
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface amorphized in situ RuO-NiFeOOH/Au islands for electrocatalytic oxygen evolution reaction†
Karthick
Kannimuthu
a,
Pawan
Kumar
a,
Pooja
Gakhad
b,
Hadi Shaker
Shiran
a,
Xiyang
Wang
 c,
Ali
Shayesteh Zeraati
a,
Sangeetha
Kumaravel
d,
Shariful Kibria
Nabil
a,
Rajangam
Vinodh
a,
Md Abdullah
Al Bari
agh,
Maria
Molina
a,
George
Shimizu
a,
Yimin A.
Wu
c,
Pulickel M.
Ajayan
e,
Abhishek Kumar
Singh
b,
Soumyabrata
Roy
ef and
Md Golam
Kibria
*a
c,
Ali
Shayesteh Zeraati
a,
Sangeetha
Kumaravel
d,
Shariful Kibria
Nabil
a,
Rajangam
Vinodh
a,
Md Abdullah
Al Bari
agh,
Maria
Molina
a,
George
Shimizu
a,
Yimin A.
Wu
c,
Pulickel M.
Ajayan
e,
Abhishek Kumar
Singh
b,
Soumyabrata
Roy
ef and
Md Golam
Kibria
*a
aDepartment of Chemical and Petroleum Engineering, University of Calgary, 2500 University Drive, NW Calgary, Alberta T2N 1N4, Canada
bMaterials Research Centre, Indian Institute of Science, Bangalore 560 012, India
cDepartment of Mechanical and Mechatronics Engineering, Waterloo Institute for Nanotechnology, Materials Interface Foundry, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada
dDepartment of Chemistry, Kalasalingam Academy of Research and Education, Srivilliputhur 626126, India
eDepartment of Materials Science and Nanoengineering, Rice University, 6100 Main St., Houston, TX 77030, USA
fDepartment of Sustainable Energy Engineering, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh 208016, India. E-mail: md.kibria@ucalgary.ca
gDepartment of Mechanical Engineering, King Fahd University of Petroleum & Minerals, Dhahran, 31261, Saudi Arabia
hInterdisciplinary Research Center for Sustainable Energy Systems, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
First published on 8th May 2025
Abstract
Hydrogen production via electrocatalytic water splitting is largely impeded by the anodic oxygen evolution reaction (OER). Herein, we report surface amorphized Ru-NiFeP/Au islands as an effective electrode for the OER in 1 M KOH, reaching a current density of 10 mA cm−2 at 223 mV overpotential. The iR corrected Tafel slope was calculated to be 32 mV dec−1, while electrochemical impedance spectroscopy (EIS) studies revealed a clearly low charge transfer resistance of 0.3 Ω at 400 mV overpotential. The high electrocatalytic activity was attributed to the amorphous nature, reduced band gap, and synergism of Ru-NiFeP with Au. In situ surface-enhanced Raman scattering (SERS) revealed the role of FeOOH at lower overpotentials for facile OH adsorption. The evolution of NiOOH peaks at higher overpotentials for O2 evolution coupled with synergistic Ru–O bonds to promote the OER was studied with DFT analysis. Bader charge analysis showed that the charge transfer from Fe to O is 0.17 units greater than that from Ni to O for *OH intermediate generation at the active site, and this corroborates the results from in situ SERS studies, where FeOOH is the active site at lower overpotentials. The bond order characteristics become more pronounced when the FeOOH/NiOOH surfaces are accessible. DFT analysis revealed a low free energy change (0.12 eV) for the rate-determining step at the RuO/NiFe-OOH surface.
Introduction
The four-electron oxygen evolution reaction (OER, 4OH− → O2 + 2H2O + 4e−) is a key half-cell reaction involved in vital energy conversion processes, such as those occurring in water and CO2 electrolyzers,1 photoelectrochemical water splitting,2 and metal–air batteries.3 Water electrolyzers powered by renewable energy sources, such as wind and photovoltaic cells, can be utilized to produce green hydrogen.4 However, the energy efficiency of these devices is hindered by the kinetically sluggish M–OH, M–O formation (M: metal) and subsequent O2 evolution with 4e− transfer during anodic OER. Commercial water electrolyzers require 1.8–2.0 V. Furthermore, scarce materials like IrO2 and RuO2 are used as OER electrodes, affecting economic viability.5 Consequently, the search for non-noble metal electrodes with improved performance is anticipated to avoid the staggering price for the sustainable and widespread commercial utilization of alkaline water electrolyzers. In alkaline electrolytes, 3d transition metals (TMs: Fe, Co, and Ni) have been studied in detail for the OER with considerable enhancement in activity and stability.6,7 This is related to the low crystal field activation energy with favorable M(3d)–O(2p) overlap of these TMs for facile M–OH adsorption and O2 cleavage.8 Recently, many reports have highlighted the use of metal oxides,9–14 hydroxides,15–19 and metal chalcogenides20–23 with substantial activity. However, their applicability is limited due to low electrical conductivity. Alternatively, TM phosphides (TMPs) can deliver exceptional OER performance with superior electrical conductivity.24–27TMPs act as ‘pre-catalysts’, and in situ structural studies have revealed the operando-generated TM oxyhydroxides (TMOOHs) as the active centers that are vital for enhancing the transport of O-intermediates.28,29 Peng et al. highlighted the in situ oxidized Ni2P/Fe2P interface for the OER and elucidated the role of surface PO43− ions and conductive bulk phosphides in accumulating charges in vacant 3p and 3d orbitals.28 The nature of the covalent strength and inductive effect of metal phosphates was investigated by Siraj et al., where the lowering of the M–O antibonding energy levels induces a positive shift of the redox potential in Fe3Co(PO4)4 to modulate the OER activity.30 As shown by Panlong et al., the inclusion of Ru(4d) to 3d transition metals promotes O–O coupling at the Ru–O active site for OER in the Ru/NiFe LDH single atom sites.16 Due to the M–H bond strength being similar to that of Pt and its lower costs, Ding et al. explored Ru-doped MnFeP/NF for efficient solar-to-hydrogen (STH) fuel generation with photo-electrocatalysis.31 In another work, an amalgamation of Ag over Ni–Fe–P was successfully carried out by Zhiyuan et al., where the non-metal P was crucial to amalgamate Ag for utilization in OER and ORR (oxygen reduction reaction) studies.32 Understanding ‘active sites’ for complex OER in multi-metallic phosphides at a molecular level can bridge the gap between the catalytic structure and activity, which is crucial to develop effective catalysts.25 Similar to phosphides, inclusion of noble metals like Ir and Ru in oxides improved the OER performance because of the surface electron distribution and oxygen vacancy sites.33,34
In highly active NiFe-OOH for OER, the actual active surface is always a matter of concern.35 It was observed from in situ Raman studies that eg bending (475 cm−1) and A1g stretching vibrations (555 cm−1) confirm NiOOH as active sites.36,37 The relative intensity (I475/I555) of Ni–O in NiOOH decreases in the presence of Fe, suggesting a modified local electronic environment, and it was proven that Fe promoted the Ni(OH)2-to-NiOOH (active site) transformation.36 SERS is a potent tool for the molecular detection of intermediates under an OER bias with a high spatial resolution because of its ultra-sensitivity at the low-frequency range and non-interference with water.38 However, the sensitivity of SERS is limited to a few metals like Au and Ag. One way to overcome this limitation is a borrowing strategy, where the electrocatalyst is grown over the rough surfaces of Au.39 This strategy helps to amplify the signals of short-lifetime species and acquires the surface properties of the electrocatalytic system. Also, the electrophilic character of Au as an ‘electron sink’ stabilizes the high oxidation states of the active sites during OER. Because of its O2-repellent nature, this ensures the fast evolution of O2 from the surface. The presence of Au assisted the conversion of oxyhydroxides. In a work by Jakob et al., the Au(111) interface in cobalt oxide favored the transformation of cobalt oxyhydroxide during OER, rather than the less active bulk cobalt oxide.40 Furthermore, in NiFe LDH systems, the use of Au as single atoms resulted in an increase in the OER activity due to the formation of NiFe oxyhydroxides.41
To ensure high catalytic activity, surface amorphization is another important strategy for developing ‘dangling bonds’ to achieve effective charge transfer due to the local short-range order.42,43 Herein, we envisioned surface amorphization and synergism of Ru over the NiFeP/Au surface to enhance the OER activity, and explored in situ SERS analysis to understand the mechanisms and active sites. Initially, the Ni foam (NF) is replaced with Au via galvanic replacement, followed by electrodeposition to develop RuFeP/Au-Ni islands for OER in 1 M KOH. The optimized heterostructure catalyst (Ru15-NiFeP) requires an overpotential of just 223 mV to attain an OER current density of 10 mA cm−2, while exhibiting a low Tafel slope (32 mV dec−1). Spectroelectrochemical investigations with in situ SERS suggested the direct role of FeOOH surface-active sites at low overpotential and NiOOH at higher overpotential for OER. In situ EIS measurements in the applied potential window of 1.3–1.6 V in the OER region indicated increased O-intermediate kinetics on Ru15-NiFeP/Au compared to Au–Ni and NiFeP/Au. DFT studies confirmed that the presence of Fe induces ionic character in the Fe–O bond for the better adsorption of O-containing intermediates, while Ni (with the increased covalency) result in relatively poor O-intermediate adsorption, which is in line with in situ SERS outcomes. This work provides insight into the development of TMPs with improved synergy for OER, and further reveals a deeper understanding of the role of OER active sites via in situ SERS studies.
Results and discussion
Synthesis and characterization
Ru-NiFeP with different concentrations of Ru was developed through the electrodeposition method on a three-dimensional porous skeleton of Au–Ni formed via galvanic replacement, as portrayed in Fig. 1a. Notably, the entire synthesis and fabrication process was carried out at room temperature under ambient conditions and no harsh conditions were employed. The crystalline feature of Au–Ni was assessed via X-ray diffraction (XRD), where the peaks at 38.6° (200), 64.3° (022), and 81.4° (222) confirmed the presence of metallic Au, in addition to the metallic Ni peaks at 44.5° (111), 51.8° (200), and 76.4° (220) (Fig. S1a and b†).44 For Ru15-NiFeP/Au, no additional characteristic peaks appeared, indicating the amorphous nature of Ru15-NiFeP. Synchrotron-based grazing incidence-wide angle X-ray scattering (GI-WAXS) analysis on Au–Ni discerned the crystalline nature of Au with sharp diffraction rings and corresponding Q-values (Fig. S2a and b†). Ru15-NiFeP/Au comprises similar amorphous characteristics, and is in line with the XRD results (Fig. 1b and c). Scanning electron microscopy (SEM) elemental mapping and energy dispersive X-ray spectroscopy (EDX) of Au–Ni (Fig. S3a–d†) and Ru15-NiFeP/Au (Fig. 1d) display the homogenous distribution of Ru, Fe, Ni, and P on the Au islands. For different concentrations of Ru, the formed Ru-NiFeP showed similar morphological features (Fig. S4–S6†). To gain further structural insight, high-resolution transmission electron microscopy (HR-TEM) and high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) were employed. As shown in Fig. 1e, the HR-TEM images demonstrate the uniform sheet morphology for Au-Ni and Ru15-NiFeP over the Au-Ni layer. High-magnification TEM images of Ru15-NiFeP/Au in the nanosheets regions displayed lattice fringes with 0.14 nm d-spacing corresponding to Au(002), while Ru15-NiFeP nano islands were largely amorphous with a short-range crystallinity and reflection originating from the underlying Au/Ni layers (Fig. 1e and f). The selected area electron diffraction (SAED) pattern in Fig. 1e inset shows the diffused ring pattern due to the amorphous nature, while the fast Fourier transform (FFT) of the image in Fig. 1f inset displays sharp diffraction spots due to the Au (111) plane, which is consistent with the XRD results.43 HAADF-STEM elemental mapping of Ru15-NiFeP/Au further validated the homogeneous distribution of the constituting elements (Fig. 1g–l). The EDX spectrum and intensity profile in STEM mode confirmed the presence of Ru (0.64%), Fe (0.07%), Ni (14.26%), Au (0.03%), and P (0.51%) from the Ru-NiFeP/Au islands (Fig. 1m). The Raman spectra of NiFeP/Au and Ru-NiFeP/Au exhibited signature peaks centered between 300 and 700 cm−1, corresponding to the Fe–P and Ru–O stretching vibrations, respectively (Fig. 1n).39,45 The morphologies of Au-Ni and Ru15-NiFeP/Au at various magnifications are shown in Fig. S7–S8.† | ||
| Fig. 1 (a) Schematic of the synthesis of Ru-NiFeP/Au catalysts. (b and c) Synchrotron-based WAXS 2D map and corresponding Q-values. (d) SEM elemental mapping color composite of Ru15-NiFeP/Au islands. (e) HRTEM images of Ru15-NiFeP/Au islands, with the SAED pattern in the inset. (f) High-magnification TEM image of Ru-NiFeP/Au, with the corresponding FFT in the inset. (g–l) EDS mapping results of the mix composite, Au, Ru, Fe, Ni, and P. (m) EDS spectra in STEM mode, with the EDS intensity profile in the inset. (n) Raman spectra of NiFeP/Au and Ru15-NiFeP/Au catalysts. | ||
To unravel the electronic and chemical composition, X-ray photoelectron spectroscopy (XPS) was conducted for the Au-Ni and Ru-NiFeP/Au materials. The XPS of Au-Ni in the Au4f region shows two peaks at 83.8 and 87.5 eV assigned to 4f7/2 and 4f5/2, respectively, indicating metallic Au (Fig. S9†).41 The Ni2p peaks at 854.8 and 872.4 eV originating from the 2p3/2 and 2p1/2 transitions correspond to the +2 states of the surface oxidized Ni of the Ni foil.41 The Ru3p core level spectrum of Ru-NiFeP/Au exhibited two peaks centered at 463.1 and 485.3 eV due to intermediate oxidation between Ru(0) and Ru(IV), respectively, while the peaks at 468.2 and 488.8 eV resulted from the Ru(IV) states (Fig. S10†).31,42,46 For the Fe2p spectrum, the XPS peaks at 712 eV originated from 2p3/2 and those at 709.6, 711.4, and 713.9 eV are related to the +2, +3, and satellite peaks of Fe, respectively.43 The Ni2p XPS spectrum of Ru-NiFeP/Au shows peaks at 854.3, 856.1, 872.3, and 874.6 eV, and are indexed to +2/+3 oxidation states.47,48
The Ni2p and Au 4f peaks of Ru-NiFeP/Au were shifted 0.5 and 0.2 eV, respectively, toward low BE value compared to Au–Ni, indicating the electronic coupling of Ru and Fe with Ni. The P2p spectrum shows peaks at 131.08 and 132.5 eV corresponding to the P3− and PO43− states.28 The Ru3d spectrum shows the presence of the Ru3d3/2 and Ru3d5/2 states for the Ruδ+ states.31,42,46 The O1s core spectra display four peaks indexed to M–O, M–P, M–H2O, and a shoulder peak at 528–532 eV for the surface adsorbed –OH, respectively.28,43 XPS analysis for other concentrations of Ru in Ru-NiFeP/Au also demonstrated a similar pattern (Fig. S11†). The electronic nature and coordination environment of Ru15-NiFeP/Au were investigated using synchrotron-based soft X-ray absorption (sXAS) (TEY mode), X-ray absorption near-edge structure (XANES), and extended X-ray absorption fine structure (EXAFS) analyses. Fig. 2a and b show the Excitation-Emission Matrix Spectroscopy (EEMS) scanning profile for Ru15-NiFeP/Au. The EEMS scan clearly shows bright Ru M, Ni L (Fig. S12a†), P K (Fig. S12b†) and O K (Fig. S12c†) and Fe L edges; however, the Au edge cannot be detected due to the high energy of the Au L-edge compared to the energy range for soft X-rays (2000 keV). The Ru M-edge sXAS spectra of Ru15-NiFeP/Au displayed two signature peaks for the Ru MIII and Ru MII-edges at 485.4 and 463.5 eV, suggesting Ruδ+ ≤ 4+ oxidation state (Fig. 2c).49 The Fe L-edge spectra also show two peaks (Fe LIII and Fe LII) at 720.9 and 709.4 eV for iron present in the 3+ oxidation state (Fig. 2d).50 The Fe LII edge displayed t2g and eg peak splitting, suggesting the presence of the O coordination iron species. Fig. 2a–d reveal that Ru15-NiFeP/Au contains Ruδ+ ≤ 4+ and Fe 3+ oxidation states, respectively. The Ni K-edge XANES spectra of NiO displayed the edge energy at around 8330.5 eV with a pre-edge feature, while the Ni foil was metallic. Therefore, a positive shift was observed for Ni in Ru15-NiFeP/Au, indicating the oxidized Ni in the material (Fig. S13†).51 The Fe K-edge XANES spectra of Ru15-NiFeP/Au exhibited a sharp rising edge at 7115.6 eV, along with a pre-edge feature similar to that reported for γ-FeOOH or Fe2O3, corroborating the 3+ octahedral (Oh) environment for the Fe species.52 Furthermore, the Fe K-edge energy of Ru15-NiFeP/Au was present at relatively high energy compared to Fe2O3 (Fe3+), signifying the electron deficient under-coordinated Fe species (Fig. 2e).53 The difference between Fe2O3 and Ru15-NiFeP/Au is related to a change in coordination in the latter. The increased white line intensity for Ru15-NiFeP/Au further strengthens the assumption of a partial charge transfer from Fe3d → O2p.53 The Ru K-edge XANES spectra also displayed a similar enhancement in the white line peak intensity due to the undercoordinated state (Fig. 2f).42 The Fourier transform (FT) Fe-EXAFS spectra of Ru15-NiFeP/Au show an intense first shell scattering at 1.39 Å, originating from the Fe–P/O coordination. The presence of weak scatterings features at higher R values (2.66 and 3.29 Å) originated from the Fe–O–Fe and Fe–O–Ru coordination, suggesting partial oxidation of the Fe–P species (Fig. 2g).52,53 The EXAFS data fitting of Ru15-NiFeP/Au, considering the presence of the Fe–P/Fe–O coordination, demonstrated a Fe–P/Fe–O bond length of 2.34/1.90 Å with a coordination number of 4.4/4.5 (Table S1†). The lower CN values of Fe–P/Fe–N in Ru15-NiFeP/Au compared to that of the crystalline FeP and Fe–O (Fe3O4) structures suggest that the undercoordinated species probably originated from the unfulfilled coordination in the amorphous structure. The Ru EXAFS spectra of Ru15-NiFeP/Au shows peak features that are significantly different from those of RuO2 and RuCl3. The Ru–O/P scattering was observed at low R values compared to RuCl3, suggesting the successful transformation of ruthenium chloride to phosphide. Additionally, the Ru–O–Ru peak for the RuO2 state was absent, indicating that the Ru–O sites were not present in the oxide. Furthermore, no peak features corresponding to metallic Ru were observed, indicating that Ru was bonded with P/O (Fig. 2h).42,46 The fitting of EXAFS gives Ru–O, Ru–P and Ru–Fe bond lengths of 1.92, 2.03 and 2.59 Å, and R values with a CN of 1.3, 4.6 and 6.9, respectively. The R-values for Ru and Fe in EXAFS show that the coordination lengths of Ru–O and Fe–O were slightly shifted at higher value due to the introduction of the Ru–Fe–P species in Ru-NiFeP/Au.42,53 The Wavelet Transform (WT) EXAFS map of Ru for Ru15-NiFeP@Au and RuCl3 was further acquired to understand the coordination environment (Fig. 2i and j). The Ru WT EXAFS map for Ru15-NiFeP@Au displayed a sharp bright zone centered at K = 7.37 Å−1 and R = 1.77 Å, originating from Ru–P/O scattering. These scatterings were quite different from those of RuCl3 (K = 7.02 Å−1 and R = 1.56 Å), suggesting the complete transformation of RuCl3 to Ru–P during electrocatalytic synthesis. A very faint zone in the WT map of Ru15-NiFeP@Au at K = 10.98 Å−1 and R = 2.47 Å was assigned to an extremely small contribution of Ru–Fe/Ru–P–Ru scattering.
 | ||
| Fig. 2 Synchrotron-based soft and hard X-ray absorption spectra of the Ru15-NiFeP@Au islands. (a) and (b) EEMS scanning profiles of Ru and Fe. (c) and (d) Corresponding soft XAS spectra for the Ru M-edge and Fe L-edge. (e) and (f) Fe K-edge and Ru L-edge XANES spectra (dots–raw data and lines–fitted data). (g) and (h) FT-EXAFS spectra of Ru and Fe. (i) and (j) Ru WT EXAFS map of RuCl3 and Ru15-NiFeP@Au. | ||
Electrocatalytic OER assessment of Ru-NiFeP/Au
The electrochemical OER activity of the electrodes was assessed in an O2-saturated three-electrode setup in 1 M KOH. Initially, polarization studies were conducted at 5 mV s−1 and the backward CV response is provided to exclude any capacitive interference, as shown in Fig. 3a. Among the different electrodes, Ru15-NiFeP/Au exhibited outstanding activity, exhibiting low overpotentials (η) of 223 and 256 mV at current densities (j) of 10 and 100 mA cm−2, respectively. Other variants required comparatively higher overpotentials [e.g., Ru5-NiFeP/Au (242 mV), Ru25-NiFeP/Au (254 mV), NiFeP/Au (284 mV), NiFeP/Ni (287 mV), Au–Ni (314 mV), and pristine Ni (410 mV)] to deliver 10 mA cm−2. The carrier migration is well enriched by suppressing the Ni2+ to Ni3+ oxidation behavior on the RuFeP sites compared to only Ni, as evidenced by the positive steep increase in the current.16,54 The nature of the charge transport phenomenon derived from log j versus η and the low Tafel slope of 32 mV dec−1 obtained for Ru15-NiFeP/Au infers a 4e− transfer process. Meanwhile, others showed high Tafel values (Fig. 3b), Ru5-NiFeP/Au (47 mV dec−1), Ru25-NiFeP/Au (35 mV dec−1), NiFeP/Au (68 mV dec−1), NiFeP/Ni (55 mV dec−1), Au–Ni (120 mV dec−1) and pristine Ni (132 mV dec−1), indicating lower charge transport kinetics. The charge transfer resistance (Rct), representative of the electron transfer ability of the electrodes, was measured via EIS at 400 mV vs. RHE from the fitted electric equivalent circuit (EEC). From Fig. 3c, the Rct, measured using the corresponding equivalent circuit (inset), steadily decreased from pristine Ni (2.53 Ω), Au–Ni (2.01 Ω), and NiFeP/Au (0.752 Ω) to Ru15-NiFeP/Au (0.29 Ω), with the lowest semicircle. This agrees with the polarization studies and the sharp rise in current for Ru15-NiFeP/Au, signifying greater carrier migration. A comparison of the state-of-the-art RuO2 is shown in Fig. S14.† The electrochemical active surface area (ECSA) for the catalysts was measured from the double-layer capacitance (Cdl) (Fig. S15†). As shown in Fig. S15,† Ru15-NiFeP/Au showed a lesser slope value (7.6 mF cm−2) compared to Au–Ni (14.53 mF cm−2). This directly demonstrates the improved charge migration across the interface and the capacitance in Au–Ni being linked to the superior ‘electrophilic nature of Au’.41 Furthermore, the intrinsic specific activity from the ECSA normalized LSVs indicate the improved OER performance for the Ru15-NiFeP/Au islands, as depicted in Fig. 3d. The calculated TOF values at the potential intervals (Fig. 3e) showed that Ru15-NiFeP/Au delivers better O2 turnover (0.012 S−1 at 1.7 V). Ru15-NiFeP/Au delivered a high mass activity of 408 A g−1 at 1.7 V (Fig. 3f). This is attributable to the surface reconstruction enabling lesser active species results in low loading and improved mass activity.55 The OER performance of Ru15-NiFeP/Au is superior to many other related effective electrodes in terms of the overpotential (η10) and Tafel slope, as shown in Fig. 3g.56–64 A comparison of the catalytic performance of Ru15-NiFeP/Au with various phosphide catalysts is also tabulated in Table S2.† LSVs at different pH values of 12, 13, and 14 were analyzed to understand the onset potential of Ru15-NiFeP/Au. As shown in Fig. 3h, a linear decrease in the onset potential with increasing pH signifies the population of OH− ions at the electrode/electrolyte interface. The electrochemical stability of Ru15-NiFeP/Au was tested under potentiodynamic and potentiostatic conditions. The LSV study performed before and after cycling, as shown in Fig. S16,† demonstrates the high stability with minimum degradation. Furthermore, a constant current density of 100 mA cm−2 is retained with very low decrement in activity for 100 h, suggesting the superior stability of the Ru15-NiFeP/Au islands (Fig. 3i).26,27 To unveil the catalyst robustness stability, post-OER characterizations such as SEM with mapping (Fig. S17†), HR-TEM (Fig. S18†) and XPS (Fig. S19†) analyses were carried out. The SEM images showed the retained structural features of the Ru15-NiFeP/Au islands, and the mapping results confirmed the presence of Au, Ni, Fe, Ru, and P. The HR-TEM images demonstrate the Ru-NiFeP islands after harsh anodic conditions, indicating the substantial stability of the catalyst. The XPS high-resolution spectra of the Ru 3d, Ni 2p, Fe 2p, and P 2p demonstrate the stability and show the oxidized forms of the catalyst surface that occur due to the RuOx, Ni and Fe oxy-hydroxides, which is usually formed under anodic overpotentials in KOH.26,27 | ||
| Fig. 3 Electrocatalytic OER performance of the electrodes. (a) Backward cyclic voltammetry (CV) curves in 1 M KOH at a scan rate of 5 mV s−1. (b) Corresponding Tafel slopes. (c) EIS at 400 mV overpotential. Inset showing the Randle circuit obtained after fitting the EIS curve. (d) Specific activity. (e) Turn over frequency. (f) Mass activity. (g) Comparison of the overpotential and Tafel slope with literature. (h) Onset potential versus pH effect on Ru15-NiFeP@Au-Ni. (i) Potentiostatic stability of Ru15-NiFeP@Au-Ni for 100 h in 1 M KOH. | ||
Dynamic spectroelectrochemical investigations on electrocatalysts with in situ EIS and in situ SERS analyses
The in situ EIS method is an important tool that can be used to elucidate details on the adsorption and desorption of intermediates at the electrode/electrolyte interface.65In situ EIS between 1.3–1.6 V with 50 mV potential interval was performed on Au–Ni, NiFeP/Au, and Ru15-NiFeP/Au to assess the kinetics of the O-intermediates on the metal active sites and the resulting electrical conductivity, as depicted in Fig. 4a–c. The iR-uncorrected CV from 1.3–1.6 V chosen for EIS is given in Fig. S20,† where the chosen region couples the oxidation of the Ni2+/Ni3+, Fe2+/Fe3+ states and concomitant OER. The Nyquist plots revealed the trends in the O-intermediate kinetics with a semicircle arc from 1.3–1.6 V, which were significantly reduced with increasing potentials, showing the alleviated O-adsorption for a faster charge transport. Among them, the Ru15-NiFeP/Au islands delivered improved electrical conductivity due to the abrupt adsorption of the O-intermediates. The EEC was fitted from 1.45–1.6 V, and the resultant Rs and Rct values are shown in Fig. 4d. The OER process at this region is completely charge transfer-controlled. From a closer look at Au–Ni in Fig. S20,† the potential is exploited more for the oxidation current in the oxidation of Ni2+/Ni3+. The O-intermediate charge transport is suppressed, agreeing with the EIS results with the increased semicircle arc. In the case of NiFeP/Au and Ru15-NiFeP/Au islands, the oxidation current for the Ni2+/Ni3+, Fe2+/Fe3+ states are suppressed. The applied potential is mainly used for charge transport, and the O-intermediate adsorption is facile for Ru15-NiFeP/Au with improved charge transport (Fig. 4d).66 These results match nicely with the polarization studies given in Fig. S20,† where the oxidation and OER currents are varied, and the oxidation currents are mainly reduced after coupling the Ru15-NiFeP/Au islands with the faster O-intermediate adsorption. To further investigate the nature of the actual active sites under the applied potential regime, in situ SERS studies were carried out in 1 M KOH under varying potential intervals.38,39 SERS studies mainly require Au, Ag, or Cu as coinage metals to intensify the signals of the surface adsorbed species that are relatively low in concentration and exhibit short lifetimes during OER.39 In NiFe systems, the role of the active sites was previously investigated by Cejun et al. with the Au plasmonic core for SERS.38 Interestingly, it was found that Fe atoms are the preliminary sites for the oxidation of OH− to O–O− at lower overpotentials, and that NiIII acted as a site for oxidation of O–O− to O2 at higher overpotentials. It was affirmed that FeOOH acts as a real active site at low bias, while NiOOH catalyzes O2 evolution at high bias, yielding a ‘dual metal synergistic mechanism’ in the Ni–O–O–Fe structure.38 Based on these findings, initially, Ni foam is chosen as a substrate and in situ SERS studies were performed between 1.4–1.65 V at 50 mV interval (Fig. 4e). As shown in Fig. S20,† under OCP and until 1.35 V, no peaks evolved, suggesting that there was no oxidation. At 1.4 V, two new peaks were observed at 465 and 584 cm−1, corresponding to the A1g and B2g modes from NiIII–O stretching in NiOOH.38 These peaks were enhanced with increasing potentials, and started to fade at high overpotentials above 1.65 V due to the bubble formation that interferes with exposure to the laser beam (Fig. 4e). The Lorentzian fitting of Ni is shown in Fig. S21,† and the peak position and areas were plotted as a function of the applied potential. To elucidate the nature of the active species in NiFeP/Au, SERS studies were carried out under OCP, from 1.35 to 1.45 V at 5 mV intervals in the lower potential range to identify the precise changes (Fig. 4f). | ||
| Fig. 4 In situ EIS and in situ SERS results. (a–c) In situ EIS spectral studies of Au–Ni, NiFeP/Au, and Ru15-NiFeP/Au at varying potential intervals from 1.3 to 1.6 V. (d) Comparison of fitted Rs and Rct values for Au-Ni, NiFeP/Au, and Ru15-NiFeP/Au.ues (e) In situ SERS studies on Ni foam from 1.4 to 1.65 V with a 50 mV potential interval. (f and g) In situ SERS results of NiFeP/Au and Ru15-NiFeP/Au islands with 5–10 mV potential interval. (h and i) Area and peak position difference in NiFeP/Au for NiOOH A2g and FeOOH peaks. | ||
For NiFeP/Au, there was no increase in the peak intensity observed until 1.375 V. At 1.38 V, a new peak corresponding to the FeOOH sites for O–O bond formation was observed. At 1.4 V and above, a new Ni3+ peak appeared for NiOOH, which acted as an ‘active site’ for O2 desorption at the surface.28,38,67 From these observations, it is affirmed that the Ni–O–O–Fe sites are essential for OER and at low overpotentials. Fe is the ‘active site’ for adsorbing the hydroxides. At higher overpotentials, Ni3+ offers a fast generation of O2 molecules.38 Furthermore, the prepared M–P surface changed to oxides/oxyhydroxides during OER and acted as ‘real active sites’. This SERS observation reveals how the presence of both Fe and Ni controls the O2 formation. The Lorentzian fitting of NiFeP/Au (Fig. 4h) shows that the peak for FeOOH increases in intensity at 1.38 V and fades at higher bias, whereas the NiOOH peak intensity is enhanced after 1.4 V (Fig. 4i). In the case of Ru15-NiFeP/Au, Ru–O peaks are observed in addition to the peaks for NiOOH and FeOOH (Fig. 4g). This indicates the formation of Ru–O bonding over Ni–Fe–O–O–H, which alters the local charge distribution near the surface in high alkaline conditions.39,46 The Lorentzian fitting of the peak position and area is depicted in Fig. S22.† In the Ru15-NiFeP/Au islands, Ru ensures a high-rate of activity, and the presence of Ru–O bonds for the M(4d)–O(2p) synergism improves the catalytic activity of the NiFe-oxyhydroxide surface.68
Computational mechanistic studies on Ru15-NiFeP/Au
A schematic representation of the plausible electron transfer pathways is depicted in Fig. 5a, which portrays the electron transport with increased electrical conductivity in amorphous catalysts.68 The doping of Ru over FeP resulted in a decrease in the band gap, thus inducing a facile electron transfer, as shown in the histogram for FeP and RuFeP (Fig. 5b).68 The optical band gap calculated using the Tauc plot for FeP and RuFeP was found to be 2.34 eV and 1.56 eV, respectively, validating the band gap reduction (Fig. S23a and b†). Furthermore, water contact angle studies (Fig. 5c) demonstrated the superhydrophilic wettability of Ru15-NiFeP/Au compared to pristine NF (water contact angle 120°), which is expected to contribute towards the high catalytic activity in OER. This ‘superhydrophilicity’ was prompted from the conducting/hydrophilic P3− ions and PO43− groups in the bulk.44In situ SERS studies confirm that NiFeOOH/RuO are the active sites on which the O-intermediates turn into O2 molecules. Thus, we performed DFT calculations for RuO over the NiFe-OOH system to understand the OER mechanism on this catalyst. It is well known that the nature of the active sites on the surface can significantly impact the adsorption energy of the intermediates.69,70 The surface of NiFe-OOH has two types of oxygen. The type-1 configuration shows an active site where the surface oxygen is connected to two Ni atoms and one Fe atom, whereas the type-2 configuration shows the surface oxygen connectivity with two Fe atoms and one Ni atom (Fig. S24 and S25†). After identifying the two non-equivalent sites over the NiFe-OOH surface, the RuO unit is incorporated above the Fe atom and named as type-1 RuO/NiFe-OOH, and above the Ni atom as type-2 RuO/NiFe-OOH (Fig. S26 and S27†). The OER pathways in type-1 NiFe-OOH and type-2 NiFe-OOH are shown in Fig. S28,† and the OER pathways in type-1 RuO/NiFe-OOH and type-2 RuO/NiFe-OOH are provided in Fig. 5d and e. At U = 0 V, the Gibbs free energy is ascending from *OH to *OOH for all four systems (Fig. 5f, g and S29†). For type-1 NiFe-OOH, the potential limiting step is *OH formation and adsorption on the surface-active site since the ΔG value is maximum for this step, i.e., 1.94 eV. However, for type-2 NiFe-OOH, the potential limiting step is deprotonation of *OH to *O with a ΔG value of 1.82 eV. The onset potential has been reduced from type-1 to type-2 NiFe-OOH by a magnitude of 0.12 V. This change in magnitude can be attributed to the difference in the chemical environment of surface oxygen. For the type-2 case, the overall OER activity has been enhanced because of the presence of two Fe atoms. The Fe atom induces ionic character in the Fe–O bond, which helps in better absorption of the intermediates. This observation agrees with the in situ SERS results, where the O-intermediate adsorption is facile in FeOOH sites. However, for the Ni atom, the increased covalency in the Ni–O makes it relatively poor against OER activity.47 At the theoretical onset potential of OER, i.e., 1.23 V, both type-1 and type-2 NiFe-OOH exhibit ascending free energy until the *O intermediate and then descending until O2. However, the ΔG values for the PLS of type-1 and type-2 at the theoretical onset potential are 0.71 eV and 0.58 eV, respectively. Hence, to make the reaction spontaneous, an onset potential of 1.94 V must be applied for type-1 NiFe-OOH and 1.82 V for type-2 NiFe-OOH. To better understand the charge transfer, a Bader charge analysis was performed.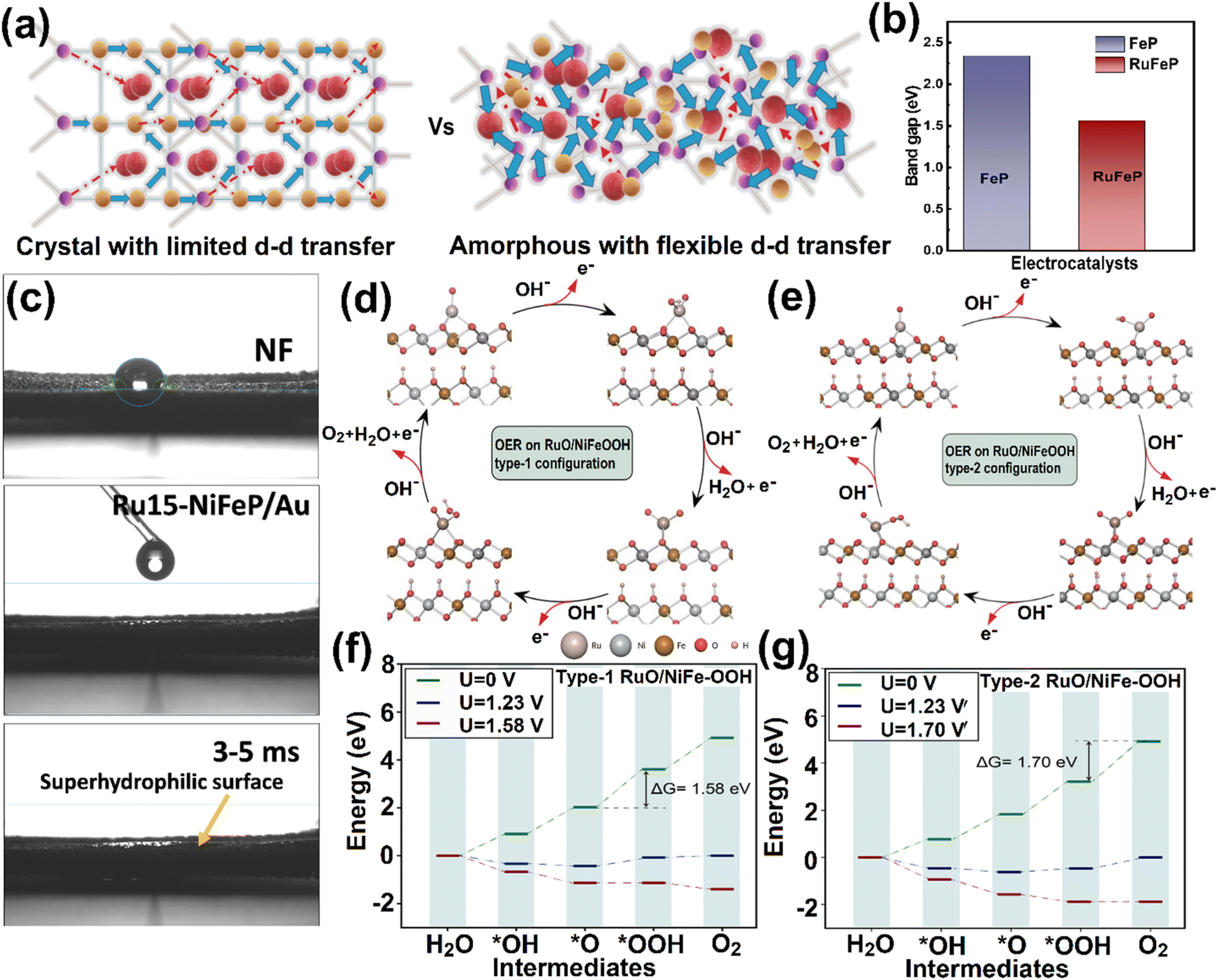 | ||
| Fig. 5 (a) Electronic structure of crystalline and amorphous RuFeP islands. (b) Band gap of the FeP and RuFeP electrocatalysts. (c) Superhydrophilic nature of Ru15-NiFeP/Au islands, on which the water droplet falls in immediately within 3–5 ms. (d) OER on the RuO/NiFeOOH type-1 configuration. (e) OER on the RuO/NiFeOOH type-2 configuration. (f) OER free energy profile on the RuO/NiFeOOH type-1 configuration and (g) OER free energy profile on the RuO/NiFeOOH type-1 configuration. | ||
For the *OH intermediate, the charge transfer from Fe to O at the active site is 0.17 units more than the charge transfer from Ni to O at the active site, further confirming the role of Fe as preliminary active sites in OER (Tables S3–S6†). Additionally, the charge separation between the O and H of *OH is less for the case of type-2 NiFe-OOH. The deprotonation step for the same becomes difficult compared to type-1. Therefore, the potential limiting step for type-2 NiFe-OOH is deprotonation of *OH to *O. Furthermore, the *OOH intermediate is weakly adsorbed on the surface for both cases (Fig. S25†). The presence of the RuO unit over NiFe-OOH has shown a significant reduction in the overpotential of OER. The oxyhydroxide intermediate is stable on the surface when Ru is the active site on RuO/NiFe-OOH (Fig. 5d and e). The type-1 RuO/NiFe-OOH requires 1.58 V for the formation of the oxyhydroxide intermediate. On the other hand, for type-2 RuO/NiFe-OOH, the formation of O2 from OOH requires 1.70 V (Fig. 5f and g). At U = 0 V, the free energy change for both cases is positive and ascending. At 1.23 V, the theoretical overpotential of water splitting, the ΔG values of all intermediates are negative unlike type-1 and type-2 NiFe-OOH (Tables S7–S10†). Both type-1 and type-2 RuO/NiFe-OOH perform better than the bare NiFe-OOH surfaces. Ru in RuO/NiFe-OOH acts as the active site because of the availability of the uncoordinated sites on the Ru atom, which is supported with the XANES results (Fig. 2). In addition, Ru atoms can provide electrons more readily to the intermediates than surface oxygen. To make the whole reaction spontaneous, theoretical overpotentials of 0.35 V and 0.47 V have to be supplied for type 1 RuO/NiFe-OOH and type-2 RuO/NiFe-OOH, respectively. Here, the synergistic effect of the Fe atom can be seen in RuO/NiFeOOH. The theoretical overpotential of type 1 RuO/NiFe-OOH is 0.12 V lower in magnitude than that of type 2 RuO/NiFe-OOH. In this study, the minimum overpotential of 0.35 V towards OER was observed in type-1 RuO/NiFe-OOH. From the overall studies, the presence of NiFe-oxyhydroxides with the RuO surface ensured fast electron transfer towards OER, and can be extended to other catalytic applications.
Conclusions
In summary, we have developed a highly active and stable OER catalyst comprising Ru-NiFeP/Au islands with optimized Ru loading that showcase very low overpotential (η10: 223 mV) and Tafel slope (32 mV dec−1). In situ SERS studies demonstrated that the presence of Ru synergistically improves the performance, with FeOOH sites acting as a promoter at lower bias for –OH adsorption and NiOOH being the active sites at higher bias, for O2 desorption. From DFT, it is confirmed that the FeOOH sites induce ionic character in the Fe–O bond for the facile absorption of the O-intermediates. DFT studies further concluded that the presence of Ru–O coordination to the NiFeOOH sites could drastically improves the activity by diminishing the free energy barrier of 0.12 eV for the OER rate-determining step. Also, amorphization gives rise to ‘dangling bonds’ on the surface to induce better charge transport, as reflected in the faster kinetics. Overall, we demonstrated that the TMPs surface with noble metals like Ru can optimize the OER performance, and the Au surface for SERS can provide a deeper understanding of the active sites during OER. The findings will reinforce research studies in the design of stable and active TMPs-based OER catalysts, and those employing SERS to understand the nature of the active site.Data availability
All data that support the findings of this study are provided within the paper, and its ESI† files and are also available from the corresponding author upon request.Author contributions
K. K. planned and prepared the electrodes, carried out the experiments and wrote the manuscript. P. K. helped with XRD, XPS, sXAS, and XANES data analysis and helped in planning electrochemical measurements. P. G. performed the DFT studies. S. R. provided HRTEM, XPS characterizations, and edited the manuscript. H. S. S. performed in situ Raman studies and A. S. Z. helped with SEM analysis. S. K. N., A. A. B. and M. M. collected contact angle measurements and performed XRD analysis data. G. S., A. K. S., P. M. A., S. R. and M. G. K. supervised the research and edited the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the financial support by the Canada First Research Excellence Fund (CFREF) at the University of Calgary. Part of the research described in this paper was performed at the Canadian Light Source (project: 35G12344), a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. S. R. acknowledges support from IIT Kanpur (Project No.: 2024098) and the Chandrakanta Kesavan Centre for Energy Policy and Climate Solutions (Project No.: 2021136H). S. K. acknowledges SEED grant from Kalasalingam Academy of Research and Education. Drs Ning Chen, Beatriz Diaz-Moreno, Jay Dynes, Tom Regier and Zachary Arthur are kindly acknowledged for their help with the hard/soft X-ray and GI-WAXS analysis.References
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2018, 3, 46–52 Search PubMed.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- H. Over, ACS Catal., 2021, 11, 8848–8871 Search PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 Search PubMed.
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun, S. J. H. Ong, S. Xi, C. Diao, Y. Du, J. O. Wang, M. B. H. Breese, S. Li, H. Zhang and Z. J. Xu, Nat. Catal., 2020, 3, 554–563 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- H. Wang, K. H. L. Zhang, J. P. Hofmann, V. A. de la Peña O'Shea and F. E. Oropeza, J. Mater. Chem. A, 2021, 9, 19465–19488 RSC.
- N. Zhang and R. Jiang, Chempluschem, 2021, 86, 1586–1601 CrossRef CAS PubMed.
- J. Kang, X. Qiu, Q. Hu, J. Zhong, X. Gao, R. Huang, C. Wan, L. M. Liu, X. Duan and L. Guo, Nat. Catal., 2021, 4, 1050–1058 CrossRef CAS.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- F. Wang, Y. Zhang, J. Zhang, W. Yuan, Y. Li, J. Mao, C. Liu, C. Chen, H. Liu and S. Zheng, ACS Sustain. Chem. Eng., 2022, 10, 5976–5985 CrossRef CAS.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 Search PubMed.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J. W. Zhang, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- S. Dutta, A. Indra, Y. Feng, T. Song and U. Paik, ACS Appl. Mater. Interfaces, 2017, 9, 33766–33774 CrossRef CAS PubMed.
- X. Xia, L. Wang, N. Sui, V. L. Colvin and W. W. Yu, Nanoscale, 2020, 12, 12249–12262 RSC.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144–152 CrossRef CAS PubMed.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 1–23 CrossRef.
- Y. Shi, M. Li, Y. Yu and B. Zhang, Energy Environ. Sci., 2020, 13, 4564–4582 RSC.
- W. Li, D. Xiong, X. Gao and L. Liu, Chem. Commun., 2019, 55, 8744–8763 RSC.
- P. F. Liu, X. Li, S. Yang, M. Y. Zu, P. Liu, B. Zhang, L. R. Zheng, H. Zhao and H. G. Yang, ACS Energy Lett., 2017, 2, 2257–2263 CrossRef CAS.
- P. Yan, Q. Liu, H. Zhang, L. Qiu, H. Bin Wu and X. Y. Yu, J. Mater. Chem. A, 2021, 9, 15586–15594 RSC.
- S. Sultan, M. Ha, D. Y. Kim, J. N. Tiwari, C. W. Myung, A. Meena, T. J. Shin, K. H. Chae and K. S. Kim, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- D. Chen, Z. Pu, R. Lu, P. Ji, P. Wang, J. Zhu, C. Lin, H. W. Li, X. Zhou, Z. Hu, F. Xia, J. Wu and S. Mu, Adv. Energy Mater., 2020, 10, 1–10 Search PubMed.
- Z. Xu, X. Zhang, X. Wang, J. Fang, Y. Zhang, X. Liu, W. Zhu, Y. Yan and Z. Zhuang, ACS Nano, 2021, 15, 7131–7138 Search PubMed.
- N. Li, L. Mao, Y. Fu, H. Wang, Y. Shen, X. Zhou, Q. Li and J. Qian, Inorg. Chem. Front., 2024, 8139–8145 RSC.
- J. Chen, J. Liu, S. Xu, Y. Wu, Y. Ye and J. Qian, Inorg. Chem. Front., 2024, 11, 4876–4885 RSC.
- D. Chen, Q. Sun, C. Han, Y. Guo, Q. Huang, W. A. Goddard and J. Qian, J. Mater. Chem. A, 2022, 10, 16007–16015 RSC.
- Z. Qiu, Y. Ma and T. Edvinsson, Nano Energy, 2019, 66, 104118 CrossRef CAS.
- Y. Li, Y. Wu, H. Hao, M. Yuan, Z. Lv, L. Xu and B. Wei, Appl. Catal., B, 2022, 305, 121033 CrossRef CAS.
- C. Hu, Y. Hu, C. Fan, L. Yang, Y. Zhang, H. Li and W. Xie, Angew. Chem., Int. Ed., 2021, 60, 19774–19778 CrossRef CAS PubMed.
- W. Q. Li, R. Y. Zhou, X. T. Wang, L. Y. Hu, X. Chen, P. C. Guan, X. G. Zhang, H. Zhang, J. C. Dong, Z. Q. Tian and J. F. Li, J. Catal., 2021, 400, 367–371 CrossRef CAS.
- J. Fester, A. Makoveev, D. Grumelli, R. Gutzler, Z. Sun, J. Rodríguez-Fernández, K. Kern and J. V. Lauritsen, Angew. Chem., 2018, 130, 12069–12073 CrossRef.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- J. Du, Y. Huang, Z. Huang, G. Wu, B. Wu, X. Han, C. Chen, X. Zheng, P. Cui, Y. Wu, J. Jiang and X. Hong, JACS Au, 2022, 2, 1078–1083 CrossRef CAS PubMed.
- F. Hu, S. Zhu, S. Chen, Y. Li, L. Ma, T. Wu, Y. Zhang, C. Wang, C. Liu, X. Yang, L. Song, X. Yang and Y. Xiong, Adv. Mater., 2017, 29, 1–9 Search PubMed.
- X. Yu, Z. Y. Yu, X. L. Zhang, Y. R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M. R. Gao, G. Wang and S. H. Yu, J. Am. Chem. Soc., 2019, 141, 7537–7543 CrossRef CAS PubMed.
- O. S. Vereshchagin, D. V. Pankin, M. B. Smirnov, N. S. Vlasenko, V. V. Shilovskikh and S. N. Britvin, J. Alloys Compd., 2021, 853, 156468 CrossRef CAS.
- Y. Zhao, M. Xi, Y. Qi, X. Sheng, P. Tian, Y. Zhu, X. Yang, C. Li and H. Jiang, J. Energy Chem., 2022, 69, 330–337 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- F. Tang, T. Liu, W. Jiang and L. Gan, J. Electroanal. Chem., 2020, 871, 114282 CrossRef CAS.
- T. Harano, G. Shibata, K. Ishigami, Y. Takashashi, V. K. Verma, V. R. Singh, T. Kadono, A. Fujimori, Y. Takeda, T. Okane, Y. Saitoh, H. Yamagami, T. Koide, H. Yamada, A. Sawa, M. Kawasaki, Y. Tokura and A. Tanaka, Appl. Phys. Lett., 2013, 102, 4 CrossRef.
- J. Everett, E. Céspedes, L. R. Shelford, C. Exley, J. F. Collingwood, J. Dobson, G. Van Der Laan, C. A. Jenkins, E. Arenholz and N. D. Telling, J. R. Soc. Interface, 2014, 11, 20140165 CrossRef CAS PubMed.
- F. Wang, J. Jiang and B. Wang, Catalysts, 2019, 9, 477 Search PubMed.
- P. Acharya, R. H. Manso, A. S. Hoffman, S. I. P. Bakovic, L. Kekedy-Nagy, S. R. Bare, J. Chen and L. F. Greenlee, ACS Catal., 2022, 12, 1992–2008 CrossRef CAS.
- J. Zhu, Z. Zeng and W. X. Li, J. Phys. Chem. C, 2021, 125, 26229–26239 CrossRef CAS.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- X. Liu, K. Ni, B. Wen, R. Guo, C. Niu, J. Meng, Q. Li, P. Wu, Y. Zhu, X. Wu and L. Mai, ACS Energy Lett., 2019, 4, 2585–2592 CrossRef CAS.
- R. A. Marquez-Montes, K. Kawashima, Y. J. Son, J. A. Weeks, H. H. Sun, H. Celio, V. H. Ramos-Sánchez and C. B. Mullins, J. Mater. Chem. A, 2021, 9, 7736–7749 RSC.
- W. Dai, X. Bai, Y. A. Zhu, Y. Zhang, T. Lu, Y. Pan and J. Wang, J. Mater. Chem. A, 2021, 9, 6432–6441 RSC.
- Z. Niu, C. Qiu, J. Jiang and L. Ai, ACS Sustain. Chem. Eng., 2019, 7, 2335–2342 CrossRef CAS.
- H. Ren, X. Sun, C. Du, J. Zhao, D. Liu, W. Fang, S. Kumar, R. Chua, S. Meng, P. Kidkhunthod, L. Song, S. Li, S. Madhavi and Q. Yan, ACS Nano, 2019, 13, 12969–12979 CrossRef CAS PubMed.
- S. A. Chala, M. C. Tsai, B. W. Olbasa, K. Lakshmanan, W. H. Huang, W. N. Su, Y. F. Liao, J. F. Lee, H. Dai and B. J. Hwang, ACS Nano, 2021, 15, 14996–15006 CrossRef CAS PubMed.
- J. Yu, T. Zhang, Y. Sun, X. Li, X. Li, B. Wu, D. Men and Y. Li, ACS Appl. Mater. Interfaces, 2020, 12, 12783–12792 CrossRef CAS PubMed.
- X. Ding, Y. Xia, Q. Li, S. Dong, X. Jiao and D. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 7936–7945 CrossRef CAS PubMed.
- Y. N. Wang, Z. J. Yang, D. H. Yang, L. Zhao, X. R. Shi, G. Yang and B. H. Han, ACS Appl. Mater. Interfaces, 2021, 13, 8832–8843 CrossRef CAS PubMed.
- F. Yang, X. Chen, Z. Li, D. Wang, L. Liu and J. Ye, ACS Appl. Energy Mater., 2020, 3, 3577–3585 CrossRef CAS.
- K. Zhu, X. Zhu and W. Yang, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef CAS PubMed.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W. X. Li, J. Greeley, B. Roldan Cuenya and P. Strasser, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- S. Chen, L. Ma, Z. Huang, G. Liang and C. Zhi, Cell Rep. Phys. Sci., 2022, 3, 100729 CrossRef CAS.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- Y. Guan, W. Suo, Z. Zhang, Y. Wang, S. Sun and G. Liu, Mol. Catal., 2021, 511, 111725 CrossRef CAS.
- M. M. J. Li, Z. Zeng, F. Liao, X. Hong and S. C. E. Tsang, J. Catal., 2016, 343, 157–167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00958h |
| This journal is © The Royal Society of Chemistry 2025 |