electrochemical measurements. Y. C. and G. K. performed the catalyst synthesis, conducted structural characterizations, and wrote the initial draft. D. K. and J. N. analysed the measured data. K.-S. K. conducted ICP-MS measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2024-00338686, No. NRF-2022M3H4A1A02074314, No. 2019M3D1A1079306, and No. RS-2025-00554594).References
- M. A. Rosen and S. Koohi-Fayegh, Energy Ecol. Environ., 2016, 1, 10–29 CrossRef.
- S.-Y. Ahn, K.-J. Kim, B.-J. Kim, G.-R. Hong, W.-J. Jang, J. W. Bae, Y.-K. Park, B.-H. Jeon and H.-S. Roh, Renewable Sustainable Energy Rev., 2023, 186, 113635 CrossRef.
- J. Kim, H. Jung, S.-M. Jung, J. Hwang, D. Y. Kim, N. Lee, K.-S. Kim, H. Kwon, Y.-T. Kim and J. W. Han, J. Am. Chem. Soc., 2020, 143, 1399–1408 CrossRef PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Cao, ACS Nano, 2021, 15, 11014–11039 CrossRef.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef PubMed.
- I. S. Amiinu, Z. Pu, X. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. Zhang and S. Mu, Adv. Funct. Mater., 2017, 27, 1702300 CrossRef.
- X. Wang, Y. Chen, B. Zheng, F. Qi, J. He, Q. Li, P. Li and W. Zhang, J. Alloys Compd., 2017, 691, 698–704 CrossRef.
- I. H. Kwak, T. T. Debela, I. S. Kwon, J. Seo, S. J. Yoo, J.-G. Kim, J.-P. Ahn, J. Park and H. S. Kang, J. Mater. Chem. A, 2020, 8, 25131–25141 RSC.
- S. Tongay, H. Sahin, C. Ko, A. Luce, W. Fan, K. Liu, J. Zhou, Y.-S. Huang, C.-H. Ho and J. Yan, Nat. Commun., 2014, 5, 3252 CrossRef PubMed.
- Q. Sun, B. Zhang, L. Diao, B. Chen, K. Song, L. Ma and F. He, J. Mater. Chem. A, 2020, 8, 11607–11616 RSC.
- Q. Zhang, S. Tan, R. G. Mendes, Z. Sun, Y. Chen, X. Kong, Y. Xue, M. H. Rümmeli, X. Wu and S. Chen, Adv. Mater., 2016, 28, 2616–2623 CrossRef PubMed.
- Q. Zhang and L. Fu, Chem, 2019, 5, 505–525 Search PubMed.
- Q. Q. Pang, Z. L. Niu, S. S. Yi, S. Zhang, Z. Y. Liu and X. Z. Yue, Small, 2020, 16, 2003007 Search PubMed.
- K. Wu, B. Chen, S. Yang, G. Wang, W. Kong, H. Cai, T. Aoki, E. Soignard, X. Marie and A. Yano, Nano Lett., 2016, 16, 5888–5894 CrossRef PubMed.
- Y. Zhao, J. Li, J. Huang, L. Feng, L. Cao, Y. Feng, Z. Zhang, Y. Xie and H. Wang, Adv. Mater. Interfaces, 2020, 7, 2001196 CrossRef.
- G. Kim, S.-M. Jung, A. Giri, J.-S. Kim, Y.-W. Kim, K.-S. Kim, D. Kim, Y. Choi, B.-J. Lee and Y.-T. Kim, J. Power Sources, 2023, 579, 233287 CrossRef.
- Y. Liu, H. Li, J. Li, X. Ma, Z. Cui, D. Gao and Z. Tang, J. Mater. Chem. A, 2021, 9, 14451–14458 RSC.
- A. Tahira, Z. H. Ibupoto, R. Mazzaro, S. You, V. Morandi, M. M. Natile, M. Vagin and A. Vomiero, ACS Appl. Energy Mater., 2019, 2, 2053–2062 CrossRef.
- S. Nong, W. Dong, J. Yin, B. Dong, Y. Lu, X. Yuan, X. Wang, K. Bu, M. Chen and S. Jiang, J. Am. Chem. Soc., 2018, 140, 5719–5727 CrossRef CAS PubMed.
- J. X. Feng, H. Xu, Y. T. Dong, X. F. Lu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2017, 56, 2960–2964 CrossRef CAS PubMed.
- D. C. Nguyen, T. L. L. Doan, S. Prabhakaran, D. T. Tran, D. H. Kim, J. H. Lee and N. H. Kim, Nano Energy, 2021, 82, 105750 CrossRef CAS.
- Y. Chen, Y. Wang, L. Zheng, Y. Chang, S. Xu, Y. Wu, W. Zhou, Y. Lu, J. Wang and H. Li, Mater. Today Energy, 2022, 27, 101042 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- M. Nie, H. Sun, J. Liao, Q. Li, Z. Xue, F. Xue, F. Liu, M. Wu, T. Gao and L. Teng, Int. J. Hydrogen Energy, 2021, 46, 6441–6447 CrossRef CAS.
- C. Park, E. Lee, G. Lee and Y. Tak, Appl. Catal., B, 2020, 268, 118414 CrossRef CAS.
- S.-H. You, S.-M. Jung, K.-S. Kim, J. Lee, J. Park, H. Y. Jang, S. Shin, H. Lee, S. Back and J. Lee, ACS Energy Lett., 2023, 8, 2201–2213 CrossRef CAS.
- J. Sun, L. Liu and F. Yang, Appl. Catal., B, 2022, 308, 121215 CrossRef CAS.
- J. Park, S. Kim, G. Lee and J. Choi, ACS Omega, 2018, 3, 10205–10210 CrossRef PubMed.
- Y. Xu and G. Zangari, Coatings, 2021, 11, 931 CrossRef.
- P. Si, Z. Zheng, Y. Gu, C. Geng, Z. Guo, J. Qin and W. Wen, Materials, 2023, 16, 3864 Search PubMed.
- R. R. Mishra and A. K. Sharma, Composites, Part A, 2016, 81, 78–97 Search PubMed.
- L. Wang, Z. Sofer, J. Luxa, D. Sedmidubský, A. Ambrosi and M. Pumera, Electrochem. Commun., 2016, 63, 39–43 CrossRef.
- Y. Zhou, E. Song, J. Zhou, J. Lin, R. Ma, Y. Wang, W. Qiu, R. Shen, K. Suenaga and Q. Liu, ACS Nano, 2018, 12, 4486–4493 CrossRef PubMed.
- M. Aparna and R. Chatanathodi, ACS Appl. Nano Mater., 2022, 5, 2385–2394 CrossRef.
- W. H. Lee, Y.-J. Ko, J. H. Kim, C. H. Choi, K. H. Chae, H. Kim, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Nat. Commun., 2021, 12, 4271 CrossRef PubMed.
- G.-Y. Kim, J. Lee, Y.-J. Rho, W.-H. Kim, M. Kim, J.-H. Ahn and W.-H. Ryu, Chem. Eng. J., 2022, 446, 136951 CrossRef.
- S. M. Jung, Y. Kim, B. J. Lee, H. Jung, J. Kwon, J. Lee, K. S. Kim, Y. W. Kim, K. J. Kim and H. S. Cho, Adv. Funct. Mater., 2024, 2316150 CrossRef.
- B. Martín-García, D. Spirito, S. Bellani, M. Prato, V. Romano, A. Polovitsyn, R. Brescia, R. Oropesa-Nuñez, L. Najafi and A. Ansaldo, Small, 2019, 15, 1904670 CrossRef PubMed.
- Y. Liu, J. Li, W. Huang, Y. Zhang, M. Wang, X. Gao, X. Wang, M. Jin, Z. Hou and G. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 33586–33594 CrossRef PubMed.
- C. Pei, M. C. Kim, Y. Li, C. Xia, J. Kim, W. So, X. Yu, H. S. Park and J. K. Kim, Adv. Funct. Mater., 2023, 33, 2210072 CrossRef.
- K.-R. Yeo, K.-S. Lee, H. Kim, J. Lee and S.-K. Kim, Energy Environ. Sci., 2022, 15, 3449–3461 RSC.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu and L. Zheng, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S.-A. Park, K.-S. Kim, H. D. Jung, S.-M. Jung, Y. H. Moon, S. Back and Y.-T. Kim, ACS Appl. Energy Mater., 2021, 4, 4173–4180 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01077b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |