
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the structure and mechanism of Na+ diffusion in NASICON solid-state electrolytes and the effect of Sc- and Al/Y-substitution†
Ivana
Pivarníková
 *ad,
Stefan
Seidlmayer
aef,
Martin
Finsterbusch
b,
Gerald
Dück
b,
Niina
Jalarvo
c,
Peter
Müller-Buschbaum
d and
Ralph
Gilles
a
*ad,
Stefan
Seidlmayer
aef,
Martin
Finsterbusch
b,
Gerald
Dück
b,
Niina
Jalarvo
c,
Peter
Müller-Buschbaum
d and
Ralph
Gilles
a
aHeinz Maier-Leibnitz Zentrum (MLZ), Technical University of Munich, Lichtenbergstr.1, 85748 Garching, Germany
bForschungszentrum Jülich GmbH, Institute of Materials and Devices, Materials Synthesis and Processing (IMD-2), Wilhelm-Johnen-Strasse, 52425 Jülich, Germany
cNeutron Sciences Directorate, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37861-6475, USA
dTechnical University of Munich, TUM School of Natural Sciences, Department of Physics, Chair for Functional Materials, James-Franck-Str. 1, 85748 Garching, Germany
eDepartment of Chemistry, Universität Bayreuth, Universitätsstrasse 30, 95447 Bayreuth, Germany
fBavarian Center for Battery Technology (BayBatt), Weiherstrasse 26, 95448 Bayreuth, Germany
First published on 7th April 2025
Abstract
NASICON (sodium superionic conductor) based ceramics are one of the most promising classes of solid-state electrolytes for all-solid-state batteries. However, the mechanism of sodium ion diffusion is not understood in great detail since there is still a discrepancy between reported average structure models, local structures, and the number and position of sodium sites. To close this gap, we investigate the underlying diffusion mechanism and structural changes governing the Na+ transport in Na3.4Zr2Si2.4P0.6O12 using quasielastic neutron scattering (QENS) and powder X-ray diffraction (XRD). In the temperature range from 298 K to 640 K, the correlations between structural changes of a monoclinic C2/c to rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c phase transition and the result of ion diffusion are investigated. The analysis of the quasielastic neutron scattering data reveals two quasielastic components corresponding to the Chudley-Elliott jump-diffusion model. It clearly shows two different Na+ diffusion processes, local and long-range, on two different time and length scales and allows calculations of their corresponding activation energies. Additionally, the effects of Sc3+ and Al3+/Y3+ aliovalent substitution of Zr4+ ions on the crystal structure and Na+ diffusion are also studied. We can distinguish a local, chain, and cross-chain diffusion mechanism based on correlated QENS and XRD comparison of relevant nearest crystallographic Na–Na distances. The results reveal that the Na+ diffusion in these NASICONs is three-dimensional and can provide guidelines on how dopants and changes in the crystal structure can affect the Na+ conductivity.
c phase transition and the result of ion diffusion are investigated. The analysis of the quasielastic neutron scattering data reveals two quasielastic components corresponding to the Chudley-Elliott jump-diffusion model. It clearly shows two different Na+ diffusion processes, local and long-range, on two different time and length scales and allows calculations of their corresponding activation energies. Additionally, the effects of Sc3+ and Al3+/Y3+ aliovalent substitution of Zr4+ ions on the crystal structure and Na+ diffusion are also studied. We can distinguish a local, chain, and cross-chain diffusion mechanism based on correlated QENS and XRD comparison of relevant nearest crystallographic Na–Na distances. The results reveal that the Na+ diffusion in these NASICONs is three-dimensional and can provide guidelines on how dopants and changes in the crystal structure can affect the Na+ conductivity.
1. Introduction
Extensive research efforts over the past few years focused on exploring sodium-ion batteries (SIBs) as a potential alternative to the widely used lithium-ion batteries (LIBs), particularly because of sodium's low costs and its high natural abundance.1,2 Numerous studies investigated SIBs with a liquid electrolyte, however, due to the safety concerns, there is a growing trend towards the exploration and utilization of all-solid-state batteries (ASSBs).3 The usage of solid electrolytes can bring several important advantages because they are non-flammable, non-toxic, non-volatile, and thermally stable.4 Moreover, replacing liquid electrolytes with solid electrolytes has the potential to facilitate the implementation of high-voltage cathode materials and Li/Na metal anodes for future high-energy-density ASSBs.5,6 The challenge is to find a highly conductive (>10−3 S cm−1) solid electrolyte material with a large electrochemical window and low production costs. It has been reported that there are a few Li+/Na+-ion solid electrolytes with ionic conductivities approaching or even surpassing the conductivity of liquid electrolytes, such as LISICON, Li-argyrodites and borohydrides (e.g., NaCBn−1Hn (n = 10 and 11), but sufficiently high conductivity is reached in many more material classes like garnets (e.g., LLZO), and NASICON (both Na and Li).7,8One of the most promising classes of sodium-based solid-state electrolytes, with its excellent chemical and thermal stability, is called NASICON, which is short for sodium (NA) SuperIonic CONductor. A crystal structure range with exceptionally high ionic conductivity, Na1+xZr2SixP3−xO12 (0 ≤ x ≤ 3), was discovered in 1976 by Hong et al. and Goodenough et al.9,10 Within this range, 1.8 ≤ x ≤ 2.2, it crystallizes in a monoclinic phase with space group symmetry at room temperature. For x < 1.8 and x > 2.2, the rhombohedral structure with space group symmetry can be obtained at room temperature,11,12 though some reports suggest that at 2.3 ≤ x ≤ 2.5, the system is a mixture of both phases at room temperature.13 While it is generally agreed in the literature that all compositions undergo a phase transition to the rhombohedral structure above ≈200 °C (≈473 K), the correct assignment of rhombohedral versus monoclinic phase, particularly for subtle splittings, remains still challenging.13–16
The NASICON materials belong to the material family AxM2(TO4)3.17,18 Both rhombohedral and monoclinic phases consist of corner-shared larger M-site ions (+3, +4, +5) in MO6 octahedra and smaller-sized, higher charged T-site (+4, +5, +6) ions in TO4 tetrahedra. Together, they form a three-dimensional framework into which they accommodate mobile A-site cations with the lowest charges (+1, +2, +3). Depending on the choice of M and T, the amount of mobile A-site cations is somewhat variable in these structures and can range from 1 to 5, while the best ionic conductors lie in the range from 3 to 4.17 In our case, A = Na, M = Zr, Sc, Al, Y and T = Si, P.
The monoclinic and rhombohedral structure models for NASICON are shown in Fig. 1. There are mainly two types of sodium sites present in the rhombohedral structure, which are usually referred to as Na1 (6b positions in Rc) and Na2 (18e positions in Rc). There is a third middle Na3 position (36f position of Rc).21,22 All Na positions in the rhombohedral structure model are typically only partially occupied in AxM2(TO4)3 because the distance between Na2 (18e) and Na3 (36f) is small, as well as the distance between Na3 (36f) and the O1 (36f) position. Thus, rhombohedral structure models with Na2 (18e) and Na3 (36f) sites are always disordered structure models. The phase transition following the monoclinic distortion Rc → C2/c (a − b, a + b, −a/3 + b/3 + c/3), allows to further differentiate the positions of the Na+ sites, Na2 (18e) and Na3 (36f). The site splitting and the notation changes for the monoclinic distortion are shown in Table 1. The rhombohedral Na2 (18e) site splits into the monoclinic Na2 (4e) and Na3 (8f), while the rhombohedral Na3 (36f) splits into three sites, Na4 (8f), Na5 (8f), Na6 (8f). As can be seen from the Rc → C2/c subgroup transition (see details in Fig. S1†), there are, in total, up to 6 possible sodium sites that are symmetry-related to the rhombohedral space group in any monoclinic structure description.
 | ||
| Fig. 1 NASICON structure: Na1+xZr2SixP3−xO12 (0 ≤ x ≤ 3) consisting of corner-sharing ZrO6 octahedra and PO4/SiO4 tetrahedra and mobile Na positions: (a) single unit cell of the rhombohedral structure (Rc f3e2cb); (b) two unit cells along b of the monoclinic structure (C2/c f12e2a) in order to clarify the similarity to the rhombohedral model. Both are based on refined structures in this paper for x = 2.4 at 640 K. Visualized with the Endeavour software.19,20 | ||

|
Among these different NASICON Zr-based electrolytes, Na3.4Zr2Si2.4P0.6O12 has been reported to be one of the best available sodium ionic conductors, for which a total conductivity of 5 × 10−3 S cm−1 was achieved at 25 °C.13 The remarkable conductivity is typically attributed to the high concentration of carrier ions and their high mobility, but it is also presumed to be influenced by other factors, as discussed in the following. The size of the oxygen triangles, through which Na+ moves, determines the ease of Na+ conduction and is presumed to be a bottleneck for Na+ conduction in the system.12,23 The bottleneck size is sensitive to both sodium content and temperature changes.24 The site occupancy of Na sites at which the migration channels connect influences the diffusion events as well.25 A direct correlation between activation energy for ionic conduction and the bottleneck size has been confirmed.12,26
Exploring and comprehending the rhombohedral-monoclinic phase transition, the underlying diffusion mechanism, and their impact on ionic conductivity in NASICON solid-state materials is crucial for its successful application and performance of ASSBs. This is particularly important because such a phase transition can cause volume changes, alter ionic pathways, modify bottleneck size and shape, impact sodium ion mobility, and potentially reduce conductivity and mechanical stability. Despite a large amount of available literature sources, the sodium ion transport mechanism and structure are not fully understood today. Moreover, there have been discrepancies between the proposed structure models. In literature, models have been proposed and tested with different amounts of Na sites (three,22,27 four,21 five,28,29 and six25 sites) with various Na site distribution, occupancies, and identified diffusion channels.
These big discrepancies found in the literature and the lack of experiment-based studies rather than modeling-based ones require further experimental examination of the phase transition and sodium transport in the crystal structure. The present study aims to elucidate the sodium diffusion mechanism in the structure at a microscopic level obtained via quasielastic neutron scattering (QENS) and to correlate them with the X-ray diffraction (XRD) studies and electrochemical impedance spectroscopy (EIS) data on ionic conductivity to reveal the activation energy of ionic conduction and to support the QENS experiment.
The overall transport of Na+ ions is a complex phenomenon governed by a broad range of local migration and site energies and energy barriers, which are critically influenced by factors such as Na+–Na+ and Na+-cation interactions, local charge, Coulomb repulsion effects, local lattice distortion effects, the degree of elemental substitution, etc. Depending on the experimental method, different time and length scales of this complex process can be probed.30 These parameters collectively determine the dominant diffusion pathways and mechanisms, as also demonstrated in prior modeling-based studies using ab initio molecular dynamics (AIMD), density functional theory (DFT), and kinetic Monte Carlo (kMC) simulations.31–33
QENS is a powerful neutron scattering-based technique, which is the only experimental method that can probe diffusion both spatially and temporally.34–36 There have been a few successful QENS studies that investigated sodium ion diffusion in sodium-based solid electrolytes such as NaAlSiO4,37 Na3ZnGaX4 (X = S, Se),38 Na2.9Sb0.9W0.1S4,39 in bi-functional electrode P2-type layered material Na2/3[Ni1/3Ti2/3]O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 or in cathode materials such as sodium cobaltate (Na0.8CoO2).41
40 or in cathode materials such as sodium cobaltate (Na0.8CoO2).41
One of the strategies for altering and improving the properties of NASICON materials is by introducing suitable substituents. For example, a partial substitution of Zr4+ by a trivalent cation generates a deficiency of a positive charge.42 This deficiency is then compensated by additional Na+ ions which leads to an increased ionic carrier concentration and improved ionic mobility and thus higher conductivity. According to the optimization studies on Na3+xScxZr2−x(SiO4)2(PO4) materials with 0 ≤ x ≤ 0.6, the Na3.4Sc0.4Zr1.6Si2PO12 (x = 0.4) have been reported to achieve the highest total conductivity of 4.0 × 10−3 S cm−1 at 25 °C.43 The conductivity of Na3Sc2(PO4)3 at 573 K has been reported to be 10−2 S cm−1.44 Because of similar ionic radii of Sc3+(r = 0.745 Å) and Zr4+ (r = 0.72 Å), the steric effect and structural changes during substitution should be minor. The higher ionic conductivity has been mainly attributed to the differences in Na+ concentration with the optimal ratio between occupied and vacant Na sites. The maximum number of Na occupancies in one unit cell is 4. However, it is crucial that some Na vacancies are present in the structure to allow Na+ diffusion. According to the previous reports, the ideal ratio between occupied and unoccupied sites for the fastest sodium transport is 3.4:0.6.12,31,43
To further investigate the effects of substituting cations, a double partial substitution of Zr4+ by equimolar amounts of Al3+ (r = 0.53 Å) and Y3+ (r = 0.90 Å) has been reported to get Na3+2xAlxYxZr2−2x(SiO4)2(PO4)3 series, where the unsubstituted sample (x = 0) exhibited the highest conductivity in comparison to Al/Y substituted, despite the increasing Na+ concentration. The total conductivity was in the range of 0.5–0.06 mS cm−1 for 0 ≤ x ≤ 0.3. This suggested that the ionic diffusion properties in NASICON materials can be affected not only by Na+ concentration and steric interactions but also by electrostatic interactions, such as the charge or bond strength of substituting elements.23
In this study, we also investigate scandium- and aluminum/yttrium-substituted NASICON materials, namely Na3.4Sc0.4Zr1.6Si2PO12 (referred to as NaSc) and Na3.4Al0.2Y0.2Zr1.6Si2PO12 (referred to as NaAlY) in order to elucidate the differences in conductivity and diffusion mechanism, as well as structural changes in comparison to the unsubstituted Na3.4Zr2Si2.4P0.6O12 (referred to as NaZr), where the concentration of Na+ charge carriers is unchanged.
2. Experimental
2.1 Synthesis of materials
All NASICON materials were synthesized via the SA-SSR method as described by Ma et al.43 Stoichiometric amounts of NaNO3 (Merck, ACS), ZrO(NO3)2·xH2O (Sigma-Aldrich, techn. grade), and where applicable, Sc2O3 (Projector GmbH, 99.5%), Al(NO3)2·9H2O (VWR, 99.2%) and Y(NO3)2·6H2O (VWR, 99.8%), were dissolved in diluted HNO3 (Aldrich, ACS grade), followed by tetraethyl orthosilicate (Merck, ≥99%). After complete hydrolyzation of the orthosilicate, NH4H2PO4 (Merck, ACS) was added. The solution was dried at 80 °C, and the precipitates were calcined at 800 °C for 4 hours in air. Finally, the calcined powder was ball milled with a mixture of 3 and 5 mm zirconia balls (50:50 by weight) in ethanol for 72 h. Dense ceramic membranes were produced by uniaxial pressing of the calcined powder in 13 mm diameter molds and sintering at 1260 °C for 6 hours or at 1150 °C for 6 hours in the case of the NaAlY sample. All NASICON powder samples that were investigated in this study were only sintered for 4 hours but otherwise treated at the same temperatures as their respective dense membrane equivalents. The chemical composition of each sample, which was determined by ICP-OES measurements, is listed in Table 2. Longer heat treatment and higher temperatures are required to achieve the highest possible densification in sintered pellets. However, lower temperatures or less exposure time is more than sufficient to achieve full crystallization of the NASICON phase. Due to the higher surface area in loose powder and, therefore, potentially faster loss of volatile species like sodium, minor changes were made to the thermal treatment parameters for the powder samples.
| Sample | Composition (theoretical) | Composition (ICP-OES) | Heat treatment |
|---|---|---|---|
| NaZr | Na3.4Zr2Si2.4P0.6O12 | Na3.32(11)Zr1.98Si2.37(9)P0.60(2)O12 | 4 h at 1260 °C |
| NaSc | Na3.4Sc0.4Zr1.6Si2PO12 | Na3.33(14)Sc0.39(2)Zr1.58Si1.95(9)P0.98(5)O12 | 4 h at 1260 °C |
| NaAlY | Na3.4Al0.2Y0.2Zr1.6Si2PO12 | Na3.42(4)Al0.19(1)Y0.21(1)Zr1.58Si1.99(3)P0.96(2)O12 | 4 h at 1150 °C |
2.2 Impedance measurements
For impedance measurements, both sides of the dense ceramic membrane were polished with SiC sandpaper up to a grit of P400, and a fine gold layer was sputtered (Cressington 108 auto coater, TESCAN GmbH, Dortmund, Germany) on both sides. Temperature-dependent impedance data were collected using an Alpha-A High Performance Modular Measurement System and a Quatro Cryostem System (Novocontrol Technologies GmbH & Co. KG, Germany). Measurements were conducted in the frequency range of 10 MHz–1 Hz. Nyquist plots of EIS data were fitted using an equivalent circuit of the type shown in Fig. S2.†2.3 Differential scanning calorimetry (DSC)
DSC measurements were performed using a METTLER TOLEDO DSC 3 thermal analysis instrument. The measurements were performed on powder samples (around 10 mg) in a standard Al sample holder (40 μL) at heating/cooling rates of 10 K min−1 in a temperature range of 298–573 K. During the measurement, samples were heated from 298 K to 573 K, held for 5 minutes at 573 K and then cooled down to 298 K in two cycles.2.4 Quasielastic neutron scattering (QENS)
With QENS, neutron scattering intensity is measured as a function of momentum (Q) and energy (E) transfer. The resulting QENS peak broadening is caused by scattering from atoms that are moving in a stochastic manner with a small energy transfer. QENS data were collected on the backscattering spectrometer (BASIS) at the spallation neutron source (SNS) at Oak Ridge National Laboratory (ORNL), USA.45,46 Si111 analyzers were used with wavelength centered at 6.4 Å. This configuration results in an energy resolution (FWHM) of 3.6 μeV, Q values ranging from 0.3 to 1.9 Å−1, and the dynamic range was selected from −100 to 100 μeV. The QENS spectra were collected at temperatures from 297 K to 640 K in 50 K increments. The resolution function was measured at 20 K. The QENS analysis was performed using the data analysis and visualization environment (DAVE) package.472.5 X-ray powder diffraction (XRD) measurements
Temperature-dependent XRD patterns were collected from the same powder batch as the samples used for QENS measurements using an Empyrean X-ray powder diffractometer (Malvern Panalytical, Almelo, Netherlands). The measurements were carried out with Cu-Kα1/Kα2 radiation (λKα1 = 1.5406 Å, 45 kV, 40 mA) in a reflection geometry using a PIXcel-detector. A fixed 1/4° divergence slit, a 1/2° anti-scatter slit, and a 0.04 rad Soller slit were used for the primary beam optics. A fixed 1/4° anti-scatter slit and a 0.04 rad Soller slit, and a Kβ-filter were used for the secondary beam optics. An incident beam mask of 10 mm was used to limit the illuminated sample area. Samples were mounted in an Al2O3 sample holder of 16 mm diameter for HT-XRD. Data were recorded in the range 10–130 ° 2θ with a step size of 0.026° and a counting time of 350 s using a 255-channel PIXcel-detector.To obtain the instrument resolution function, a reference sample of CeO2 powder was measured in the range from 23 to 100° 2θ with a counting time of 55 s per step in the same instrument setup. All peaks in the standard measurement were refined with a Pawley-Fit to obtain instrument peak shape parameters, Kα2/Kα1 ratio, and remaining small sample displacement. Peak asymmetry was calculated with the Finger-Cox-Jephcoat model,48 and these instrument setup-related parameters were then taken as fixed values for all Rietveld refinements of the measured samples.
2.6 X-ray powder diffraction (XRD): rietveld refinements
Rietveld refinements were carried out using HighScore (Malvern Panalytical) software.49 The refinements were based on ICSD# 62386 rhombohedral Rc structure by Boilot et al.22 The details of the refinement procedure and all refined parameters are described and tabulated in a separate supplement of refined crystallographic XRD data.†
In the following, we explain the site annotations and the approach we took for the refinements. Between the two space groups, Rc and C2/c, a group-subgroup relationship17 exists as depicted in Fig. S1.† The subgroup relation between the rhombohedral and monoclinic structure, the Wyckoff site splitting, and our Na site labeling are shown in Table 1. Keeping maximum comparability between the refinements in the rhombohedral and monoclinic structure models in mind, we labeled our Na atoms consistently throughout the paper and between the different structure models, correspondingly following site symmetry and the Wyckoff splitting from the lowest number equalling the highest symmetry. Thus there are in the rhombohedral case Na1 on 6b, Na2 on 18e and Na3 on 36f, corresponding to the monoclinic f12e2a structure derivation with Na1 (4a), Na2 (4e), Na3 (8f) and Na4, Na5, Na6 (all 8f).
Furthermore, what should be mentioned here is that all of the published NASICON monoclinic structure models are described in a crystallographic setting with a monoclinic cell with a ≈ 15.74 Å, b ≈ 9.1 Å, c ≈ 9.2 Å and β ≈ 124.3° with six O, Zr and one Si and one Na (here Na3) atom on 8f, one Na (here Na2) and one Si on 4e and the last Na (here Na1) on 4d-site leading to the f9e2d short-hand notation. However, the monoclinic setting we use throughout this manuscript is a ≈ 16.1 Å, b ≈ 9.1 Å, c ≈ 22.8 Å, and β ≈ 161.0° and is the one directly obtained from applying the subgroup relation as depicted in Fig. S1† from the rhombohedral Rc f3e2cb structure model. The three different monoclinic structure starting models for the refinements were then successively obtained by removing the 8f Na4, Na5, and Na6 atoms from the refinement. The equivalence of the different settings can be shown by the Compstru tool of the Bilbao Crystallographic Server.50,51 The relation between the two different monoclinic settings and the corresponding transformation matrix is also depicted in Fig. S1.† The subgroup relation shows that the monoclinic f9e2a and rhombohedral f2e2cb structure models are similar, except for additional degrees of freedom and the additional Na3 site, which is split from the Na2 site and allows more choice for Na ordering. Similar effects explain the relationship between rhombohedral f3e2cb models and monoclinic f10e2a and f12e2a models. We did not refine a separate f11e2a model as the Na-ordering refinement leaves exactly one position (Na6) empty in the f12e2a model.
3. Results and discussion
3.1 Ionic conductivity
The effective ion mobility across different length scales can be assessed depending on the used measurement technique. Electrochemical impedance spectroscopy (EIS) is a powerful and widely used method for studying the ionic conductivity in ceramic solid-state electrolyte materials.52,53 By analyzing the impedance data, valuable insights can be gained into the macroscopic transport properties of Na+ ions within the NASICON structure, which is important for the effective operation of a solid-state battery. The total (σtotal), bulk (σbulk), and grain boundary (σgb) conductivity values obtained from impedance spectra of NaZr, NaSc, and NaAlY samples at room temperature (297 K) are shown in Table 3. However, it is also important to mention that the conductivity is strongly temperature-dependent. The list of all values at all measured temperatures is provided in Table S1 in the ESI.†| Sample | σ bulk (mS cm−1) | σ gb (mS cm−1) | σ total (mS cm−1) |
|---|---|---|---|
| NaZr | 8.5 | 6.5 | 3.7 |
| NaSc | 4.9 | 6.2 | 2.7 |
| NaAlY | 2.5 | 0.2 | 0.2 |
The unsubstituted NaZr shows the highest σbulk, σgb, and σtotal compared to NaSc and NaAlY at 297 K. The amount of ionic charge carriers Na+ is here the same, so the differences are attributed mainly to the nature of substituted elements. The NaZr and NaSc samples have more comparable conductivities, which might be linked to comparable ionic radii of Zr and Sc, as mentioned in the Introduction. The decrease in ionic conductivity with increasing Sc3+ substitution on the Zr4+ site has been previously confirmed by AC impedance measurements and MD simulations.54 It has been demonstrated that the crystal structure is more disordered due to the random distribution of Zr/Sc on MO6 sites and Si/P on TO4 sites, which impedes sodium-ion diffusivity.54–56
On the other hand, NaZr shows much higher conductivity than the NaAlY sample at 297 K. This was confirmed by the previous studies where the unsubstituted NASICON exhibited the highest σtotal, σgb, and σbulk conductivity in comparison to Al/Y substituted.23 In the Y-only substituted case, the improvement in total conductivity was observed, but it was minor.57 In another study, the conductivity decreases as the content of Y3+ dopants increases.58 In this case, the ionic size of Y3+ is larger than Zr4+.
These observations point out that the radii of elements play an important role and influence the ionic conductivity and, therefore, potentially alter diffusion pathways, but the effects of grain boundary, microstructure, and porosity also have a significant impact on conductivity as well.57,59 The Sc- and Al/Y- substitution did not directly improve the EIS-measured conductivity. One possible explanation is that subvalent dopants (Sc3+, Al3+, Y3+) introduce local electrostatic distortions due to their lower charge compared to the host ion (Zr4+), which can cause a stronger trapping effect that decreases the Na+ ion mobility.32
However, there are several other reasons for doping strategies, such as improvement in structural stability and phase transition behavior, which is later discussed in Chapter 3.4. Some other aspects and importance of doping in NASICONs have previously been discussed in the literature.32,60–63 They show that doping has a strong influence on Na+ site occupancy and sample density and that the choice of the right doping elements can modify the microstructure, thereby affecting grain boundary conductivity. The dopant size affects the size of the migration bottlenecks, and the electronic structure of the dopant affects the flexibility of the structural framework. Additionally, since undoped NASICONs often suffer from mechanical degradation during cycling, doping can strengthen the NASICON framework by reducing lattice strain and improving cyclability, chemical stability, and interfacial stability with electrodes.
The Arrhenius plots (ln(σT) vs. 1/T) of σbulk and σtotal of NaZr, NaSc, and NaAlY samples in the temperature range of 173 K to 423 K are shown in Fig. 2a and b. The obtained corresponding activation energies are also shown in the plots. The NaZr possesses the highest conductivity, so consequently, it shows the lowest activation energy (Ea (total, NaZr) = 0.324(1), Ea (bulk, NaZr) = 0.285(1)) compared to NaSc (Ea (total, NaSc) = 0.333(1), Ea (bulk, NaSc) = 0.315(1)) and NaAlY (Ea (total, NaAlY) = 0.344(1), Ea (bulk, NaAlY) = 0.313(6)) samples. The activation energy of Al/Y substituted NASICON has been previously reported to be ≈0.3 eV.23 For the Sc-substituted NASICON system it has been reported as 0.26 eV.43 In general, all the Ea values derived from EIS are in the range of 0.2–0.4 eV, which are typical values for solid-state electrolytes.64,65 Many aspects of differences in ionic conductivities and activation energy barriers based on NASICON composition have previously been well discussed in the literature.12,31 For completeness, the Arrhenius plot (ln(σT) vs. 1/T) of σgb of NaZr, NaSc, and NaAlY samples in the temperature range of 173 K to 423 K are shown in Fig. S3 in the ESI.† The grain boundary features have a significant impact on EIS-measured conductivity, while they have no impact on QENS measurements. Therefore, grain boundary contributions do not directly correlate with the QENS microscopic processes discussed further in this work.
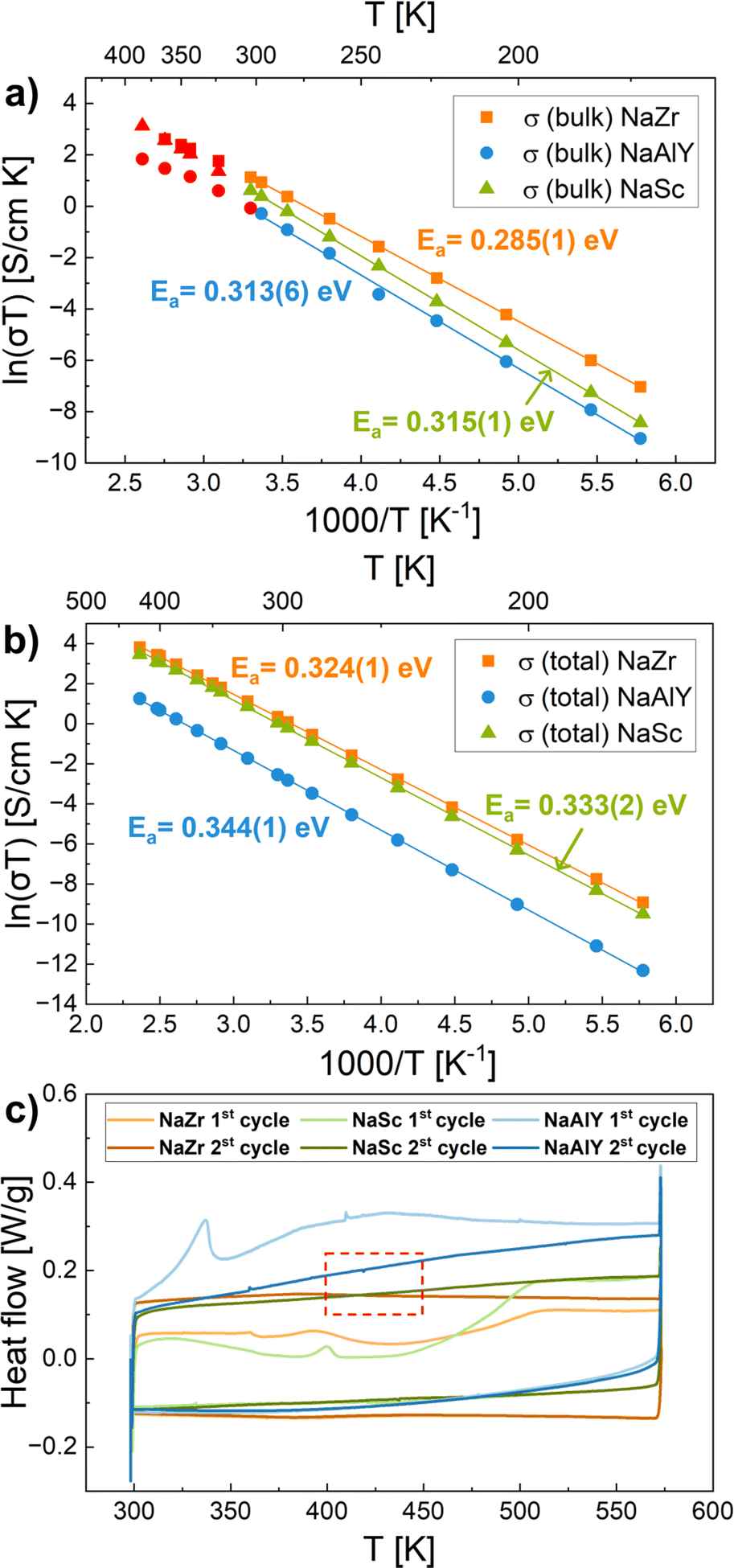 | ||
| Fig. 2 Arrhenius plots of (a) bulk and (b) total conductivities of Na3.4Zr2Si2.4P0.6O12 (NaZr), Na3.4Sc0.4Zr1.6Si2PO12 (NaSc) and Na3.4Al0.2Y0.2Zr1.6Si2PO12 (NaAlY) samples and the corresponding activation energies (Ea). Red data points in (a) are derived from less reliable fit parameters of impedance measurements. They were not considered to determine Ea. (c) DSC curves for NaZr, NaSc, and NaAlY: there are no observable phase transition peaks for the temperature range of ≈400–450 K, as marked with a red square. | ||
3.2 Sodium ion dynamics and jump-diffusion mechanism
Temperature-dependent QENS measurements were performed to obtain information about the sodium diffusion mechanism within the crystal structure of our NASICON materials. The QENS spectra consist of two major parts: the elastic peak and the quasielastic broadening of the elastic peak. Analysis of the quasielastic broadening enables the determination of Na+ self-diffusion coefficients, jump distances, and relaxation times. The corresponding activation energies can be extracted by investigating the temperature dependence of the observed motion. A direct correlation between QENS and XRD results, providing a detailed understanding of the sodium ion diffusion mechanism and elucidating migration pathways, will be presented later in Chapter 3.5.Sodium has an incoherent scattering cross-section of ≈1.62 barns but also an equally big coherent scattering cross-section (≈1.66 barns). Therefore, the measured QENS signal contains contributions from both incoherent (uncorrelated) and coherent (correlated) diffusive motions. However, previous simulation-based studies on Na+ ionic conductors showed that the incoherent scattering signal typically dominates the QENS signal, as the coherent dynamical structure factor shows significantly lower intensity than the incoherent one.33,66 Our analysis, therefore, assumes incoherent scattering as dominant, allowing us to probe uncorrelated single-particle diffusion.
In Fig. 3a, the QENS spectra for NaZr at all measured temperatures (20–640 K) are shown at selected Q = 0.7 Å−1. It can be noted that the elastic peak intensity is decreasing and the quasielastic broadening is increasing with increasing temperatures. This indicates apparent ion dynamics and demonstrates enhanced diffusion at higher temperatures.
 | ||
| Fig. 3 Selected QENS spectra for Na3.4Zr2Si2.4P0.6O12 (NaZr): (a) dynamic structure factor S(Q, E) as a function of energy transfer at selected Q = 0.7 Å−1 for different temperatures. (b) Representative fit: dynamic structure factor S(Q, E) as a function of energy transfer from the QENS experiment at selected T = 450 K and Q = 0.7 Å−1. The black triangles represent the experimental data and the red line represents the total fit of eqn (1) with a χ2 = 1.527. (c) Dynamic structure factor S(Q, E) as a function of both – energy transfer and Q at selected T = 400 K. | ||
The experimentally measured dynamic structure factor S(Q, E) can be well described by a delta function δ(E) (elastic peak contribution from immobile species with an amplitude x(Q)), and single or multiple Lorentzian functions Lj(Q, E) (quasielastic contribution arising from species that undergo diffusive motions), convoluted with the instrumental resolution function R(Q, E), F is the scaling factor and background B(Q, E) is fitted by a linear function:
| S(Q, E) = F[x(Q)δ(E) + (1 − x)(Q)Lj(Q, E)] ⊗ R(Q, E) + B(Q, E) | (1) |
An example of the fit to the data for NaZr with χ2 = 1.527 is shown for Q = 0.7 Å−1 at 450 K in Fig. 3b. The peak broadening is defined as half-width at half maximum (HWHM). The dependence of the HWHM on Q provides critical information into the temporal (time-scale) and spatial (length-scale) characteristics of the diffusion processes. In Fig. 3c, the data and the fits are shown for NaZr at selected T = 400 K for the whole range of measured Q values from 0.3 to 1.9 Å−1.
A gradual temperature-dependent increase of quasielastic broadening has been observed in previous studies on Na-based ionic conductors.37–39 The increase in jump frequency with temperature, thus enhanced ionic mobility, has also been observed by several molecular dynamics simulation studies on NASICON27 and other ionic conductors, e.g., β-Li3PS4.67 Some of the remaining QENS spectra are shown in ESI (Fig. S4†).
Two Lorentzian components (narrow and broad) are required to describe the quasielastic broadening in the temperature range from 400 K to 640 K. Fit results for NaZr are shown in Fig. 4a and b. The HWHM of both Lorentzian functions with clear Q dependence describes Na+ diffusion and follows a so-called Chudley-Elliott (Ch-E) jump-diffusion model.68 This model is regularly used to describe ion diffusion in solid crystalline matrices.
 | ||
| Fig. 4 HWHM of the Lorentzians as a function of Q at different temperatures. Solid lines represent fits of the Chudley-Elliott model of eqn (2): Na3.4Zr2Si2.4P0.6O12 (NaZr): (a) narrow Lorentzian function at 400–640 K, (b) broad Lorentzian function at 298–640 K; Na3.4Sc0.4Zr1.6Si2PO12 (NaSc) at 298–400 K: (c) narrow Lorentzian function, (d) broad Lorentzian function; Na3.4Al0.2Y0.2Zr1.6Si2PO12 (NaAlY) at 298–400 K: (e) narrow Lorentzian function, (f) broad Lorentzian function. | ||
The model is characterized as follows:
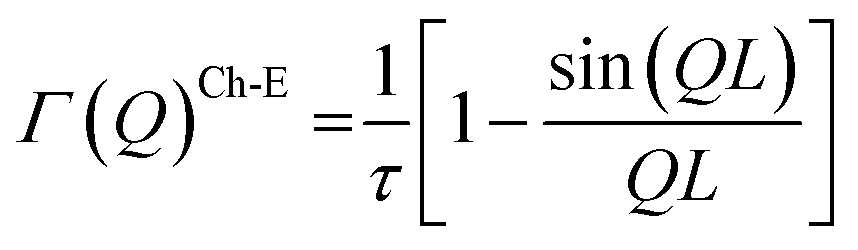 | (2) |
The Ch-E model assumes that diffusion occurs as a series of successive jumps in random directions from one available crystallographic site to another, with a jump length, L, a residence (also known as relaxation) time, τ, and that the time of the jump process is negligible. The self-diffusion coefficient is calculated as:68
 | (3) |
We can observe a clear temperature and Q dependence. The observed Q dependence aligns well with the characteristics described by the chosen translational diffusion model. Each Lorentzian corresponds to a dynamical process observable on the measured time scale. At temperatures below 400 K, the narrow component was not resolvable within the instrumental time scale. Therefore, only the broad Lorentzian function is used to interpret the data. This result shows a change in the mechanism of Na+ diffusive motion above the temperature of 400 K, which corresponds well to the previously reported phase transition at ≈200 °C (≈473 K).13–16
In Fig. 4c and d, the QENS results for the NaSc sample are shown. We can observe a clear Q dependence, however, not a very strong temperature dependence, showing no apparent change in diffusion mechanism at temperatures below 400 K. Similarly, the results for the NaAlY sample are shown in Fig. 4e and f. Unfortunately, due to the beamtime time restrictions, these samples were not measured at temperatures above 400 K.
3.3 Key parameters of sodium dynamics: diffusion coefficient, jump length, residence time, and activation energy
Key diffusion parameters, such as residence times and jump lengths, are extracted here from the analyzed and modeled QENS data to provide a quantitative understanding of the sodium ion transport process. In Fig. 5a, residence times τ for NaZr are shown. The residence times extracted from the narrow Lorentzian function (right green) are on the ns scale and have a clear temperature dependence, increasing until 450 K and then decreasing above 450 K. The residence time τ on the ps scale (left blue) extracted from the broad Lorentzian function appears to be more constant. But similarly, we can observe a slight increase as a function of temperature up to 450 K and a slight decrease afterward up to 640 K. This time scale is in agreement with previous studies on Na+ hoping in NASICON via molecular dynamics simulations that determined that long-range diffusion occurs on time scales smaller than 1 ns.54 | ||
| Fig. 5 (a) Residence (relaxation) times and (b) jump lengths of Na+ ionic diffusion extracted from QENS data for Na3.4Zr2Si2.4P0.6O12 (NaZr). Solid lines are guides to the eye. (c) Arrhenius plot of diffusion constants and corresponding activation energies (Ea) obtained from the fits of eqn (4). Only the positive error values are shown in this case because of the logarithmic scale used. The broad Lorentzian function (blue) describes a Na+ diffusion mechanism with more frequent jumps with a smaller residence time (≈7–12 ps) and a shorter jump length. The narrow Lorentzian function (green) describes a Na+ diffusion in which jumps are less frequent with a higher residence time (≈48–157 ps) and longer jump length. | ||
In Fig. 5b, the jump length L extracted from the narrow Lorentzian increases with higher T up to a maximum of 3.38(34) Å at 490 K and then decreases slightly. The jump length L extracted from the broad Lorentzian increases with the increasing T, revealing that longer and more frequent jumps can be activated by higher thermal energy and diffusion processes become faster. The jump length ranges from 1.38(59) Å to 3.15(13) Å for the data fit for the broad Lorentzian function and from 1.81(36) Å to 3.38(34) Å for the narrow Lorentzian function.
The use of two Lorentzian components in this QENS analysis reflects the presence of two distinct physically reasonable diffusion processes contributing to the total quasielastic signal. The results from the broad Lorentzian function reveal a Na+ diffusion mechanism with more frequent jumps, smaller residence times (≈7–12 ps), and shorter jump lengths corresponding to a local Na+ diffusion. Meanwhile, the results obtained from the narrow Lorentzian function demonstrate a Na+ diffusion mechanism in which jumps are less frequent with a higher residence time (≈48–157 ps) and a longer jump length that reflects single jumps in a long-range diffusion process through extended pathways in the NASICON structure. Similar studies of ionic conductors have previously also used two Lorentzians for the description of localized and long-range motion.69–71 These results are consistent with the structural XRD results, validating the proposed Na+ diffusion mechanism, detailed in Chapter 3.5.
All values of determined residence times and jump lengths for all three samples NaZr, NaSc and NaAlY at all measured temperatures are listed in Table S2.† For NaSc, the jump length ranges from 2.09(48) Å to 2.75(14) Å for the broad Lorentzian function. We can observe a slight increase as temperature increases, similarly as for NaZr sample. For the narrow Lorentzian function, the jump length ranges from 2.64(46) Å to 2.43(35) Å, which is a slight decrease but comparable within the errors. For NaAlY, the jump length ranges from 5.63(88) Å to 2.48(30) Å for the broad Lorentzian function and from 2.54(55) Å to 2.47(36) Å for the narrow Lorentzian function, both as temperature increases.
To compare the three samples directly at 400 K, the jump distances are shorter for NaZr, 1.70(41) Å and 1.81(36) Å, than for NaSc, 2.75(14) Å and 2.43(35) Å and for NaAlY, 2.48(30) Å and 2.47(36) Å (values extracted from narrow and broad Lorentzian respectively). This may be caused by different ionic radii of substituted elements that influence structural changes and, therefore, changes in the sodium diffusion mechanism in the material, as discussed in Chapter 3.1. The NaZr sample exhibits the highest conductivity and lowest activation energy, which is the result of shorter jump distances allowing faster diffusion, as we demonstrate here by QENS.
The Na+ self-diffusion in NASICON can be well-described by the self-diffusion coefficients D (calculated by eqn (3)) shown in Fig. 5c. The Na+ diffusion coefficients are in the order of 10−6 to 10−5 cm2 s−1, which confirms the superionic properties of studied NASICONs. As can be observed, the diffusion coefficients increase as a function of temperature. We obtain two sets of diffusion coefficients derived from broad and narrow Lorentzian function fits, describing a slower and a faster process within the material. All values of diffusion coefficients and their associated calculated errors for all three samples, NaZr, NaSc, and NaAlY, are listed in Table S2.†
Comparison of unsubstituted and substituted samples at 400 K shows that the diffusion coefficients for the faster process are 4.79 × 10−6 cm2 s−1 (NaZr), 7.68 × 10−6 cm2 s−1 (NaAlY), and 12.04 × 10−6 cm2 s−1 (NaSc), so DNaZr < DNaAlY < DNaSc. For the slower diffusion process at 400 K, the following values are determined: 0.35 × 10−6 cm2 s−1 (NaZr), 0.49 × 10−6 cm2 s−1 (NaAlY), and 0.59 × 10−6 cm2 s−1 (NaSc), so they follow the same trend DNaZr < DNaAlY < DNaSc. Both the Sc- and AlY-substituted samples show slightly higher diffusivity than unsubstituted NaZr at 400 K. This effect of an increase of diffusion coefficients with aliovalent elemental substitution has previously been observed on different types of Na+ ionic conductors, e.g., Na3−xSb1−xWxS4, x = 0; 0.05; 0.1.39 However, this outcome might appear in disagreement with the ionic conductivity trend discussed in Chapter 3.1, where we observe that the doping does not improve the conductivity, so the unsubstituted NaZr sample shows the highest ionic conductivity values. The diffusion coefficients derived from the bulk conductivity (Dσ) are also shown here for the comparison with QENS. All these values, which are calculated using the Nernst–Einstein equation, are listed in ESI in Table S1.† The Dσ values at 363 K are 5.9 × 10−8 cm2 s−1 (NaZr), 5.6 × 10−8 cm2 s−1 (NaSc), and 2.6 × 10−8 cm2 s−1 (NaAlY), so they show the same trend as ionic conductivity DσNaAlY < DσNaSc < DσNaZr. The values are also 1–2 orders of magnitude lower than the diffusion coefficient extracted from the QENS measurements.
All the discrepancies can be explained by the fact that EIS- and QENS-derived parameters are based on different measuring principles and it's important to consider this when discussing properties of ionic conductors. The differences can arise from the fact that impedance spectroscopy represents rather macroscopic length scales. In comparison, QENS probes the microscopic diffusion of the materials.30 The features of the materials, like grain boundary, microstructure, and porosity, as well as electrode interfaces during the EIS measurements, have a significant impact on EIS-measured conductivity,59 whereas no impact or role in QENS measurements.
In Fig. 5c, the temperature dependence of diffusion coefficients for NaZr follows the Arrhenius law:
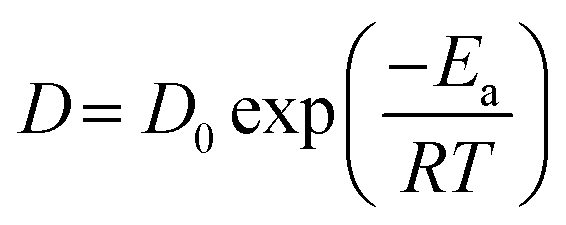 | (4) |
The activation energy barrier of ionic jumps for NaZr obtained from the Arrhenius fit for the narrow Lorentzian is 0.18(1) eV in the temperature range 400–640 K. An inflection point can be observed for the broad Lorentzian fits around ≈440–490 K. The activation energies obtained from the Arrhenius fit for the broad Lorentzian are 0.03(1) eV below 490 K and 0.29(3) eV above 490 K. This rapid change in activation energy reveals a change in diffusion mechanism somewhere in the temperature range from 400 to 450 K. Above this temperature range a second type of diffusion process starts taking place. Once Na+ ions escape their local cages, long-range diffusion (narrow Lorentzian) involves jumping between sites with lower energy barriers. Similarly, it has been previously observed that the unsubstituted NASICON, Na3Zr2(SiO4)2(PO4), showed a change in activation energy between low-temperature and high-temperature measurements (>400 ± 200 °C) to be 0.33 eV and 0.27 eV, respectively.9,10,12
As mentioned, the closest EIS-derived parameter to compare to QENS is the bulk conductivity and its corresponding activation energy. The value 0.18(1) eV from the narrow Lorentzian is around 33% lower than the activation energy values Ea (bulk) = 0.285(1) eV obtained from the impedance measurements from Chapter 3.1. The value 0.29(3) eV from broad Lorentzian is comparable within the errors at higher T ≥ 490 K. The differences can arise from the fact that impedance spectroscopy represents rather macroscopic length scales, thus the averaged migration of Na+ over the bulk solid electrolyte. In comparison, the QENS measurement probes the microscopic self-diffusion of Na+ in the material, i.e., single particle motion. That means that the reported jump distances and both local and long-range diffusion mechanisms from QENS reflect elementary steps in the translational diffusion process, so they cannot be comprehended exactly in the same way as long-range cumulative migration observed in EIS.
Nonetheless, almost identical values obtained from QENS (at T ≥ 490 K) and EIS (at T ≤ 300 K) mean that the average macroscopic transport is similar to the underlying microscopic diffusivity, but it is difficult to directly match one of the long-range and local QENS processes to macroscopic EIS processes. One of the possible explanations for the agreement in activation energies is that the local Na+ jumping, i.e., short-range jumps, influences the temperature-dependent resistance measured by EIS. Because localized motion can be a rate-limiting step (at higher T with high activation energy) and the initial energy barrier for ion migration, it is a contributor to both EIS macroscopic and QENS microscopic activation energies. At temperatures above room temperature, we cannot safely separate bulk and grain boundary contributions in the EIS data, so the information about the possible changes in activation energy might be missing, while in QENS, we observe a temperature-dependent change in activation energy. There are also no changes in activation energy in the Arrhenius fit of σtotal, Ea (total) = 0.324(1) eV, which provides more reliable data at higher temperatures.
To summarize, both methods show a reasonably low activation energy barrier that is comparable to previous studies on NASICON ceramic ionic conductors, as discussed in Chapter 3.1. The activation energy for NaSc and NaAlY samples could not be determined based on the QENS experiment due to a lack of measured data points at different temperatures. The overall analysis revealed that the QENS spectra could be reproduced with two quasielastic components, corresponding to distinct Na+ diffusion processes with associated activation energies, representing Na+ dynamics on two different time and length scales, slower long-range diffusion (narrow Lorentzian) and faster local diffusion (broad Lorentzian). In the following chapters, we examine how these results correlate with the observed structural changes.
3.4 Phase transition analysis and structure determination
The heat effects of the phase transitions in NASICON were previously investigated with DSC measurements in several studies, where DSC shows a distinct peak at ≈160 °C (≈433 K) for Na3Zr2Si2PO12,72 at ≈150 °C (≈423 K) for Na3.4Zr2Si2.4P0.6O12 (ref. 73) and at ≈140 °C (≈413 K) for Na3.12Zr1.88Y0.12Si2PO12.73 The peaks were reproducible for both heating and cooling and were attributed to the second-order phase transition, which is close to the reported structural phase transition temperature. The DSC analysis for our NaZr, NaSc, and NaAlY samples at temperatures 298–573 K with a heating rate of 10 K min−1 is shown in Fig. 2c. The peak at 393 K in the first cycle for NaZr is attributed to the release of H2O because the samples were stored in air and contained some moisture. After the complete evaporation of H2O, the DSC results for the second cycle show no distinct phase transition peaks during either heating or cooling. Similarly, for the NaSc and NaAlY samples, the DSC analysis shows a peak in the first cycle at 398 K and 348 K, respectively, but no peak in the second cycle. All peaks in the first cycles are attributed to moisture loss. The increase in heating/cooling rate (20 K min−1 and 30 K min−1) is tried out to enhance the sensitivity and identify the possible weak transformation, but no distinct peaks have been visible. No clear phase transition is observed from our data in the subsequent DSC cycles. Similarly, in the studied temperature range of 173 K to 423 K for EIS measurements shown in Fig. 2a and b, there is no inflection point observed in the Arrhenius plot of σtotal, and no inflection point observed in the Arrhenius plot of σbulk in the reliable temperature range of 173 K to 300 K, thus no modifications of activation energy, which would be linked to a phase transition. However, the QENS results reveal a diffusion mechanism change happening in the temperature range 400–450 K, as shown in Fig. 5c and discussed in the previous chapter.These observations motivate us to investigate the structural changes in NASICON, mainly in unsubstituted NaZr, in greater detail to help us elucidate the connection between temperature-dependent activation energy changes from QENS and the absence of DSC peaks. The question arises if there really is a previously reported phase transition from monoclinic to rhombohedral structure. To further understand this behavior, the XRD measurements are performed in the same broad temperature range, 297 K to 640 K, as for the QENS measurements. Here, the jump length results from QENS are compared with interatomic distances from the XRD Rietveld refined structural data. The Bragg reflections from XRD detect average structural properties within the material. On the other hand, diffusion models used on QENS data are based on local jumps from one empty lattice site to the neighboring one. This local behavior might differ from the average crystal structure obtained by XRD. Since the Ch-E diffusion model from QENS is based on atom-to-atom jumps, we take into consideration the nearest crystallographic Na–Na distances obtained from the monoclinic and rhombohedral structure models.
In this work, we compare five structural models with different amounts of available Na+ positions within the crystal structure, which are listed in Table 4. Their Wyckoff sequence is listed as well. The f11e2a structure model was not explicitly used in the refinements. For temperatures below 400 K, the refined f12e2a model results in the f11e2a model because the site occupancy of one of the Na positions is refined to zero. For temperatures above 400 K, it becomes non-zero.
| Crystal system | Space group | Wyckoff sequence | Number of Na positions |
|---|---|---|---|
| Rhombohedral |
Rc |
f2e2cb | 2 |
| Rhombohedral |
Rc |
f3e2cb | 3 |
| Monoclinic | C2/c | f9e2a | 3 |
| Monoclinic | C2/c | f10e2a | 4 |
| Monoclinic | C2/c | f12e2a | 5/6 |
A few typical XRD patterns and corresponding Rietveld refinement results at selected temperatures (lowest and highest measured temperatures, 298 K and 640 K) are shown in Fig. S5.† All refined parameters for each material and measurement at each temperature are listed in a separate supplement of refined crystallographic XRD data.
In Fig. 6a and b, the goodness of fit values Rwp (weighted profile R factor) and RBragg (the R factor measuring the agreement between the reflection intensities calculated from a crystallographic model and those measured experimentally) are shown, which characterize the suitability of the selected structure models used for our XRD data for the NaZr sample. In case of a clear phase transition, it is expected that the RBragg values in an XRD analysis of the correct structure model decrease as a function of T after the expected phase transition point from monoclinic to rhombohedral at ≈400 K. However, we observe an increase in RBragg values for both rhombohedral models above 450 K, which points out that the models are less correct and disagree with the expected clear phase transition.
 | ||
| Fig. 6 Fit residuals values: Rwp and RBragg for 5 different rhombohedral and monoclinic structural models: (a) and (b) Na3.4Zr2Si2.4P0.6O12 (NaZr), (c) and (d) Na3.4Sc0.4Zr1.6Si2PO12 (NaSc), (e) and (f) Na3.4Al0.2Y0.2Zr1.6Si2PO12 (NaAlY). | ||
However, we observe that according to Rwp values, the structure is clearly monoclinic below the temperature of 400 K. Above 400 K, both monoclinic and rhombohedral fits seem to be suitable at first sight. From the three monoclinic models, the f12e2a with A3-type sodium sites (Na4, Na5, and Na6) is the best fit in all cases. Additionally, at temperatures 400 K and below, the occupancy of the Na6 site is zero. The refinements thus confirm the f11e2a structure type with only five occupied Na sites.
This result fits well with the change in activation energies from QENS measurements (Chapter 3.3). Thus, the observed changes in the mechanism of diffusive motions prove a change in crystal structure, such as a phase transition. However, since both monoclinic and rhombohedral models fit comparably over 400 K, assigning the correct space groups is challenging. What we observe is rather a continuous change towards higher symmetry, possibly still unfinished at 640 K. It can be interpreted as a second-order phase transition according to the modern phase transition classification based on the original Ehrenfest scheme.74 The transformation from high-symmetry (Rc) to low symmetry (C2/c) requires no sudden appearance of reflections in the XRD pattern. It is caused by a gradual shifting of the reflection pattern and gradual intensity changes. However, a missing DSC signal, as shown in Fig. 2c, shows that no latent heat accompanies the process. At first sight, this would indicate that no phase transition happens at all or that it is “smeared out” over the whole measured temperature range.
The phase transition might still be incomplete even at our highest measured experimental temperature (640 K) as the site occupancy factors (SOFs) of the Na4/Na5/Na6 sites in the monoclinic f12e2a remain asymmetric (see Fig. S6†). At temperatures 400 K and below, where the Na6 site occupancy refines to 0, the structure clearly cannot be characterized as rhombohedral. We consider, in general, that the phase transition we observe here is of the type of a second-order phase transition, which has both displacive and order-disorder-related effects. The order parameter seems to be the occupancy of the Na6 site of the monoclinic structure model or the “breaking” of the symmetry in the occupational arrangement between the Na4, Na5, and Na6 sites. If the rhombohedral model was correct, the site occupancies of Na4, Na5, and Na6 sites would be equal if the symmetry is transformed to the higher symmetry structure model in the rhombohedral Rc, as the three sites merge into the Na3 site.
In general, we observe that the Na+ ions in NaZr are disordered across multiple crystallographic sites for both rhombohedral and monoclinic models, as shown by their partial occupancies refined for these positions (SOFs < 1) in Fig. S6.† This disorder also strongly supports the assumption that Na+ diffusion occurs via uncorrelated, single-ion jumps. The only ordered site is the Na2 site in the monoclinic models, which is nearly fully occupied (SOF ≈ 1) at lower temperatures (297–350 K) and then it drops to ≈0.8 and lower at temperatures above 400 K. Thus, at higher temperatures, Na+ ions partially vacate the Na2 site, increasing structural disorder and migrating to other sites. While the Na2 site provides a more stable position and might cause partly correlated motion, the low-occupancy sites still mostly enable uncorrelated jumps. The drop in Na2 occupancy above 400 K suggests thermally activated disorder, which is enhancing ionic mobility.
In Fig. 7, selected angular regions are shown to further emphasize the differences between rhombohedral and monoclinic fits on chosen angles. In (a) and (b), a stack of X-ray diffraction patterns collected during temperature-dependent measurements is shown at selected angular 2θ range of 18.5–20° and 29.5–31.5°, where their intensity of reflections is represented by color. At the lowest (298 K) temperature shown in (c)–(f), the structure is clearly monoclinic because a much more reliable fit is obtained at selected reflections. A reflection at around 2θ = 19.3° is not present in the rhombohedral model but can be fitted using the monoclinic model. In contrast, at the highest measured temperature (640 K) shown in (g)–(j), both rhombohedral and monoclinic structural fits are more comparable, but monoclinic is still more suitable. The full patterns for 298 K and 640 K of Rc f3e2cb and of C2/c f12e2a are shown in Fig. S5.†
 | ||
| Fig. 7 Na3.4Zr2Si2.4P0.6O12 (NaZr): (a) and (b) mapping of X-ray diffraction patterns collected during temperature-dependent measurements, shown at selected angular range with intensity represented in color; (c) and (d) monoclinic (C2/c f12e2a) fit at 298 K; (e) and (f) rhombohedral (Rc f3e2cb) fit at 298 K; (g) and (h) monoclinic (C2/c f12e2a) fit at 640 K; (i) and (j) rhombohedral (Rc f3e2cb) fit at 640 K. | ||
The question about the number of sodium sites present in the structure needs to be addressed as well. Here, we can again look at the SOFs of Na extracted from the Rietveld-refined XRD data, as shown in Fig. S6.† As mentioned, the site occupancies of the different Na sites show that almost all the Na sites are partially occupied at all temperatures. However, for the C2/c f12e2a structure, we can observe that a 6th Na position Na6 is partially occupied at 450 K and above. Below 400 K, the occupancy of Na6 is always refined to 0. This also confirms a change in the Na+ diffusion mechanism through an additional Na6 site. Overall, this proves that previously reported 3- and 4-Na site models are not sufficient to describe the NaZr structure, especially at higher temperatures. However, as previously mentioned, all tested models provide plausible descriptions of the average structure, with only minor differences in fit residual values above 400 K (Fig. 6a). As the XRD refinements show, the changes in partial Na occupancy, especially for the A3 site-related positions (Na4, Na5, Na6), drive the phase transition from monoclinic to rhombohedral.
In Fig. S7,† all Na–Na distances extracted from the refined XRD data for NaZr are shown. What we observe for all models is the existence of a longer and shorter set of Na–Na distances, which correlate with jump distances extracted from QENS. For the first four models I–IV, the interatomic distances remain constant as a function of temperature. Again, for the C2/c f12e2a structure, we can observe a change in the interatomic distances at 400 K.
To explain the changes in interatomic and jump distances, we can examine the lattice parameters a and c, as shown in Fig. S8a.† The data reveal the anisotropic thermal expansion, where the lattice parameter c expands more significantly than a, leading to a greater increase in the average distance between neighboring atoms along the c-direction. The expansion in the c-direction may influence the ionic transport pathways. At the same time, the anisotropy of the expansion could also affect the material's mechanical stability and overall performance as a solid-state electrolyte. The anisotropic expansion in NASICON has previously been reported in several studies.13,75 Additionally, there are differences in temperature-dependent lattice parameter a between the rhombohedral and monoclinic structural fits (Fig. S8b†). Again, the change in the temperature range of 400–450 K is consistent with the phase transition temperature.
We can take a similar approach to discuss the structure analysis of NaSc and NaAlY samples. In Fig. 6c–f, their fit values Rwp and RBragg are shown. For NaSc, at room temperature, the structure is clearly monoclinic; however, at temperatures 350 K and 400 K, all the Rietveld fits are almost comparable, with the lowest Rwp for the C2/c f12e2a structure. For the NaAlY structure, the situation is similar; however, for both measured temperatures, 298 K and 400 K, all monoclinic and rhombohedral models fit comparably, with the lowest Rwp for the C2/c f12e2a structure. In the same manner as for NaZr and for the completeness of data, all the refined occupancies and Na–Na distances for NaSc and NaAlY are shown in Fig. S9–S12.† The SOFs of the different Na sites also show that almost all the Na sites are partially occupied at all temperatures. For the C2/c f12e2a structure, the Na6 position for NaSc becomes occupied at 400 K, whereas for NaAlY, the Na6 is partially occupied at both measured temperatures. These observations show differences in diffusion mechanisms between the unsubstituted and substituted NASICON materials, which are strongly dependent on Na+ site occupancy. As shown here, this doping strategy is also designed to address the structural stability and phase behavior of NASICONs. The unsubstituted NaZr undergoes a gradual monoclinic to rhombohedral phase transition starting at ≈400 K, which significantly impacts ionic transport. While the Sc- and Al/Y-substitutions partly shift the onset of the monoclinic distortion and show better suitability of the rhombohedral phase fits at lower temperatures. In other words, the results indicate that the start of the phase transition is pushed towards lower temperatures, starting at 350 K for the NaSc sample and even at 298 K for the NaAlY sample.
All the above-mentioned observations point out that the continuous monoclinic to rhombohedral symmetry change has an influence on the structural parameters, such as interatomic distances, occupancies, and lattice parameters, and therefore, influences the overall Na+ transport mechanism in NASICONs. The high-temperature phase shows enhanced long-range mobility and increased jump lengths. In the next Chapter 3.5, we show how the monoclinic structure model allows for the clear distinction of the reported diffusion pathways in NaZr material and emphasizes the significance of changes in Na site occupancy, e.g., for the Na6 site, which becomes occupied only above 400 K. The observed anisotropic site occupancy indicates that the rhombohedral symmetry remains broken even at very high temperatures. Although the cell metrics and other parameters no longer exhibit monoclinic distortion, the Na site occupancy factors continue to deviate from true rhombohedral symmetry.
3.5 Proposed Na+ diffusion pathways in Na3.4Zr2Si2.4P0.6O12 (NaZr)
To further reveal the migration channels in the most suitable monoclinic structure model C2/c f12e2a for NaZr, we propose a combined approach and a closer look at experimentally measured Na–Na distances by XRD and QENS. In Fig. 8, we selected the relevant interatomic distances extracted from Rietveld refined XRD data to be directly compared with QENS jump distances. The green and blue areas mark the lower and upper error limits of QENS-determined jump distances at each temperature extracted from narrow and broad Lorentzian, respectively (see Table S2†). Fig. 8a shows that below the temperature of 400 K (blue area), the diffusion mainly occurs locally between the Na1–Na4, Na1–Na5, Na3–Na4, Na3–Na5, and Na4–Na5 sodium sites. We can attribute these XRD distances (≈1.2–2.2 Å) to the jump distances obtained from the broad Lorentzian QENS fits, which describe the local in-cage diffusion mode, as these Na–Na distances are localized within a particular structural unit, which we call a local cage, around the Na1 position (see depiction in Fig. 9a-I and b-I). According to QENS, the Na2 site does not participate in the local diffusion at these lower temperatures below 400 K.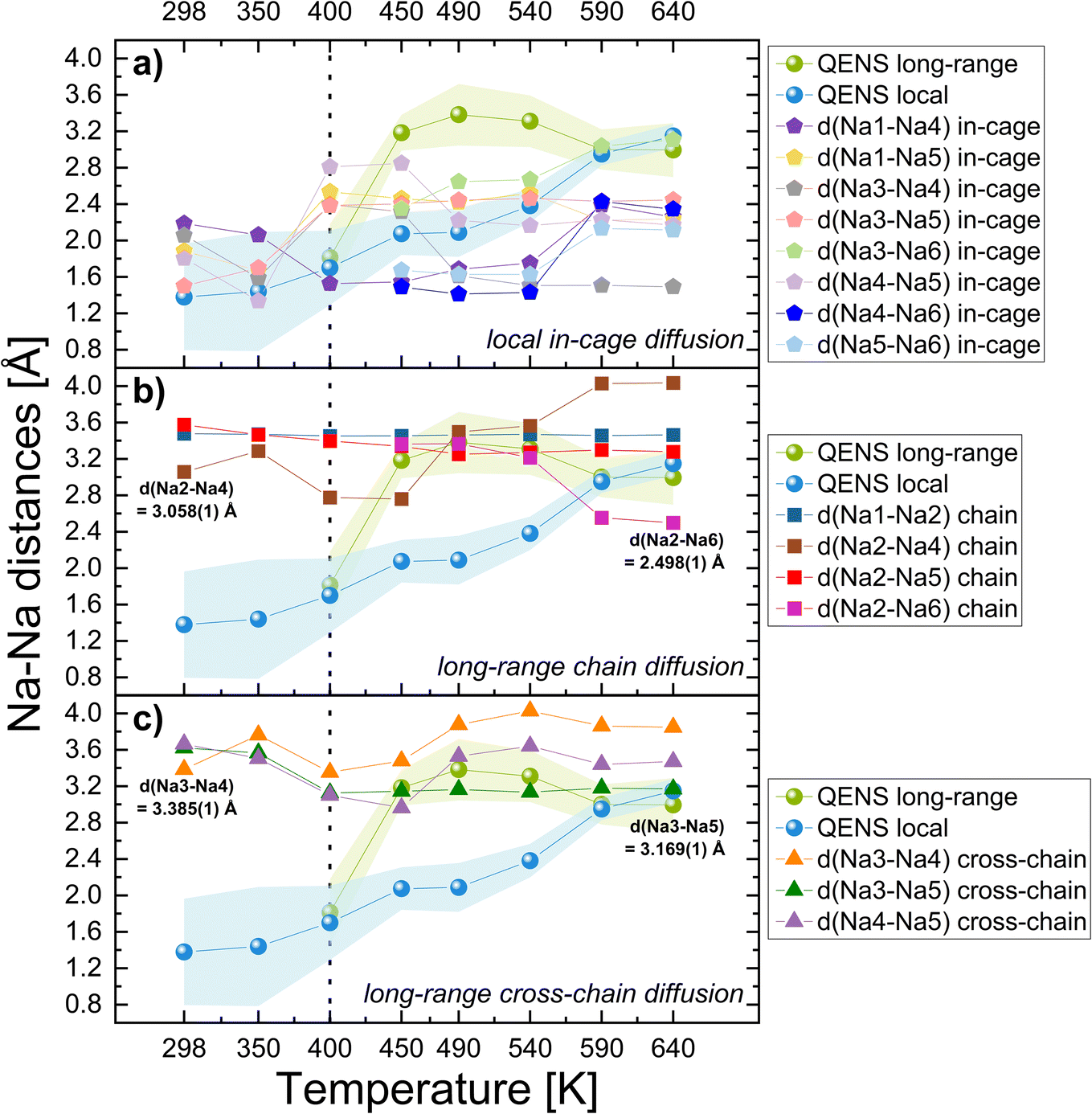 | ||
| Fig. 8 Comparison of all relevant interatomic Na–Na distances from the monoclinic C2/c f12e2a model with QENS jump distances for Na3.4Zr2Si2.4P0.6O12 (NaZr). The plotted distances correspond to (a) local in-cage, (b) long-range chain, and (c) long-range cross-chain diffusion pathways. The green (jump distances from narrow Lorentzian) and blue (jump distances from broad Lorentzian) areas mark the upper and lower error limits for distances derived from QENS. The dashed line shows that at ≈400 K, the diffusion mechanism changes according to QENS results. In (b) and (c), the values of Na–Na distances are displayed at the lowest (298 K) and highest (640 K) measured T. They indicate the shortest Na–Na distance through which chain diffusion and, afterward, cross-chain Na+ diffusion occurs. Solid lines are a guide to the eye. | ||
Above 400 K, the local diffusion, as shown in Fig. 8a, is still present. However, two separate long-range diffusion processes start to take place, as shown in Fig. 8b and c. Firstly, in the long-range chain diffusion (Fig. 8b), the diffusion takes place through the additional Na2 site and a newly occupied Na6 site. The Na2-sites create a chain between different localized Na1-cages (see depiction in Fig. 9a-II and b-II). The Na+ ions migrate along a chain-like pathway within the crystal structure. We can attribute the jump distances obtained from the narrow Lorentzian QENS fits (green area in Fig. 8) to the diffusion through the Na2 site, thus allowing long-range chain diffusion (≈2.5–4 Å). The long-range jump distances from QENS also correlate with the XRD Na–Na distances (≈3.2–4 Å) responsible for long-range cross-chain diffusion, as shown in Fig. 8c. Long-range cross-chain diffusion refers to the Na+ transport between neighboring chain-like pathways in a crystal lattice, enabling movement across multiple chains and contributing to three-dimensional transport properties (see depiction in Fig. 9a-III and b-III). The existence of local transport around the Na1 site, as well as the existence of anisotropic diffusion pathways that contribute to the overall diffusion, has previously been proposed by AIMD simulations and BVEL analysis.25
In Fig. 9, we show a depiction of the diffusion pathways, the unit cell view, and corresponding relevant close-ups of the local structures from the XRD data at the lowest (298 K) and highest (640 K) measured temperatures. The Ch-E diffusion model determined by QENS is based on single atom-to-atom jumps. Therefore, we consider here that most of the Na+ diffusion occurs through the nearest (shortest) crystallographic Na–Na distance determined by XRD, which are depicted together with their values for each corresponding diffusion process in Fig. 9. The QENS results confirm that there are two kinds of conduction pathways: one for the long-range diffusion of Na+ ions and one for the short-range local diffusion. However, we can further differentiate the long-range diffusion pathways with XRD: chain and cross-chain. Since the chain and cross-chain diffusion happen on a similar length and time scale, they are not further distinguishable by QENS. The QENS method and the used Ch-E model provide only an effective jump distance, which is the average distance of similar jump distances when Na+ migrates via multiple pathways in the structure. Thus, the narrow Lorentzian contains information about both chain and cross-chain long-range processes.
 | ||
| Fig. 9 Potential diffusion pathways for local in-cage, long-range, and long-range cross-chain diffusion model in Na3.4Zr2Si2.4P0.6O12 (NaZr): C2/c f12e2a monoclinic structure model close-ups showing the mechanism and relevant Na sites responsible for the respective diffusion types at (a) T = 298 K and at (b) T = 640 K. Visualized with the Endeavour software.19,20 | ||
For the long-range diffusion at 298 K, we observed that the diffusion first happens through individual chains through Na2–Na4 sites (3.058(1) Å). The diffusion through cross-chains happens through Na3–Na4 sites (3.385(1) Å), as can be observed in Fig. 9a-III. Some interatomic distances are shortened at higher temperatures and the diffusion mechanism changes. The diffusion at 640 K now goes through the additional newly occupied Na6 site, through Na2–Na6 (2.498(1) Å), to create individual chain-like pathways. The cross-chain diffusion happens through the Na3–Na5 sites (3.169(1) Å), as can be seen in Fig. 9b-III. From the unit cell view, it becomes evident that the orientation of the diffusion pathways also changes with increasing temperature.
As mentioned, according to QENS, the Na2 site does not participate in the diffusion at lower temperatures below ≈400 K. However, we know that NASICON solid-state electrolytes are good room-temperature ionic conductors, so there must be some kind of long-range diffusion present, but the local diffusion process might be dominant. At higher temperatures, Na+ ions gain more energy and may access longer pathways more frequently (e.g., chain jumps through Na2-NaX), which are less used at lower temperatures. It is possible that the QENS data for narrow Lorentzian could not be resolved for lower temperatures, which is why this information is potentially missing.
However, by combining insights from XRD structural analysis (Fig. 8 and 9) and activation energy barriers from QENS (Fig. 5c), the temperature-dependent diffusion mechanism in our NASICON system can be summarized as follows:
•Below ≈400 K: Diffusion is dominated by localized jumps with a very low activation energy of 0.03(1) eV (broad Lorentzian), reflecting Na+ motion concentrated primarily in a local cage around the Na1 site. Long-range chain diffusion is negligible, i.e., the Na2 site does not participate in the diffusion. Long-range diffusion might be limited to the cross-chain pathway via Na3 side jumps out of the cage. The escape from local cage motion is the limiting step to long-range diffusion.
• In the range of ≈400–490 K: long-range diffusion increases as the monoclinic to rhombohedral phase transition begins. However, local diffusion remains critical as Na+ ions must first overcome local energy environments to escape.
• Above ≈490 K: the energy barrier for local jumps increases to 0.29(3) eV (broad Lorentzian) due to structural changes as some of the Na–Na distances within the local cage increase (Na3–Na6, see Fig. 8a). At the same time the fastest local processes within the cage may become too fast to be detectable by QENS resolution. Long-range diffusion benefits from reduced activation energy of 0.18(1) eV (narrow Lorentzian), enabling Na+ migration through interconnected chain and cross-chain pathways.
4. Conclusions
In this work, the Na+ ion self-diffusion in Na3.4Zr2Si2.4P0.6O12 and Sc- and Al/Y-substituted Na3.4Sc0.4Zr1.6Si2PO12 and Na3.4Al0.2Y0.2Zr1.6Si2PO12 NASICON-type solid-state electrolytes are investigated by multimethod approach combining backscattering QENS experiments and XRD measurements. Together with complementary methods such as EIS and DSC, detailed information on the diffusion mechanism, crystal structure, phase transition, and local atomic environments is obtained. The results from the QENS experiment show that Na+ motion follows a jump-diffusion mechanism well described by the Chudley- Elliott model. In this model, Na+ migrates between crystallographic sites by a series of successive jumps. The jump lengths are correlated with interatomic distances extracted from the Rietveld refined XRD data to reveal 3D diffusion pathways. This comparison reveals the existence of two different diffusion processes, local and long-range diffusion, representing Na+ dynamics on two different time and length scales in Na3.4Zr2Si2.4P0.6O12. The localized short-range faster motion (≈7–12 ps) describes local in-cage diffusion concentrated around Na1 site through Na1–Na4, Na1–Na5, Na3–Na4, Na3–Na5, and Na4–Na5 sites. The slower long-range diffusion process (≈48–157 ps) can be further distinguished by XRD analysis of the nearest crystallographic distances into two mechanisms. Firstly, it allows chain diffusion through the cage-connecting Na2 site, where individual chain-like pathways are not connected. Secondly, the cross-chain long-range diffusion that connects these individual chains occurs through Na3–Na4 sites at room temperature. As the temperature increases, the interatomic distances are shortened, and the diffusion mechanism changes and occurs through Na3–Na5 sites. The structural analysis using XRD and Rietveld refinements confirmed the existence of the Na6 site for temperatures ≥450 K in Na3.4Zr2Si2.4P0.6O12, which contributes to both local and long-range diffusion processes. The findings presented here confirm the monoclinic C2/c f12e2a structure as the most suitable model for Na3.4Zr2Si2.4P0.6O12, not only at room temperature but also at elevated temperatures ≥450 K. However, the observations confirm the existence of continuous monoclinic to rhombohedral symmetry transformation, which has an influence on the structural parameters, such as interatomic distances, occupancies, and lattice parameters, and therefore, influences the overall Na+ transport mechanism in NASICONs. For Sc- and Al/Y-dopped samples, the onset of phase transition is shifted towards lower temperatures, starting at 350 K for the Na3.4Sc0.4Zr1.6Si2PO12 sample and even at 298 K for the Na3.4Al0.2Y0.2Zr1.6Si2PO12 sample. The systematic study presented here offers significant insights into the family of NASICON ceramic materials, aiming to improve its application for next-generation solid-state batteries.Data availability
The data supporting this article have been included in the ESI.† Further supporting data are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the German Federal Ministry of Education and Research (BMBF) for funding the project ExZellTUM III (03XP0255) within the ExcellBattMat cluster, as well as funding within the MEET-HiEnD III project (13XP0258B) and the project MiTemp (13XP0183B). Furthermore, the authors acknowledge the German Federal Ministry of Economic Affairs and Climate Action (BMWK) for funding the project CAESAR (03EI3046F). P.M.-B. acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2089/1 – 390776260 (e-conversion) and via the International Research Training Group 2022 Alberta/Technical University of Munich International Graduate School for Environmentally Responsible Functional Materials (ATUMS). Furthermore, the authors thank Pablo A. Alvarez Herrera (TUM) for his support with the DSC measurements. Part of this research used resources at the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. The beam time was allocated to BASIS on proposal number IPTS-26517.References
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- C. Yang, S. Xin, L. Mai and Y. You, Adv. Energy Mater., 2021, 11, 2000974 CrossRef CAS.
- Y. Lu, L. Li, Q. Zhang, Z. Niu and J. Chen, Joule, 2018, 2, 1747–1770 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- X. Y. Yao, B. X. Huang, J. Y. Yin, G. Peng, Z. Huang, C. Gao, D. Liu and X. X. Xu, Chinese Phys. B, 2016, 25, 6007–6018 Search PubMed.
- J.-F. Wu, R. Zhang, Q.-F. Fu, J.-S. Zhang, X.-Y. Zhou, P. Gao, C.-H. Xu, J. Liu and X. Guo, Adv. Funct. Mater., 2021, 31, 2008165 CrossRef CAS.
- H. Y. P. Hong, Mater. Res. Bull., 1976, 11, 173–182 CrossRef CAS.
- J. B. Goodenough, H. Y. P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- Z. Jian, Y.-S. Hu, X. Ji and W. Chen, Adv. Mater., 2017, 29, 1601925 CrossRef PubMed.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- Q. Ma, C.-L. Tsai, X.-K. Wei, M. Heggen, F. Tietz and J. T. Irvine, J. Mater. Chem. A, 2019, 7, 7766–7776 RSC.
- U. von Alpen, M. F. Bell and W. Wichelhaus, Mater. Res. Bull., 1979, 14, 1317–1322 CrossRef CAS.
- J. P. Boilot, J. P. Salanié, G. Desplanches and D. Le Potier, Mater. Res. Bull., 1979, 14, 1469–1477 CrossRef CAS.
- H. Kohler, H. Schulz and O. Melnikov, Mater. Res. Bull., 1983, 18, 1143–1152 CrossRef CAS.
- M. Avdeev, Chem. Mater., 2021, 33, 7620–7632 CrossRef CAS.
- B. Singh, Z. Wang, S. Park, G. S. Gautam, J.-N. Chotard, L. Croguennec, D. Carlier, A. K. Cheetham, C. Masquelier and P. Canepa, J. Mater. Chem. A, 2021, 9, 281–292 RSC.
- H. Putz and K. Brandenburg, Endeavour – Structure Solution from Powder Diffraction, https://www.crystalimpact.de/endeavour, accessed 10 August 2024 Search PubMed.
- H. Putz, J. Schön and M. Jansen, J. Appl. Crystallogr., 1999, 32, 864–870 Search PubMed.
- J. P. Boilot, G. Collin and P. Colomban, Mater. Res. Bull., 1987, 22, 669–676 CrossRef CAS.
- J. P. Boilot, G. Collin and P. Colomban, J. Solid State Chem., 1988, 73, 160–171 CrossRef CAS.
- S. Naqash, F. Tietz and O. Guillon, Solid State Ionics, 2018, 319, 13–21 CrossRef CAS.
- H. Kohler and H. Schulz, Mater. Res. Bull., 1985, 20, 1461–1471 CrossRef CAS.
- Z. Zou, N. Ma, A. Wang, Y. Ran, T. Song, B. He, A. Ye, P. Mi, L. Zhang and H. Zhou, Adv. Funct. Mater., 2021, 31, 2107747 CrossRef CAS.
- E. R. Losilla, M. A. G. Aranda, S. Bruque, M. A. París, J. Sanz and A. R. West, Chem. Mater., 1998, 10, 665–673 CrossRef CAS.
- Z. Zou, N. Ma, A. Wang, Y. Ran, T. Song, Y. Jiao, J. Liu, H. Zhou, W. Shi and B. He, Adv. Energy Mater., 2020, 10, 2001486 CrossRef CAS.
- Z. Zhang, Z. Zou, K. Kaup, R. Xiao, S. Shi, M. Avdeev, Y. S. Hu, D. Wang, B. He and H. Li, Adv. Energy Mater., 2019, 9, 1902373 CrossRef CAS.
- E. A. Cheung, H. Nguyen, M. Avdeev, N. R. de Souza, Y. S. Meng and N. Sharma, Chem. Mater., 2021, 33, 8768–8774 CrossRef CAS.
- Y. Gao, A. M. Nolan, P. Du, Y. Wu, C. Yang, Q. Chen, Y. Mo and S.-H. Bo, Chem. Rev., 2020, 120, 5954–6008 Search PubMed.
- Z. Deng, T. P. Mishra, E. Mahayoni, Q. Ma, A. J. K. Tieu, O. Guillon, J.-N. Chotard, V. Seznec, A. K. Cheetham, C. Masquelier, G. S. Gautam and P. Canepa, Nat. Commun., 2022, 13, 4470 CrossRef CAS PubMed.
- J. Schuett, A. S. Kuhn and S. Neitzel-Grieshammer, J. Mater. Chem. A, 2023, 11, 9160–9177 RSC.
- M. K. Gupta, J. Ding, N. C. Osti, D. L. Abernathy, W. Arnold, H. Wang, Z. Hood and O. Delaire, Energy Environ. Sci., 2021, 14, 6554–6563 RSC.
- M. Bée, Quasielastic Neutron Scattering, 1988 Search PubMed.
- R. Hempelmann, Quasielastic Neutron Scattering and Solid State Diffusion, Oxford University Press, 2000 Search PubMed.
- J. P. Embs, F. Juranyi and R. Hempelmann, Z. Phys. Chem., 2010, 224, 5–32 CrossRef CAS.
- M. K. Gupta, R. Mittal, S. Kumar, B. Singh, N. H. Jalarvo, O. Delaire, R. Shukla, S. N. Achary, A. I. Kolesnikov and A. K. Tyagi, J. Mater. Chem. A, 2021, 9, 16129–16136 RSC.
- S. Kumar, M. K. Gupta, R. Mittal, S. Sundaramoorthy, A. Choudhury, N. C. Osti, A. I. Kolesnikov, M. B. Stone, Y. Cheng and S. L. Chaplot, J. Mater. Chem. A, 2023, 11, 23940–23949 RSC.
- O. Maus, M. T. Agne, T. Fuchs, P. S. Till, B. Wankmiller, J. M. Gerdes, R. Sharma, M. Heere, N. Jalarvo, O. Yaffe, M. R. Hansen and W. G. Zeier, J. Am. Chem. Soc., 2023, 145, 7147–7158 CrossRef CAS PubMed.
- Q. Chen, N. H. Jalarvo and W. Lai, J. Mater. Chem. A, 2020, 8, 25290–25297 Search PubMed.
- T. J. Willis, D. G. Porter, D. J. Voneshen, S. Uthayakumar, F. Demmel, M. J. Gutmann, M. Roger, K. Refson and J. P. Goff, Sci. Rep., 2018, 8, 3210 CrossRef CAS PubMed.
- Y. Saito, K. Ado, T. Asai, H. Kageyama and O. Nakamura, Solid State Ionics, 1992, 58, 327–331 Search PubMed.
- Q. Ma, M. Guin, S. Naqash, C.-L. Tsai, F. Tietz and O. Guillon, Chem. Mater., 2016, 28, 4821–4828 CrossRef CAS.
- N. I. Sorokin, Phys. Solid State, 2014, 56, 678–681 CrossRef CAS.
- E. Mamontov and K. W. Herwig, Rev. Sci. Instrum., 2011, 82, 085109 CrossRef CAS PubMed.
- N. C. Osti, N. Jalarvo and E. Mamontov, Mater. Horiz., 2024, 11, 4535–4572 RSC.
- R. T. Azuah, L. R. Kneller, Y. Qiu, P. L. W. Tregenna-Piggott, C. M. Brown, J. R. D. Copley and R. M. Dimeo, J. Res. Natl. Inst. Stand Technol., 2009, 114, 341–358 CrossRef CAS PubMed.
- L. Finger, D. Cox and A. Jephcoat, J. Appl. Crystallogr., 1994, 27, 892–900 CrossRef CAS.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, Powder Diffr., 2014, 29, S13–S18 CrossRef CAS.
- G. de la Flor, D. Orobengoa, E. Tasci, J. M. Perez-Mato and M. I. Aroyo, J. Appl. Crystallogr., 2016, 49, 653–664 CrossRef CAS.
- E. Tasci, G. de la Flor, D. Orobengoa, C. Capillas, J. Perez-Mato and M. Aroyo, EPJ Web Conf., 2012, 9 CrossRef.
- B. Zhang, R. Tan, L. Yang, J. Zheng, K. Zhang, S. Mo, Z. Lin and F. Pan, Energy Storage Mater., 2018, 10, 139–159 CrossRef.
- P. Vadhva, J. Hu, M. J. Johnson, R. Stocker, M. Braglia, D. J. L. Brett and A. J. E. Rettie, ChemElectroChem, 2021, 8, 1930–1947 CrossRef CAS.
- Y. Deng, C. Eames, L. H. Nguyen, O. Pecher, K. J. Griffith, M. Courty, B. Fleutot, J.-N. l. Chotard, C. P. Grey and M. S. Islam, Chem. Mater., 2018, 30, 2618–2630 CrossRef CAS.
- S. Roy and P. P. Kumar, Phys. Chem. Chem. Phys., 2013, 15, 4965–4969 RSC.
- S. Roy and P. P. Kumar, Solid State Ionics, 2013, 253, 217–222 CrossRef CAS.
- Y. Ruan, S. Song, J. Liu, P. Liu, B. Cheng, X. Song and V. Battaglia, Ceram. Int., 2017, 43, 7810–7815 CrossRef CAS.
- D. Chen, F. Luo, W. Zhou and D. Zhu, J. Alloys Compd., 2018, 757, 348–355 CrossRef CAS.
- S. Naqash, D. Sebold, F. Tietz and O. Guillon, J. Am. Ceram. Soc., 2019, 102, 1057–1070 CrossRef CAS.
- Y. B. Rao, K. K. Bharathi and L. N. Patro, Solid State Ionics, 2021, 366–367, 115671 CrossRef CAS.
- N. Zhang, H. Di, B. Wen, J. Luo, L. Zhang and S. Zhang, Nano Energy, 2024, 130, 110175 CrossRef CAS.
- H. Tian, S. Liu, L. Deng, L. Wang and L. Dai, Energy Storage Mater., 2021, 39, 232–238 CrossRef.
- P. W. Jaschin, C. R. Tang and E. D. Wachsman, Energy Environ. Sci., 2024, 17, 727–737 RSC.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart and P. Lamp, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- J. A. S. Oh, L. He, A. Plewa, M. Morita, Y. Zhao, T. Sakamoto, X. Song, W. Zhai, K. Zeng and L. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 40125–40133 CrossRef CAS PubMed.
- M. K. Gupta, J. Ding, H.-M. Lin, Z. Hood, N. C. Osti, D. L. Abernathy, A. A. Yakovenko, H. Wang and O. Delaire, Chem. Mater., 2024, 36, 11377–11392 CrossRef CAS.
- N. J. J. de Klerk, E. van der Maas and M. Wagemaker, ACS Appl. Energy Mater., 2018, 1, 3230–3242 CrossRef CAS PubMed.
- C. T. Chudley and R. J. Elliott, Proc. Phys. Soc., 1961, 77, 353–361 CrossRef.
- N. Jalarvo, L. Stingaciu, D. Gout, Z. Bi, M. P. Paranthaman and M. Ohl, Solid State Ionics, 2013, 252, 12–18 CrossRef CAS.
- N. Jalarvo, O. Gourdon, Z. Bi, D. Gout, M. Ohl and M. P. Paranthaman, Chem. Mater., 2013, 25, 2741–2748 CrossRef CAS.
- M. K. Gupta, J. Ding, D. Bansal, D. L. Abernathy, G. Ehlers, N. C. Osti, W. G. Zeier and O. Delaire, Adv. Energy Mater., 2022, 12, 2200596 CrossRef CAS.
- N. Bukun, Ionics, 1996, 2, 63–68 CrossRef CAS.
- A. Ignaszak, P. Pasierb, R. Gajerski and S. Komornicki, Thermochim. Acta, 2005, 426, 7–14 CrossRef CAS.
- G. Jaeger, Arch. Hist. Exact Sci., 1998, 53, 51–81 CrossRef.
- T. Oota and I. Yamai, J. Am. Ceram. Soc., 1986, 69, 1–6 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00826c |
| This journal is © The Royal Society of Chemistry 2025 |