DOI:
10.1039/D5TA00827A
(Paper)
J. Mater. Chem. A, 2025,
13, 11445-11457
Hierarchically porous Co–N–C electrocatalysts with enhanced mass transport and cobalt utilization efficiency for oxygen reduction reaction in high-performance PEMFCs†
Received
31st January 2025
, Accepted 14th March 2025
First published on 14th March 2025
Abstract
Cobalt-coordinated nitrogen-doped carbon (Co–N–C) materials have emerged as promising alternatives to platinum-based catalysts for proton exchange membrane fuel cells (PEMFCs) due to their cost-effectiveness and durability. However, conventional Co–N–C catalysts exhibit limitations in mass transport as the active Co–Nx sites are often embedded within a dense carbon matrix, reducing their site accessibility. This study introduces a melamine-assisted synthesis approach to develop Co–N–C catalysts with a hierarchical porous structure that significantly enhances the accessibility of Co–Nx active sites. By incorporating melamine with zeolitic imidazolate frameworks (ZIFs) during synthesis, an optimized pore architecture is achieved, facilitating efficient mass transport of reactants (H+ and O2) to active sites and enabling effective water removal. This unique structure yields a high density of accessible active sites, resulting in superior oxygen reduction reaction (ORR) activity. XPS and electrochemical measurements confirm the increased density of Co–Nx species, establishing a robust structure–property correlation. In membrane electrode assembly (MEA) integration for PEMFC applications, the synthesized Co–N–C catalyst exhibits excellent performance with enhanced stability and reduced mass transfer overpotential. This work highlights a scalable strategy for developing durable, highly active non-precious metal catalysts, advancing the practical viability of PEMFC technology.
1. Introduction
The rapid depletion of fossil fuels, coupled with rising global energy demands, has escalated both the urgency of the energy crisis and concerns about environmental degradation.1 As a promising solution, hydrogen fuel with a high energy density (120–140 kJ kg−1) has emerged as a critical alternative energy source.2 Proton exchange membrane fuel cells (PEMFCs), which convert hydrogen into electricity through a sustainable, clean process, are at the forefront of next-generation energy technology, drawing considerable attention and extensive research investment.3 However, the slow kinetics of the oxygen reduction reaction (ORR) at the cathode remains a major bottleneck, making it the rate-determining step (RDS) in PEMFCs and necessitating highly active catalysts.3,4 Platinum (Pt)-based catalysts, known for their superior kinetic performance, are currently the benchmark for ORR. Unfortunately, the high cost and limited supply of Pt severely restrict the widespread commercialization of PEMFCs, underscoring the urgent need for alternative, low-cost, non-precious metal catalysts (NPMCs).5 Transition metals embedded within nitrogen-doped carbon matrices (M–N–C, where M = Fe, Co, Mn, Cu, etc.) have emerged as a promising class of NPMCs with nitrogen-coordinated metal centers (M–Nx), playing a key role in enhancing ORR activity.6,7 Among these, Fe–N–C electrocatalysts typically demonstrate the highest ORR activity.8 However, their susceptibility to producing reactive oxygen species (ROS), such as hydroxyl (·OH) and hydroperoxyl (·OOH) radicals via the Fenton reaction, presents a significant challenge for PEMFC applications.9 These ROS degrade membrane and ionomer structures, quickly diminishing PEMFC performance and lifespan.10,11 By contrast, Co–N–C catalysts not only exhibit superior ORR activity among other transition metals (Fe > Co > Mn > Cu > Ni) but are also immune to Fenton reactions, offering improved stability under PEMFC operation conditions.12 Consequently, Co–N–C materials hold substantial promise as alternatives to traditional PGM- and Fe-based catalysts, balancing ORR activity with long-term durability.
Recently, zeolitic imidazolate frameworks (ZIFs), a unique subclass of metal–organic frameworks (MOFs), have garnered significant attention as promising precursors for the synthesis of advanced M–N–C catalysts.13 ZIF precursors with their distinct structure of organic ligands coordinated to metal-containing nodes, can be transformed through pyrolysis into N-doped porous carbon materials enriched with carbon, nitrogen, and transition metals.14,15 These ZIF-derived M–N–C catalysts maintain their inherent high porosity, large surface area, and incorporation of heteroatomic dopants while exhibiting atomically dispersed active sites that are essential for superior electrochemical performance.16 The controlled thermal decomposition of ZIF precursors yields catalysts that retain the desirable textural properties of the parent materials and introduce active sites critical for catalytic activity, such as single-atomic metal centers. This structural transformation endows the materials with high catalytic efficiency, particularly for the ORR. To harness the full potential of ZIF-derived Co–N–C catalysts, various design strategies have been explored. Han and co-workers, for example, synthesized high-performance Co–N–C catalysts by engineering atomic-scale isolation of Co species through the pyrolysis of bimetallic Zn/Co ZIFs, fine-tuning the Zn dopant content to achieve optimal dispersion.17 Similarly, Qu and colleagues developed composite catalysts featuring highly graphitized carbon nanotubes (CNTs) densely populated with Co–N4 sites.18 This was accomplished by modulating the ratio of 2-methylimidazole to metal ions (M = Zn, Co) during the synthesis of bimetallic ZIFs (BMZIFs), which effectively anchored Co–N sites on the CNT structure. He and colleagues recently introduced a carbon-shell confinement strategy to address the issue of cobalt aggregation by coating a surfactant layer onto Co-doped ZIF precursors, thereby enhancing the density of exposed active sites.11 Despite significant advances in the development of Co–N–C catalysts, such as those described above, their performance often falls short in membrane electrode assemblies (MEAs) with proton exchange membrane fuel cells (PEMFCs) although comparable to commercial Pt/C catalysts in rotating disk electrode (RDE) tests. This discrepancy arises because only Co–Nx sites located at three-phase interfaces, where electrons, protons (H+), and oxygen (O2) converge, actively contribute to the ORR.5,19 The microporous nature of ZIF-derived structures can encapsulate a substantial portion of active sites, which suffers from steric hindrance and water flooding, leading to diminished catalytic efficiency.20,21 Addressing these limitations requires a synthesis strategy that prioritizes enhanced mass transport, thereby improving the accessibility of active sites rather than merely increasing their quantity. In this context, Li and co-workers reported Fe–N–C electrocatalysts with superior performance by incorporating carboxylate during the ZIF-8 precursor synthesis. This approach induced mesoporosity and a high Fe doping content, resulting in an abundance of accessible Fe–Nx active sites entangled CNTs.8 Similarly, Wu and co-workers devised an efficient active site rearrangement strategy for Fe–N–C catalysts by applying disorder and recrystallization processes to a Fe-doped ZIF precursor, followed by direct pyrolysis. This method substantially enhanced both mesoporosity and the density of active sites.20 In another pioneering approach, Wan and colleagues synthesized concave-shaped Fe–N–C single-atom catalysts characterized by an extensive external surface area and increased mesoporosity. This was achieved through the combined use of electrostatic adsorption, space-confinement pyrolysis, and anisotropic thermal microstress, facilitated by incorporating a mesoporous SiO2 coating layer during synthesis.4 Recently, Zhu and co-workers developed a core–shell structured Fe–N–C catalyst comprising an N-doped porous carbon core surrounded by a shell derived from conjugated microporous polymers. This architecture was designed to reduce the diffusion distance for H+ and O2, thereby enhancing H2O removal. During thermal activation, the polymer shell reorganized to form dense, accessible, and highly active Fe–N4 sites, while the ZIF-8-derived carbon core prevented Fe aggregation and facilitated the uniform dispersion of N-coordinated Fe sites.5 Despite the exceptional ORR performance demonstrated by MEAs incorporating these advanced Fe–N–C electrocatalysts, their long-term operational stability remains a significant challenge. The rapid performance degradation observed during prolonged fuel cell operation highlights the need for further research to enhance the durability of these systems.
This study introduces a robust and efficient synthetic approach for developing a highly durable Co–N–C catalyst with a high density of accessible Co–Nx active centers using a melamine-assisted strategy. Through this strategy, we achieved outstanding PEMFC performance despite using Co, a relatively low-activity metal, as the active center. Even with an ultra-low cathode loading of 0.5 mg cm−2, the catalyst exhibited peak power densities of 0.327 W cm−2 and 0.159 W cm−2 under 1 bar H2–O2 and H2–air conditions, respectively. Moreover, it demonstrated excellent durability, retaining 80.6% of its initial activity over 100 hours at 0.6 V. This superior performance is attributed to the advanced Co–N–C catalyst, in which the hierarchical porous structure enhances mass transport and maximizes the accessibility of active sites. During high-temperature thermal activation, melamine undergoes co-pyrolysis with ZIF precursors, enhancing the inherent microporous structure of the ZIF and resulting in an extended external surface area conducive to mass transport. The superior ORR activity of the synthesized Co–N–C catalyst is attributed to the shallow dispersion of Co–Nx moieties, which exhibit increased utilization due to their enhanced surface exposure. XPS analysis confirms the elevated proportion of Co–Nx active centers, while quantitative evaluations using reversible nitrite reduction methods reveal that the active site density (SD), influenced by the tailored pore structure modifications, plays a critical role in catalytic activity. In practical PEMFC assessments, MEAs incorporating this advanced Co–N–C catalyst demonstrate notable performance with a reduction in mass transfer overpotential (ηmt) highlighting the significant improvement in fundamental mass transport achieved through this approach. This work underscores the importance of strategic material design to optimize active site accessibility and mass transport, providing a promising pathway for developing durable and efficient non-precious metal catalysts for PEMFC applications.
2. Experimental details
2.1 Materials
Cobalt nitrate hexahydrate (Co(NO3)2·6H2O) and zinc nitrate hexahydrate (Zn(NO3)2·6H2O) were procured from Sigma-Aldrich along with 2-methylimidazole (2-mIm, C4H6N2), sodium nitrite (NaNO2), sodium acetate (C2H3NaO2), acetic acid (C2H4O2), and Aquivion D79-25BS solution. Perchloric acid (HClO4, 70%), isopropyl alcohol (IPA; C3H8O), and methyl alcohol (CH3OH) were obtained from Daejung Chemicals and Metals Co., Ltd. Melamine (C3H6N6) and commercial Pt/C (40 wt%) were sourced from Alfa Aesar, and sulfuric acid (H2SO4) from Duksan Pure Chemicals Co., Ltd. A 5 wt% Nafion solution was acquired from Nara Cell Tech, while de-ionized (DI) water was prepared via reverse osmosis. All reagents were used as received without additional purification.
2.2 Catalysts synthesis
2.2.1 Synthesis of BMZIF.
To synthesize BMZIF, Co(NO3)2·6H2O (0.083 g) and Zn(NO3)2·6H2O (1.70 g) were dissolved in 40 mL of methanol. Subsequently, 40 mL of a methanol solution containing 2-methylimidazole (1.97 g) was added and the mixture was stirred vigorously for 24 hours. The resulting precipitate was collected by centrifugation, washed several times with methanol, and dried overnight at 80 °C.
2.2.2 Synthesis of mCo–NC-x.
The synthesized BMZIF (350 mg) was dispersed in 100 mL of methanol, followed by the addition of varying amounts of melamine (87.5, 175, 350, or 700 mg). The mixture was stirred for 6 hours and subsequently dried via rotary evaporation to obtain the melamine/BMZIF powder. The collected powder was then subjected to heat treatment at 900 °C for 3 hours under an argon atmosphere. The final product was denoted as mCo–NC-x, where x corresponds to the mass ratio of melamine to BMZIF. For comparison, a similar synthesis was conducted without melamine to produce Co–NC.
2.3 Characterizations
The morphology and structure of the samples were analyzed using field-emission transmission electron microscopy (FE-TEM; Titan G2 ChemiSTEM Cs Probe, FEI) with energy dispersive spectroscopy (EDS) for elemental mapping. Single cobalt atom images were acquired using aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM; ARM-200F, JEOL) operated at 200 kV. Thermogravimetric analysis (TGA; Q500, TA Instruments) was performed from 30 to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The crystal structure of the catalysts was examined using X-ray diffraction (XRD; XPERT PRO, Panalytical) with Cu Kα radiation. Raman spectra were recorded on a micro-Raman spectrophotometer (XploRA, Horiba). The specific surface area was measured using the Brunauer–Emmett–Teller (BET) method with a Surface Area Porosity Analyzer (Quandrasorb evo, Quantachrome), and the pore size distribution was determined using the Barrett–Joyner–Halenda (BJH) method. The external surface area, defined as the non-micropore area, was calculated by subtracting the micropore surface area from the BET surface area.4 Samples were dried under vacuum at 150 °C before N2 adsorption. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Fisher Scientific K-Alpha using Al Kα radiation, and all binding energies were calibrated using the C 1s peak at 284.6 eV. The overall cobalt content was quantified through inductively coupled plasma atomic emission spectroscopy (ICP-AES; Optima 8300, PerkinElmer). X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data were collected at the 1D XRS KIST-PAL beamline of the Pohang Accelerator Laboratory, South Korea. The normalized XANES spectra and Fourier-transformed radial distribution function of EXAFS were performed within the range of 3–11 Å−1 for Co using the IFEFFIT software package.
2.4 Electrochemical measurements
Electrochemical measurements were conducted with an Autolab potentiostat/galvanostat (PGSTAT 302N, Metrohm, Eco-Chemie, Utrecht, the Netherlands) in a standard three-electrode cell configuration. A glassy carbon (GC) electrode (0.196 cm2) served as the working electrode with a Pt sheet and an Ag/AgCl (3.0 M KCl) electrode as the counter and reference electrodes, respectively. Catalyst inks were prepared by dispersing 5 mg of the catalyst in a solution containing 480 μL of ethanol, 480 μL of IPA, and 40 μL of 5 wt% Nafion solution. After sonication for 30 min to form a homogeneous suspension, 32 μL of the dispersion was deposited on a rotating disk electrode (RDE) or rotating ring-disk electrode (RRDE), achieving a catalyst loading of 816 μg cm−2. A commercial Pt/C catalyst with a loading of 40 μgPt cm−2 was used for benchmarking. Prior to electrochemical measurements, 0.1 M HClO4 electrolyte was purged with pure O2 or N2 for 30 min. Catalyst films were activated by cyclic voltammetry (CV) between 0.1 and 1.2 V (vs. RHE) at a scan rate of 500 mV s−1 in an N2-saturated electrolyte for 500 cycles. Linear sweep voltammetry (LSV) curves for ORR activity were obtained at 1600 rpm with a scan rate of 10 mV s−1 in O2-saturated electrolyte. Electrochemical impedance spectroscopy (EIS) was conducted at 1600 rpm and the potential corresponding to 0.5 mA current for each catalyst. All applied potentials were referenced to RHE using the following equation:| | | ERHE = EAg/AgCl + 0.0592pH + 0.1976 | (1) |
Electrochemical surface area (ECSA) was estimated by evaluating the double-layer capacitance in the non-faradaic region. Accelerated stress test (AST) for the catalysts and commercial Pt/C was performed by cycling between 0.6 and 1.0 V (vs. RHE) at 50 mV s−1 for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in O2-saturated electrolyte. Chronoamperometry was conducted at 0.3 V (vs. RHE) with a rotation speed of 1600 rpm. During RRDE measurements, the Pt ring electrode was maintained at 1.2 V (vs. RHE). The hydrogen peroxide (H2O2) yield and electron transfer number (n) were calculated using the following equations:
000 cycles in O2-saturated electrolyte. Chronoamperometry was conducted at 0.3 V (vs. RHE) with a rotation speed of 1600 rpm. During RRDE measurements, the Pt ring electrode was maintained at 1.2 V (vs. RHE). The hydrogen peroxide (H2O2) yield and electron transfer number (n) were calculated using the following equations:
| | | H2O2% = 200 × Ir/(Ir + NId) | (2) |
where
Id and
Ir are the recorded disk and ring currents, respectively, and
N = 0.37 is the collection efficiency of the ring.
2.5 Active site quantification
The active site density (SD) and turnover frequency (TOF) were determined using the nitrite reduction method as described by Kucernak and co-workers.22 Electrochemically active Co–Nx sites were quantified by nitrite interaction. Extensive cycling was first performed in 0.5 M acetate buffer (pH 5.2) under alternating N2 and O2 saturation until stable CV curves were obtained in N2. Active sites were then poisoned in 125 mM NaNO2 solution by rotating the electrode at 300 rpm for 5 min, followed by nitrite stripping from −0.3 to 0.4 V (vs. RHE) at 10 mV s−1 in N2-saturated electrolyte. Excess charge (Qstrip) associated with nitrite stripping, proportional to the number of active sites, was calculated using the following equation:| | 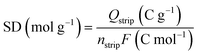 | (4) |
where nstrip = 5 (electrons for one nitrite reduction per site) and F = 96485 C mol−1 (Faraday constant). TOF at 0.8 V (vs. RHE) was derived using the difference of kinetic current density (jk) before and after nitrite poisoning and the following equation:| | 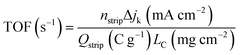 | (5) |
| | 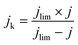 | (6) |
where LC = 0.27 mg cm−2 (catalyst loading) and jlim is the limiting current density at 0.4 V (vs. RHE).
2.6 MEA preparation and single cell tests
MEAs were fabricated by hand-spraying catalyst-coated membranes (CCM). Catalyst ink was prepared by ultrasonically dispersing 7.5 mg of catalyst in a mixture of 20 μL of DI water, 1.0 mL of IPA, and 7.89 μL of Aquivion D79-25BS solution (Solvay). The well-dispersed ink was sprayed onto a Nafion 211 membrane (5 cm2 geometric active area, DuPont) until a loading of 0.5 mg cm−2 was achieved. For the anode, commercial Pt/C (40 wt% Pt, Alfa Aesar) was used with a loading of 0.2 mgPt cm−2. The CCMs were then assembled with gas diffusion layers (GDLs; SGL). As a reference, a CCM using commercial Pt/C was prepared with loadings of 0.1 mgPt cm−2 for the anode and 0.2 mgPt cm−2 for the cathode. Single-cell performance was conducted using a fuel cell station (Nara Cell Tech) at 80 °C with 100% RH with H2 and O2 or air as reactants. Gas flow rates were set at 350 sccm for H2, 700 sccm for O2, and 1000 sccm for air. Stability was assessed by monitoring the current at a constant 0.6 V for 100 hours. The mass transport overpotential (ηmt) was determined by subtracting the conceptual polarization curve corrected for ohmic resistance and mass transport from the iR-free polarization curve.5 The iR-free polarization curves were obtained by compensating for ohmic resistance obtained at low current densities.
3. Result & discussion
3.1 Morphological elucidation of mCo–NC catalysts
Fig. 1a illustrates the melamine-assisted calcination strategy used to synthesize mCo–NC-x catalysts. The process begins with the synthesis of BMZIF by partially substituting Zn2+ with Co2+ in Zn-based ZIF-8, maintaining the optimal Zn/Co molar ratio as established in previous studies.15,17 The resulting BMZIF particles exhibit a well-defined dodecahedral morphology with an average particle size of approximately 400 nm, as shown in Fig. S1a.† The XRD analysis confirms that BMZIF matches the crystal structures of ZIF-8 and ZIF-67 structures, indicating successful Co incorporation (Fig. S1b†).14,18 Subsequently, BMZIF is converted into Co-embedded N-doped carbon (Co–NC) clusters through pyrolysis under an Ar atmosphere. During high-temperature treatment, organic linkers transform into N-doped porous carbon, while Co nodes are reduced.14,17 Meanwhile, Zn with a low boiling point (907 °C) evaporates, yielding Co-embedded porous carbon (Co–NC).15,23 Zn also serves to increase interatomic Co spacing, preventing aggregation and facilitating atomic dispersion of Co species.24 The as-obtained Co–NC morphology shows no significant changes compared to the pre-pyrolysis structure, consistent with previous observations for ZIF-8-derived porous carbon materials (Fig. 1b).25,26 Introducing melamine into BMZIF was intended to examine its impact on the catalyst morphology and ORR activity. Even small amounts of melamine introduced notable structural changes in the pyrolysis products, such as internal cavities and surface roughness (Fig. 1c and d). These changes are evident in the HAADF-STEM images of mCo–NC-0.25 and mCo–NC-0.5 (Fig. S2†). TGA conducted on melamine, BMZIF, and melamine/BMZIF with a 1:1 mass ratio shows that melamine decomposes first, followed by gradual BMZIF decomposition up to approximately 480 °C (Fig. S3†). The TGA curve for melamine/BMZIF displays a plateau between 320 and 450 °C with a 48% mass loss, aligning with the optimal melamine-to-BMZIF ratio. During decomposition, melamine releases reducing gases like NH3.16,27 These gases disrupt ligand–metal bonds and create internal cavities by altering the morphology and pore structure of the final catalyst.28,29 As the melamine-to-BMZIF ratio increased, the Co–NC-1 catalyst developed a more porous, and fluffy surface while maintaining its core structure (Fig. 1e). The Co nanoparticles (NPs) began to appear in mCo–NC-2 (Fig. 1f). Interestingly, with further melamine increase, the dodecahedral structure disappears completely, resulting in the formation of CNTs entangled with Co NPs, as observed in TEM images and EDS mapping of mCo–NC-4 (Fig. 1g and S4a, b†). High-resolution TEM (HR-TEM) images further reveal that Co NPs are encapsulated in a few-layered carbon shell (∼2.6 nm thick) at CNT tips (CNT wall thickness ∼3.2 nm), forming multi-walled features (Fig. S4c and d†). These metallic Co NPs are formed by reducing gases and then act as catalysts for CNT growth and become encapsulated within CNTs.30,31 The thickness of the carbon shell significantly influences electrocatalytic performance.26,32 However, the thick carbon shell hinders reactant access to Co NPs, potentially reducing catalytic efficiency.30 Detailed morphological characterization of mCo–NC-1, the optimized catalyst, was conducted using HR-TEM and AC-HAADF-STEM. HR-TEM images indicate that melamine introduction enhances the graphitization, explaining the excess CNT formation with higher melamine content (Fig. 1h). Numerous bright spots are clearly visible on the edge area of mCo–NC-1, which can be attributed to atomically dispersed Co species throughout the carbon frameworks (Fig. 1i). Elemental mapping further confirms the uniform distribution of N and Co with weight ratios of C, N, O, and Co corresponding to 88.86%, 5.91%, 3.03%, and 2.20%, respectively (Fig. 1j).
 |
| | Fig. 1 (a) Schematic illustration of the synthesis process for mCo–NC electrocatalysts. TEM images of (b) Co–NC, (c) mCo–NC-0.25, (d) mCo–NC-0.5, (e) mCo–NC-1, (f) mCo–NC-2, and (g) mCo–NC-4. (h) HR-TEM image, (i) AC-HAADF-TEM image, and (j) EDS elemental mapping of mCo–NC-1. | |
3.2 Characterization of the catalysts
The XRD patterns for the pyrolyzed samples, excluding mCo–NC-2, display a single broad peak between 20 and 30°, which can be assigned to the (002) crystal planes of graphitic carbon (Fig. 2a).8 The absence of peaks related to the metallic Co indicates that Co atoms are atomically dispersed within the N-doped carbon matrix. In contrast, the mCo–NC-2 exhibits diffraction peaks around 44° and 52° in correspond to the (111) and (200) crystal planes of Co, which become sharper in mCo–NC-4, suggesting numerous Co NPs encapsulated within a graphitic layer (Fig. S5†).33 These results clearly confirm Co reduction and support the presence of dispersed atomic Co, consistent with previous observations. Raman spectroscopy reveals two dominant peaks for all mCo–NC-x samples at approximately 1350 (D-band, disordered sp3 carbon) and 1590 cm−1 (G-band, graphitic sp2 carbon) (Fig. 2b).34 The D/G intensity ratio (ID/IG) decreases with increased melamine content, indicating a higher degree of graphitization. This trend is further corroborated by the more defined graphitic peak in XRD patterns of mCo–NC-4 compared to other samples (Fig. S5†). The porous structure characterization using N2 adsorption–desorption isotherms shows distinct differences between the catalysts, shown in the inset of Fig. 2c. The Co–NC catalyst exhibits a typical type-I isotherm with high adsorption volume at low pressure (P/P0 < 0.1), indicating a micropore-rich structure. In contrast, the mCo–NC-1 catalyst exhibits a pronounced hysteresis loop (0.4 < P/P0 < 1.0) that can be ascribed to a type-IV isotherm, indicating a hybrid structure with both micropores (<2 nm) and mesopores (2–50 nm) (Fig. 2c and S6†). The pore size distribution curves further demonstrate that mCo–NC-1 has fewer micropores but more mesopores compared to Co–NC. Additional information about the pore structure and pore size distribution of the entire series of mCo–NC-x catalysts is shown in Fig. S7.† As summarized in Table S1† and Fig. 2d, it is noteworthy that increasing melamine content significantly enhances the external area directly associated with mesoporosity. Although the Co–NC catalyst has the highest BET surface area (1123 m2 g−1), melamine-containing samples show a higher proportion of the external area, suggesting that some micropores were merged into mesopores, which facilitates reactant access to active sites and enhances catalytic activity.20,21 Conversely, the mCo–NC-2 exhibits the largest external area but reduced BET surface (548 m2 g−1) and is expected to show lower catalytic activity due to Co aggregation. This structural analysis demonstrates that melamine-assisted synthesis enhances the hierarchical pore network, improving active site accessibility, which is critical for optimizing catalytic performance.
 |
| | Fig. 2 (a) XRD patterns and (b) Raman spectra of the catalysts. (c) Pore size distribution of Co–NC and mCo–NC-1 with N2 adsorption–desorption isotherms in the inset. (d) Comparison of BET surface area and external surface area of all catalysts. (e) XANES spectra and (f) Fourier-transformed EXAFS (FT-EXAFS) of Co–NC and mCo–NC-1 compared with Co foil and Co(II) phthalocyanine (CoPc). | |
Fig. 2e presents the XANES spectra for Co–NC and mCo–NC-1 compared with Co foil and Co(II) phthalocyanine (CoPc). The pre-edge peak near 7710 eV observed for CoPc shifts leftward to approximately 7700 eV in Co–NC and mCo–NC-1, indicating a deviation from the Co–N4 square planar configurations.3 This shift suggests a modification in the coordination environment around cobalt, distinct from the reference CoPc. The Fourier-transformed EXAFS (FT-EXAFS) spectra in Fig. 2f further confirm this observation. The reference peaks for Co–Co (Co foil) and Co–N (CoPc) clearly distinguish the coordination states of cobalt. In both Co–NC and mCo–NC-1, the FT-EXAFS peaks closely resemble those of CoPc, meaning that the cobalt atoms are coordinated as Co–NC rather than existing in metallic Co–Co bonds. These results strongly suggest that the melamine-assisted synthesis, which enhances the hierarchical pore structure, does not alter the chemical state of Co active sites. Instead, it maintains the cobalt atoms in single-atom dispersion within the carbon matrix, as corroborated by Fig. 1i. This evidence reinforces the structural integrity of cobalt active sites during hierarchical pore structure formation, further validating their role in catalytic performance.
The chemical bonding states of the synthesized catalysts were investigated using XPS. The high-resolution N 1s spectra were deconvoluted into five components corresponding to different N species: pyridinic-N (∼398.2 eV), Co–Nx (∼399.3 eV), pyrrolic-N (∼400.2 eV), graphitic-N (∼400.8 eV), and oxidized-N (∼402.8 eV) (Fig. 3a).21,35,36 Among these species, pyrrolic-N, Co–Nx, and graphitic-N are known to facilitate the ORR process.15 with Co–Nx being particularly critical as the most active moiety for ORR.37 The Co 2p spectra reveal multiple cobalt species, including Co 2p3/2 (∼780.1 eV), Co–Nx (∼782.3 eV), Co 2p1/2 (∼796.5 eV), and satellite peaks (Fig. 3b).38 Notably, a metallic Co signal (∼778.2 eV) was detected exclusively in the mCo–NC-2, consistent with previous observations.39Fig. 3c indicates that the proportion of Co–Nx, an active site, increases with the addition of melamine. This enhancement is attributed to the transformation of micropores into mesopores during pyrolysis, which facilitates the exposure of Co–Nx species previously buried deep within the carbon matrix. However, in mCo–NC-2, excessive decomposition of ligand–metal bonds due to melamine-derived reducing gases led to Co aggregation, causing a deviation from this trend. The C 1s spectra of all catalysts exhibited consistent features, including peaks for C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (∼284.5 eV), C–N (∼285.6 eV), and CO (∼287.8 eV) (Fig. S8†).40,41 The overall and surface Co contents were quantified using XPS and ICP-AES, respectively (Fig. 3d). While the overall Co content increased modestly with the melamine addition due to the partial decomposition of the carbon structure, the surface Co content showed a more pronounced increase. This result further supports the hypothesis that melamine promotes the exposure of Co–Nx species by enhancing pore structure and converting micropores into mesopores. These findings demonstrate that melamine-assisted synthesis effectively optimizes the distribution and accessibility of Co–Nx active sites, thereby enhancing the ORR activity of the Co–NC catalysts. This process underscores the critical role of pore structure modification in improving catalytic performance.
C (∼284.5 eV), C–N (∼285.6 eV), and CO (∼287.8 eV) (Fig. S8†).40,41 The overall and surface Co contents were quantified using XPS and ICP-AES, respectively (Fig. 3d). While the overall Co content increased modestly with the melamine addition due to the partial decomposition of the carbon structure, the surface Co content showed a more pronounced increase. This result further supports the hypothesis that melamine promotes the exposure of Co–Nx species by enhancing pore structure and converting micropores into mesopores. These findings demonstrate that melamine-assisted synthesis effectively optimizes the distribution and accessibility of Co–Nx active sites, thereby enhancing the ORR activity of the Co–NC catalysts. This process underscores the critical role of pore structure modification in improving catalytic performance.
 |
| | Fig. 3 High-resolution (a) N 1s and (b) Co 2p XPS spectra of the catalysts. (c) Comparison of Co–Nx contents from N 1s and Co 2p spectra. (d) Comparison of surface Co content and overall Co content of all catalysts. | |
3.3 Electrocatalytic performance for the ORR
The ORR activity of the catalysts was evaluated using rotating disk electrode (RDE) measurements in O2-saturated 0.1 M HClO4 electrolyte at 1600 rpm. As shown in Fig. 4a and summarized in Table S2,† the mCo–NC-1 catalyst, synthesized with optimized melamine content, demonstrated superior electrocatalytic performance. It exhibited an onset potential (Eonset) of 0.82 V (vs. RHE) and a half-wave potential (E1/2) of 0.77 V (vs. RHE), which are significantly higher than those of the other catalysts and approach the performance of commercial Pt/C (E1/2 = 0.775 V vs. RHE). Furthermore, the limiting current density (JL) at 0.4 V (vs. RHE) for mCo–NC-1 was 5.29 mA cm−2, comparable to that of Pt/C (JL = 5.28 mA cm−2). In contrast, mCo–NC-2, which contains Co NPs, displayed poor ORR activity (Eonset = 0.757 V, E1/2 = 0.696 V vs. RHE), indicating the superior performance of Co–Nx active sites over metallic Co species. The Tafel slopes, which indicate the ORR kinetics, were determined for all catalysts and are presented in Fig. 4b. The values were 90 mV dec−1 (Co–NC), 80 mV dec−1 (mCo–NC-0.25), 88 mV dec−1 (mCo–NC-0.5), 76 mV dec−1 (mCo–NC-1), 62 mV dec−1 (mCo–NC-2), and 70 mV dec−1 (Pt/C). Among these, mCo–NC-1 exhibited one of the lowest Tafel slopes, indicating improved ORR kinetics except for mCo–NC-2. Although mCo–NC-2 demonstrated the lowest Tafel slope, its large overpotential and lower catalytic performance are attributed to the increased graphic carbon content and Co aggregation, which limit the effectiveness of active sites.42 EIS measurements further validate these findings (Fig. 4c and Table S3†). While the electrolyte resistance (Rs) was similar across all catalysts, mCo–NC-1 exhibited the lowest charge transfer resistance (Rct), indicating enhanced charge transfer properties due to the melamine-assisted synthesis. Meanwhile, mCo–NC-2 displayed an additional semi-circle in the low-frequency region, associated with O2 diffusion limitations caused by Co metal aggregation.43,44
 |
| | Fig. 4 Electrochemical performance of the catalysts and Pt/C: (a) ORR polarization curves in O2-saturated 0.1 M HClO4 solution at a rotation speed of 1600 rpm with a scan rate of 10 mV s−1. (b) Tafel plots derived from polarization curves. (c) EIS Nyquist plots. (d) Electrical double layer capacitance (Cdl). (e) CV curves of nitrite adsorption/desorption for active site density (SD) quantification of mCo–NC-1. (f) Comparison of SD and Co utilization of all catalysts. | |
The electrochemically active surface area (ECSA) of the catalyst was estimated by analyzing the double-layer capacitance (Cdl) through cyclic voltammetry (CV) measurements at varying scan rates within the non-faradaic potential range. As shown in Fig. 4d, the Cdl value for mCo–NC-1 is notably higher than that of other catalysts, indicating that the balanced coexistence of micropores and mesopores in mCo–NC-1 provides enhanced electrochemical accessibility and active surface properties. The accessible active site density (SD) of the catalysts was further quantified using the nitrite reduction method to investigate the correlation between Co–Nx active sites and the pore structure.3,22 This method measures the charge generated by the reversible adsorption and desorption of nitrite anions during electrochemical CV treatment. As shown in Fig. 4e and S9† (summarized in Table S4†), the mCo–NC-1 catalyst exhibited the highest active site density of 76.93 μmol g−1, calculated based on the five electrons involved in the reduction of nitrite anions adsorbed on Co–Nx moieties. These findings indicate that the enhanced hierarchical pore structure induced by the melamine-assisted synthesis facilitates greater exposure of Co–Nx active sites, directly influencing the intrinsic ORR activity of the catalyst. The nitrite reduction results were further validated by LSV curves recorded before and after nitrite poisoning, as well as in the recovered state for each catalyst (Fig. S10†). These measurements confirm the reversible blocking and restoration of Co–Nx sites, proving that the number of active sites corresponds to the amount of nitrite anions adsorbed. Additionally, Fig. 4f plots the specific SD values and cobalt utilization for all catalysts. Cobalt utilization, calculated from SD values and the overall Co content determined by ICP-AES, highlights that mCo–NC-1 achieves the most efficient activation of atomically dispersed Co–Nx active sites. These findings demonstrate that previously inaccessible Co–Nx moieties are exposed by the optimized pore structure, allowing them to actively participate in the ORR.
Fig. 5 compares the electrocatalytic performance of mCo–NC-1 with commercial Pt/C for the ORR. The electron transfer number (n) and hydrogen peroxide (H2O2) yield were evaluated using rotating ring disk electrode (RRDE) tests to elucidate the ORR pathway of mCo–NC-1. As shown in Fig. 5a, mCo–NC-1 exhibits an exceptionally low H2O2 yield (<0.5%) across the potential range of 0.3–0.7 V (vs. RHE), corresponding to an n value exceeding 3.99. This behavior demonstrates that mCo–NC-1 predominantly follows the highly efficient four-electron pathway (eqn (8)), generating water molecules instead of hydrogen peroxide (eqn (7)). In comparison, commercial Pt/C shows a slightly higher H2O2 yield (∼1.24%) and a lower n value (∼3.97), indicating reduced selectivity. The superior performance of mCo–NC-1 is attributed to its ability to overcome the energy barrier for ·OH separation, which is more favorable compared to breaking the O–O bond.8,45 This finding confirms that the mCo–NC-1 catalyst effectively follows the four-electron reaction route (eqn (8)) to generate H2O, avoiding the sluggish two-electron reaction pathway that compromises power density in fuel cell applications.46
| | | O2 + 2H+ + 2e− → H2O2, E = 0.700 V (vs. RHE) | (7) |
| | | O2 + 4H+ + 4e− → 2H2O, E = 1.229 V (vs. RHE) | (8) |
 |
| | Fig. 5 (a) H2O2 yield and electron transport number (n) of mCo–NC-1 and Pt/C in O2-saturated 0.1 M HClO4 solution at a rotation speed of 1600 rpm with a scan rate of 10 mV s−1. ORR polarization curves of (b) mCo–NC-1 and (c) Pt/C before and after 10000 cycles between 0.6 and 1.0 V (vs. RHE) at 50 mV s−1. (d) Chronoamperometry and (e) methanol tolerance measurement of mCo–NC-1 and Pt/C at 0.3 V (vs. RHE). | |
The stability of mCo–NC-1 was evaluated using an accelerated stress test (AST) with potential cycling between 0.6 and 1.0 V (vs. RHE) for 10000 cycles in an O2-saturated electrolyte. As shown in Fig. 5b and c, mCo–NC-1 demonstrates remarkable durability with negligible change in the half-wave potential (ΔE1/2 = 8.36 mV) and limiting current density (ΔJL = 0.05 mA cm−2). In contrast, Pt/C suffers severe degradation (ΔE1/2 = 204.23 mV, ΔJL = 1.57 mA cm−2), attributed to Pt nanoparticles aggregation, as confirmed by post-AST morphological analysis (Fig. S11†).
To further confirm the long-term stability, chronoamperometric measurements were conducted at 0.3 V (vs. RHE) for 36000 s in an O2-saturated electrolyte (Fig. 5d). The current retention of mCo–NC-1 decreased by only 2.9%, significantly outperforming Pt/C, which exhibited continuous current degradation. Furthermore, methanol tolerance tests highlight the robustness of mCo–NC-1 (Fig. 5e). Upon injecting 2 mL of 1.0 M methanol into the electrolyte, mCo–NC-1 showed only slight attenuation in current, followed by rapid recovery. Conversely, Pt/C experienced a sharp decline in current retention with no recovery under the same conditions. These results establish mCo–NC-1 as a highly durable and selective ORR catalyst with excellent methanol tolerance, making it a promising candidate for direct methanol fuel cell (DMFC) applications.
3.4 Fuel cell performance
The MEAs were fabricated to evaluate the practical performance of Co–N–C catalysts with and without melamine as cathodes in PEMFCs (Fig. 6a). These MEAs were prepared with an ultra-low catalyst loading of 0.5 mg cm−2. Initial PEMFC tests were conducted under H2–O2 conditions at 80 °C and 100% relative humidity (RH) to assess the actual activity of catalysts loaded on solid-state MEA cathodes. Based on the aforementioned results, the mesoporous model of mCo–NC, which exhibits higher utilization of Co active sites and facilitates good gas and liquid mass transfer, is expected to demonstrate superior performance in actual fuel cells compared to the conventional microporous model of Co–NC. As shown in Fig. 6b, the current densities at 0.6 V for Co–NC and mCo–NC-1 cathodes were 0.364 and 0.415 A cm−2, respectively, demonstrating a 14% improvement for mCo–NC-1. To further analyze the overall catalyst performance, the peak power density (Pmax) was measured, reflecting the combined effects of kinetic activity, internal resistance, and mass transfer.47 The Pmax values for Co–NC and mCo–NC-1 cathodes were 0.266 and 0.327 W cm−2, respectively, representing a 23% improvement for mCo–NC-1. This enhancement in Pmax was more pronounced under a back pressure of 2 bar, achieving values of 0.327 W cm−2 for Co–NC and 0.442 W cm−2 for mCo–NC-1, corresponding to a 35% increase (Fig. 6c). These results highlight the impact of sufficient reactant supply and modified pore structure on catalytic performance at high current densities despite previously reported challenges such as oxygen resistance and water flooding in the catalyst layer.5,48 Further testing under air conditions, a more practical scenario than oxygen (Fig. S12†), revealed similar performance trends. The Pmax values for mCo–NC-1 were 0.159 W cm−2 and 0.286 W cm−2 at 1 bar and 2 bar, respectively, showing improvements of 13% and 25% compared to Co–NC (0.141 W cm−2 and 0.229 W cm−2, respectively). These results confirm that the structural modifications enhancing ORR activity in half-cell tests translate effectively to MEA-based PEMFCs. The mass-specific activity was evaluated by normalizing the polarization curves to the cobalt in mCo–NC-1 and platinum in Pt/C (Table S5†). While the areal-specific performance of commercial Pt/C remained the highest, the mass-specific performance of mCo–NC-1 was significantly superior (Pmax = 26.27 W g−1, i0.6 V = 33.73 A g−1) compared to Pt/C (Pmax = 4.00 W g−1, i0.6 V = 4.65 A g−1). This highlights the exceptional efficiency of mCo–NC-1 achieved through mass transport and low cobalt loading, making it a highly competitive alternative to commercial Pt/C for PEMFC applications.
 |
| | Fig. 6 (a) Schematic illustration of improved mass transport in single-stack PEMFC using mCo–NC. H2/O2 PEMFC performance with Co–NC and mCo–NC-1 cathodes: polarization curves (b) at 1 bar and (c) at 2 bar. Mass transport overpotential (ηmt) under 1 bar H2/O2 conditions for (d) Co–NC and (e) mCo–NC-1. (f) Durability test of mCo–NC-1 at 0.6 V over 100 hours. | |
The mass transfer properties of the Co–N–C catalysts were evaluated by calculating the mass transfer overpotential (ηmt) from the polarization curves measured at a high current density of 0.5 A cm−2 under H2–O2 conditions. Under 1 bar pressure without back pressure, the MEAs assembled with Co–NC and mCo–NC-1 cathodes exhibited ηmt values of 130 mV and 89 mV, respectively, as shown in Fig. 6d and e. This substantial difference demonstrates that the hierarchical pore structure of mCo–NC-1 facilitates more efficient mass transport by improving reactant accessibility to the active sites. When a back pressure of 2 bar was applied to enhance reactant diffusion and activate previously inaccessible Co–Nx sites buried within the catalyst layer, ηmt for mCo–NC-1 decreased slightly to 84 mV, whereas Co–NC showed a more significant reduction to 119 mV (Fig. S13†). The relatively small change in ηmt for mCo–NC-1 indicates that most Co–Nx sites in mCo–NC-1 are already highly accessible under atmospheric pressure due to their well-balanced coexistence of micropores and mesopores. These results highlight the superior mass transfer efficiency of mCo–NC-1, which arises from its structural optimization. The ability of catalysts to maintain low ηmt across different pressures underscores the effectiveness of the melamine-assisted synthesis in creating a porous architecture that exposes more Co–Nx active sites for catalytic reactions.
The durability of the mCo–NC-1 catalyst was evaluated by applying a constant cell voltage of 0.6 V in H2–O2 PEMFC, and the results were compared with those of commercial Pt/C. As shown in Fig. 6f, mCo–NC-1 exhibits superior stability with only a 19.4% loss in activity over the initial 100 hours, compared to a significantly higher 61.7% activity loss for Pt/C (Fig. S14†). Furthermore, a single-stack cell test conducted after the 100 hour durability test demonstrated that the performance retention of mCo–NC-1 was substantially better than that of Pt/C with less structural degradation observed in mCo–NC-1 (Fig. S15†). The enhanced durability of mCo–NC-1, compared to Fe-based catalysts known for their high activity but poor stability, can be attributed to the redox stability of cobalt. This relationship is explained using the standard reduction potential (E0) for Co, Fe, and the H2O2/·OH redox pair along with thermodynamic interpretations of the Fenton reaction:3,49
| | | Co3+ + e− ↔ Co2+, E0 = 1.920 V | (9) |
| | | Fe3+ + e− ↔ Fe2+, E0 = 0.770 V | (10) |
| | | H2O2 + H+ + e− ↔ ·OH + H2O, E0 = 0.880 V | (11) |
| | | Fe2+ + H2O2 + H+ → Fe3+ + ·OH + H2O | (12) |
| | | Co3+ + e− ↔ Co2+, E0 = 1.920 V | (13) |
According to previous studies,50 metal cations act as Fenton reagents only if their redox potential is lower than that of the H2O2/·OH pair (E0 = 0.880 V). The redox potential of Fe3+/Fe2+ (E0 = 0.770 V) is lower, making it thermodynamically favorable for H2O2 to oxidize Fe2+via the Fenton reaction (eqn (12)), generating highly reactive hydroxyl radicals (·OH). These free radicals attack the membrane, active sites, and carbon support, limiting long-term stability. In contrast, the relatively higher standard reduction potential of Co3+/Co2+ (E0 = 1.920 V) enhances the resistance of cobalt-based catalysts to degradation pathways.51,52 Meanwhile, Pt NPs have been identified as effective scavengers of ROS, effectively neutralizing free radicals.53,54 Based on these findings, the degradation of commercial Pt/C-based MEAs does not appear to be associated with the Fenton reaction but rather results from mass transport limitations, leading to flooding. This flooding behavior is evident from the oscillations in current density observed during continuous operation, as shown in Fig. S14.†55 Additionally, the hierarchical pore structure of mCo–NC-1 facilitates the formation of numerous three-phase boundaries (TPBs), enhancing the exposure of buried Co–Nx active sites, improving accessibility to H+ and O2, and mitigating water flooding. This structural advantage ensures sustained catalytic activity even under demanding conditions. Although the overall performance of mCo–NC-1 falls slightly below that of some state-of-the-art Co-based catalysts (Table S6†), its low catalyst loading and superior stability make it highly competitive. Unlike Fe-based PGM-free catalysts that suffer from durability issues due to the Fenton reaction, mCo–NC-1 offers reliable long-term performance. This positions it as a viable replacement for Fe-based and PGM catalysts in PEMFC applications. Additionally, several studies have reported that the incorporation of hetero-dopants or additional transition metals into M–N–C catalysts can generate new active moieties or dual-site configurations, further improving electrocatalytic performance. Therefore, future research should aim to bridge the performance gap with advanced Co-based catalysts by integrating these effective strategies, including the approach presented in this study. With further refinement, mCo–NC-1 holds significant promise for widespread adoption in sustainable fuel cell technologies.
4. Conclusion
This study demonstrates the successful synthesis of hierarchically porous Co–N–C electrocatalysts with enhanced active site density using a melamine-assisted co-pyrolysis strategy. The introduction of melamine significantly improved the external surface area and graphitic characteristics, enabling precise control over the pore structure and facilitating the formation of atomically dispersed Co–Nx moieties. The optimized mCo–NC-1 catalyst exhibited remarkable PEMFC performance, achieving a peak power density of 0.327 W cm−2 under H2–O2 and 0.159 W cm−2 under H2-air at 1 bar, alongside excellent durability with only a 19.4% activity loss over 100 hours at 0.6 V. These results highlight the catalyst's robustness and efficiency, even with an ultra-low cathode catalyst loading. Quantification of active sites underscored the strong structure–performance correlation with the high density of exposed Co–Nx sites enabling efficient reactant transport and water removal. Notably, mCo–NC-1 delivered exceptional mass-specific performance (Pmax = 26.27 W g−1, i0.6 V = 33.73 A g−1), far surpassing commercial Pt/C (Pmax = 4.00 W g−1, i0.6 V = 4.65 A g−1), offering a significant cost advantage for PEMFC commercialization. The high durability and scalability of mCo–NC-1 position as a viable alternative to Pt/C for continuous-operation fuel cells. This work provides a robust design framework for developing low-loaded non-precious metal catalysts, particularly ZIF-derived Co–N–C materials. By incorporating melamine as a structural modifier, we successfully enhance active site accessibility and mass transport properties, overcoming conventional limitations of Co-based catalysts. This advancement significantly improves ORR performance and durability, paving the way for sustainable and cost-effective PEMFC technologies.
Data availability
The data that support the findings of this study are available within the paper and its ESI,† and all data are also available from the corresponding authors upon request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2019R1A5A8080290).
References
- S. M. N. Jeghan, B. Seo, H. Son, S. W. Joo, M. Kim and G. Lee, Chem. Eng. J., 2024, 487, 150576 CAS.
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS.
- L. Chen, X. Liu, L. Zheng, Y. Li, X. Guo, X. Wan, Q. Liu, J. Shang and J. Shui, Appl. Catal., B, 2019, 256, 117849 CrossRef CAS.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- J. Zhu, Z. Fang, X. Yang, M. Chen, Z. Chen, F. Qiu, M. Wang, P. Liu, Q. Xu, X. Zhuang and G. Wu, ACS Catal., 2022, 12, 6409–6417 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CAS.
- A. Zitolo, N. R. -Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrignton, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef PubMed.
- Q. Zou, F. Xu, J. Ma, H. Zhang and Y. Wang, J. Alloys Compd., 2022, 923, 166393 CAS.
- C. Walling, Acc. Chem. Res., 1975, 8, 125 CAS.
- X. X. Wang, D. A. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 CAS.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CAS.
- Z. Yang, C. Zhao, Y. Qu, H. Zhou, F. Zhou, J. Wang, Y. Wu and Y. Li, Adv. Mater., 2019, 31, 1808043 Search PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CAS.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CAS.
- K. Im, J.-H. Jang, J. Heo, D. Kim, K.-S. Lee, H.-K. Lim, J. Kim and S. J. Yoo, Appl. Catal., B, 2022, 308, 121220 CrossRef CAS.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- X.-M. Qu, S.-H. Yin, Y.-N. Yan, J. Yang, Y.-R. Li, X.-Y. Chen, F. Lu, C.-T. Wang, Y.-X. Jiang and S.-G. Sun, Chem. Eng. J., 2023, 461, 142054 CrossRef CAS.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- Y. Wu, M. Yuan, X. Li, R. Ding, X. Duan, J. Li, Y. Wang, X. Li, Y. Zhang and J. Liu, Appl. Catal., B, 2022, 312, 121365 CrossRef CAS.
- H. Xu, D. Wang, P. Yang, L. Du, X. Lu, R. Li, L. Liu, J. Zhang and M. An, Appl. Catal., B, 2022, 305, 121040 CAS.
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 13285 CAS.
- Z. Wang, T. Yan, J. Fang, L. Shi and D. Zhang, J. Mater. Chem. A, 2016, 4, 10858–10868 CAS.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, X. Ren, J. Zhang and M. An, J. Mater. Chem. A, 2020, 8, 23187–23201 CAS.
- C. Young, R. R. Salunkhe, J. Tang, C.-C. Hu, M. Shahabuddin, E. Yanmaz, M. S. A. Hossain, J. H. Kim and Y. Yamauchi, Phys. Chem. Chem. Phys., 2016, 18, 29308–29315 CAS.
- Y. Xue, H. Li, X. Ye, S. Yang, Z. Zheng, X. Han, X. Zhang, L. Chen, Z. Xie, Q. Kuang and L. Zheng, Nano Res., 2019, 12, 2490–2497 CAS.
- S. Wang, J. Qin, T. Meng and M. Cao, Nano Energy, 2017, 39, 626–638 CAS.
- T. Wang, R. Jin, Y. Wu, J. Zheng and X. Li, Nanoscale Horiz., 2018, 3, 218 CAS.
- X. Li, Q. Sun, J. Liu, B. Xiao, R. Li and X. Sun, J. Power Sources, 2016, 302, 174–179 CAS.
- B. Y. Xia, Y. Yan, H. B. Wu, X. W. (D. ) Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CAS.
- J. Meng, C. Niu, L. Xu, J. Li, X. Liu, X. Wang, Y. Wu, X. Xu, W. Chen, Q. Li, Z. Zhu, D. Zhao and L. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CAS.
- J. Deng, L. Yu, D. Deng, X. Chen, F. Yang and X. Bao, J. Mater. Chem. A, 2013, 1, 14868–14873 CAS.
- Y. Tan, Z. Zhang, Z. Lei, W. Wu, W. Zhu, N. Cheng and S. Mu, J. Power Sources, 2020, 473, 228057 Search PubMed.
- G. Chao, Y. Zhang, L. Zhang, W. Zong, N. Zhang, T. Xue, W. Fan, T. Liu and Y. Xie, J. Mater. Chem. A, 2022, 10, 5930–5936 CAS.
- C. Guo, Y. Wu, Z. Li, W. Liao, L. Sun, C. Wang, B. Wen, Y. Li and C. Chen, Nanoscale Res. Lett., 2017, 12, 144 Search PubMed.
- T. Cheng, H. Yu, F. Peng, H. Wang, B. Zhang and D. Su, Nanoscale Res. Lett., 2016, 6, 1007–1015 CAS.
- X. Zhang, S. Zhang, Y. Yang, L. Wang, Z. Mu, H. Zhu, X. Zhu, H. Xing, H. Xia, B. Huang, J. Li, S. Guo and E. Wang, Adv. Mater., 2020, 32, 1906905 CrossRef CAS PubMed.
- W. Wu, Q. Zhang, X. Wang, C. Han, X. Shao, Y. Wang, J. Liu, Z. Li, X. Lu and M. Wu, ACS Catal., 2017, 7, 7267–7273 CrossRef CAS.
- C. Zhang, H. Yang, D. Zhong, Y. Xu, Y. Wang, Q. Yuan, Z. Liang, B. Wang, W. Zhang, H. Zheng, T. Cheng and R. Cao, J. Mater. Chem. A, 2020, 8, 9536–9544 RSC.
- Y. Meng, Y. Huang, G. Huang and Y. Song, RSC Adv., 2023, 13, 28148–28157 RSC.
- O. Pop-Georgievski, D. Kubies, J. Zemek, N. Neykova, R. Demianchuk, E. M. Chánová, M. Šlouf, M. Houska and F. Rypáček, Beilstein J. Nanotechnol., 2015, 6, 617–631 CrossRef CAS PubMed.
- T. Feng, W. Liao, Z. Li, L. Sun, D. Shi, C. Guo, Y. Huang, Y. Wang, J. Cheng, Y. Li and Q. Diao, Nanoscale Res. Lett., 2017, 12, 595 Search PubMed.
- K. Qiu, G. Chai, C. Jiang, M. Ling, J. Tang and Z. Guo, ACS Catal., 2016, 6, 3558–3568 CAS.
- K. Huang, P. Xu, X. He, R. Wang, Y. Wang, H. Yang, R. Zhang, M. Lei and H. Tang, ChemElectroChem, 2020, 7, 3341–3346 CAS.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 CAS.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CAS.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 Search PubMed.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618–621 CAS.
- S.-F. Kang and H.-M. Chang, Water Sci. Technol., 1997, 36, 215–222 CAS.
- D. Banham, S. Ye, K. Pei, J.-I. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CAS.
- M. Zhu, D. Guan, Z. Hu, H.-J. Lin, C.-T. Chen, H.-S. Sheu, S. Wang, J. Zhou, W. Zhou and Z. Shao, Appl. Catal., B, 2021, 297, 120463 CAS.
- R. Sgarbi, K. Kumar, F. Jaouen, A. Zitolo, E. A. Ticianelli and F. Maillard, J. Solid State Electrochem., 2021, 25, 45–46 CAS.
- M. Nomura, Y. Yoshimura, T. Kikuiri, T. Hasegawa, Y. Taniguchi, Y. Deyama, K.-I. Koshiro, H. Sano, K. Suzuki and N. Inoue, J. Pharmacol. Sci., 2011, 117, 243–252 CAS.
- M. Kajita, K. Hikosaka, M. Iitsuka, A. Kanayama, N. Toshima and Y. Miyamoto, Free Radical Res., 2007, 41, 615–626 CAS.
- J. Wang, G. Wu, W. Xuan, J. Jiang, J. Wang, L. Li, W.-F. Lin, W. Ding and Z. Wei, J. Mater. Chem. A, 2019, 7, 19786 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Sung Jong
Yoo
b,
Sung Jong
Yoo








