
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bond-guided micellar self-assembly-directed carbon superstructures for high-energy and ultralong-life zinc-ion hybrid capacitors†
Shreeti
Jha
a,
Yang
Qin
a,
Yumin
Chen
a,
Ziyang
Song
a,
Ling
Miao
*a,
Yaokang
Lv
 c,
Lihua
Gan
*ab and
Mingxian
Liu
*ab
c,
Lihua
Gan
*ab and
Mingxian
Liu
*ab
aShanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Rd, Shanghai, 200092, P. R. China. E-mail: 22169@tongji.edu.cn; ganlh@tongji.edu.cn; liumx@tongji.edu.cn
bState Key Laboratory of Cardiovascular Diseases and Medical Innovation Center, Shanghai East Hospital, School of Medicine, Tongji University, 150 Jimo Rd, Shanghai 200120, P. R. China
cCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China
First published on 22nd April 2025
Abstract
Well-intricate carbon superstructures have tremendous potential as cathode materials in zinc-ion hybrid capacitors (ZHCs) owing to their maximum accessible surface, superior structural integrity and robustness. However, significant challenges still remain in simplifying the synthesis process. Herein, a hydrogen-bond-guided micellar self-assembly strategy was leveraged to construct flower-like carbon superstructures for targeting high-energy and ultralong-life ZHCs. This approach employed aggregated micelles to serve as structural guides facilitating the controlled growth of oligomers into intricate polymer superstructures with a distinct flower-like morphology and architecture. The subsequent hierarchical porous carbon superstructures exhibited a considerable surface area (2824 m2 g−1) with efficient charge-transport pathways. Remarkably, the synergistic combination of dual-ion storage, ion-accessible pore structures, and endogenous zincophilic sites enabled exceptional performance. This included a remarkable energy density of 160.8 W h kg−1 at 112.1 W kg−1, extraordinary cycling stability of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 20 A g−1, and a high specific capacity of 262.8 mA h g−1 at 0.2 A g−1. The exceptional electrochemical performances were primarily attributed to the effective and alternating Zn2+/CF3SO3− adsorption at the abundant, easily accessible zincophilic sites within the superstructures. Additionally, proton-assisted zinc-ion storage through H+ adsorption onto the carbon matrix significantly enhanced the capacity and rate performance. This work paves a novel pathway for the exploration and design of well-intricate carbon superstructure cathodes for enabling superior performance in ZHCs.
000 cycles at 20 A g−1, and a high specific capacity of 262.8 mA h g−1 at 0.2 A g−1. The exceptional electrochemical performances were primarily attributed to the effective and alternating Zn2+/CF3SO3− adsorption at the abundant, easily accessible zincophilic sites within the superstructures. Additionally, proton-assisted zinc-ion storage through H+ adsorption onto the carbon matrix significantly enhanced the capacity and rate performance. This work paves a novel pathway for the exploration and design of well-intricate carbon superstructure cathodes for enabling superior performance in ZHCs.
1 Introduction
Zinc-ion hybrid capacitors (ZHCs) have garnered substantial attention as highly promising energy-storage equipment, captivating researchers with their alluring combination of the electrochemical properties of batteries and supercapacitors.1,2 Specifically, the highly reversible adsorption/desorption process of zinc ions on the cathode offers a supercapacitor-like output, while the rapid plating/stripping of zinc ions promises battery-level energy density.3 Zinc anodes with a remarkable theoretical capacity (820 mA h g−1) and an inherently low redox potential (−0.762 V vs. the standard hydrogen electrode) can provide sufficient charge in the energy-storage process.4–8 Therefore, numerous studies have been devoted to developing carbon cathodes to match the performance of zinc anodes, with the goal of achieving the desired zinc-ion-storage capacity.9–13 Nevertheless, the development of high-performance ZHCs remains constrained due to the insufficient zincophilic sites and limited accessibility to these sites, which renders carbon cathodes incapable of affording high capacity, triggering a low energy density and cycle durability.14–20 Addressing this challenge requires substantial progress in the development of highly electroactive cathode materials with well-organized configurations for ZHCs with enhanced electrochemical metrics.To date, a wide range of carbon materials with various dimensions and morphologies as capable cathode materials have been reported for ZHCs.21–23 These includes nanostructured carbon-based materials, including 0D carbon nanoparticles,24 carbon spheres,25 1D carbon nanotubes,26 carbon nanorods,27 2D carbon nanosheets, graphene28,29 and 3D porous carbon.30,31 Low-dimensional carbon-based materials (0–1D) possess a high chemical stability, adjustable porosity, and exceptional flexibility, which can enable better electrolyte accessibility and facilitate the ion-transportation pathways,32,33 while on the contrary 2D carbon-based materials with enlarged interlayer spacing demonstrate a large surface area and great number of accessible electrochemical active sites with an improved charge-storage capability.18,34,35 However, low-dimensional materials (0–2D) cannot avoid stacking and aggregation, which will sacrifice some of the accessible surface area. Additionally, their irregular morphology restricts the kinetics of electrolyte ions, limiting the volumetric ion-storage capacity and slowing the reaction kinetics, thereby diminishing the overall energy-storage performance.22,36,37 By contrast, porous carbons with a 3D structure as cathode materials exhibit superior electrochemical performance for ZHCs.38 This can be attributed to their structural interconnectivity and hierarchical porous channels, which allow more active sites, which enhances the electrochemical activity also increases the charge-storage activity.39–41 However, the potential structural breakdown and dissolution of active sites limit their energy-storage capacity.42 Notably, three-dimensional (3D) carbon superstructures meticulously assembled from well-coordinated low-dimensional components can effectively harness the benefits, while reducing the energy barriers to achieve the targeted energy-storage objectives.43,44 This is due to the fact that carbon superstructures not only retain the advantageous properties of their individual components but can also achieve ultra-stable, well-organized structures with an increased exposure of surface-active sites and enhanced pathways for rapid electron migration.45,46 Therefore, it is imperative to design carbon superstructures characterized by hierarchical porous designs and organized morphologies to improve the accessibility of the electroactive active sites and facilitate uninterrupted charge-transport pathways with enhanced electrochemical performance and low energy barriers. However, realizing the controllable construction of superstructures exhibiting diverse and well-organized morphologies is a great challenge because of their unprecedented architectural complexity, which can lead to random orientation. Therefore, it is still an intriguing and ongoing task to understand the self-assembly behavior in constructing these superstructures to attain well-organized structures with a controllable morphology. Thus, the construction of carbon superstructures with a specific morphology is of great interest but still a daunting challenge.
Herein, we report a hydrogen-bond-guided micellar self-assembly strategy to synthesize flower-like carbon superstructures. The self-assembled micelles function as a structural guide to direct the superstructures' formation process. These polymer superstructures can thus be efficiently converted to highly porous carbons while preserving their inherent architecture. The resultant flower-like carbon superstructures with numerous interconnected carbon nanosheets allow exceptional accessibility to abundant active sites, facilitating ion/electron transfer with plentiful ion interactions. By exploiting the optimal carbon cathode, an assembled ZHCs exhibited a remarkable capacity of 262.8 mA h g−1 and an exceptionally high energy density of 160.8 W h kg−1. Ex situ characterization results combined with the theoretical findings indicated that the outstanding electrochemical performance arose from the synergetic interplay of two key factors: the alternating absorption of Zn2+ and CF3SO3− at zincophilic sites and the strong redox interactions between Zn2+ and carbonyl/pyridine motifs leading to the formation of O–Zn–N bonds. Thus, this work introduces a novel approach for designing and fabricating efficient carbon superstructures, further facilitating the development of high-performance ZHCs.
2 Results and discussion
2.1 Construction and structural characterization
The construction process of a typical carbon superstructure (CS-750) is illustrated in Fig. 1a. In brief, (1) tetrachlorobenzoquinone–4,4′-methylenedianiline (TM) oligomers interact with the hydrophilic segment of F127 resulting in the formation of F127/oligomer composite micelles; (2) when subjected to a temperature of 100 °C under hydrothermal conditions, the composite micelles aggregate with each other through hydrogen bonding; (3) the micelles act as structure-directing agents leading the TM oligomers to grow a flower-like morphology and form polymer superstructures; and (4) the subsequent carbonization and activation steps convert the polymer superstructures into well-organized highly hierarchical porous flower-like carbon superstructures. The synthesis route for the TM oligomers via the polymerization reaction is shown in Fig. S1† and involved a nucleophilic reaction between the –Cl and –NH2 groups, as confirmed by the Fourier-transform infrared spectroscopy (FT-IR) spectra shown in Fig. S2.† In accordance with the scanning electron microscopy (SEM) observations (Fig. 1b and c), CS-750 comprised a flower-like superstructure with a hierarchical arrangement of nanosheets and an open porous structure that ensured abundant accessible active sites while creating a well-developed network of pores. Transmission electron microscopy (TEM) observations further validated the above conclusion about the interconnecting channels, consisting of nanosheets connecting towards the surface with open pores (Fig. 1d). Further observation by TEM (Fig. S3†) offered a more detailed view of the structural features, revealing a disordered carbon lattice. Moreover, elemental mappings (Fig. 1e and S4†) demonstrated the consistent and highly uniform distribution of C, N, O elements on the surface. The pyrolysis temperature significantly influences the surface area and porosity of the resulting carbon products, critically impacting the electrochemical performance. Thus, the carbon products are designated as CS-x (where x denotes the pyrolysis temperature). The N2 adsorption/desorption isotherms, displaying a sharp rise at a relatively low pressure (P/P0 < 0.1) and mild hysteresis loop at a relatively elevated pressure (0.45 < P/P0 < 0.95) as well as very minor rise at (P/P0 > 0.95), revealed the hierarchical porous properties of CS-x (Fig. 1f).47,48 The above mentioned results indicate that CS-x displayed a more micropore and mesopore dominated hierarchal architecture, which was further supported by the pore-size distribution curves (Fig. 1g). These findings show that plentiful micropores and mesopores were derived from the polymer decomposition and from the cavities of the open flower-like structure.49 Moreover, the mesopores of CS-750 were primarily distributed in the 2–10 nm size range, with a prominent peak appearance compared with in CS-700 and CS-800. Meanwhile, CS-750 was endowed with a larger specific surface area of 2824 m2 g−1 compared to that of CS-700 (954 m2 g−1) and CS-800 (1431 m2 g−1) (Table S1†). The selection of an appropriate pyrolysis temperature and hold time (750 °C, 2 h) allowed the functional segments to further decompose and craft the immature porous structure (CS-700) into a well-developed porous architecture with significantly increased micropores and a correspondingly high surface area. Moreover, the mesoporous volume ratio (Vmeso/Vtotal) of CS-750 was observed to be 75.6%, which was overwhelmingly higher than that of CS-700 and CS-800 (34.7% and 50.6%, respectively). Thus, suitable pyrolysis temperatures for CS-x are optimal for achieving well-defined and controlled morphologies with a good balance between pore development and structural integrity. | ||
| Fig. 1 (a) Schematic of the synthesis of carbon superstructures. (b and c) SEM images of CS-750. (d) TEM images of CS-750. (e) EDS mapping images of CS-750. (f) Nitrogen adsorption/desorption isotherms of CS-x. (g) Pore-size distribution curves of CS-x. | ||
Robust carbon superstructures with highly interconnected channels and abundant adsorption sites help fast ion migration and superior charge-storage capability, ensuring high energy density. The formation of flower-like superstructures is briefly discussed below. Initially the TM oligomers assemble with F127 micelles to generate F127/TM oligomer composite micelles. In this process, the –NH– in TM oligomers connect with the hydrophilic domain of F127 micelles through hydrogen bonds. Subsequently, under hydrothermal treatment, the interaction between the oligomer and F127 micelles becomes more dynamic and aggregated oligomer composite micelles are formed. The oligomer composite micelles tend to join together primarily due to hydrogen-bond interactions and the increased kinetic energy. As the reaction proceeds with a continuous rise in temperature to about 100 °C under the hydrothermal conditions, the system tends to equilibrate and the aggregated oligomer micelles act as nucleation sites for the formation of nanosheets. This transformation into a nanosheet morphology occurs due to the hydrophobic nature of the oligomers, which tend to flatten and spread out to minimize the surface area.
Thus, the resulting nanosheets exhibit enhanced growth activity, facilitating the incorporation of additional nanosheets and binding to each other, thereby promoting self-assembly and polymerization, which are ultimately stabilized by various intermolecular forces. As more nanosheets aggregate, they start to arrange in a hierarchical manner, giving rise to a robust intricate flower-like morphology. Thus, the aggregated micelles act as structure-directing agents leading the TM oligomers to grow a sophisticated and distinct flower-like morphology. This results in a stable and intricate architecture derived from the oligomers. Further to elucidate the role of F127 in the formation of the flower-like carbon superstructures, different analysis were conducted. In the absence of F127, an aggregated sphere-like carbon structure was formed, demonstrating the critical role of F127 in creating well-defined structures (Fig. S5†). Additionally, the Tyndall effect was employed to assess the aggregation state of the micelles. Results indicate that increasing the concentration of F127 enhanced the intensity of scattered light, suggesting a higher degree of micelle assembly (Fig. S6†).
The increase in F127 concentration led to the micelles assembling together through hydrogen bonding under specific temperatures to form a larger structure.50,51 Moreover, the Tyndall effect of the 1,4 dioxane–F127 solution was demonstrated by the increase in scattered light intensity with the rise in temperature (Fig. S7†), this suggests that the F127 micelles underwent a temperature-dependent aggregation, forming a flower-like structure. Overall, a new strategy was proposed to create a flower carbon superstructure.
Further, X-ray photoelectron spectroscopy (XPS) analysis was performed to gain valuable insights into the chemical composition of the CS-x carbon materials. Upon examining the XPS spectra of samples CS-700, CS-750 and CS-800, distinct peaks could be observed at 284.4, 400.2 and 531.9 eV, which were the core level signals for C 1s, N 1s and O 1s, respectively (Fig. S8†). The O 1s spectra could be resolved into three distinct peaks positioned at 530.4 eV for O–N, 532.1 eV for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and 533.2 eV for C–O (Fig. S9a†). Besides, the N 1s spectra revealed three distinct peaks, namely N-5, N-6 and N-Q, with the peaks positioned at 399.9, 398.2 and 401.3 eV, respectively (Fig. S9b†). Also, the chemical composition summary confirmed the presence of C, O, N species (Fig. S10†). CS-750 showed a high O content due to its more stable CO bonds, which resisted the release of O species. However, CS-700 and CS-800, even with a high N content compared to CS-750, delivered an inadequate capacitance due to their limited accessible surface areas.52 In addition, Raman spectroscopy was employed to investigate the disorder within the carbon lattice. The degree of disorder was quantified by the intensity ratio of the D band (at approximately 1306 cm−1), indicative of sp3-hybridized or defective carbon, to the G band (at around 1530 cm−1), corresponding to sp2-bonded carbon in the graphitic structure. The ID/IG ratio ranged from 0.83 to 0.85. Generally, a higher ID/IG ratio reflects a greater degree of disorder or a higher concentration of defects, whereas a lower ratio suggests a more ordered graphitic structure. This ratio provides valuable insights into a material's structural integrity and overall quality (Fig. S11a†). The CS-750 sample showed the best compromise between defects and graphitization, with an ID/IG ratio of 0.85. Next, X-ray diffraction (XRD) patterns of the CS-x materials were recorded and they displayed diffraction peaks attributable to the (002) and (100) planes of the carbon lattice, respectively, indicating the amorphous nature of the CS-x materials (Fig. S11b†).
O and 533.2 eV for C–O (Fig. S9a†). Besides, the N 1s spectra revealed three distinct peaks, namely N-5, N-6 and N-Q, with the peaks positioned at 399.9, 398.2 and 401.3 eV, respectively (Fig. S9b†). Also, the chemical composition summary confirmed the presence of C, O, N species (Fig. S10†). CS-750 showed a high O content due to its more stable CO bonds, which resisted the release of O species. However, CS-700 and CS-800, even with a high N content compared to CS-750, delivered an inadequate capacitance due to their limited accessible surface areas.52 In addition, Raman spectroscopy was employed to investigate the disorder within the carbon lattice. The degree of disorder was quantified by the intensity ratio of the D band (at approximately 1306 cm−1), indicative of sp3-hybridized or defective carbon, to the G band (at around 1530 cm−1), corresponding to sp2-bonded carbon in the graphitic structure. The ID/IG ratio ranged from 0.83 to 0.85. Generally, a higher ID/IG ratio reflects a greater degree of disorder or a higher concentration of defects, whereas a lower ratio suggests a more ordered graphitic structure. This ratio provides valuable insights into a material's structural integrity and overall quality (Fig. S11a†). The CS-750 sample showed the best compromise between defects and graphitization, with an ID/IG ratio of 0.85. Next, X-ray diffraction (XRD) patterns of the CS-x materials were recorded and they displayed diffraction peaks attributable to the (002) and (100) planes of the carbon lattice, respectively, indicating the amorphous nature of the CS-x materials (Fig. S11b†).
2.2 Electrochemical performances
To assess the electrochemical performances of CS-x in ZHCs, the as-designed CS-x material was fabricated as the cathode, Zn foil as the anode and 3 M Zn(OTF)2 as the electrolyte for ZHCs (Fig. 2a). The cyclic voltammetry (CV) profiles of the as-fabricated ZHCs at the scan rate of 10 mV s−1 displayed quasi-rectangular shapes, indicating a combined contribution of an electrical double-layer behaviour plus pseudo-capacitance response, which was related to the electrochemically active motifs on the carbon skeleton (Fig. 2b).53,54 The CV curves for the CS-x devices at different scan rates are provided in Fig. S12.† Furthermore, the specific capacities were determined by galvanostatic charge–discharge (GCD) tests. CS-750 showed a superior discharge capacity of 262.8 mA h g−1, which exceeds that of CS-700 (132.2 mA h g−1) and 800 (168.1 mA g−1), respectively (Fig. 2c). In addition, CS-750 exhibited a well-developed porous architecture characterized by a high surface area and optimized pore distribution. Its abundant micropores and a correspondingly high surface area (2824 m2 g−1) provide more active sites for electrochemical reactions, while its multi-level pore architecture (Vmeso/Vtotal = 75.6%) facilitates enhanced charge-storage capacity through conductive pathways for electron and ion transport. Conversely, CS-700 possessed an immature porous structure with limited porosity (SBET = 954 m2 g−1, Vmeso/Vtotal = 34.7%). This limits the availability of active sites and hinders the transport of electrons and ions, leading to a reduced charge-storage capacity. Meanwhile, the GCD curves of the CS-750 device demonstrated symmetrical shapes at a wide range of current densities ranging from 0.2 to 20 A g−1 (Fig. 2d). The GCD curves of the CS-800 device and CS-700 device at different current densities are provided in Fig. S13.† Moreover, the CS-750 cathode showed high capacity retention, even at 20 A g−1, reflecting its good rate capability and excellent structural stability (Fig. S14†), which was 54.6% for CS-750, whereas the capacity retention was 32.6% and 28.3% for CS-800 and CS-700, respectively. The CS-x electrodes were tested at every single current density for a continuous 10 cycles ranging from 0.2 to 20 A g−1, with the results showing that the devices could maintain their initial capacities, even after cycling back to 0.2 A g−1. This indicates that the devices possessed good reversibility (Fig. 2e). Moreover, the high capacity of the as-designed Zn//CS-750 device yielded a high energy density of 160.8 W h kg−1, demonstrating its great energy/power delivery compared with CS-800 (120.3 W h kg−1) and CS-700 (110.6 W h kg−1) (Fig. 2f). Among the three prepared CS-x materials, CS-750 with a flower-like superstructure possessed a large surface area, which provided sufficient space for accommodating the volume changes that occur during charge/discharge cycles, leading to improved cycling stability. The robust carbon superstructures guarantee quick ion penetration into the core zincophilic sites, which ultimately induces efficient charge adsorption and N/O redox reactions, providing an extraordinary capacity as well as ultra-long stability.55,56 Significantly, the CS-750 device demonstrated a notable capacity retention of 93.2% across 200000 cycles at 20 A g−1 and a 99.1% coulombic efficiency, showcasing its excellent long-term cycling performance (Fig. 2g). The remarkable stability of CS-750 was due to its improved structural integrity, which reduces the loss of active sites and structural collapse during repeated charge and discharge cycles. Moreover, in CS-750, the combination of an improved microstructure and enhanced porosity also relieve capacity loss and structural breakdown over time, which contributes to its minimal degradation over prolonged cycling. These factors collectively contribute to the exceptional long-term stability of CS-750, enabling it to retain 93.2% of its initial capacity. The cycling performances of the CS-700 and CS-800 devices are shown in Fig. S15.†
 | ||
| Fig. 2 Electrochemical performances of Zn//CS-x ZHCs device. (a) Working principal of ZHCs. (b) CV curves of CS-x devices at 10 mV s−1. (c and d) GCD profiles. (e) Rate capability of CS-x-based devices. (f) Ragone plot. (g) Cycling stability of a CS-x based device. | ||
Furthermore, the morphology of CS-750 did not show any specific alterations after extensive cycling, revealing the stable and robust structure of the CS-750 cathode (Fig. S16†). Besides, GCD tests were performed on CS-750-based ZHCs with a high mass loading from 10 to 30 mg cm2, which showed capacities of 216.7, 199.2, 175.3 and 142.5 mA h g−1, respectively (Fig. S17†). Compared with recent reports, these results show the significant potential of the CS-750 cathode, with superior electrochemical performances for use in practical ZHCs (Table S2†).
2.3 Reaction kinetics
A more in-depth investigation was conducted to gain a comprehensive understanding of the charge-storage kinetics and electron/ion-transport properties within the CS-x based devices (i.e. ZHCs). All of the three assembled ZHCs exhibited semicircle curves in the higher frequency domain together with a linear tail in the lower frequency domain, as can be seen in the Nyquist plots of the CS-x cathodes (Fig. 3a). This displays the dual features of the pseudocapacitive process and fast kinetics diffusion-controlled process.25 An analogous circuit model for EIS was used to fit the curves, which is shown in Fig. S18.† The CS-x electrodes showed smaller equivalent resistances (Rs), which were 2.8 Ω for CS-700, 1.8 Ω for CS-750 and 2.2 Ω for CS-800. Of note, the charge-transfer resistance (Rct) was 9.2 Ω for CS-750, which was significantly less than that of other CS-x-based devices, suggesting the optimized structure with a rapid ion-transfer rate (Table S3†). Furthermore, the ion-diffusion resistance (σ) was quantitatively determined based on the relation between the angular frequency (ω) and the real part of the impedance (Z′).57 The CS-750 device showed a lower steepness of the linear tendency of 12.1 Ω s−0.5 compared with 22.4 Ω s−0.5 for the CS-700 device and 18.8 Ω s−0.5 for the CS-800 device, verifying the intermediate access for ion migration during the charging and discharging process (Fig. 3b). Moreover, the variation in capacitance change along with the wide frequency change were studied in an elaborated capacitance mode, C(ω) = C′(ω) + jC′′(ω), where C′(ω) symbolizes the real capacitance of the device and C′(ω) represents the imaginary part, corresponding to irreversible energy-dissipation processes.58 The relaxation time constant τ0 = 1/f could be extracted from the frequency at which the maximum C′′(ω) was observed, indicating the temporal sustainability affording the power and saved energy.59 Of note, CS-750 delivered an optimal τ0 value of 22.7 s (Fig. 3c and d) verifying its rapid reaction kinetics and higher capability compared to CS-700 for 41.6 s and CS-800 for 30.3 s. Additionally, the CS-x electrodes all displayed the smallest voltage drop, showing they had the best ion-transport capability (Fig. S19†). The CV curves displayed a quasi-rectangular shape across the scan rates ranging from 5 to 200 mV s−1, suggesting a combined contribution of electric double-layer capacitance and pseudo-capacitance (Fig. 3e).60,61 Furthermore, the power law exponent b was determined using the equation i = kvb, and it was found that typically b lies within a range of 0.5–1. A value of 0.5 generally indicates a diffusion-controlled process whereas b value of 1 signifies a surface-controlled process.62 The b value of CS-750 was 0.93/0.92 for the cathodic and anodic peaks (Fig. 3f) which is larger than the 0.83/0.82 of CS-700 and 0.85/0.84 of CS-800, indicating an ideal surface-governed process (Fig. S20†).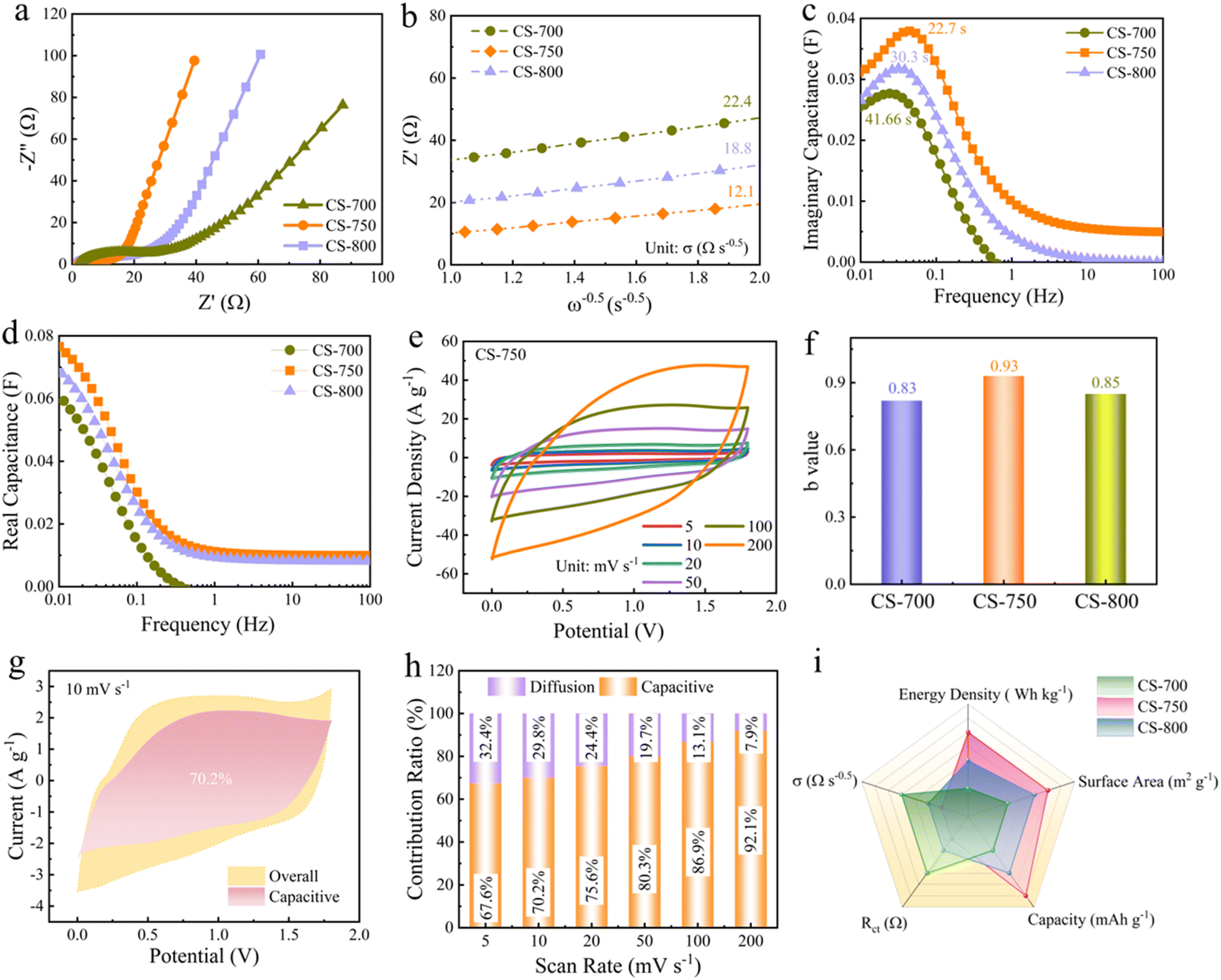 | ||
| Fig. 3 (a) Nyquist plots of the assembled ZHCs. (b) Relationship between Z′ and ω−0.5. (c) Imaginary capacitance versus frequency. (d) Real capacitance versus frequency. (e) CV profile of the CS-750-based device. (f) b-values for the CS-x devices. (g) Capacitive contribution of CS-750 at 10 mV s−1. (h) Capacitive and diffusion-controlled contribution ratios of CS-x based devices. (i) Comparison chart of five parameters of CS-x-based devices. | ||
Further the quantitative contributions were calculated using Dunn's method, which involves the equation: k1v + k2v0.5, where k2v0.5 and k1 correspond to the capacitive and diffusion-controlled contributions, respectively.63,64 Based on the above, the rectangular CV curves at 10 mV s−1 of CS-750 suggested a synergetic contribution from diffusion-limited and ultrafast surface capacitance-controlled processes, with an obvious significant capacitive contribution of 70.2% (Fig. 3g). As the scan rate was increased, a pronounced dominance of capacitive processes over diffusion-controlled processes was clearly evident, with the contribution increasing significantly from 67.6% to 92.1% (Fig. 3h and S21†), suggesting an excellent rate capability. The superior and enduring charge-storage kinetics of the zincophilic flower-like superstructures was primarily due to their advantageous morphology, which facilitated ion migration with a low energy barrier, thereby optimizing the dynamic capacity. A comparative analysis of various parameters (Fig. 3i) further revealed that the outstanding energy-storage performance of the CS-750 cathode was due to the synergistic interplay of multiple factors, like it maximizing its accessible surface area, its well-optimized hierarchical pore structure and robust carbon framework with numerous zincophilic sites, which all contributed to its exceptional electrochemical energy-storage capabilities.
2.4 Charge-storage mechanism
To enhance our comprehension of the charge-storage mechanism, a series of ex situ characterizations was conducted. The GCD profile (Fig. 4a) identified five critical points (A, B, C, D, and E) for characterization corresponding to 1.8, 0.9 and 0 V during the discharge cycle, and 0.9 and 1.8 V during the charge cycle. Ex situ XRD analysis was then employed to investigate the modifications in structure of the CS-750 cathode material. A pronounced peak at ∼26.4° was observed in the XRD pattern (Fig. S22†). The 2D contour plot of the XRD patterns substantiated this conclusion (Fig. 4b). There was an enhancement of the peak signal with the progression of the discharge process due to ion uptake, which gradually diminished following the charging process. This was accompanied by a shift towards lower angle, ultimately returning to the initial state, thereby highlighting the dynamic structural changes associated with the charge and discharge cycles.65 This process refers to the reversible origination and dissolution of Zn(CF3SO3) [Zn(OH)2]3·xH2O on the cathode surface throughout the charging and discharging processes. It was driven by the coordination of OH− with Zn(CF3SO3)2 and H2O (Fig. S23†). Furthermore, the 2D XPS contour map of Zn 2p revealed a progressive intensification of the Zn signal during the discharging process, indicating a substantial incorporation of Zn2+ ions into the cathode structure (Fig. 4c). This phenomenon was reversed during the charging phase, wherein the Zn signal showed a notable decrease, suggesting the reversible expulsion of Zn2+ ions to their initial state. Alternatively, the intensity of the S 2p peak exhibited an inverse fluctuation trend compared to the Zn signal throughout the charging and discharging cycles (Fig. 4d). This demonstrates the cathodes' capability to alternately accommodate Zn2+/CF3SO3− charge carriers. Moreover, in the ex situ FT-IR analysis, a new signal was observed at 2923 cm−1, which was attributed to the generation of –OH species, indicating the coordination between H+ ions and the CO functional group (Fig. S24†). This suggests that during discharge the CO group underwent a reduction reaction, facilitating proton transfer, which resulted in the formation of C–OH bonds. This gradually intensified during discharging and weakened during charging, which was also consistent with the ex situ XPS and XRD results. Further, insights into the chemical conversion mechanism within the CS-750 cathode were obtained through detailed analysis of the O 1s, C 1s and N 1s XPS spectra (Fig. S25†). Regarding the O 1s spectra (Fig. 4e), peaks for quinone-type O (532.4 eV) and phenol/carboxyl O (532.5 eV) were obtained by curve fitting. Notably, the carbonyl O (CO) exhibited electrochemical redox responses to produce strong binding interactions with electrolyte ions. The intensity of the C–O peak demonstrated a significant increase during discharge while the CO peak was diminished continuously, while conversely in the charge process, the CO peak showed a reverse trend. The changes in the ex situ spectra of C 1s were compatible with the variations in the C–O signal (Fig. 4f). In the discharged state, an emergent peak signal indicative of C–O–Zn bonds could be observed, which denotes the occurrence of chemical interactions between Zn2+ ions and C–OH groups, which resemble the reaction between carbonyl and Zn ions. During the discharge process, the CO unit in the carbon cathode undergoes a reduction reaction and forms C–OH through the addition of protons and electrons. Concurrently, H+ and Zn2+ ions migrate to the cathode, forming C–Zn. As discharge progresses, the C–Zn species undergo metathesis with additional CO groups, releasing protons back into the electrolyte and resulting in the generation of the C–O–Zn configuration. Regarding the N species (Fig. S26†), the pyridinic N peak at 398.8 eV, pyrrolic N peak at 400.5 eV and quaternary N peak at 401.7 eV could be obtained from the deconvoluted XPS spectra. Notably, the pyridinic N peak gradually declined during discharge process indicating an ongoing zincation reaction. Conversely, this peak increased during the charging process, reflecting the elimination of Zn2+ ions from the electrochemically active sites. Notably, the emergence of a new peak at 401.3 eV for O–Zn–N suggests that pyridinic N was capable of uptake of Zn2+ ions. This observation suggests the synergetic interplay between nitrogen and oxygen functional groups markedly enhances the ability to accommodate Zn2+ ions, thereby offering insightful implications for the optimization of the electrochemical performance.66,67 Further to authenticate the incorporation of H+ ions throughout the charging and discharging cycles, DMF, a solvent devoid of protonic constituents, was selected as a medium for the Zn(CF3SO3)2 electrolyte. This approach was chosen to mitigate the influence of H+ ions present in H2O. Consequently, in the Zn(CF3SO3)2–DMF electrolyte, the CS-750 cathodes demonstrated a specific capacity of 183.5 mA h g−1 at 0.2 A g−1 (Fig. S27†), which was conspicuously lower compared to the capacity observed in a Zn(CF3SO3)2·H2O electrolyte. Various comparative tests were performed to verify the possible chance for participation of H+ ions during Zn2+ storage in the CS-750 cathode, which were conducted in aqueous HCF3SO3 electrolyte at various pH values with the same pH value of 3 M Zn(CF3SO3)2 electrolyte used. At different corresponding pH values (1.01, 3.28, and 5.71), HCF3SO3 showed capacities of 41.1, 49.1, and 66.4 mA h g−1 (Fig. S28†), respectively, clearly indicating the participation of H+ in cathodic electrochemical reactions. The enhancement of the capacity was influenced by the elevation of the hydrogen-ion concentration during the discharge phase from 1.8 to 0 V. This was also evidenced by SEM, which revealed the emergence of a flake-like structure on the CS-750 cathode surface (Fig. S29†). Remarkably, these structures vanished upon recharging (Fig. S30†), demonstrating the reversible nature of H+-ion corporation. Additionally, the smallest size of proton facilitated the rapid redox dynamics, addressing the interfacial charge-transfer limitations of Zn2+. This minimized the mass load and structural growth, resulting in faster charge and discharge rates. Consequently, the cathode material stability was enhanced during cycling, leading to long-term cycling stability and an improved rate performance. However, the proton insertion/extraction process can lead to structural degradation and subsequent capacity fading during long -term cycling. Furthermore, at high current densities, the accumulation of protons within the electrode structure can impede ion-diffusion, thereby limiting the rate capability. The entire proton-assisted zinc-ion-storage mechanism within the superstructure cathode is illustrated in Fig. 4g. This process involves reversible electrochemical hydrogen-ion adsorption/desorption and redox reactions of oxygen species.68 During every charge–discharge cycle, the CS-750 material undergoes a series of oxidation–reductions, facilitating efficient and rapid ion transport and charge storage. A combined experimental and theoretical investigation elucidated the energy-storage process of the CS-750 cathodes in ZHCs, as shown in Fig. 4h. The charge-storage process of the carbon cathode with Zn(SO3CF3)2 electrolyte involves an alternative physical uptake of Zn2+ ions, mediated by OTF− anions, which occurs alternatively within the superstructure framework. This process is accompanied by reversible electrochemical reactions involving Zn2+ ions and nitrogen/oxygen motifs, leading to the formation of O–Zn–N bonds throughout whole charge and discharge process. The charging process is characterized by the physical adsorption of OTF− anions at the cathode surface, which is primarily related to charge balancing and maintaining electrochemical reversibility. The OTF− ions play a crucial role in stabilizing the overall electrochemical system by maintaining charge neutrality throughout the intercalation and de-intercalation of Zn2+ ions into and out of the cathode. This reversible interplay of the anion and cation within the cathode structure is the critical aspect for achieving long-cycling stability and high performance. The overall reaction mechanism
involved in zinc-ion storage is depicted in Fig. 4i. Leveraging theoretical and practical findings, the mechanism of proton-assisted Zn2+ storage within the cathodes can be mainly characterized into two pivotal stages. First, the combined action of carbonyl and pyridine structures facilitates the chemical adsorption of zinc ions, leading to the formation of O–Zn–N bonds. Second, the adsorption of H+ ions onto the carbon cathode, which is a crucial factor that significantly contributes to the energy-storage capacity.69,70 These synergistic interactions underpin propelling the electrochemical energy-storage efficiency observed in the CS-750 model, showcasing the essential influence of the proton-facilitated zinc-ion charge-storage mechanism in advancing the device performance.
 | ||
| Fig. 4 (a) GCD curves for the Zn//CS-750 device. (b) Ex situ XRD characterization. (c) Ex situ XPS high-resolution spectra of Zn 2p and (d) S 2p. (e) Ex situ XPS spectra of O 1s and (f) C 1s. (g) Redox reaction mechanism. (h) Charge-storage mechanism of the CS-750 cathode in ZHCs. (i) Charge-storage reaction mechanism. | ||
3 Conclusion
In conclusion, a hydrogen-bond-induced micellar aggregation-driven self-assembly approach is developed to synthesize flower-like carbon superstructures with exceptional structural robustness. The well-optimized architecture enhances the availability of ample sites for electrolyte interaction and charge storage, leading to superior specific capacitance and outstanding electrochemical responses. Specially, CS-750 device-based carbon superstructures delivered a remarkable energy density of 160.8 W h kg−1 with a high discharge capacity of 262.8 mA h g−1 and consistent cycle life with a 93.2% capacity retention after 200000 cycles at 20 A g−1. The superior electrochemical properties could be ascribed primarily to the efficient alternating adsorption of Zn2+ and CF3SO3− ions at zincophilic sites within the superstructures and the Zn2+-linked redox reactions involving the pyridine/carbonyl motifs. Additionally, the adsorption of H+ ions onto the carbon framework significantly contributed to enhancing the capacity and rate performance. These synergetic interactions underscore the crucial role of the proton-assisted zinc-ion-storage process. Overall, the as-constructed carbon superstructures as cathodes offer several advantages and characteristics that enhance the capacitive activity and durability of zinc-ion capacitors. This approach provides a novel paradigm for the design of advanced carbon superstructures for use in energy-storage system.
Data availability
The data that support the findings of this study are available upon reasonable request from the corresponding author.Author contributions
Shreeti Jha: methodology, conceptualization, formal analysis, data curation, and writing-original draft. Yang Qin: visualization, writing-review, and conceptualization. Yumin Chen: visualization and methodology. Ziyang Song: visualization, methodology, funding acquisition, and writing-review & editing. Ling Miao: visualization and writing-review & editing. Yaokang Lv: project administration and investigation. Lihua Gan: supervision, funding acquisition, project administration, visualization, and writing review & editing. Mingxian Liu: methodology, supervision, funding acquisition, and writing-review & editing.Conflicts of interest
The authors confirm they have no conflicts of interest to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 22272118, 22172111, and 22309134), Science and Technology Commission of Shanghai Municipality, China (No. 22ZR1464100, 20ZR1460300, and 19DZ2271500), China Postdoctoral Science Foundation (2022M712402), and Fundamental Research Funds for the Central Universities (2023-3-YB-07), Shanghai Rising-Star Program (23YF1449200), and Zhejiang Provincial Science and Technology Project (2022C01182).Notes and references
- H. Li, J. Wu, L. Wang, Q. Liao, X. Niu, D. Zhang and K. Wang, Chem. Eng. J., 2022, 428, 131071 CrossRef CAS.
- C. Wang, Z. Pei, Q. Meng, C. Zhang, X. Sui, Z. Yuan, S. Wang and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 990–997 CrossRef CAS PubMed.
- S. Zuo, X. Xu, S. Ji, Z. Wang, Z. Liu and J. Liu, Chem.–Eur. J., 2020, 27, 830–860 CrossRef PubMed.
- X. Chen, H. Zhang, Y. Gao, J. H. Liu, X. Cao, C. Zhan, S. Wang, J. Wang, S. X. Dou and D. Cao, Carbon Neutraliz., 2022, 1, 159–188 CrossRef.
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2022, 34, 2206963 CrossRef CAS PubMed.
- Y. Li, J. Huang, L. Kang, Z. Tian, F. Lai, D. J. L. Brett, T. Liu and G. He, Sci. China Mater., 2022, 65, 1495–1502 CrossRef CAS.
- Y. Zhang, Z. Song, Q. Huang, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202423936 CrossRef CAS PubMed.
- Y. Chen, L. Miao, Z. Song, H. Duan, Y. Lv, L. Gan and M. Liu, Adv. Funct. Mater., 2024, 34, 2409428 CrossRef CAS.
- J. Jin, X. Geng, Q. Chen and T.-L. Ren, Nano-Micro Lett., 2022, 14, 64 CrossRef CAS PubMed.
- H. Tang, J. Yao and Y. Zhu, Adv. Energy Mater., 2021, 11, 2003994 CrossRef CAS.
- Q. Liu, H. Zhang, J. Xie, X. Liu and X. Lu, Carbon Energy, 2020, 2, 521–539 CrossRef CAS.
- L. Liu, Y. Lu, D. Qiu, D. Wang, Y. Ding, G. Wang, Z. Liang, Z. Shen, A. Li, X. Chen and H. Song, J. Colloid Interface Sci., 2022, 620, 284–292 CrossRef CAS PubMed.
- H. Li, P. Su, Q. Liao, Y. Liu, Y. Li, X. Niu, X. Liu and K. Wang, Small, 2023, 19, 2304172 CrossRef CAS PubMed.
- J. R. Loh, J. Xue and W. S. V. Lee, Small Methods, 2023, 7, 2300101 CrossRef CAS PubMed.
- G. Zampardi and F. L. Mantia, Nat. Commun., 2022, 13, 687 CrossRef CAS PubMed.
- Z. Li, Y. An, S. Dong, C. Chen, L. Wu, Y. Sun and X. Zhang, Energy Storage Mater., 2020, 31, 252–266 CrossRef.
- J. Chen, L. Wang, M. Peng, T. Hu, K. Yuan and Y. Chen, Chem. Mater., 2023, 35, 4089–4099 CrossRef CAS.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- H. Wang, W. Ye, Y. Yang, Y. Zhong and Y. Hu, Nano Energy, 2021, 85, 105942 CrossRef CAS.
- C. Wang, X. Zeng, J. Qu, J. M. Cairney, Q. Meng, P. J. Cullen and Z. Pei, Matter, 2023, 6, 3993–4012 CrossRef CAS.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Adv. Energy Mater., 2021, 11, 2100201 CrossRef CAS.
- Y. Wang, S. Sun, X. Wu, H. Liang and W. Zhang, Nano-Micro Lett., 2023, 15, 78 CrossRef CAS PubMed.
- G. Yang, Q. Zhang, C. He, Z. Gong, Z. Liu, J. Song, S. Jiang, J. Han, H. Yang and X. Li, Angew. Chem., 2025, 137, e202421230 CrossRef.
- R. Fei, H. Wang, Q. Wang, R. Qiu, S. Tang, R. Wang, B. He, Y. Gong and H. J. Fan, Adv. Energy Mater., 2020, 10, 2002741 CrossRef CAS.
- P. Liu, W. Liu, Y. Huang, P. Li, J. Yan and K. Liu, Energy Storage Mater., 2020, 25, 858–865 CrossRef.
- J. Li, L. Yu, W. Wang, X. He, G. Wang, R. Liu, X. Ma and G. Zhang, J. Mater. Chem. A, 2022, 10, 9355–9362 RSC.
- M. S. Choi, M. K. Jung, H. H. Rana and H. S. Park, Carbon Lett., 2024, 34, 25–39 CrossRef CAS.
- F. Wei, H. Tian, P. Chen, Y. Lv and J. Huang, Appl. Surf. Sci., 2023, 613, 156021 CrossRef CAS.
- W. Zhang, H. Kang, Z. Gu, H. Liu, Z. Li, X. Li and B. Yang, J. Energy Storage, 2023, 61, 106715 CrossRef.
- D. Wang, S. Wang and Z. Lu, Int. J. Energy Res., 2021, 45, 2498–2510 CrossRef CAS.
- K. Shang, Y. Liu, P. Cai, K. Li and Z. Wen, J. Mater. Chem. A, 2022, 10, 6489–6498 RSC.
- B. Wang, P. Zhao, J. Feng, D. Chen, Y. Huang, L. Sui, H. Dong, S. Ma, L. Dong and L. Yu, J. Colloid Interface Sci., 2021, 588, 184–195 CrossRef CAS PubMed.
- L. Wu and Y. Dong, Energy Storage Mater., 2021, 41, 715–737 CrossRef.
- X. Li and J. Wang, InfoMat, 2019, 2, 3–32 CrossRef.
- Z. Xiong, W. Jin, H. Liu and H. Liu, Adv. Energy Sustainability Res., 2022, 3, 2100194 CrossRef CAS.
- Y. Zhao, Y. Zhang, Y. Wang, D. Cao, X. Sun and H. Zhu, Carbon Energy, 2021, 3, 895–915 CrossRef CAS.
- Y. Lv, L. Zhang, X. Wei, B. Qiu, W. Zhang, Q. Qin, D. Jia, X. He, Z. Liu and F. Wei, Sustain. Mater. Technol., 2023, 35, e00536 CAS.
- Q. Chu, Z. Chen, C. Cui, Z. Li, X. Li, Y. Xu, Y. Li, Y. Cui and Q. Liu, Carbon, 2024, 227, 119283 CrossRef CAS.
- Y. Chen, M. Zhou, J. Wu, X. Xiao and L. Xu, J. Mater. Sci. Technol., 2024, 178, 155–161 CrossRef CAS.
- L. Yang, X. He, Y. Wei, H. Li, Y. Yu and J. Qiu, J. Power Sources, 2022, 542, 231743 CrossRef CAS.
- S. J. Patil, N. R. Chodankar, S.-K. Hwang, P. A. Shinde, G. S. R. Raju, K. S. Ranjith, S. V. Karekar, Y.-S. Huh and Y.-K. Han, J. Mater. Chem. A, 2023, 11, 5112–5126 RSC.
- N. R. Chodankar, S. J. Patil, S. Lee, J. Lee, S. K. Hwang, P. A. Shinde, I. V. Bagal, S. V. Karekar, G. S. R. Raju and K. S. Ranjith, InfoMat, 2022, 4, e12344 CrossRef CAS.
- H. Fan, X. Hu, S. Zhang, Z. Xu, G. Gao, Y. Zheng, G. Hu, Q. Chen, T. S. AlGarni and R. Luque, Carbon, 2021, 180, 254–264 CrossRef CAS.
- Z. Yang, X. Chang, H. Mi, Z. Wang, J. Gao, X. Xiao, F. Guo, C. Ji and J. Qiu, J. Colloid Interface Sci., 2024, 658, 506–517 CrossRef CAS PubMed.
- P. Liu, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Small, 2024, 20, 2400774 CrossRef CAS.
- C. C. Hou, L. Zou, Y. Wang and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 21360–21366 CrossRef CAS PubMed.
- K. Yang, L. Hu, Y. Wang, J. Xia, M. Sun, Y. Zhang, C. Gou and C. Jia, J. Mater. Chem. A, 2022, 10, 12532–12543 RSC.
- C. Hu, P. Liu, Z. Song, Y. Lv, H. Duan, L. Xie, L. Miao, M. Liu and L. Gan, Chin. Chem. Lett., 2025, 36, 110381 CrossRef CAS.
- H. Tang, J. Yao and Y. Zhu, Adv. Energy Mater., 2021, 11, 2003994 CrossRef CAS.
- T. N. Gao, T. Wang, W. Wu, Y. Liu, Q. Huo, Z. A. Qiao and S. Dai, Adv. Mater., 2019, 31, 1806254 CrossRef.
- F. Fu, D. Yang, B. Zhao, Y. Fan, W. Liu, H. Lou and X. Qiu, J. Colloid Interface Sci., 2023, 640, 698–709 CrossRef CAS.
- L. Yu, J. Li, N. Ahmad, X. He, G. Wan, R. Liu, X. Ma, J. Liang, Z. Jiang and G. Zhang, J. Mater. Chem. A, 2024, 12, 9400–9420 RSC.
- H. Gupta, Y. Dahiya, H. K. Rathore, K. Awasthi, M. Kumar and D. Sarkar, ACS Appl. Mater. Interfaces, 2023, 15, 42685–42696 CrossRef CAS.
- X. Zheng, L. Miao, Z. Song, W. Du, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, J. Mater. Chem. A, 2022, 10, 611–621 RSC.
- Y. Yang, D. Chen, H. Wang, P. Ye, Z. Ping, J. Ning, Y. Zhong and Y. Hu, Chem. Eng. J., 2022, 431, 133250 CrossRef CAS.
- Y. Li, K. Xiao, C. Huang, J. Wang, M. Gao, A. Hu, Q. Tang, B. Fan, Y. Xu and X. Chen, Nano-Micro Lett., 2021, 13, 1–14 CrossRef CAS PubMed.
- H. Wang, X. Chen, J. Zhang, Z. Yuan, P. Ye, J. Shen, Y. Zhong and Y. Hu, Appl. Surf. Sci., 2022, 598, 153819 CrossRef CAS.
- D. Jia, Z. Shen, W. Zhou, Y. Li, J. He, L. Jiang, Y. Wei and X. He, Chem. Eng. J., 2024, 485, 149820 CrossRef CAS.
- H. Zhang, Q. Liu, Y. Fang, C. Teng, X. Liu, P. Fang, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1904948 CrossRef CAS PubMed.
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- Z. Li, D. Chen, Y. An, C. Chen, L. Wu, Z. Chen, Y. Sun and X. Zhang, Energy Storage Mater., 2020, 28, 307–314 CrossRef.
- Q. Zhang, J. Wang, Y. Zhang, F. Deng, J. Shang, R. Li and B. Xing, Electrochim. Acta, 2023, 467, 143135 CrossRef CAS.
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202203453 CrossRef CAS PubMed.
- Q. Q. Sun, J. Y. Du, T. Sun, Z. B. Zhuang, Z. L. Xie, H. M. Xie, G. Huang and X. B. Zhang, Adv. Mater., 2024, 36, 2313388 CrossRef CAS.
- P. Shang, M. Liu, Y. Mei, Y. Liu, L. Wu, Y. Dong, Z. Zhao and J. Qiu, Small, 2022, 18, e2108057 CrossRef PubMed.
- F. Xie, H. Li, X. Wang, X. Zhi, D. Chao, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS.
- X. Yang, C. Hu, Y. Chen, Z. Song, L. Miao, Y. Lv, H. Duan, M. Liu and L. Gan, J. Energy Storage, 2024, 104, 114509 CrossRef.
- C. C. Hou, Y. Wang, L. Zou, M. Wang, H. Liu, Z. Liu, H. F. Wang, C. Li and Q. Xu, Adv. Mater., 2021, 33, 2101698 CrossRef CAS PubMed.
- Y. Chen, Z. Song, Y. Lv, L. Gan and M. Liu, Nano-Micro Lett., 2025, 17, 117 CrossRef CAS PubMed.
- D. Zhang, L. Miao, Z. Song, X. Zheng, Y. Lv, L. Gan and M. Liu, Energy Fuels, 2024, 38, 12510–12527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00357a |
| This journal is © The Royal Society of Chemistry 2025 |