
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Potential dependent degradation of spinel LiMn2O4 (LMO) and related structures assessed via manganese- and oxygen-sensitive scanning electrochemical microscopy†
Dong Ok
Kim
 a,
Abhiroop
Mishra
a,
Michelle
Zorigt
a,
Yichen
Li
a,
Richard T.
Haasch
b and
Joaquín
Rodríguez-López
*abc
a,
Abhiroop
Mishra
a,
Michelle
Zorigt
a,
Yichen
Li
a,
Richard T.
Haasch
b and
Joaquín
Rodríguez-López
*abc
aDepartment of Chemistry, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
bMaterials Research Laboratory, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA. E-mail: joaquinr@illinois.edu
cBeckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
First published on 6th March 2025
Abstract
Manganese dissolution has been a long-standing problem that limits the widespread application of Mn-based Li-ion battery (LIB) cathodes, despite their low cost and high Li storage capacity. The accurate detection and quantification of species generated during the degradation of Mn-based cathodes, such as dissolved Mn and evolved lattice oxygen as a function of potential and/or state of charge, are essential for designing better cathode materials and interfaces. Here, we utilize mercury-based scanning electrochemical microscopy (SECM) probes that enable the real-time quantitative investigation (∼1 μM limit of detection) of Mn dissolution near the surface of spinel LiMn2O4 cathodes. Combined with SECM oxygen detection using Au probes, we characterize both oxygen and Mn loss from the cathode as a function of cathode potential. Our study reveals two distinct potential regions for Mn dissolution, where the degradation in the latter region is accelerated by both Mn and oxygen loss from the cathode. Our methodology also demonstrates that an electrolyte additive, tributyl phosphate (TBP), successfully suppresses Mn dissolution in the first region at low cathode potential, further supporting the idea of distinct degradation mechanisms in each region. This work elucidates the complex interplay of acid-base, interphase formation, and oxygen loss in Mn dissolution mechanisms in operating cathodes as a function of potential. It also establishes a methodology to investigate degradation processes in a variety of existing and future Mn-based cathodes and their related structures.
1. Introduction
Li-ion batteries (LIBs) are the favored technology driving the market toward electrified transportation.1 Cathode materials are critical components of LIBs because they determine both energy storage capacity and cost-effectiveness.2–4 Manganese (Mn)-based oxide cathodes are particularly attractive due to their low toxicity and cost, compared to other transition metal oxide (TMO) materials.3,5,6 Furthermore, there is increasing interest in Li-, Mn-rich cathodes, which can deliver specific capacities of over 250 mA h g−1.7–9 However, Mn dissolution in these cathodes is a problem that limits their performance and cycle life.10–13 Mn dissolution not only leads to the loss of the cathode active material but also accompanies several detrimental outcomes. For example, dissolved ions migrate and deposit on the anode, accelerating the fading of LIB performance.13–16Until now, researchers have relied on bulk methods of analysis to study dissolved transition metals (TMs) from LIB cathodes in non-aqueous electrolytes. Inductively coupled plasma optical emission spectroscopy (ICP-OES), electron paramagnetic resonance (EPR) spectroscopy, and nuclear magnetic resonance spectroscopy (NMR) are a few examples that are commonly employed to study dissolved metals.17–19 However, these methods are typically ex situ in nature, lacking temporal resolution. Ongoing efforts have focused on developing in situ configurations that use ultraviolet-visible spectroscopy or ICP as detection methods, enabling imaging capabilities and detection of low levels of Mn dissolution.20,21 A few in situ studies using electrochemical methods have reported the potential or cycle number dependence of Mn dissolution, as assessed in the bulk electrolyte.22–24
Here, we propose that improvements in electrochemical quantification at low state-of-charge, along with the ability to detect species related to degradation (such as oxygen) in addition to Mn, would significantly enhance our mechanistic understanding of Mn dissolutions from LIB cathodes. Extensive studies have been devoted to investigating the role of oxygen loss in the degradation of Mn-based cathodes.25–28 Irreversible lattice oxygen loss during electrochemical charging degrades cathode performance while accompanying deleterious surface reconstruction and Mn dissolution.28,29 Studies using X-ray absorption spectroscopy (XAS) revealed that TM composition and the oxidation states within TMO cathodes affect the lattice oxygen stability.30,31 Therefore, understanding the complex interplay between evolved oxygen and Mn as a function of cathode potential, with high temporal resolution, is key to understanding Mn-based cathode degradation.
Spinel LiMn2O4 (LMO) is one of the most classical Mn-based cathodes which suffers from pronounced Mn dissolution.32,33 The dissolution process from LMO has been intensively investigated via both experiments and theoretical calculations.34–38 It is generally believed that Jahn–Teller distortion of high-spin Mn3+ drives the dissolution within LMO in the lower charging state.10,14 Trace amounts of water in the electrolyte and the decomposition of the electrolyte result in the generation of hydrofluoric acid (HF). This aggravates the disproportionation of Mn3+ to Mn2+ and Mn4+, where Mn2+ is readily soluble in the conventional carbonate electrolyte.
Countless studies also support that the Mn dissolution rate increases with decreasing Li content within the LMO structure, i.e. at a highly charged state.13,22,23 Mn dissolution at a highly charged state can be associated with surface reconstruction of the delithiated LMO, λ-MnO2. Specifically, Tang et al. proposed that unstable λ-MnO2 forms Mn3O4 at the surface upon the release of oxygen as shown in eqn (1).39,40
| 3λ-MnO2 → Mn3O4 + O2 | (1) |
Since 1/3 of Mn-ions in Mn3O4 are Mn2+ ions which are soluble in the conventional carbonate electrolyte, this phase can contribute to Mn dissolution from LMO. These previous studies all indicate that dissolution and degradation of LMO depend on the state-of-charge and involve the complex interplay of Mn, oxygen loss, and interface formation.
Scanning electrochemical microscopy (SECM) is a powerful tool to probe various processes taking place at the interface of battery electrode–electrolyte.17,41–43 SECM positions an ultramicroelectrode (UME) tip close to the substrate, i.e., the battery material of interest, and measures various transient processes happening at the interface between the substrate and the electrolyte.42,44–46 Its versatility lies in the fact that one can choose an appropriate UME as a probe depending on the phenomenon of interest. Previously, researchers have employed mercury (Hg)-based SECM probes to study the kinetics of ion (de)intercalation and quantify ionic species.45,47,48 Gossage et al. correlated Li+ flux with SEI formation at the HOPG interface at varying (de)intercalation potentials.46 Hatami et al. used a Hg SECM probe to quantify Mn2+ species in non-aqueous solution using anodic stripping voltammetry.49
In this work, we employ SECM as an in situ tool to enable real-time quantitative investigation of the degradation of LMO as a LIB cathode. Hg-based SECM probes allowed selective investigation of the Mn dissolved from the cathode with exceptional sensitivity and temporal resolution (Fig. 1a), while Au SECM probes were used to detect oxygen loss from the cathode. Furthermore, we illustrate the suppression of Mn dissolution upon the addition of an electrolyte additive, TBP, highlighting the versatility of the SECM method in investigating various parameters and processes that affect the Mn dissolution. While the additive was effective at mitigating Mn dissolution at low state-of-charge, the dissolution was accelerated as charging proceeded. By quantitatively analyzing the degradation products from LMO, we elucidate the potential dependent degradation of Mn-based cathodes, identifying distinct regimes and their relation to oxygen loss.
 | ||
Fig. 1 Mn detection using Hg UMEs. (a) Hg UME based-SECM setup in non-aqueous electrolyte which allows selective detection of Mn2+ from the operating LMO cathode. (b) CV of Mn2+ ions in 1 M LiPF6 PC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EC (1:1) solution. The cathodic peak observed during the initial reductive sweep corresponds to the amalgamation of Mn2+ while the anodic peak corresponds to the stripping of Mn2+ back to the bulk solution. (c) Calibration curve obtained from ASV measurements with a 5-minute amalgamation time. The error bars and LOD are derived from the standard deviation of the three measurements. The stripping current for each concentration is shown in the inset. :EC (1:1) solution. The cathodic peak observed during the initial reductive sweep corresponds to the amalgamation of Mn2+ while the anodic peak corresponds to the stripping of Mn2+ back to the bulk solution. (c) Calibration curve obtained from ASV measurements with a 5-minute amalgamation time. The error bars and LOD are derived from the standard deviation of the three measurements. The stripping current for each concentration is shown in the inset. | ||
2. Experimental
2.1 Materials
LiMn2O4 (LMO, electrode sheet, aluminum substrate) was purchased from NEI Corporation. All chemicals were purchased from commercial sources. Ethylene carbonate (EC, anhydrous, 99%), propylene carbonate (PC, anhydrous, 99.7%), lithium hexafluorophosphate (LiPF6, battery grade, 99.99%), tetrabutylammonium hexafluorophosphate (TBAPF6, 98%), 7,7,8,8-tetracyanoquinodimethane (TCNQ, 98%), tributyl phosphate (TBP, 99%), hydroxymethylferrocene (FcMeOH, 97%), mercury(II) nitrate monohydrate (99.99% trace metal basis), manganese bis(trifluoromethanesulfonate) (Mn(TFSI)2), and manganese(II, III) oxide were all purchased from Sigma-Aldrich. Manganese(IV) oxide (activated) was purchased from Alfa Aesar.2.2 Working electrode fabrication
Hg UMEs were fabricated following the procedure described in the previous report.48 In short, platinum (Pt) disc UMEs were made by sealing a Pt wire (radius = 12.5 μm) in a borosilicate glass capillary. Then, the Pt wire was etched electrochemically by applying an AC waveform of 2.70 V with a variac between a Pt UME and a carbon rod for 20 seconds. The etching solution was 30 v% sat. CaCl2 in 10 v% HCl in H2O and the etching process was carried out under ultrasonic agitation. The etching depth of the discwell formed was characterized by measuring changes in the limiting current for electrolysis of hydroxymethylferrocene solution via chronoamperometry and cyclic voltammetry (CV) of the UME before and after the etching process (Fig. S1†).50 Then, the Hg deposition was done potentiostatically at +0.1 V (vs. Ag/AgCl, 3 M KCl) in 10 mM Hg(NO3)2·H2O + 0.1 M KNO3 in H2O until the deposition current reached 120 nA. The excess Hg was removed using a glass coverslip which results in a leveled Hg discwell ultramicroelectrode (Hg UME). It is worth noting that the amount of Hg deposited on the UME is minimal due to its small size, typically in the order of ∼500 pL for the electrodepositions in this study.2.3 Electrochemical measurements
For the Hg UME fabrication process, a CHI760D bipotentiostat (CH Instruments, Inc.) was used with Ag/AgCl as a reference electrode and a tungsten (W) wire as a counter electrode. All other electrochemical measurements were conducted using a CHI920D scanning electrochemical microscope (CH Instruments, Inc.) inside a glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm).For SECM measurements, a standard Teflon SECM cell was assembled with a fresh cathode and transferred inside the glovebox. A Li metal strip and Pt wire were used as a reference electrode and a counter electrode, respectively. The fabricated Hg UME was positioned ∼15 μm away from the cathode by applying a constant potential (3.0 V vs. Li+/Li) to obtain an approach curve with a TCNQ redox mediator. Then, the UME was retracted 1.0 mm and the cell was rinsed ten times with fresh propylene carbonate (PC) solution to remove any TCNQ left in the cell. For the Mn dissolution experiment, 1 M LiPF6 PC:EC (1:1 v/v) solution with and without the TBP additive was added to the cell and the UME was returned to the position close to the cathode. For oxygen detection, 0.1 M TBAPF6 in PC:EC (1:1 v/v) solution was added to the cell.
2.4 X-ray photoelectron spectroscopy (XPS) measurements
XPS measurements were performed with a Kratos Supra+ X-ray photoelectron spectrometer (Kratos Analytical, Inc., Manchester, U.K), using monochromatic Al Kα radiation (1486.6 eV). Cycled electrodes were mounted on the XPS sample holder in an Ar filled glovebox (O2, H2O ≤ 0.1 ppm) and then taken to the instrument in an air-tight container. Spectra were calibrated against the sp2 carbon peak at 284.5 eV associated with the Super P carbon additive within the composite electrode. Then, they were fitted with Gaussian–Lorentzian (GL) line shapes. Atomic concentrations within samples were determined via integration of fitted XPS spectra that were adjusted for atomic relative sensitivity factors (RSFs).2.5 Scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX), and X-ray diffraction (XRD) measurements
SEM/EDX was conducted with a Hitachi S4800 at an accelerating voltage of 5 kV. EDX was performed in the same system with an Oxford Instruments Ultim Max 100 mm2 large silicon area drift detector. Ex situ XRD was conducted with a Rigaku MiniFlex 600 X-ray diffractometer. The X-ray source was Cu Kα (λ = 1.5418 Å) operated at 40 kV and 15 mA. The measurements were conducted with 0.02 degree step increments with 4-second acquisition per step. All charged cathode samples were rinsed with PC to remove the residual electrolyte and then dried under vacuum overnight before the characterization.3. Results and discussion
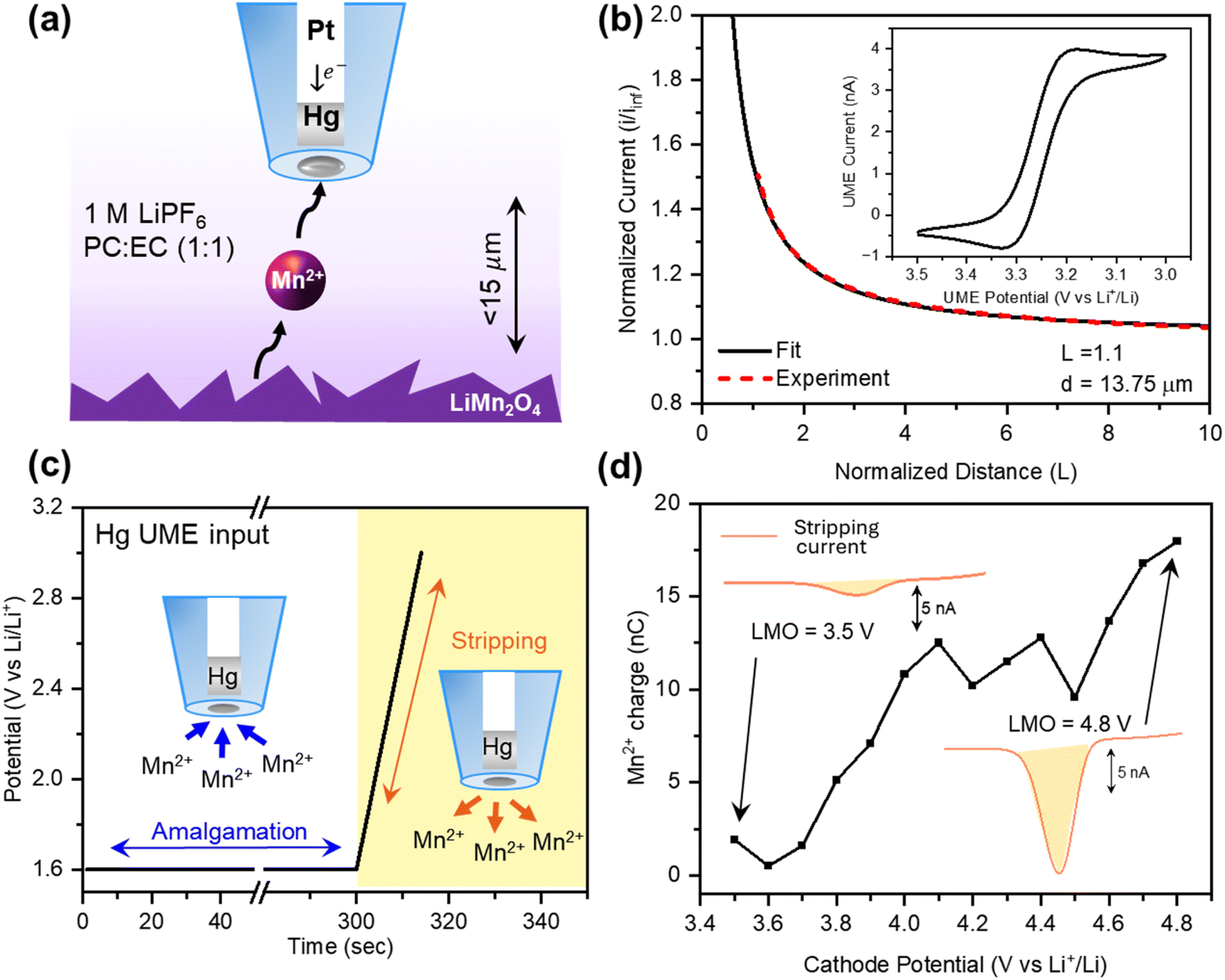
First, we evaluated the sensitivity of the Hg UME in detecting Mn-ion in LIB electrolyte. Fig. 1b illustrates a typical CV in a three-electrode cell configuration, consisting of a 12.5 μm radius Hg UME as a working electrode, Pt wire as a counter, and Li-metal strip as a reference. For this study, various concentrations of Mn(TFSI)2 were dissolved in 1 M LiFP6 PC:EC. When a reductive potential (1.6 V vs. Li+/Li) is applied, Mn2+ ions amalgamate into the Hg phase of the UME (Fig. 1b). Upon sweeping back to the oxidative potential, Mn2+ ions are stripped back into the solution, with a peak centered at around 2.05 V. While released products from the cathode, such as Li ions, are expected, this strategy provides a selective way to detect only the amalgamated Mn2+ (Fig. S2a†).
We carried out anodic stripping voltammetry (ASV) by amalgamating Mn2+ at 1.6 V for several minutes, followed by stripping of the ions back to the solution. Both 4 μM and 8 μM Mn2+ solutions showed a good linear relationship (R2 > 0.99) between the amalgamation time and the stripping charge (Fig. S2b†). This linear relationship confirms that higher sensitivity for trace amounts of Mn2+ ions in the solution can be achieved by extending the amalgamation time. Considering that the expected amount of Mn2+ ions dissolving from the LMO cathode is in the order of μM,20,51,52 further ASV measurements were done using 5 minutes of amalgamation time in 2.25, 4.5, 9, 12, and 16 μM Mn(TFSI)2 solutions. ASV with 5 minutes of amalgamation time resulted in a limit of detection 1 μM (Fig. 1c), which agrees with the previous ASV study done with Hg UMEs for lead (Pb) and cadmium (Cd).53 Calibration curves with three Hg UMEs fabricated on different days yielded a similar slope (m = 0.31 ± 0.02 nC μM−1), confirming the consistency of the method (Fig. S3†).
Incorporating the Hg UME in an SECM setup allowed us to detect low Mn2+ concentrations generated locally at the cathode–electrolyte interface. Fig. 2a illustrates the SECM setup for Mn detection from the LMO cathode. It consists of the Hg UME characterized in Fig. 1 as the working electrode, a commercial LMO cathode as the second working electrode (substrate electrode), a Li-metal strip as a reference, and a Pt wire as a counter electrode. The commercial LMO cathode used in this experiment was further characterized in the ESI† using XRD, SEM, and CV (Fig. S4–S6†), confirming its expected structure, morphology, and activity. The positioning of the Hg UME was done by obtaining probe approach curves of 2 mM TCNQ dissolved in 0.1 M TBAPF6 in PC (Fig. 2b). The Hg UME was positioned approximately 15 μm away from the LMO cathode. After obtaining the approach curve, the solution was replaced with 1 M LiPF6 PC:EC (1:1 v/v) electrolyte. The cathode was held at the charging potential from 3.5 V to 4.8 V while the Hg UME was held at the Mn2+ amalgamation potential (1.6 V) for 5 minutes for the ASV measurement. Then, the stripping peak was obtained by sweeping the Hg UME potential from 1.6 V to 3.0 at a scan rate of 0.1 V s−1 (Fig. 2c). ASV at the Hg UME was repeated at each cathode potential (from 3.5 V to 4.8 V) at intervals of 0.1 V. Each set of ASV measurement provided us with Mn2+ stripping current that was then integrated to obtain Mn2+ charge collected at the specific charging potential for LMO (Fig. 2d).
 | ||
| Fig. 2
In situ Mn detection in the SECM configuration. (a) Depiction of the SECM setup for Mn2+ detection with the Hg UME and LMO cathode in 1 M LiPF6 PC:EC (1:1) electrolyte. (b) Probe approach curve using TCNQ as a redox mediator. The CV of TCNQ is depicted in the inset. (c) The potential input at the Hg UME for ASV measurement. (d) Representative final output of the SECM experiment, obtained by integrating the area under the curve of the Hg UME stripping current. | ||
Fig. 3a depicts the average Mn2+ charge measured via SECM for Mn dissolution as a function of potential. The standard deviation in these experiments, shown as the shaded areas, can be primarily attributed to the inherent heterogeneity of the composite cathodes, which includes inconsistencies in the mixture of the binder, the active material, and conductive additives, as well as variations in topology. This variability in compositions is reflected in the EDX mapping at two different sites (Fig. S8 and Table S2†). In addition, the SECM mapping technique allowed us to probe the spatial heterogeneity of Mn dissolution from LMO (Fig. S9†). Quantification of Mn dissolution was done by correlating the SECM experimental results (i.e., current or charge) with finite element COMSOL simulations reflecting the flux of Mn2+ required to elicit such a response (Fig. S7 and Table S1†). The COMSOL model allowed us to correlate the Mn2+ flux from the cathode and the corresponding Hg UME stripping current from the 5-minute ASV measurement.
 | ||
| Fig. 3 Potential dependent Mn dissolution from LMO and the Mn suppression effect of TBP. (a) Average Mn2+ charge at each LMO potential throughout the 1st charging cycle. Corresponding Mn2+ flux was quantified using COMSOL simulations. The shaded region around each point represents the standard deviation of three samples. (b) XPS F 1s spectra of LMO electrodes charged up to 4.0 V with and without TBP. (c) Mn dissolution as a function of the cathode potential with and without TBP. The shaded region around each point represents the standard deviation of the three samples. A comparison of cumulative Mn2+ charge during the 1st charging process is depicted in the inset. (d) XPS P 2p spectra of LMO electrodes charged up to 4.0 V with and without TBP. | ||
The results in Fig. 3a distinctly resolve two potential regions where Mn dissolution accelerates, depicted as Region 1 and Region 2 for low and high potentials, respectively. These results led us to suspect that different chemical mechanisms are associated with each of these regions, so we turned to explore factors that would explicate these behaviors.
To further investigate the potential dependent behavior of Mn dissolution, we employed an electrolyte additive that is known to form a cathode–electrolyte interphase and change the reactions happening at the electrode interface. Recent studies on electrolyte additives suggest that they mitigate the Mn dissolution from TMO cathodes.54–59 Li et al. reported TBP as an electrolyte additive that improves the performance of the LiNi0.5Mn1.5O4 (LNMO) cathode by hindering the attack of HF and potentially suppressing the Mn dissolution.60 However, the direct evidence of it mitigating Mn dissolution and the mechanism behind it are lacking. Our method of quantification supports the previous study by demonstrating the decrease of Mn dissolution from LMO in the presence of the additive during the initial charging process. The potential dependent study illustrates that 1 wt% TBP is effective in suppressing Region 1 dissolution, resulting in a significant decrease of the cumulative Mn2+ charge throughout the 1st charging cycle (Fig. 3c and S10†).
XPS measurements on LMO charged to 4.0 V, at just the onset of Mn dissolution, indicate that the mitigation of Region 1 dissolution is indeed related to the effect of the additive. F 1s spectra of LMO cycled in TBP electrolyte demonstrate diminished LiF (685.4 eV)61,62 compared to that of an electrolyte without TBP (Fig. 3b). LiF is the main product of LiPF6 hydrolysis, along with HF, which aggravates Mn3+ disproportionation. The diminished LiF suggests that the additive effectively reduces HF levels within the electrolyte. The P–O bond in phosphate preferentially breaks upon electrochemical oxidation.54,60 The resulting oxidized molecule, which is Lewis-basic, serves as an HF scavenger. In addition, P 2p spectra of the TBP sample charged to 4.0 V demonstrate more P–O, P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O compositions (135 eV), suggesting the incorporation of TBP at the interphase instead of LiPF6 (Fig. 3d). Owing to the existence of P–O and PO bonds, TBP has a high HOMO level, which makes it preferentially oxidized prior to carbonate solvents.60 The formation of a TBP-derived interphase minimizes the direct contact of the electrolyte components with the cathode, ultimately diminishing undesired electrolyte oxidation (i.e., degradation of LiPF6). This result is also in agreement with O 1s XPS spectra (Fig. S11†). O 1s spectra of LMO cycled with TBP exhibit a higher O-cathode peak (529.8 eV) than those of LMO cycled without it. This suggests that the TBP treated sample either has lost less lattice oxygen during charging or presents a thinner cathode–electrolyte interphase as a result of minimized electrolyte degradation. Meanwhile, the XPS spectra of LMOs charged to 4.8 V are similar regardless of the presence of the additive (Fig. S11, S12 and Table S3†). This agrees with the SECM result that demonstrates that the presence of the additive does not successfully suppress Mn dissolution in Region 2.
O compositions (135 eV), suggesting the incorporation of TBP at the interphase instead of LiPF6 (Fig. 3d). Owing to the existence of P–O and PO bonds, TBP has a high HOMO level, which makes it preferentially oxidized prior to carbonate solvents.60 The formation of a TBP-derived interphase minimizes the direct contact of the electrolyte components with the cathode, ultimately diminishing undesired electrolyte oxidation (i.e., degradation of LiPF6). This result is also in agreement with O 1s XPS spectra (Fig. S11†). O 1s spectra of LMO cycled with TBP exhibit a higher O-cathode peak (529.8 eV) than those of LMO cycled without it. This suggests that the TBP treated sample either has lost less lattice oxygen during charging or presents a thinner cathode–electrolyte interphase as a result of minimized electrolyte degradation. Meanwhile, the XPS spectra of LMOs charged to 4.8 V are similar regardless of the presence of the additive (Fig. S11, S12 and Table S3†). This agrees with the SECM result that demonstrates that the presence of the additive does not successfully suppress Mn dissolution in Region 2.
The SECM experiment with the electrolyte additive again suggests two different processes for Mn dissolution. Region 1 lies within the potential window where the nominal oxidation state of Mn-ions is +3.5. While Region 1 dissolution is more likely to be correlated with the disproportionation of Mn3+ in the existence of HF in the electrolyte, Region 2 dissolution may be governed by a different process. According to the literature, delithiated LMO, which is also known as λ-MnO2, goes under phase transformation into Mn3O4 on the surface upon charging.11,38,39 As mentioned earlier, the Mn3O4 phase contains 1/3 Mn2+ ions that are soluble in the electrolyte, causing the dissolution of Mn-ions from the cathode at high charging potentials. The formation of the Mn3O4 phase accompanies the release of oxygen according to eqn (1).
Quantification of lattice oxygen loss during charging of LMO provides us with better insight into dissolution at Region 2. Investigation of lattice oxygen loss was done within the SECM setup following the methodology developed by Mishra et al.63 For this specific measurement, a 12.5 μm radius Au UME was utilized to detect oxygen released by the substrate via the oxygen reduction reaction (ORR) at 1.2 V on the UME tip (Fig. 4d). The cathode was immersed in 0.1 M TBAPF6 electrolyte to avoid the loss of the UME signal in the presence of Li+, as Mishra et al.63 previously reported. The cathode was potentiostatically biased from 3.5 V to 4.8 V while the Au UME was used to perform a 20-second chronoamperometric measurement at each cathode potential (Fig. 4a and b). Integration of the ORR current at the UME tip, followed by dividing it by the reduction step time (10 seconds) provided us with the average ORR current at the specific cathode potential (Fig. 4c). To quantify the oxygen loss, a COMSOL model from the previous work on oxygen detection via the ORR was used.63 The increase in the Au UME signal beyond 4.0 V supports that the degradation of LMO at Region 2 is accelerated by both Mn and oxygen loss (Fig. 4e). Furthermore, phase transformation of λ-MnO2 into Mn3O4 correlates with the increase of both Mn and oxygen loss at high charging potential.
 | ||
| Fig. 4
In situ oxygen detection via SECM and the comparison of Mn and oxygen dissolved from LMO. (a) Input potential waveform at the Au UME and (b) the current output at the Au UME. (c) The final output of the SECM experiment obtained by integrating the charge and then dividing by the reduction step time. The values are averaged over three sets of experiments, and the shaded area represents the standard deviation of the measurements. (d) SECM setup for oxygen detection with the Au UME and LMO cathode in 0.1 M TBAPF6 PC:EC electrolyte. (e) Average Mn2+ charge and ORR current at each LMO potential throughout the 1st charging cycle. | ||
To confirm that phase transformation of λ-MnO2 contributes to oxygen and Mn loss in Region 2, we quantified both species (i.e. oxygen and Mn) from Mn3O4 and λ-MnO2 using SECM. We should note, however, that these samples were prepared using a slurry method with vendor-bought active particles, which are distinct from the commercial LMO and the phases formed during the subsequent charging process. Furthermore, despite our efforts, significant heterogeneity in the response of these samples for both oxygen and Mn detection was observed, meaning that our results primarily indicate trends. Au UME oxygen measurements from Mn3O4 held at 4.2 V resulted in a negligible response, compared to λ-MnO2, which significantly evolved oxygen for the first 10 minutes (Fig. S13 and Table S4†). The decrease in UME current over time can be due to the limited amount of oxygen released from this phase at 4.2 V. Thus, we can confirm that the transformation of delithiated LMO (λ-MnO2) mainly contributes to the oxygen measured at the Au UME from Region 2. Mn dissolution measurements on λ-MnO2 and Mn3O4 showed distinctly opposite trends (Fig. S14 and Table S4†). Although both electrodes displayed inconsistencies in the amount of Mn dissolved under open circuit conditions, likely due to Mn2+ formed during slurry preparation, Mn3O4 showed increasing dissolution beyond 4.2 V. On the other hand, there was no measurable Mn dissolving from the λ-MnO2 electrode with applied potentials between 4.2 and 4.6 V. This set of SECM experiments provides insight into the LMO degradation taking place in Region 2 and the distinct contributions of relevant phases of LMO to oxygen and Mn loss at high state-of-charge.
4. Conclusions
In summary, we developed a SECM method to quantify Mn dissolution in situ from LMO as a model Mn-based LIB cathode in non-aqueous electrolyte. Our study demonstrates high temporal resolution and the excellent detection sensitivity (∼1 μM limit of detection) allowed by the SECM setup and Hg UME. We demonstrate SECM as a versatile tool for investigating various parameters to effectively mitigate the dissolution, as specifically shown with the use of an electrolyte additive such as TBP. Comparing Hg UME measurements with an additional SECM method for oxygen detection in nonaqueous media, we identified potential dependent oxygen loss from the cathode as an additional mechanism of sample degradation, alongside Mn dissolution. Our results revealed two distinct potential regimes for Mn dissolution from LMO, termed Region 1 and Region 2, which suggests two different mechanisms driving the cathode degradation: one driven by Mn3+ disproportionation catalyzed by HF generation and another driven by the phase transformation of delithiated LMO accompanied by oxygen evolution. This distinction was further supported by the mitigated Mn loss from Region 1 upon the addition of TBP, which suppresses the HF generation at the interface, as well as the direct observation of increasing oxygen loss in Region 2. Overall, our in situ method sheds light on elucidating the dissolution mechanism in an operating battery electrode and holds possibility in investigating degradation mechanisms in various Mn-based LIB cathodes including Li-, Mn-rich cathodes. Furthermore, this method can be extended to new and existing materials in which the versatility of Hg UMEs could offer answers to transition metal dissolution under various conditions and in different media.Data availability
Most data supporting the findings of this study are available from the main text of this article and its ESI.† All experimental data can be found in the ESI file ‘Raw Data.rar’.Author contributions
D. K., A. M. and J. R.-L. conceptualized the idea. D. K. planned, executed the experiments, contributed to the validation, and wrote the manuscript. A. M. contributed to data curation and formal analysis. M. Z. and Y. L. contributed to investigation and visualization. R. T. H. executed XPS measurements. J. R.-L. supervised the work, procured funding, and edited and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon the work supported by the National Science Foundation under CHE Award No. 2404245. Portions of this work were carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois. The authors acknowledge the use of facilities and instrumentation at theGeorge L. Clark X-ray Facility, University of Illinois. D. K. acknowledges Yunkyu Choi for his assistance with the electrode preparation.References
- M. Winter, B. Barnett and K. Xu, Before Li Ion Batteries, Chem. Rev., 2018, 118(23), 11433–11456, DOI:10.1021/acs.chemrev.8b00422.
- S. Lee and A. Manthiram, Can Cobalt be Eliminated from Lithium-Ion Batteries?, ACS Energy Lett., 2022, 7(9), 3058–3063, DOI:10.1021/acsenergylett.2c01553.
- J. Ma, T. Liu, J. Ma, C. Zhang and J. Yang, Progress, Challenge, and Prospect of LiMnO2: An Adventure toward High-Energy and Low-Cost Li-Ion Batteries, Adv. Sci., 2024, 11(2), 2304938, DOI:10.1002/advs.202304938.
- A. Manthiram, A Reflection on Lithium-Ion Battery Cathode Chemistry, Nat. Commun., 2020, 11(1), 1550, DOI:10.1038/s41467-020-15355-0.
- S. Liu, B. Wang, X. Zhang, S. Zhao, Z. Zhang and H. Yu, Reviving the Lithium-Manganese-Based Layered Oxide Cathodes for Lithium-Ion Batteries, Matter, 2021, 4(5), 1511–1527, DOI:10.1016/j.matt.2021.02.023.
- M. M. Thackeray and K. Amine, LiMn2O4 Spinel and Substituted Cathodes, Nat. Energy, 2021, 6(5), 566, DOI:10.1038/s41560-021-00815-8.
- P. K. Nayak, E. M. Erickson, F. Schipper, T. R. Penki, N. Munichandraiah, P. Adelhelm, H. Sclar, F. Amalraj, B. Markovsky and D. Aurbach, Review on Challenges and Recent Advances in the Electrochemical Performance of High Capacity Li- and Mn-Rich Cathode Materials for Li-Ion Batteries, Adv. Energy Mater., 2018, 8(8), 1702397, DOI:10.1002/aenm.201702397.
- H. Li, R. Fong, M. Woo, H. Ahmed, D.-H. Seo, R. Malik and J. Lee, Toward High-Energy Mn-Based Disordered-Rocksalt Li-Ion Cathodes, Joule, 2022, 6(1), 53–91, DOI:10.1016/j.joule.2021.11.005.
- Z. Lun, B. Ouyang, Z. Cai, R. J. Clément, D.-H. Kwon, J. Huang, J. K. Papp, M. Balasubramanian, Y. Tian, B. D. McCloskey, H. Ji, H. Kim, D. A. Kitchaev and G. Ceder, Design Principles for High-Capacity Mn-Based Cation-Disordered Rocksalt Cathodes, Chem, 2020, 6(1), 153–168, DOI:10.1016/j.chempr.2019.10.001.
- H. Y. Asl and A. Manthiram, Reining in Dissolved Transition-Metal Ions, Science, 2020, 369(6500), 140–141, DOI:10.1126/science.abc5454.
- T. Liu, A. Dai, J. Lu, Y. Yuan, Y. Xiao, L. Yu, M. Li, J. Gim, L. Ma, J. Liu, C. Zhan, L. Li, J. Zheng, Y. Ren, T. Wu, R. Shahbazian-Yassar, J. Wen, F. Pan and K. Amine, Correlation between Manganese Dissolution and Dynamic Phase Stability in Spinel-Based Lithium-Ion Battery, Nat. Commun., 2019, 10(1), 4721, DOI:10.1038/s41467-019-12626-3.
- A. Bhandari and J. Bhattacharya, Review—Manganese Dissolution from Spinel Cathode: Few Unanswered Questions, J. Electrochem. Soc., 2017, 164(2), A106–A127, DOI:10.1149/2.0101614jes.
- J. Wandt, A. Freiberg, R. Thomas, Y. Gorlin, A. Siebel, R. Jung, H. A. Gasteiger and M. Tromp, Transition Metal Dissolution and Deposition in Li-Ion Batteries Investigated by Operando X-Ray Absorption Spectroscopy, J. Mater. Chem. A, 2016, 4(47), 18300–18305, 10.1039/c6ta08865a.
- C. Zhan, T. Wu, J. Lu and K. Amine, Dissolution, Migration, and Deposition of Transition Metal Ions in Li-Ion Batteries Exemplified by Mn-Based Cathodes – a Critical Review, Energy Environ. Sci., 2018, 11(2), 243–257, 10.1039/c7ee03122j.
- D. R. Vissers, Z. Chen, Y. Shao, M. Engelhard, U. Das, P. Redfern, L. A. Curtiss, B. Pan, J. Liu and K. Amine, Role of Manganese Deposition on Graphite in the Capacity Fading of Lithium Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8(22), 14244–14251, DOI:10.1021/acsami.6b02061.
- Z. Zhang, S. Said, A. J. Lovett, R. Jervis, P. R. Shearing, D. J. L. Brett and T. S. Miller, The Influence of Cathode Degradation Products on the Anode Interface in Lithium-Ion Batteries, ACS Nano, 2024, 18(13), 9389–9402, DOI:10.1021/acsnano.3c10208.
- D. Huang, C. Engtrakul, S. Nanayakkara, D. W. Mulder, S.-D. Han, M. Zhou, H. Luo and R. C. Tenent, Understanding Degradation at the Lithium-Ion Battery Cathode/Electrolyte Interface: Connecting Transition-Metal Dissolution Mechanisms to Electrolyte Composition, ACS Appl. Mater. Interfaces, 2021, 13(10), 11930–11939, DOI:10.1021/acsami.0c22235.
- J. C. Hestenes, J. T. Sadowski, R. May and L. E. Marbella, Transition Metal Dissolution Mechanisms and Impacts on Electronic Conductivity in Composite LiNi0.5Mn1.5O4 Cathode Films, ACS Mater. Au, 2023, 3(2), 88–101, DOI:10.1021/acsmaterialsau.2c00060.
- J. P. Allen, C. A. O'Keefe and C. P. Grey, Quantifying Dissolved Transition Metals in Battery Electrolyte Solutions with NMR Paramagnetic Relaxation Enhancement, J. Phys. Chem. C, 2023, 127(20), 9509–9521, DOI:10.1021/acs.jpcc.3c01396.
- L. Zhao, Ö. Ö. Çapraz and N. R. Sottos, Probing In Operando Manganese Dissolution and Associated Mechanical Deformation in LiMn2O4 Cathodes, ACS Appl. Energy Mater., 2024, 7(6), 2142–2152, DOI:10.1021/acsaem.3c02820.
- S. J. Wachs, C. Behling, J. Ranninger, J. Möller, K. J. J. Mayrhofer and B. B. Berkes, Online Monitoring of Transition-Metal Dissolution from a High-Ni-Content Cathode Material, ACS Appl. Mater. Interfaces, 2021, 13(28), 33075–33082, DOI:10.1021/acsami.1c07932.
- D. H. Jang, Y. J. Shin and S. M. Oh, Dissolution of Spinel Oxides and Capacity Losses in 4 V Li/LixMn2O4 Cells, J. Electrochem. Soc., 1996, 143(7), 2204–2211, DOI:10.1149/1.1836981.
- L.-F. Wang, C.-C. Ou, K. A. Striebel and J.-S. Chen, Study of Mn Dissolution from LiMn2O4 Spinel Electrodes Using Rotating Ring-Disk Collection Experiments, J. Electrochem. Soc., 2003, 150(7), A905–A911 CrossRef CAS.
- P. P. Lopes, M. Zorko, K. L. Hawthorne, J. G. Connell, B. J. Ingram, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, Real-Time Monitoring of Cation Dissolution/Deintercalation Kinetics from Transition-Metal Oxides in Organic Environments, J. Phys. Chem. Lett., 2018, 9(17), 4935–4940, DOI:10.1021/acs.jpclett.8b01936.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Surface Reconstruction and Chemical Evolution of Stoichiometric Layered Cathode Materials for Lithium-Ion Batteries, Nat. Commun., 2014, 5(1), 3529, DOI:10.1038/ncomms4529.
- H. Chen and M. S. Islam, Lithium Extraction Mechanism in Li-Rich Li 2 MnO 3 Involving Oxygen Hole Formation and Dimerization, Chem. Mater., 2016, 28(18), 6656–6663, DOI:10.1021/acs.chemmater.6b02870.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, The Structural and Chemical Origin of the Oxygen Redox Activity in Layered and Cation-Disordered Li-Excess Cathode Materials, Nat. Chem., 2016, 8(7), 692–697, DOI:10.1038/nchem.2524.
- X. Gao, Y. H. Ikuhara, C. A. J. Fisher, R. Huang, A. Kuwabara, H. Moriwake, K. Kohama and Y. Ikuhara, Oxygen Loss and Surface Degradation during Electrochemical Cycling of Lithium-Ion Battery Cathode Material LiMn2O4, J. Mater. Chem. A, 2019, 7(15), 8845–8854, 10.1039/C8TA08083F.
- S. Jung, H. Gwon, J. Hong, K. Park, D. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Understanding the Degradation Mechanisms of LiNi0.5Co0.2Mn0.3O2 Cathode Material in Lithium Ion Batteries, Adv. Energy Mater., 2014, 4(1), 1300787, DOI:10.1002/aenm.201300787.
- T. Nakamura, H. Gao, K. Ohta, Y. Kimura, Y. Tamenori, K. Nitta, T. Ina, M. Oishi and K. Amezawa, Defect Chemical Studies on Oxygen Release from the Li-Rich Cathode Material Li1.2Mn0.6Ni0.2O2−δ, J. Mater. Chem. A, 2019, 7(9), 5009–5019, 10.1039/c8ta12484a.
- S. Hwang, W. Chang, S. M. Kim, D. Su, D. H. Kim, J. Y. Lee, K. Y. Chung and E. A. Stach, Investigation of Changes in the Surface Structure of LixNi0.8Co0.15Al0.05O2 Cathode Materials Induced by the Initial Charge, Chem. Mater., 2014, 26(2), 1084–1092, DOI:10.1021/cm403332s.
- W. Choi and A. Manthiram, Comparison of Metal Ion Dissolutions from Lithium Ion Battery Cathodes, J. Electrochem. Soc., 2006, 153(9), A1760, DOI:10.1149/1.2219710.
- R. J. Gummow, A. De Kock and M. M. Thackeray, Improved Capacity Retention in Rechargeable 4 V Lithium/Lithium-Manganese Oxide (Spinel) Cells, Solid State Ionics, 1994, 69(1), 59–67, DOI:10.1016/0167-2738(94)90450-2.
- R. Benedek and M. M. Thackeray, Reaction Energy for LiMn2O4 Spinel Dissolution in Acid, Solid-State Lett., 2006, 9, A265 CrossRef CAS.
- S. Li, Y. Xue, X. Cui, S. Geng and Y. Huang, Effect of Sulfolane and Lithium Bis(Oxalato)Borate-Based Electrolytes on the Performance of Spinel LiMn2O4 Cathodes at 55 °C, Ionics, 2016, 22(6), 797–801, DOI:10.1007/s11581-015-1611-z.
- J. Park, J. H. Seo, G. Plett and W. Lu, Sastry Numerical Simulation of the Effect of the Dissolution of LiMn2O4 Particles on Li-Ion Battery Performance, Electrochem. Solid-State Lett., 2011, 14, A14 CrossRef CAS.
- X. Sun, R. Xiao, X. Yu and H. Li, First-Principles Simulations for the Surface Evolution and Mn Dissolution in the Fully Delithiated Spinel LiMn2O4, Langmuir, 2021, 37(17), 5252–5259, DOI:10.1021/acs.langmuir.1c00197.
- Y. Zhang, A. Hu, D. Xia, S. Hwang, S. Sainio, D. Nordlund, F. M. Michel, R. B. Moore, L. Li and F. Lin, Operando Characterization and Regulation of Metal Dissolution and Redeposition Dynamics near Battery Electrode Surface, Nat. Nanotechnol., 2023, 18(7), 790–797, DOI:10.1038/s41565-023-01367-6.
- D. Tang, Y. Sun, Z. Yang, L. Ben, L. Gu and X. Huang, Surface Structure Evolution of LiMn2O4 Cathode Material upon Charge/Discharge, Chem. Mater., 2014, 26(11), 3535–3543, DOI:10.1021/cm501125e.
- D. Tang, L. Ben, Y. Sun, B. Chen, Z. Yang, L. Gu and X. Huang, Electrochemical Behavior and Surface Structural Change of LiMn2O4 Charged to 5.1 V, J. Mater. Chem. A, 2014, 2(35), 14519–14527, 10.1039/c4ta02109f.
- S. Liu, D. Liu, S. Wang, X. Cai, K. Qian, F. Kang and B. Li, Understanding the Cathode Electrolyte Interface Formation in Aqueous Electrolyte by Scanning Electrochemical Microscopy, J. Mater. Chem. A, 2019, 7(21), 12993–12996, 10.1039/c9ta03199e.
- J. Hui, M. Burgess, J. Zhang and J. Rodríguez-López, Layer Number Dependence of Li + Intercalation on Few-Layer Graphene and Electrochemical Imaging of Its Solid-Electrolyte Interphase Evolution, ACS Nano, 2016, 10(4), 4248–4257, DOI:10.1021/acsnano.5b07692.
- B. Krueger, L. Balboa, J. F. Dohmann, M. Winter, P. Bieker and G. Wittstock, Solid Electrolyte Interphase Evolution on Lithium Metal Electrodes Followed by Scanning Electrochemical Microscopy Under Realistic Battery Cycling Current Densities, ChemElectroChem, 2020, 7(17), 3590–3596, DOI:10.1002/celc.202000441.
- A. Mishra, M. Zorigt, D. O. Kim and J. Rodríguez-López, Voltammetric Detection of Singlet Oxygen Enabled by Nanogap Scanning Electrochemical Microscopy, J. Am. Chem. Soc., 2024, 146(13), 8847–8851, DOI:10.1021/jacs.4c00414.
- D. Sarbapalli, Y.-H. Lin, S. Stafford, J. Son, A. Mishra, J. Hui, A. Nijamudheen, A. I. B. Romo, Z. T. Gossage, A. M. Van Der Zande, J. L. Mendoza-Cortes and J. Rodríguez-López, A Surface Modification Strategy Towards Reversible Na-Ion Intercalation on Graphitic Carbon Using Fluorinated Few-Layer Graphene, J. Electrochem. Soc., 2022, 169(10), 106522, DOI:10.1149/1945-7111/ac9c33.
- Z. T. Gossage, J. Hui, Y. Zeng, H. Flores-Zuleta and J. Rodríguez-López, Probing the Reversibility and Kinetics of Li + during SEI Formation and (de)Intercalation on Edge Plane Graphite Using Ion-Sensitive Scanning Electrochemical Microscopy, Chem. Sci., 2019, 10(46), 10749–10754, 10.1039/c9sc03569a.
- Z. J. Barton and J. Rodríguez-López, Lithium Ion Quantification Using Mercury Amalgams as in situ Electrochemical Probes in Nonaqueous Media, Anal. Chem., 2014, 86(21), 10660–10667, DOI:10.1021/ac502517b.
- Z. J. Barton and J. Rodríguez-López, Fabrication and Demonstration of Mercury Disc-Well Probes for Stripping-Based Cyclic Voltammetry Scanning Electrochemical Microscopy, Anal. Chem., 2017, 89(5), 2716–2723, DOI:10.1021/acs.analchem.6b04022.
- M. Hatami, D. Polcari, M. S. Hossain, M. Z. Ghavidel, J. Mauzeroll and S. B. Schougaard, Square Wave Anodic Stripping Voltammetry for Localized Detection of Mn 2+ in Li-Ion Battery Environments, J. Electrochem. Soc., 2022, 169(4), 040526, DOI:10.1149/1945-7111/ac63f9.
- J. Guo and E. Lindner, Cyclic Voltammetry at Shallow Recessed Microdisc Electrode: Theoretical and Experimental Study, J. Electroanal. Chem., 2009, 629(1–2), 180–184, DOI:10.1016/j.jelechem.2009.01.030.
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, Nickel, Manganese, and Cobalt Dissolution from Ni-Rich NMC and Their Effects on NMC622-Graphite Cells, J. Electrochem. Soc., 2019, 166(2), A378–A389, DOI:10.1149/2.1151902jes.
- G. Zhou, X. Sun, Q.-H. Li, X. Wang, J.-N. Zhang, W. Yang, X. Yu, R. Xiao and H. Li, Mn Ion Dissolution Mechanism for Lithium-Ion Battery with LiMn2O4 Cathode: In Situ Ultraviolet-Visible Spectroscopy and Ab Initio Molecular Dynamics Simulations, J. Phys. Chem. Lett., 2020, 11(8), 3051–3057, DOI:10.1021/acs.jpclett.0c00936.
- M. A. Alpuche-Aviles, J. E. Baur and D. O. Wipf, Imaging of Metal Ion Dissolution and Electrodeposition by Anodic Stripping Voltammetry-Scanning Electrochemical Microscopy, Anal. Chem., 2008, 80(10), 3612–3621, DOI:10.1021/ac702568c.
- Y.-M. Song, J.-G. Han, S. Park, K. T. Lee and N.-S. Choi, A Multifunctional Phosphite-Containing Electrolyte for 5 V-Class LiNi0.5Mn1.5O4 Cathodes with Superior Electrochemical Performance, J. Mater. Chem. A, 2014, 2(25), 9506–9513, 10.1039/c4ta01129e.
- Y. Zhu, Y. Li, M. Bettge and D. P. Abraham, Positive Electrode Passivation by LiDFOB Electrolyte Additive in High-Capacity Lithium-Ion Cells, J. Electrochem. Soc., 2012, 159(12), A2109–A2117, DOI:10.1149/2.083212jes.
- Y. Dong, B. T. Young, Y. Zhang, T. Yoon, D. R. Heskett, Y. Hu and B. L. Lucht, Effect of Lithium Borate Additives on Cathode Film Formation in LiNi0.5Mn1.5O4/Li Cells, ACS Appl. Mater. Interfaces, 2017, 9(24), 20467–20475, DOI:10.1021/acsami.7b01481.
- K. Amine, J. Liu, S. Kang, I. Belharouak, Y. Hyung, D. Vissers and G. Henriksen, Improved Lithium Manganese Oxide Spinel/Graphite Li-Ion Cells for High-Power Applications, J. Power Sources, 2004, 129(1), 14–19, DOI:10.1016/j.jpowsour.2003.11.007.
- S. S. Zhang, A Review on Electrolyte Additives for Lithium-Ion Batteries, J. Power Sources, 2006, 162(2), 1379–1394, DOI:10.1016/j.jpowsour.2006.07.074.
- B. G. Nicolau, A. Petronico, K. Letchworth-Weaver, Y. Ghadar, R. T. Haasch, J. A. N. T. Soares, R. T. Rooney, M. K. Y. Chan, A. A. Gewirth and R. G. Nuzzo, Controlling Interfacial Properties of Lithium-Ion Battery Cathodes with Alkylphosphonate Self-Assembled Monolayers, Adv. Mater. Interfaces, 2018, 5(10), 1701292, DOI:10.1002/admi.201701292.
- C. Li, S. Wu, Y. Qiu and D. Lu, Phosphorus-Containing C12H27O4 P as Functional Electrolyte Additives for High-Voltage LiNi0.5Mn1.5O4/Graphite Li-Ion Batteries with Excellent Electrochemical Performance, Adv. Mater. Interfaces, 2021, 8(3), 2001588, DOI:10.1002/admi.202001588.
- B. Philippe, R. Dedryvère, J. Allouche, F. Lindgren, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Nanosilicon Electrodes for Lithium-Ion Batteries: Interfacial Mechanisms Studied by Hard and Soft X-Ray Photoelectron Spectroscopy, Chem. Mater., 2012, 24(6), 1107–1115, DOI:10.1021/cm2034195.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Improved Performances of Nanosilicon Electrodes Using the Salt LiFSI: A Photoelectron Spectroscopy Study, J. Am. Chem. Soc., 2013, 135(26), 9829–9842, DOI:10.1021/ja403082s.
- A. Mishra, D. Sarbapalli, Md. S. Hossain, Z. T. Gossage, Z. Li, A. Urban and J. Rodríguez-López, Highly Sensitive Detection and Mapping of Incipient and Steady-State Oxygen Evolution from Operating Li-Ion Battery Cathodes via Scanning Electrochemical Microscopy, J. Electrochem. Soc., 2022, 169(8), 086501, DOI:10.1149/1945-7111/ac857e.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08967g |
| This journal is © The Royal Society of Chemistry 2025 |