
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Densification of cellulose acetate-derived porous carbons for enhanced volumetric hydrogen adsorption performance†
Hiroshi
Matsutaka
 *abc,
Aya
Kashifuku
ab,
Takaaki
Orii
a,
Daigo
Miyajima
ad,
Naoki
Uchiyama
abe,
Satoshi
Wada
b and
Hirotomo
Nishihara
cf
*abc,
Aya
Kashifuku
ab,
Takaaki
Orii
a,
Daigo
Miyajima
ad,
Naoki
Uchiyama
abe,
Satoshi
Wada
b and
Hirotomo
Nishihara
cf
aRIKEN Baton Zone Program, 2-1 Hirosawa, Wako, 351-0198, Japan
bRIKEN Center for Advanced Photonics, 2-1 Hirosawa, Wako, 351-0198, Japan. E-mail: hiroshi.matsutaka@riken.jp
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
dSchool of Science and Engineering, The Chinese University of Hong Kong, 2001 Longxiang Boulevard, Longgang District, Shenzhen, 518172, China
eAtsumitec Co., Ltd, 7111 Ubumi, Yuto-cho, Nishi-ku, Hamamatsu, 431-0192, Japan
fAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 13th June 2025
Abstract
There is a significant demand for hydrogen gas storage technology, particularly with high volumetric storage density, across various industries as an alternative to current high-pressure compression methods. In this study, cellulose acetate (CA) was used as a precursor to adjust the KOH activation conditions, synthesizing porous carbons with a high hydrogen adsorption capacity (approximately 2.8 wt% at −196 °C and 1 bar). By applying very high pressure to pelletize the porous carbon powder with a binder to a high density (0.8 g cm−3), a carbon pellet with a large volumetric hydrogen density was achieved (20.3 g-H2 per L at −196 °C and 1 bar). For comparison, MOF-derived carbon with a hydrogen adsorption capacity equivalent to that of CA-derived carbon was synthesized and pelletized using the same method. The BET specific surface area was significantly reduced when the CA-derived carbon powder and the MOF-derived carbon powder were pelletized. As a result, the amount of hydrogen adsorption was considerably reduced in the MOF-derived carbon pellet, while the hydrogen adsorption level in the CA pellet remained high. Pore size distribution analysis revealed that in the case of the CA-derived carbon, the proportion of small pores (<0.8 nm) with high hydrogen adsorption efficiency increased more with pelletizing than that of the MOF-derived carbon pellet. Therefore, for CA-derived hydrochar, optimizing the KOH activation conditions through chemical treatment and applying physical compression densification at a high pressure of 800 MPa altered the pore size distribution within a narrow range of micropores. This resulted in constructing a pore structure that is favorable for hydrogen adsorption and achieved a high volumetric storage density. This research approach demonstrates that porous carbon pellets with a large volumetric hydrogen storage density can be produced by increasing the pore ratio favorable for hydrogen adsorption due to pelletizing porous materials with high gravimetric hydrogen storage capacity while effectively suppressing any loss of adsorption properties as much as possible.
1. Introduction
High-density pellets made from porous materials with a large surface area have attracted great interest for various applications such as electrode materials,1,2 catalysts,3 adsorbents,4 and gas separation.5 In particular, high-density pellets (monoliths) of adsorbents with a large amount of hydrogen adsorption are expected to be a practical alternative to compressed hydrogen gas storage using high-pressure tanks in fuel cell vehicles.6–8 So far, MOFs, COFs, zeolites, activated carbons, and other porous materials with large specific surface areas have been reported.9–13 However, when these materials are used as hydrogen adsorbents in tanks, their volumetric storage density is surprisingly low,8,14,15 so there is a problem that the tank volume becomes too large when a sufficient amount of hydrogen is stored. On the other hand, various different approaches to gas storage for high volumetric capacity have been proposed.16–18 For instance, if metal hydrides such as MgH2 are placed in a tank, MgH2 can store hydrogen at levels of up to 7.6 wt% and 110 g L−1, making it one of the most promising candidates.16,19 However, the need for high-temperature heating (>300 °C) during the desorption of hydrogen and the slow kinetics limit hydrides to practical use.20 Therefore, rather than chemisorption that strongly interacts with hydrogen, there is a significant demand for adsorbents due to physisorption that can reversibly adsorb and desorb hydrogen gas at high density only by pressure changes at ambient temperature.To effectively use conventional porous materials for automotive and stationary storage tanks, it is crucial to improve density through pelletizing using pressure molding.21,22 However, current pelletizing methods report that as density increases, there is a significant reduction in surface area and pore volume due to processes such as compression and the addition of binder materials.4,14,23–25 This means that the proportion of pores decreases with pelletizing. Therefore, it is necessary to develop a pelletizing method that does not impair the adsorption characteristics of porous materials or to develop a porous material that enhances volumetric hydrogen adsorption capacity through pelletizing.
In this study, cellulose acetate (CA), typically derived from renewable sources such as wood and cotton pulp,26 was used as a precursor for the preparation of porous materials. The processing conditions for the activation of the precursor, a crude carbide, were optimized by adjusting the temperature, heating time, gas flow rate, and other variables to ensure that the activation treatment conditions were suitable for hydrogen adsorption. By applying a higher pressure (800 MPa) to the obtained porous carbon powders than is typical in general pelletizing methods, a novel approach that combines chemical and physical treatments was adopted. This process resulted in the production of high-density pellets through compression molding. Consequently, we successfully produced porous carbon pellets with a high volumetric hydrogen storage density compared to previously reported porous pellets. This was achieved by effectively preventing a reduction in the pore ratio, which is essential for hydrogen adsorption during the compression of the porous carbon powders.
2. Results and discussion
2.1 Preparation of cellulose acetate-derived porous carbons suitable for hydrogen adsorption
CA-derived porous carbon, known as CA-4700, has reported a significant ability to adsorb hydrogen at low temperatures and atmospheric pressure (3.9 wt% at −196 °C and 1 bar).27 This study investigates the effects of activation on hydrochar (Char) synthesized hydrothermally using CA as a precursor to achieve higher hydrogen performance. To account for heterogeneity in hydrothermal synthesis, alkaline activation treatment was performed on a single batch of Char obtained under consistent hydrothermal synthesis conditions (see the Experimental section for details, ESI†), using potassium hydroxide (KOH) as an activating reagent. The Char sample was activated under various conditions, including different Char/KOH mixing ratios, heating durations, and N2 flow rates. In the following sections, we examine and discuss the effects of three types of activation. In particular, the resulting activated porous carbons were evaluated to understand the relationship between their porous characteristics and hydrogen adsorption capacity.2.2 Effect of Char/KOH mixing ratio during KOH activation
First, the results of varying the mixing ratio of KOH to Char, with ratios ranging from Char/KOH = 1/0.3 to 1/4, are presented in Fig. 1a–d and Table 1. Porous structures are generally formed by activating carbon-based materials using KOH as the chemical reagent, a method widely reported in previous studies.28–31 Our experimental results also confirm that porosity develops through KOH activation. The BET specific surface area (SSA) and pore volume were significantly influenced by varying Char/KOH mixing ratios (Fig. 1a and b and Table 1). The increase in the relative amount of KOH to the Char primarily drives porosity development, indicating that the micropore structure is formed through KOH activation, consistent with previous studies.29,32 On the other hand, the amount of hydrogen adsorption increased with the increase in the KOH amount until the ratio was Char/KOH = 1/2. However, subsequently, even if the amount of KOH increased in the Char/KOH ratio, the BET SSA increased slightly, and the amount of hydrogen adsorption did not increase but rather decreased.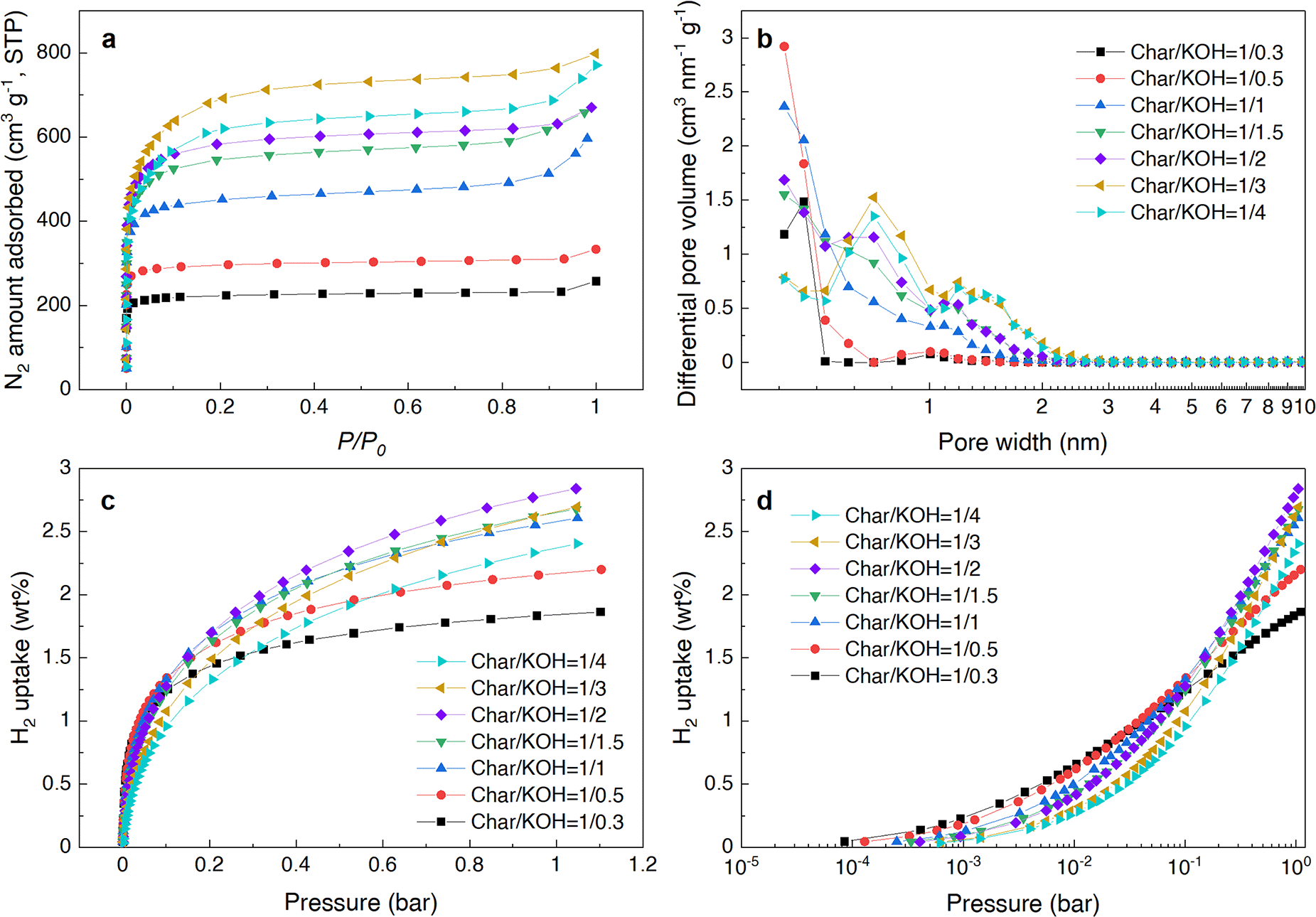 | ||
| Fig. 1 Adsorption behavior of CA-derived carbon powders prepared under different KOH activation conditions ranging from Char/KOH = 1/0.3 to 1/4 (w/w). (a) N2 adsorption isotherms measured at −196 °C and at STP (standard temperature and pressure), (b) pore size distribution calculated from N2 adsorption isotherms in (a) using the QSDFT method, (c) H2 adsorption isotherms at −196 °C, and (d) H2 adsorption isotherms with the horizontal axis of (c) displayed logarithmically. | ||
Char/KOH mixing ratio [Char![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KOH, w/w] :KOH, w/w] |
S BET [m2 g−1] | V total [cm3 g−1] | Yieldc [%] | H2 uptaked [wt%] |
|---|---|---|---|---|
| a Gravimetric specific surface area calculated by the Brunauer–Emmett–Teller (BET) method from the N2 adsorption isotherms at −196 °C in the range P/P0 = 0.05–0.3. b Total pore volume calculated at P/P0 = 0.96. The values in parentheses are cumulative micropore volumes calculated by the QSDFT method from the N2 adsorption isotherms. c Calculated from the ratio of the final KOH-activated product mass to the raw material of CA. d Gravimetric H2 storage density at −196 °C and 1 bar. | ||||
| 1:0.3 |
745 | 0.38 (0.33) | 9.1 | 1.84 |
| 1:0.5 |
963 | 0.49 (0.43) | 13.3 | 2.17 |
| 1:1 |
1506 | 0.87 (0.65) | 11.8 | 2.58 |
| 1:1.5 |
1879 | 1.01 (0.80) | 9.4 | 2.65 |
| 1:2 |
2013 | 1.01 (0.86) | 10.6 | 2.80 |
| 1:3 |
2400 | 1.21 (1.02) | 8.0 | 2.66 |
| 1:4 |
2157 | 1.13 (0.92) | 8.5 | 2.37 |
Why did the amount of hydrogen adsorption decrease despite the increase in the SSA? To answer this question, we investigated the relationship between the BET SSA, pore volume, hydrogen adsorption, and pore size distribution. In general, it has been reported that the larger the SSA, the greater the amount of hydrogen adsorption, and typically, as in “Chahine's rule”, it is supposed that the amount of hydrogen adsorption increases by 1 wt% for every increase in the SSA by 500 m2 g−1.33 Indeed, for example, certain types of MOFs (such as NU-1501-Al) have reported a high BET SSA of more than 7000 m2 g−1,34 which shows a large hydrogen adsorption performance. Additionally, as discussed by other research groups,35 there is a linear increase in the amount of hydrogen adsorption with an increase depending on the BET SSA range.
From a different perspective on the factors that influence hydrogen adsorption, it has been reported that the amount of hydrogen adsorption is more strongly dependent on pore size distribution than on surface area.36,37Fig. 1b shows the pore size distribution determined using a quenched solid density functional theory (QSDFT) method from the N2 adsorption isotherms shown in Fig. 1a. From this analysis, it is evident that when the KOH amount is less than Char/KOH = 1/1, the proportion of small pores (<1 nm) is significant. However, as the KOH amount increases, the pores around 1 nm or below decrease, while the pores around 1–2 nm increase. This change is accompanied by a corresponding increase in the BET SSA, indicating that as the KOH amount increases, the degree of activation intensifies, causing pores of 1 nm or below to connect and/or merge, resulting in larger pores exceeding 1 nm.
This change in the pore structure is also supported by the hydrogen adsorption isotherms, as shown in Fig. 1d. In the low-pressure region (<10−2 bar), the lower the KOH amount, the greater the hydrogen adsorption. This indicates that with a low KOH amount, relatively many small pores are formed, which are favorable for hydrogen adsorption, thereby resulting in higher hydrogen uptake. On the other hand, in the high-pressure region (>10−1 bar), an increase in the amount of KOH results in a steep slope in hydrogen adsorption, which significantly enhances hydrogen uptake. This indicates that, within this pressure range, higher amounts of KOH result in the formation of relatively larger pores due to intensified KOH activation.
More importantly, it should be noted that at Char/KOH = 1/4, the SSA is large, but the amount of hydrogen adsorbed is lower than that of Char/KOH = 1/2, reversing the above-mentioned trend such as “Chahine's rule”. As previously mentioned, hydrogen adsorption strongly depends on the pore size distribution, particularly pores of 2–3 nm or below, rather than solely on the specific surface area.38,39 Other research groups reported that the optimal pore size for hydrogen adsorption is theoretically around 0.6–0.7 nm,40,41 and pores about 1.3 nm or larger experimentally contribute little or nothing to hydrogen adsorption.34 Therefore, it is reasonable that the pore size distribution, as shown in Fig. 1b, explains why the total amount of hydrogen adsorption is largest at Char/KOH = 1/2. This is because the pores of 0.7 nm or smaller are significantly reduced in the sample with Char/KOH = 1/4, while they are not as reduced in the sample with Char/KOH = 1/2. Based on this difference in the narrow micropore range, although the sample with Char/KOH = 1/4 has a relatively large SSA, it has few pores with high hydrogen adsorption efficiency. Consequently, the total amount of hydrogen adsorption for Char/KOH = 1/4 is lower than that for the sample with Char/KOH = 1/2.
2.3 Effect of heating duration during KOH activation
Next, we examined whether it was possible to increase only the proportion of narrow micropores (<1 nm) preferred for hydrogen adsorption. For the milder KOH activation treatment, the KOH amount ratio was fixed at Char/KOH = 1/2 at 600 °C, which is lower than the above-mentioned temperature of 700 °C. Only the heating time varied from 30 minutes to 12 hours in a N2 flow atmosphere. The results are shown in Fig. 2a–d and Table 2.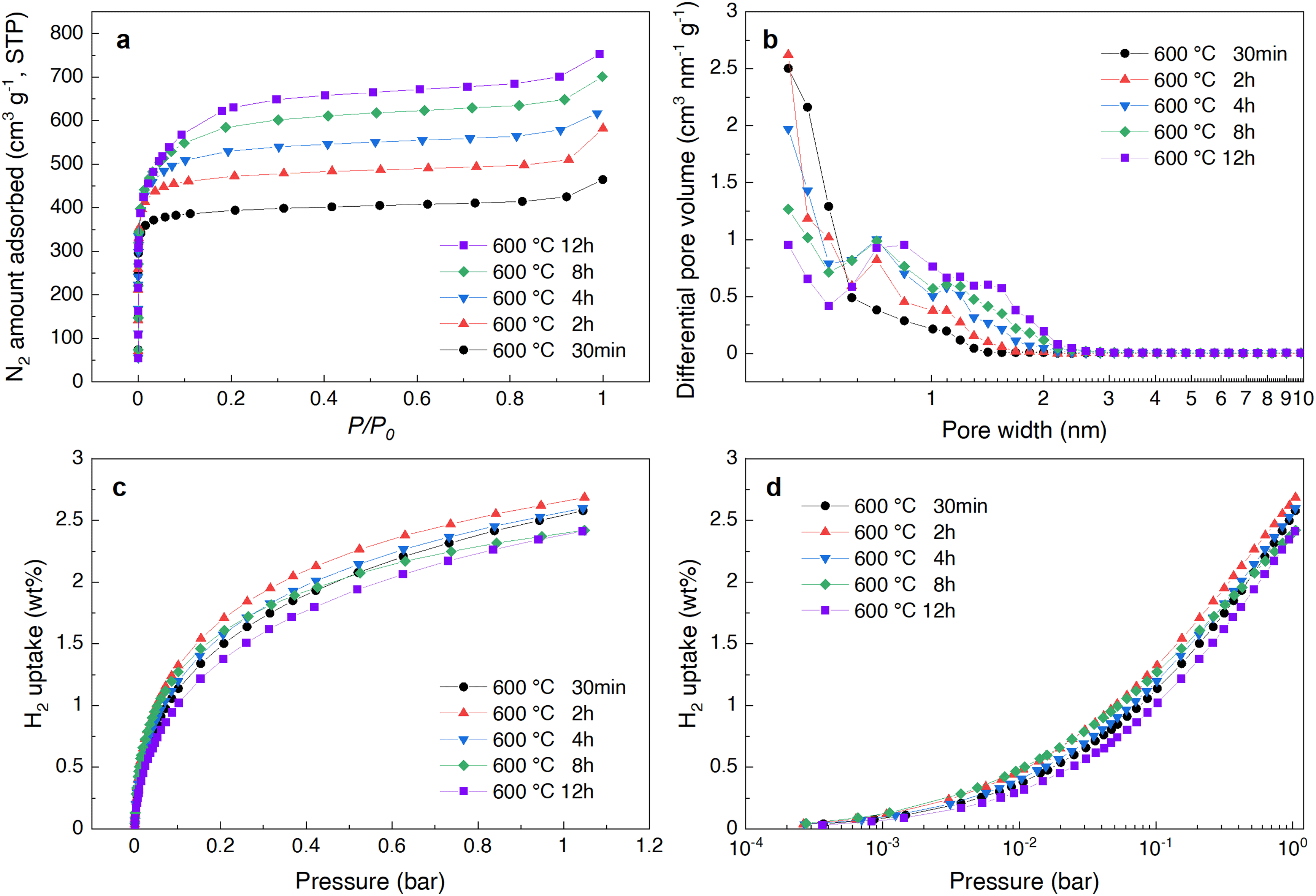 | ||
| Fig. 2 Adsorption behavior of CA-derived carbon powders prepared under different KOH activation conditions, with varied heating times ranging from 30 minutes to 12 hours at 600 °C. (a) N2 adsorption isotherms at −196 °C. (b) Pore size distribution calculated from N2 adsorption isotherms in (a) using the QSDFT method. (c) H2 adsorption isotherms at −196 °C. (d) H2 adsorption isotherms with the horizontal axis of (c) displayed logarithmically. | ||
| Heating time@600 °C [h] | S BET [m2 g−1] | V total [cm3 g−1] | Yieldc [%] | H2 uptaked [wt%] |
|---|---|---|---|---|
| a Gravimetric specific surface area calculated by the BET method from the N2 adsorption isotherms at −196 °C in the range P/P0 = 0.05–0.3. b Total pore volume calculated at P/P0 = 0.96. The values in parentheses are cumulative micropore volumes calculated by the QSDFT method from the N2 adsorption isotherms. c Calculated from the ratio of the final KOH-activated product mass to the raw material of CA. d Gravimetric H2 storage density at −196 °C and 1 bar. | ||||
| 0.5 | 1300 | 0.68 (0.51) | 14.4 | 2.40 |
| 2 | 1569 | 0.84 (0.58) | 14.4 | 2.65 |
| 4 | 1812 | 0.93 (0.74) | 10.9 | 2.57 |
| 8 | 2055 | 1.05 (0.79) | 7.3 | 2.54 |
| 12 | 2202 | 1.13 (0.88) | 4.6 | 2.38 |
As expected, increasing the heating time led to a decrease in yield due to the promotion of pyrolysis and gasification, while the BET SSA and pore volume showed an increasing trend (see Table 2), indicating that porosity develops as activation heating time increases as well. Additionally, the pore size distribution, as shown in Fig. 2b, was estimated using the N2 adsorption isotherms via the QSDFT method. This analysis revealed that with extended heating time (30 min to 12 h), the proportion of pores with 0.6–0.7 nm, which initially formed during 30 minutes of heating at 600 °C, was high even after 2 hours of heating but was not retained, gradually decreasing over 12 hours. On the other hand, the pores of approximately 0.8 nm and 1.4 nm showed a significant increase with longer heating times. This indicates that as the KOH activation progresses, it leads to a wider distribution of pore sizes. This effect resembles what was observed when the KOH amount was increased relative to the Char/KOH ratio (see Fig. 1b and 2b for comparison). This change in pore size distribution is consistent with other activated carbons reported in the literature.42 In general, during KOH activation, the chemical reaction between KOH and carbon primarily proceeds through the following redox reaction as follows:38,42,43
| 6KOH + 2C → 2K + 3H2 + 2K2CO3 | (1) |
| K2CO3 → K2O + CO2 | (2) |
For temperatures below 700 °C, the decomposition reaction of K2CO3, as shown in (2), is unlikely to occur, and the reaction leading up to (2) is limited. This indicates that the formation of microporosity caused by CO2 in (2) and H2O from the KOH dehydration reaction is hindered by the slow kinetics of K2CO3 decomposition. However, changes in micropore development were observed in this study even at a temperature of 600 °C with prolonged heating time. Therefore, it is believed that the reaction between KOH and the carbon fosters micropore formation at 600 °C, depending on the degree of KOH activation.
On the other hand, it was observed that the amount of hydrogen adsorption tended to decline with longer heating times after peaking at 2 hours of heating (see Fig. 2c). This indicates that porosity for hydrogen adsorption effectively develops as heating time increases from 30 minutes to 2 hours. However, pores formed at later times (4 h–12 h) become less effective for hydrogen adsorption. As shown in Fig. 2d, the amount of hydrogen adsorption for each sample decreases with increasing heating time as the pressure increases overall. This observed trend indicates that the total number of adsorption pore sites significantly contributing to hydrogen adsorption has either decreased or disappeared at almost the same degree, due to extended thermal decomposition under mild activation with KOH. This behavior contrasts markedly with the changes observed when the Char/KOH ratio is altered (especially a change of Char/KOH = 1/0.3 to 1/1.5), as shown in Fig. 1d. In that case, the total number of hydrogen adsorption pore sites is reconstructed randomly, leading to significant alterations in the micropore structure. Consequently, the changes in the pore structure based on observed H2 adsorption isotherms will be considerable in the ultra-micropore range.
2.4 Analysis of the ultra-micropore structure using CO2 gas in CA-derived carbons
To verify this presumption, adsorption isotherm measurements were conducted using CO2 gas, which is a suitable probe for analyzing the pore state of ultra-micropores (<0.7 nm). This is because ultra-micropores can correlate more positively with hydrogen adsorption compared to micropores (<2 nm).44 Additionally, as CO2 gas has a faster diffusion rate than N2 gas and is strongly adsorbed on carbon materials with ultra-micropores,45 changes in pore size distribution were analyzed using a non-local density functional theory (NLDFT) method. The results are shown in Fig. 3a–d.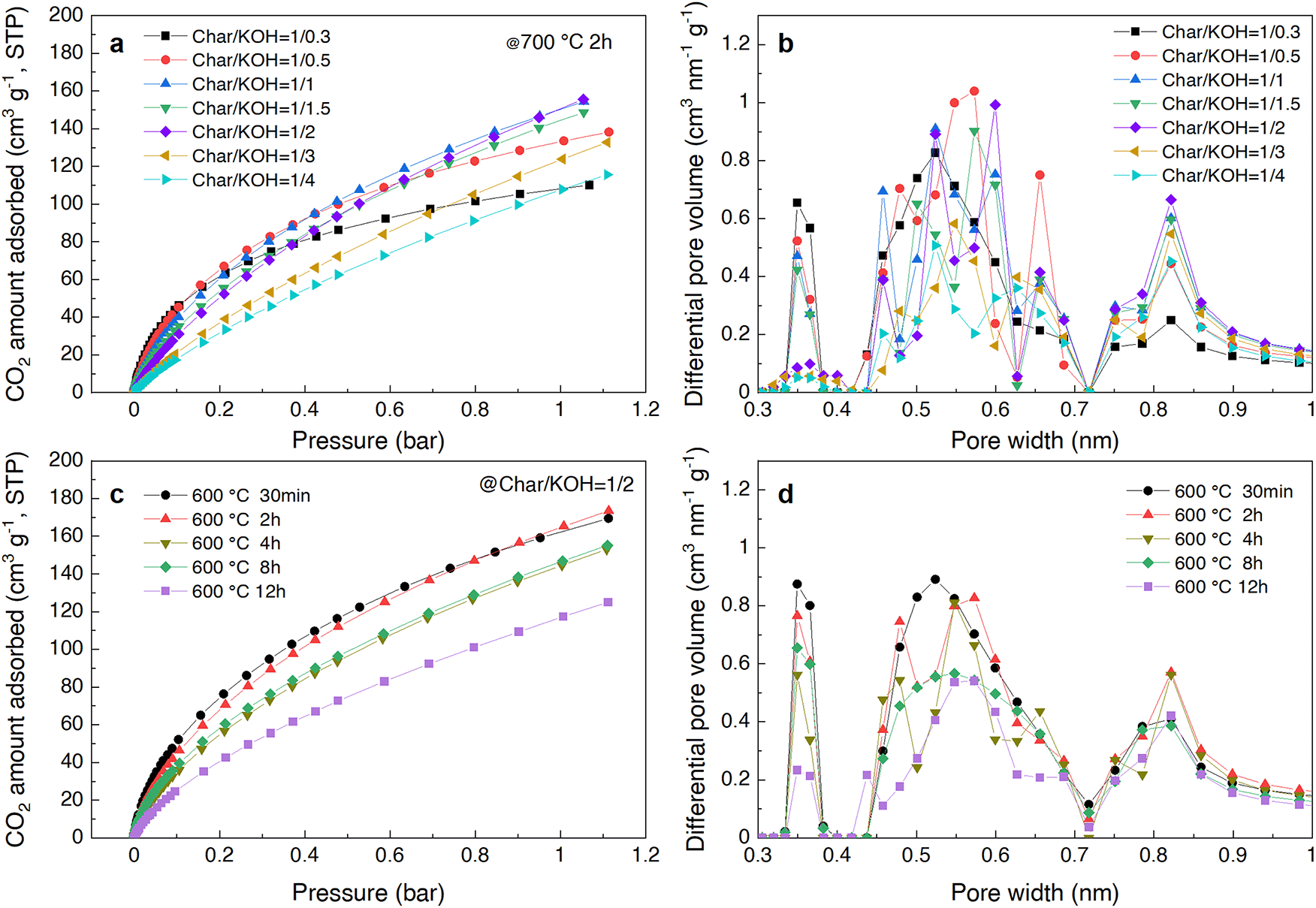 | ||
| Fig. 3 Analysis of the ultra-micropore structure using CO2 gas in CA-derived carbon powders. (a) CO2 adsorption isotherms at 0 °C for samples prepared with different Char/KOH ratios at 700 °C. (b) Pore size distribution calculated from the CO2 adsorption isotherms in (a) using the NLDFT method. (c) CO2 adsorption isotherms at 0 °C for samples prepared with a Char/KOH ratio of 1/2 at various heating times at 600 °C. (d) Pore size distribution calculated from the CO2 adsorption isotherms in (c) using the NLDFT method. | ||
From the CO2 adsorption isotherms shown in Fig. 3a and c, it was observed that the behavior is similar to the H2 adsorption isotherms when varying the Char/KOH ratio at 700 °C (Fig. 1c) and the heating time at 600 °C (Fig. 2c), respectively. Notably, when the amount of H2 adsorption at −196 °C and 1 bar was plotted against the amount of CO2 adsorption at 0 °C and 1 bar (Fig. 4), it was observed that the amount of CO2 adsorbed was significantly high across most samples, especially in those with high H2 uptake. This trend indicates that, although there is a relatively weak correlation between CO2 and H2 adsorption, the ultra-micropores contribute significantly. Moreover, the relationship between the adsorbed amounts of CO2 and H2 under both activation conditions is not completely linear (see Fig. S3 for details, ESI†). This can be attributed to two factors: (1) H2 molecules tend to be more adsorbed in pores smaller than 0.5 nm compared to CO2 molecules in the ultra-micropore region, as indicated by previous studies46 and (2) CO2 is not predominantly adsorbed in pores larger than 0.8 nm,45,47 which may result in an underestimation of pore volume.
 | ||
| Fig. 4 Relationship between CO2 adsorption at 0 °C and 1 bar and H2 adsorption at −196 °C and 1 bar for the CA-derived carbon powders. The straight dotted line represents the approximate trend for the plotted data. | ||
Furthermore, when the pore size distribution was analyzed by the NLDFT method from the CO2 adsorption isotherm at 0 °C (Fig. 3b), it was confirmed that as the Char/KOH amount ratio was varied, the pores of 0.3–0.4 nm decreased with an increase in the KOH amount and the pores around 0.4–0.7 nm changed significantly. This indicates that KOH penetrated deeply into the char and formed pores randomly, resulting in significant structural changes. On the other hand, when only the heating time was varied at a constant temperature of 600 °C, the pores of 0.3–0.4 nm were most developed with a heating duration as short as 30 minutes (Fig. 3d), while the pores around 0.4–0.7 nm were similarly reduced at long heating durations, and the pore size distribution did not change drastically in shape compared to the varying Char/KOH ratio. That is, as the heating time increased, only the overall pore volume decreased, indicating that the pore structure of ultra-micropores underwent relatively uniform changes in shape, and gradually, the pores disappeared.
Thus, when the Char/KOH ratio is altered, pores of about 0.3–0.4 nm, once formed through KOH activation at a small amount of KOH, are consumed by merging or connecting with other pores, transforming into larger pores and undergoing drastic reconstitution resulting in high disorder as the KOH amount increases. In contrast, when mildly activated by changing only the heating time at 600 °C, the pores of 0.3–0.4 nm formed most at the first short time of activation (30 min) and gradually disappeared as the heating time prolonged. However, these two activation conditions did not facilitate the selective formation of pores with high hydrogen adsorption efficiency in the 0.6–0.7 nm pore range.
From the pore size analyses using CO2 as a probe, it is suggested that pores of 0.3–0.4 nm decrease as the degree of KOH activation strengthens, and therefore, the pore size can be controlled even within a narrow range of ultra-micropores. However, it should be noted that in the pore size distribution analysis using the slit-shaped pore model in the NLDFT method, there are some parts where the actual pore state is not properly reflected due to artifacts,48 resulting in discrepancies from the actual pore structure.
2.5 Effect of N2 flow rate during KOH activation
Finally, as another approach through mild activation, we examined whether only uniform small pores of 1 nm or below were formed. An experiment was performed in which only the N2 gas flow rate was changed while the other activation conditions remained fixed: specifically, the Char/KOH ratio was set at 1/2, the temperature was fixed at 700 °C, the heating time was fixed at 2 hours, and only the gas flow rate varied from 0.06 L min−1 to 1.5 L min−1.From the pore size distribution calculated from the N2 adsorption isotherms (Fig. 5), it was revealed that it was possible to successfully change only small pores of about 1 nm below. When the gas flow rate was very slow (0.06–0.1 L min−1), there were many pores of 1 nm or below, and the amount of hydrogen adsorption at −196 °C and 1 bar was high (2.68–2.80 wt%, Fig. S4, Table S1, ESI†). On the other hand, when the gas flow rate was relatively fast (0.3–1.5 L min−1), the number of pores of around 1 nm or below decreased, the amount of hydrogen adsorption was relatively low (when the N2 flow rate was at its highest (1.5 L min−1), and the H2 uptake amount was at its lowest (2.35 wt%), Table S1, ESI†). These results indicate that pores of 1 nm or below significantly influence the experimentally measured amount of hydrogen adsorbed, even when varying the N2 flow rate. According to a previous report,49 a faster N2 flow rate leads to an increase in BET SSA and larger porosity. However, our results indicated the opposite: a faster N2 flow rate led to slightly lower SSA and reduced H2 uptake (see Table S1, ESI†). This discrepancy may be attributed to a difference in the optimal activation conditions, which are influenced by the physical properties of the precursors, the surface conditions of the samples, the furnace system, the amount of the sample on the boat, and other related factors.
 | ||
| Fig. 5 Adsorption behavior of CA-derived porous carbons prepared under different KOH activation conditions, with the N2 flow rate ranging from 0.06 to 0.1 L min−1 activated at 700 °C and 2 hours. (a) N2 adsorption isotherms at −196 °C. (b) Pore size distribution calculated from N2 adsorption isotherms in (a) using the QSDFT method. | ||
It is noteworthy that even when the N2 gas flow rate was adjusted, the shape of the pore size distribution for pores larger than 1 nm remained nearly unchanged. This robustness is due to the fact that, within the range of gas flow rates examined, the degree of activation with KOH did not significantly enhance by altering the gas flow rate enough to reconstruct the pore structure. Additionally, once larger pores (>1 nm) were formed, their size remained unaffected by these changes. In general, the gas flow rate during activation influences the mass transfer diffusion of KOH during pore formation.29 It is assumed that when the gas flow rate exceeds a certain threshold, the diffusion and evaporation rate of potassium (K) gas, derived from KOH, becomes the dominant factor. This would weaken the effectiveness of KOH activation into the char and hinder the formation of pores. In other words, when the gas flow rate is very slow (at 0.06–0.1 L min−1), the K gas derived from KOH sufficiently reaches the intra-pores and the depths of the char, enhancing the effect of KOH activation and developing small pores (<0.8 nm). However, even at the slowest gas flow rate (0.06 L min−1) that would have stronger activation conditions among them, no significant increase in hydrogen adsorption was observed (see Table S1, ESI†). This indicates the difficulty of forming a large number of small pores with selectively high hydrogen adsorption efficiency (e.g., 0.6–0.7 nm pores) while suppressing the formation of relatively larger pores (∼1.3 nm or more) that contribute little to hydrogen adsorption. Conversely, when the gas flow rate is high (0.3 L min−1 or above), it surpasses the threshold, causing the K gas in the N2 flow to not sufficiently reach the intra-pores and depths of the char, as it can be carried away by diffusion. This would result in the minimal formation of narrow micropores (1–2 nm) under fast N2 flow rate conditions.
2.6 The trade-off between the specific surface area and the pore ratio
The above-mentioned results and analysis of the pore formation effect suggest that the small micropore size of 1 nm or below can be controlled experimentally by adjusting the KOH activation conditions. That is when the degree of KOH activation is low (i.e., temperature lower than 700 °C, the KOH amount ratio is small, or the heating time is short), a large number of small pores (<1 nm) favorable for hydrogen adsorption are formed. However, the overall volume of these small pores is still relatively low, leading to limited SSA and, consequently, low hydrogen adsorption at −196 °C and 1 bar. On the other hand, when the degree of KOH activation is high (i.e., temperature above 700 °C, the KOH amount ratio is high, or the heating time is long), the SSA increases but the obtained activated carbons have pores larger than 1 nm, resulting in pore size broadening. In other words, the proportion of pores 1 nm or below, which are effective for hydrogen adsorption, disappears due to the merger or fusion between pores. Consequently, despite the large SSA, the total amount of adsorbed hydrogen is less than what would be expected from the surface area. Therefore, it can be concluded that there is a trade-off between the SSA (as well as pore volume) and the pore ratio of 1 nm or less, influenced by the level of KOH activation, as shown in Fig. 6.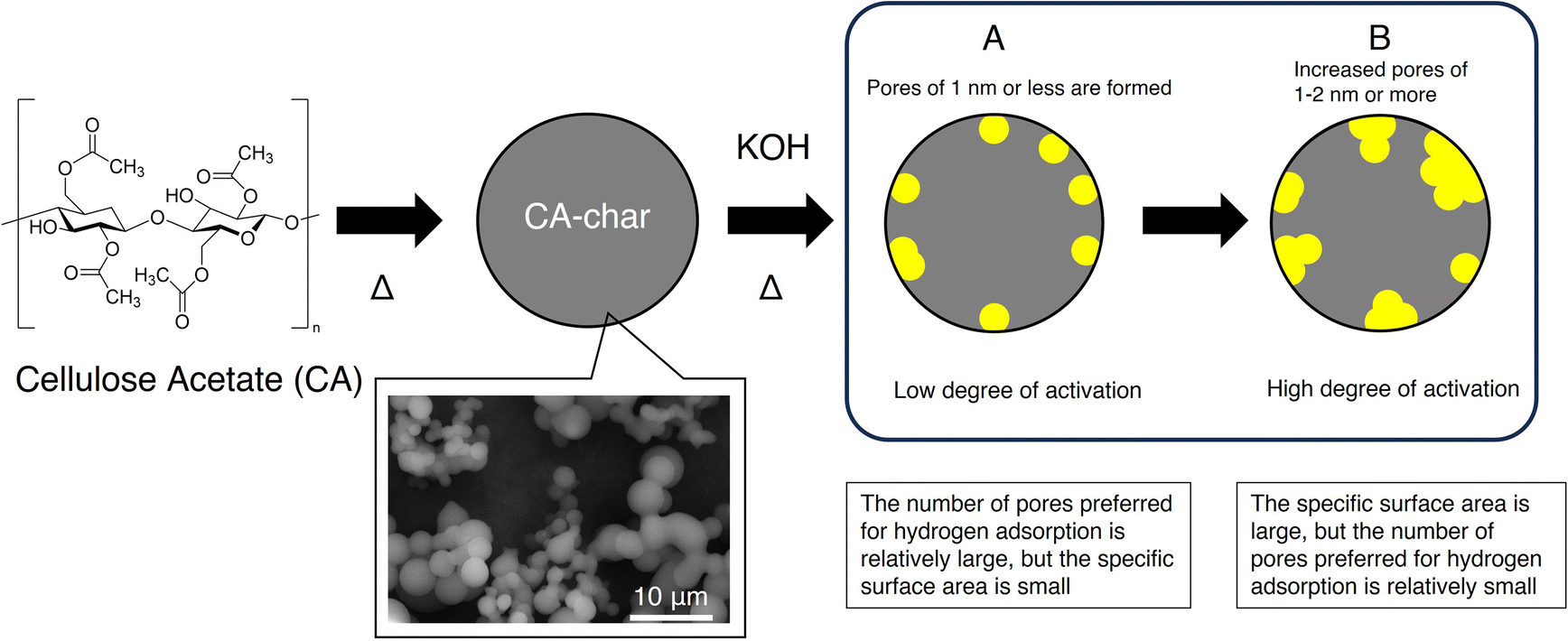 | ||
| Fig. 6 Schematic illustration of pore formation. The CA precursor undergoes hydrothermal synthesis, followed by KOH activation. (A) Under low degree of activation conditions such as a lower concentration of KOH in the CA-char/KOH ratio and/or lower temperatures, a relatively large number of small pores (<1 nm), which are preferred for hydrogen adsorption, are moderately formed, but total pore volume (specific surface area) is small. (B) Under high degree of activation conditions such as a higher concentration of KOH in the CA-char/KOH ratio and/or higher temperatures, large pores (>1–2 nm or more) are formed abundant, leading to an increase of specific surface area, but a number of small pores preferred for hydrogen adsorption are relatively small. | ||
Additionally, a similar change in pore size distribution was observed under activation conditions where the activation temperature varied from 500 °C to 800 °C (see Fig. S5 and Table S2, ESI†). Micropore analysis based on pore size sections from N2 adsorption isotherms using the QSDFT method (Fig. S6, ESI†) found that optimal KOH activation conditions, which selectively form only pores of 0.6–0.7 nm without forming any pores of 1.3 nm or larger, while maintaining a high SSA, were not identified. Additionally, we have not found activation conditions that correspond to the highest level of hydrogen adsorption, as previously reported.27 However, it was experimentally confirmed that only narrow micropores (<∼0.8 nm) were influenced by the N2 flow rate. This indicates that the micropore size favorable for hydrogen adsorption can be altered by adjusting the activation conditions using N2 gas as a probe. However, note that it is reported that evaluating pore sizes of approximately 0.5 nm or below using N2 gas is difficult due to the measurement pressure limitations of ordinary instruments.44
Furthermore, the ideal conditions for forming micropore structures suitable for hydrogen adsorption can be fine-tuned not only by adjusting the gas flow rate and the previously mentioned factors but also by considering other variables such as alternative activating chemical reagents besides KOH and raw cellulose acetate with different degrees of acetylation. For instance, there has been a report indicating that using K2CO3 as an activating reagent results in the formation of more micropores compared to KOH.50 We also examined microporosity change for the same batch of CA-derived char activated using K2CO3. However, as the activation temperature increased from 600 °C to 800 °C, the BET SSA estimated from N2 adsorption isotherms increased, while the pores (<∼0.7 nm) contributing to hydrogen adsorption became smaller and followed a similar trend to those activated with KOH (see Fig. S7 and Table S3, ESI†). Thus, even when activated under similar conditions with K2CO3, selectively forming pores that significantly enhance hydrogen adsorption was not achievable. On the other hand, different kinds of cellulose acetate with a higher degree of acetylation (so-called CTA) were used as precursor raw materials instead of the above-examined CA, and the same activation treatment was performed with KOH. However, even when the activation conditions were adjusted, the change in pore size distribution showed almost the same behavior, and the amount of hydrogen adsorption was not significantly increased, as well (see Fig. S8 and Table S4, ESI†). Therefore, as an alternative strategy, adjusting the activation parameters—such as the type of flow gas, pressure changes during activation, temperature rise rates, and the types of precursors used—might increase the proportion of pores that are particularly favorable for hydrogen adsorption, allowing for the identification of optimal activation conditions. Nevertheless, these approaches, as far as we investigated, did not successfully overcome the inherent trade-off relationship.
2.7 Preparation of cellulose acetate-derived carbon pellets
Based on the study of optimal activation conditions, a sample of CA-derived porous carbon with high hydrogen adsorption capacity was prepared (2.80 wt% in Table 1, Table S5, ESI†). Although the hydrogen adsorption amount obtained was not as high as the value reported in the previous study,27 the use of CA-derived porous carbons, which demonstrated a relatively high H2 uptake (2.55 wt% at −196 °C and 1 bar), allowed for the successful preparation of CA-derived carbon pellets. This was achieved using PVA as a binder and applying a high-pressure treatment, as shown in Fig. 7 (see the preparation method for details, ESI†). For comparison, a porous carbon with an equally high amount of hydrogen adsorption, which is MOF (ZIF-8)-derived carbon powder (H2 uptake: 2.54 wt% at −196 °C and 1 bar), was prepared based on the previous literature,51 and the pellet was prepared by the same method (ESI†). The adsorption characteristics of the powder and the pellet for CA-derived porous carbon and ZIF-8-derived carbon are shown in Fig. 8 and Table 3. Additionally, it is important to acknowledge that stable pellet molding cannot be achieved when a small amount of PVA binder is added. In contrast, pellets with a specific amount of PVA can be effectively produced at temperatures ranging from 75 °C to 150 °C during the pelletizing process, resulting in a high bulk density. However, at excessively high temperatures (>150 °C), the level of hydrogen adsorption at −196 °C and 1 bar was significantly reduced to below approximately 1.8 wt% (see Table S5, Fig. S9 and S10 for more details, ESI†). This suggests that high-density pellet processing technology, which balances pellet formability with high hydrogen adsorption performance, is crucial. | ||
| Fig. 7 Scheme for producing CA-derived carbon pellets from its powder. Photos of CA carbon before and after compaction at 800 MPa through the pelletization process using the die molding technique. | ||
 | ||
| Fig. 8 Comparison of adsorption properties of powders and pellets for CA-derived carbon and MOF(ZIF-8)-derived carbon. (a) N2 adsorption–desorption isotherms of ZIF-8-derived carbon at −196 °C, (b) N2 adsorption–desorption isotherms of CA carbon at −196 °C, (c) pore size distribution of ZIF-8 carbon calculated from N2 adsorption isotherms in (a) using the QSDFT method, (d) pore size distribution of CA carbon calculated from N2 adsorption isotherms in (b) using the QSDFT method, (e) H2 adsorption–desorption isotherms of ZIF-8 carbon at −196 °C, and (f) H2 adsorption–desorption isotherms of CA carbon at −196 °C. | ||
| Sample | S BET [m2 g−1] | V total [cm3 g−1] | V micro [cm3 g−1] | ρ [cm3 g−1] | Vol. SBETe [m2 cm−3] | Gravimetric H2 uptakef [wt%] | Volumetric H2 uptakeg [g-H2 per L] |
|---|---|---|---|---|---|---|---|
| a Gravimetric specific surface area (SBET) calculated by the BET method from N2 adsorption isotherms at −196 °C in the range P/P0 = 0.05–0.3. b Total pore volume (Vtotal) calculated at P/P0 = 0.96. c Micropore volume (Vmicro) represents a cumulative pore volume in the range of 0.6 nm–2.0 nm calculated by the QSDFT method (Table S6, ESI). d Bulk density of the pellet. e Volumetric BET specific surface area (vol. SBET) calculated by dividing the SBET by the bulk density (ρ). f Gravimetric hydrogen storage density at −196 °C and 1 bar. g Volumetric hydrogen storage density at −196 °C and 1 bar. Note that the values in parentheses are percentage reductions after pelletizing to its powder. | |||||||
| ZIF-8 carbon powder | 3008 | 1.65 | 1.12 | — | — | 2.54 | — |
| ZIF-8 carbon pellet | 2051 (−32%) | 1.27 (−23%) | 0.73 (−35%) | 0.54 | 1107 | 2.15 (−15%) | 11.6 |
| CA carbon powder | 2312 | 1.17 | 0.93 | — | — | 2.55 | — |
| CA carbon pellet | 1724 (−45%) | 0.96 (−18%) | 0.60 (−36%) | 0.80 | 1091 | 2.53 (−1%) | 20.3 |
2.8 Analysis of N2 and H2 adsorption behaviors for powder and pellets
The N2 adsorption isotherm results of the synthesized CA-derived carbon pellets and ZIF-8-derived carbon pellets are shown in Fig. 8b and a, respectively. From the comparison with each powder, it can be seen that the amount of adsorption is significantly reduced overall after pelletizing. This means that the pelletizing process has significantly reduced the surface area and pore volume (see Table 3). As is obvious, in addition to containing about 10 wt% of PVA as a binder, it can be inferred that the SSA and pore volume will be significantly reduced by at least 10%. Moreover, due to the collapse of the pore structure and the closure of the pores by heat compression and the addition of the binder, porosity would seem to be lost, as in the previous study example.24,52 In fact, based on the N2 and H2 adsorption isotherms of ZIF-8-derived carbon as shown in Fig. 8a and e, the BET SSA and amount of hydrogen adsorption decreased significantly after pelletizing (BET SSA: −32% and H2 uptake: −15 wt%, Table 3). In contrast, for CA-derived carbon, however, the reduction in H2 uptake was relatively minor (−1 wt%) and retained almost the same level despite the significant decrease in BET SSA (−45%) after pelletizing.Why was the reduction rate in hydrogen adsorption lower in CA carbon pellets compared to ZIF-8 carbon pellets? Notably, a steep increase in H2 adsorption in the low-pressure region (<∼0.3 bar) after pelletizing the CA carbon pellets, as shown in Fig. 8f, was observed, indicating that relatively small pores were newly formed during the pelletizing process. Additionally, from the analysis of the pore size distribution (Fig. 8c and d) calculated by the QSDFT method from the corresponding N2 adsorption isotherms (Fig. 8a and b), respectively, it is evident that CA carbon has a narrower pore size distribution compared to ZIF-8-derived carbon. In other words, CA carbon powder originally has many pores with relatively uniform pore sizes. This is supported by the fact that the shape of the N2 adsorption isotherm of CA carbon powder closely resembles type I (a) isotherms (pore width < ∼1 nm) according to the IUPAC classification of physisorption isotherms.53 In contrast, the N2 adsorption isotherm of ZIF-8-derived carbon powder is closer to the classification of type I (b) isotherms (pore width < ∼2.5 nm). This uniformity of the small pore size is one reason why the hydrogen adsorption amount remains high after pelletizing.
Moreover, after pelletizing CA carbon powder, the pores around 1 nm decrease, while the pores around 0.6–0.7 nm significantly increase (Fig. 8d and Table S7, ESI†). On the other hand, for ZIF-8 carbon powder, the pores around 0.6–0.7 nm do not change much even after pelletizing compared to the CA carbon. Therefore, despite the significant reduction in SSA and micropore volume by pelletizing (Table 3), the amount of hydrogen adsorbed in the CA carbon pellet did not decrease as significantly as expected based on the reduction rate in the BET SSA and pore volume. That is, this maintenance of a high level of hydrogen adsorption is due to the increase in pores of 0.6–0.7 nm, which are preferred for hydrogen adsorption.
Furthermore, the volumetric hydrogen density of the ZIF-8 carbon pellet was approximately half that of the CA carbon pellet (see Table 3). In contrast, the volumetric BET SSA of both pellet samples was equivalent, and the reduction rate of micropore volume was almost the same (Table S7, ESI†). This difference between the CA carbon pellet and the ZIF-8 carbon pellet arises primarily from two main factors. Firstly, the resistance to deformation, and secondly, the surface conditions of the pore structure. The ZIF-8 carbons have a highly hydrophobic surface and a rigid carbon framework derived from the MOF template, making significant deformation of the pore structure unlikely under the conditions of heat and strong compression. As a result, the bulk density of the ZIF-8 carbon pellet is lower than that of the CA carbon pellet. In contrast, CA carbon has a greater affinity for PVA due to its hydrophilic surface. Additionally, it contains very few large pores such as mesopores (2–50 nm), and the pore sizes are relatively uniform as mentioned above. Therefore, it is presumed that the pore structure is easily subjected to the force of compression and becomes highly packed (0.80 g cm−3) compared to the ZIF-8 carbon pellet (0.54 g cm−3), resulting in high volumetric hydrogen density in the case of the CA carbon pellet. This assumption is supported by the fact that it was successively possible to produce CA carbon pellets using highly hydrophilic polymers other than PVA, such as PVP and HPC, and still achieve a high volumetric hydrogen density (approximately 14–17 g-H2 per L, Table S5, ESI†). In the low-pressure region (around 10−2 bar) of the H2 adsorption isotherms in Fig. S11d (ESI†), the amount of hydrogen adsorption is inverted between the pellet and the powder, as similarly shown in Fig. 8f, which uses a PVA binder in the CA carbon pellet. This indicates that pelletizing has increased the number of small pores that are favorable for hydrogen adsorption, as shown by the change in the pore size distribution in Fig. S11b.† Moreover, in the examined binders, when a hydrophilic binder with relatively low water solubility, such as PEO, is used, both the BET SSA and H2 uptake of the CA carbon pellet significantly decrease (by −61% and −43 wt%, respectively) compared to other binders, indicating that pore blockage occurs at levels higher than the added binder amount (approximately 10–14 wt%). The observed difference suggests that the strong affinity at the carbon-binder interface, along with the binders' high water solubility, plays a crucial role in maintaining the powder's high porosity. In other words, this achievement would result from the absence of pore blockage caused by the hydrophobic components of the binders (see the PEO binder pellet for comparison in Fig. S11, ESI†). Additionally, the ZIF-8 carbon might also enhance the volumetric hydrogen storage density by utilizing an appropriate hydrophobic binder. However, under the pellet preparation conditions examined for ZIF-8 carbons, achieving a higher packed density and greater volumetric hydrogen uptake than CA carbon pellets appears to be difficult. This is clearly illustrated in the plots of sample data prepared under various conditions, as shown in Fig. 9, which includes CA and ZIF-8 carbon pellet samples from different batches and varying preparation conditions (see Tables S5 and S6, ESI†).
 | ||
| Fig. 9 Plots of the bulk density of pellets vs. corresponding volumetric hydrogen uptake at −196 °C and 1 bar for the CA-derived carbon pellets and ZIF-8-derived carbon pellets, respectively. Note that the data are based on Tables S5 and S6 (ESI†). | ||
2.9 Practical application of pellets and applicability to other materials
In this way, we produced a high-density porous carbon pellet by applying CA-derived carbon powder to compression using the PVA binder. The combination of adjusting the KOH activation conditions and this pelletizing method changed the pore structure of the porous carbons having micropores. It succeeded in increasing the pore ratio preferred for hydrogen adsorption. In previous studies,54 pelletizing porous materials using high-pressure treatment has altered the pore size distribution and the amount of hydrogen adsorption. However, to the best of our knowledge, such an exceptionally high volumetric density has not been reported (20.3 g-H2 per L at −196 °C and 1 bar), as shown in Table 4 (see Table S8 for more details, ESI†). It has been reported that there is a strong correlation between the amount of hydrogen adsorption at low temperatures and atmospheric pressure (−196 °C and 1 bar) and at room temperature and high pressure (25 °C and 100 bar).59,60 Therefore, it is assumed that the high volumetric hydrogen storage density achieved under the conditions of this study is likely valid even at room temperature and high pressure. For this reason, it is expected that these CA carbon pellets will demonstrate high hydrogen adsorption performance, even under practical conditions.| Pellet | Volumetric H2 uptake [g-H2 per L] | Conditions | Ref. |
|---|---|---|---|
| a Total volumetric hydrogen storage capacity. The values in ref. 14 are based on calculated data. | |||
| SNU-70 | 9.0a | 5 bar, −196 °C | 14 and 55 |
| ZTC/rGO | 22.4a | 5 bar, −196 °C | 13 and 14 |
| MOF-5/ENG | 12.9a | 5 bar, −196 °C | 14 and 56 |
| MOF-5 | 16.5a | 5 bar, −196 °C | 14 and 56 |
| MOF-177 | 13.1a (25.8) | 5 bar, −196 °C (∼60 bar, −196 °C) | 14 and 57 |
| UiO-66 | 39 | 100 bar, −196 °C | 54 |
| HKUST-1 | 15.3 | 1 bar, −196 °C | 58 |
| CA carbon | 20.3 | 1 bar, −196 °C | This work |
From an industrial application perspective, however, establishing pelletizing technology capable of handling applied pressures as high as 800 MPa on a laboratory scale is challenging in terms of process feasibility and cost efficiency. Since only a limited number of materials can withstand such high pressures, these materials tend to be expensive. Furthermore, high-pressure molding often leads to longer cycle times, rendering it unsuitable for mass production because of low manufacturing efficiency. Consequently, the findings of this study indicate a need for a pellet molding method that employs physical and/or chemical treatments to achieve energy levels comparable to those attained with an applied pressure of up to 800 MPa. Examples of such methods include powder compression or densification techniques such as spark plasma sintering (SPS),61 cold/hot isostatic pressing (CIP/HIP),62,63 or chemical densification processes such as chemical vapor infiltration (CVI) with reaction gases.64 The integration of these densification methods and related technologies has the potential to improve the practicality and scalability of industrial applications.
Recently, several MOFs with precisely adjusted pore sizes, incorporating three-dimensional organic linkers and/or multiple hetero-molecules, have been reported as hydrogen and methane adsorbents with record-high capacities.34,65 So far, to enhance the volumetric storage density using conventional MOFs, applying strong compressive forces during pelletization generally reduces porosity significantly as bulk density increases, resulting in the collapse of the MOF's pore structure.4,66 However, using these new types of MOFs in densified forms—which combine highly flexible organic linkers with diverse reactivities—may enhance the pore ratio suited for optimal hydrogen adsorption. This study shows that pelletizing CA-derived carbons decreases larger pores (1–2 nm or more) while forming smaller ones. If this change in the pore structure is applied to advanced MOFs, it could result in the formation of densified MOFs that offer new adsorption sites due to the close proximity of linkers caused by compressive deformation, making them potential candidate materials. This is an entirely new strategy for producing high-density porous materials that were previously unattainable in the studies of MOF pellets. Such an approach could foster the development of innovative MOF adsorbents with high volumetric storage densities.
In the future, by using various porous materials with a high gravimetric storage capacity and low bulk density, derived from common polymers, biomass materials, and more, the combination of the pelletizing method and optimization of chemical treatments is expected to pave the way for establishing a novel technology that produces volumetric high-performance hydrogen-adsorbing materials that effectively balance density and porosity. These porous materials will be used not only for hydrogen adsorbents but also for other gases such as CO2 and methane and further for applications such as gas separation across a wide range of industrial fields.
3. Conclusions
In this study, the dependence of hydrogen adsorption capacity on the adjustment of KOH activation conditions was experimentally investigated in the micropore range for a single batch of char obtained by hydrothermal synthesis of CA. The results of this study indicate that the degree of KOH activation (low or high) applied to the char significantly alters the pore structure favorable for hydrogen adsorption even within the narrow micropore range (<1 nm), and it was experimentally clarified that there is a trade-off relationship between the specific surface area (total pore volume) and the pore ratio of about 1 nm or less. Moreover, the pore size distribution analysis using CO2 gas indicates that pores of 1 nm or smaller (particularly 0.3–0.4 nm) diminish as the degree of activation intensifies, resulting in changes to the ultra-micropore structure.To enhance hydrogen storage density per volume, high-density pellets were prepared using PVA as a binder. This pellet was made from CA-derived carbon powder, which exhibited a relatively high hydrogen adsorption capacity per weight at −196 °C and 1 bar. For comparison, the MOF-derived carbon powder, which exhibited the same hydrogen adsorption capacity per weight as the CA carbon powder, was also successfully pelletized. In the CA carbon pellets, the hydrogen adsorption level remained significantly high even after pelletizing. From the analysis of the pore size distribution from the N2 adsorption isotherm, the pores of around 0.6–0.7 nm, preferable for hydrogen adsorption, are significantly increased in CA carbon pellets. Moreover, the bulk density of the prepared CA carbon pellet was high (0.80 g cm−3), which was greater than that of the MOF carbon pellet. Consequently, the volumetric hydrogen storage capacity at −196 °C and 1 bar was significantly higher than that of previously reported porous materials. From the above, it follows that the pelletizing technology developed in this study is expected not only to advance the production of hydrogen adsorbents but also to serve as a promising technology for volumetric high-density porous pellet production, applicable to a wide range of future industrial applications.
Data availability
The data that support the findings of this study are available from the corresponding author, Hiroshi Matsutaka (E-mail: E-mail: hiroshi.matsutaka@riken.jp), upon reasonable request.Author contributions
H. M. and D. M. conceived the idea and designed the experiments. H. M. conducted the material synthesis, experiments, and data analysis. A. K. performed the adsorption characterization and data analysis. A. K. and T. O. set up the electric furnace system. T. O. supported the development of a pellet molding experimental system. H. M. wrote the manuscript. D. M., N. U., S. W., and H. N. helped revise the manuscript and contributed to the final version. All authors discussed the results and provided comments on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, CREST Grant Number JP MJCR24S6, Japan. The authors gratefully acknowledge technical support from the Materials Characterization Support Team at the RIKEN Center for Emergent Matter Science for the SEM observation.References
- H. Li, Y. Tao, X. Zheng, J. Luo, F. Kang, H.-M. Cheng and Q.-H. Yang, Energy Environ. Sci., 2016, 9, 3135–3142 RSC.
- V. Ruiz, C. Blanco, R. Santamaría, J. M. Ramos-Fernández, M. Martínez-Escandell, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, Carbon, 2009, 47, 195–200 CrossRef CAS.
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38–52 CrossRef CAS.
- F. Rezaei and P. Webley, Sep. Purif. Technol., 2010, 70, 243–256 CrossRef CAS.
- M. Bastos-Neto, C. Patzschke, M. Lange, J. Möllmer, A. Möller, S. Fichtner, C. Schrage, D. Lässig, J. Lincke, R. Staudt, H. Krautscheid and R. Gläser, Energy Environ. Sci., 2012, 5, 8294 RSC.
- D. G. Madden, et al. , J. Am. Chem. Soc., 2022, 144, 13729–13739 CrossRef CAS PubMed.
- J. J. Purewal, D. Liu, J. Yang, A. Sudik, D. J. Siegel, S. Maurer and U. Müller, Int. J. Hydrogen Energy, 2012, 37, 2723–2727 CrossRef CAS.
- A. L. Sutton, J. I. Mardel and M. R. Hill, Chem.–Eur. J., 2024, 30, e202400717 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- D. W. Kim, Y. Chen, H. Kim, N. Kim, Y. H. Lee, H. Oh, Y. G. Chung and C. S. Hong, Adv. Mater., 2024, 36, 2401739 CrossRef CAS PubMed.
- M. D. Allendorf, Z. Hulvey, T. Gennett, A. Ahmed, T. Autrey, J. Camp, E. S. Cho, H. Furukawa, M. Haranczyk, M. Head-Gordon, S. Jeong, A. Karkamkar, D.-J. Liu, J. R. Long, K. R. Meihaus, I. H. Nayyar, R. Nazarov, D. J. Siegel, V. Stavila, J. J. Urban, S. P. Veccham and B. C. Wood, Energy Environ. Sci., 2018, 11, 2784–2812 RSC.
- A. Gabe, M. Ouzzine, E. E. Taylor, N. P. Stadie, N. Uchiyama, T. Kanai, Y. Nishina, H. Tanaka, Z.-Z. Pan, T. Kyotani and H. Nishihara, J. Mater. Chem. A, 2021, 9, 7503–7507 RSC.
- S. S. Samantaray, S. T. Putnam and N. P. Stadie, Inorganics, 2021, 9, 45 CrossRef CAS.
- Z. Chen, K. O. Kirlikovali, K. B. Idrees, M. C. Wasson and O. K. Farha, Chem, 2022, 8, 693–716 CAS.
- Y. Wang, Y. Xue and A. Züttel, Chem. Soc. Rev., 2024, 53, 972–1003 RSC.
- S. Ghotia, T. Rimza, S. Singh, N. Dwivedi, A. K. Srivastava and P. Kumar, J. Mater. Chem. A, 2024, 12, 12325–12357 RSC.
- E. Boateng and A. Chen, Mater. Today Adv., 2020, 6, 100022 CrossRef.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- N. S. Norberg, T. S. Arthur, S. J. Fredrick and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 10679–10681 CrossRef CAS PubMed.
- P. Benard and R. Chahine, Int. J. Hydrogen Energy, 2001, 26, 849–855 CrossRef CAS.
- J. P. Singer, A. Mayergoyz, C. Portet, E. Schneider, Y. Gogotsi and J. E. Fischer, Microporous Mesoporous Mater., 2008, 116, 469–472 CrossRef CAS.
- B. Yeskendir, J.-P. Dacquin, Y. Lorgouilloux, C. Courtois, S. Royer and J. Dhainaut, Mater. Adv., 2021, 2, 7139 RSC.
- K. Suresh, D. Aulakh, J. Purewal, D. J. Siegel, M. Veenstra and A. J. Matzger, J. Am. Chem. Soc., 2021, 143, 10727–10734 CrossRef CAS PubMed.
- M. Jordá-Beneyto, D. Lozano-Castelló, F. Suárez-García, D. Cazorla-Amorós and Á. Linares-Solano, Microporous Mesoporous Mater., 2008, 112, 235–242 CrossRef.
- D. Araújo, M. C. R. Castro, A. Figueiredo, M. Vilarinho and A. Machado, J. Cleaner Prod., 2020, 260, 120865 CrossRef.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2021, 12, 6368 CrossRef CAS PubMed.
- W. Zhao, V. Fierro, N. Fernández-Huerta, M. T. Izquierdo and A. Celzard, Int. J. Hydrogen Energy, 2012, 37, 14278–14284 CrossRef CAS.
- D. Lozano-Castelló, J. M. Calo, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2007, 45, 2529–2536 CrossRef.
- M. Sevilla, R. Foulston and R. Mokaya, Energy Environ. Sci., 2010, 3, 223–227 RSC.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- X. Yu, S. Liu, G. Lin, Y. Yang, S. Zhang, H. Zhao, C. Zheng and X. Gao, Appl. Surf. Sci., 2019, 498, 143617 CrossRef.
- H. Kabbour, T. F. Baumann, J. H. Satcher, A. Saulnier and C. C. Ahn, Chem. Mater., 2006, 18, 6085–6087 CrossRef CAS.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gómez-Gualdrón, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- P. García-Holley, B. Schweitzer, T. Islamoglu, Y. Liu, L. Lin, S. Rodriguez, M. Weston, J. T. Hupp, D. A. Gómez-Gualdrón, T. Yildirim and O. K. Farha, ACS Energy Lett., 2018, 3, 748–754 CrossRef.
- E. Masika and R. Mokaya, J. Phys. Chem. C, 2012, 116, 25734–25740 CrossRef CAS.
- Y. Gogotsi, C. Portet, S. Osswald, J. M. Simmons, T. Yildirim, G. Laudisio and J. E. Fischer, Int. J. Hydrogen Energy, 2009, 34, 6314–6319 CrossRef CAS.
- Y. Xiao, H. Dong, C. Long, M. Zheng, B. Lei, H. Zhang and Y. Liu, Int. J. Hydrogen Energy, 2014, 39, 11661–11667 CrossRef CAS.
- A. Yurduşen, A. Yürüm and Y. Yürüm, Int. J. Hydrogen Energy, 2020, 45, 12394–12407 CrossRef.
- P. Kowalczyk, H. Tanaka, R. Hołyst, K. Kaneko, T. Ohmori and J. Miyamoto, J. Phys. Chem. B, 2005, 109, 17174–17183 CrossRef CAS PubMed.
- L. Zhang, M. D. Allendorf, R. Balderas-Xicohténcatl, D. P. Broom, G. S. Fanourgakis, G. E. Froudakis, T. Gennett, K. E. Hurst, S. Ling and C. Milanese, Prog. Energy, 2022, 4, 042013 CrossRef CAS.
- J. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911–13913 RSC.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 RSC.
- M. I. M. Kusdhany and S. M. Lyth, Carbon, 2021, 179, 190–201 CrossRef.
- P. Kowalczyk, C. Balzer, G. Reichenauer, A. P. Terzyk, P. A. Gauden and A. V. Neimark, Carbon, 2016, 103, 263–272 CrossRef CAS.
- J. Jagiello and M. Thommes, Carbon, 2004, 42, 1227–1232 CrossRef CAS.
- V. Presser, J. McDonough, S.-H. Yeonb and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- C. Balzer, R. T. Cimino, G. Y. Gor, A. V. Neimark and G. Reichenauer, Langmuir, 2016, 32, 8265–8274 CrossRef CAS PubMed.
- M. A. Lillo-Rodenas, D. Cazorla-Amorosa and A. Linares-Solano, Carbon, 2003, 41, 267–275 CrossRef CAS.
- E. K. Motlagh, S. Sharifian and N. Asasian-Kolur, Bioresour. Technol. Rep., 2021, 16, 100853 CrossRef.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- S. Reljic, C. Cuadrado-Collados, J. Farrando-Perez, E. O. Jardim, M. Martinez-Escandell and J. Silvestre-Albero, Fuel, 2022, 324, 124753 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- S. E. Bambalaza, H. W. Langmi, R. Mokaya, N. M. Musyoka, J. Ren and L. E. Khotseng, J. Mater. Chem. A, 2018, 6, 23569–23577 RSC.
- J. Purewal, M. Veenstra, D. Tamburello, A. Ahmed, A. J. Matzger, A. G. Wong-Foy, S. Seth, Y. Liu and D. J. Siegel, Int. J. Hydrogen Energy, 2019, 44, 15135–15145 CrossRef CAS.
- J. Purewal, D. Liu, A. Sudik, M. Veenstra, J. Yang, S. Maurer, U. Müller and D. J. Siegel, J. Phys. Chem. C, 2012, 116, 20199–20212 CrossRef CAS.
- R. Zacharia, D. Cossement, L. Lafi and R. Chahine, J. Mater. Chem., 2010, 20, 2145–2151 RSC.
- D. G. Madden, D. O'Nolan, N. Rampal, R. Babu, C. Çamur, A. N. A. Shakhs, S.-Y. Zhang, G. A. Rance, J. Perez, N. P. M. Casati, C. Cuadrado-Collados, D. O'Sullivan, N. P. Rice, T. Gennett, P. Parilla, S. Shulda, K. E. Hurst, V. Stavila, M. D. Allendorf, J. Silvestre-Albero, A. C. Forse, N. R. Champness, K. W. Chapman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2022, 144, 13729–13739 CrossRef CAS PubMed.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- S. J. Yang, J. H. Im, H. Nishihara, H. Jung, K. Lee, T. Kyotani and C. R. Park, J. Phys. Chem. C, 2012, 116, 10529–10540 CrossRef CAS.
- P. Dibandjo, L. Bois, C. Estournes, B. Durand and P. Miele, Microporous Mesoporous Mater., 2008, 111, 643–648 CrossRef CAS.
- S. Gao, L. Ge, B. S. Villacorta, T. E. Rufford and Z. Zhu, Ind. Eng. Chem. Res., 2019, 58, 4957–4969 CrossRef CAS.
- N. L. Loh and K. Y. Sia, J. Mater. Process. Technol., 1992, 30, 45–65 CrossRef.
- T. Knorr, P. Heinl, J. Schwerdtfeger, C. Körner, R. F. Singer and B. J. M. Etzold, J. Chem. Eng., 2012, 181–182, 725–733 CrossRef CAS.
- L. Shi, Y. Zhong, H. Cao, H. Wang, Z. Xiong, K. Wang, H. Shen and Z. Chen, Nat. Synth., 2024, 3, 1560–1566 CrossRef CAS.
- X. Yu, B. Li, L. Wu, D. Shi and S. Han, Energy Fuels, 2023, 37, 9938–9955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11, Tables S1–S8 and references. See DOI: https://doi.org/10.1039/d5ta00993f |
| This journal is © The Royal Society of Chemistry 2025 |