
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the hydrogen bond network effect at the electrode–electrolyte interface during the hydrogen evolution reaction†
Chengcheng
Zhong
 a,
Yuxuan
Xiao
*a,
Jinxian
Feng
a,
Chunfa
Liu
a,
Lun
Li
a,
Weng Fai
Ip
b,
Shuangpeng
Wang
*a and
Hui
Pan
*ab
a,
Yuxuan
Xiao
*a,
Jinxian
Feng
a,
Chunfa
Liu
a,
Lun
Li
a,
Weng Fai
Ip
b,
Shuangpeng
Wang
*a and
Hui
Pan
*ab
aInstitute of Applied Physics and Materials Engineering, University of Macau, Macao SAR, China
bDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Macao SAR, China
First published on 27th February 2025
Abstract
The hydrogen bond network (HBN) in the electrical double layer (EDL) at the electrode–electrolyte interface plays a crucial role in governing water migration, which directly affects the efficiency of the hydrogen evolution reaction. However, existing research has primarily focused on the connectivity of the HBN within the inner Helmholtz layer, often neglecting the water transport across the entire EDL. In this study, we develop a high-performance Aermet100 steel-derived catalyst, which achieves an overpotential of 307 mV for the hydrogen evolution reaction at a current density of 500 mA cm−2 under industrial conditions. Using this catalyst, we investigate the migration of water in KOH solutions with varying concentrations. Our findings show that water migration is inhibited in 1 M KOH due to the relatively stronger HBN, whereas it is enhanced in 3 M and 6 M KOH solutions. These results provide new insights into the kinetics of water transport and offer a potential pathway for optimizing industrial water electrolysis processes.
Green hydrogen is increasingly poised to replace traditional fossil fuels due to its cleanness and renewability.1–3 Currently, water electrolysis has been recognized as an efficient technology to produce green hydrogen with high purity. Therefore, extensive research has been focused on enhancing the efficiency of the catalysts for water electrolysis.4 Materials based on noble metals such as platinum (Pt) and ruthenium (Ru) are acknowledged as the best electrocatalysts for the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER), respectively, in water electrolysis because of their exceptional capacity to adsorb and recombine the reaction intermediates.5–7 However, their low storage, high cost and inadequate durability have restricted practical application. Instead, developing non-precious transition metal-based materials as catalysts for industrially applicable water electrolysis is of great interest, because the transition metal-based materials have shown promising performance due to their high affinity to water molecules.8–20
In parallel with the design of high-performance transition metal-based catalysts, understanding the mechanism of water electrolysis, particularly in the region of the electrical double layer (EDL) is significant. The EDL consists of the inner Helmholtz plane (IHP), the outer Helmholtz plane (OHP), and the diffusion layer.18 The connectivity of the hydrogen bond network (HBN) in the inner Helmholtz layer (IHP), as described in the HBN theory, has attracted significant attention as an important index for evaluating catalytic efficiency.21–26 The HBN connectivity can regulate the kinetics of interfacial proton-coupled electron transfer reactions, thereby affecting the performance of water electrolysis.19,23 However, little attention has been paid to the effect of the HBN on the transport of water molecules throughout the entire EDL. The timely transport of water molecules to the electrode surface as reactants, facilitated by the breaking of the HBN, is essential for efficient hydrogen evolution.
Herein, the migration of water molecules influenced by the HBN is thoroughly investigated. Industrial-grade Aermet100 (A100) steel was electrochemically treated in situ to form metal hydroxides on its surface in KOH solution. The A100 treated in 6 M KOH (referred to as A100-6) exhibited the best water electrolysis performance under both laboratory and industrial conditions. In situ Raman and Fourier-transform infrared (FTIR) spectroscopies were employed to explore the distinct effects of the HBN on the mobility of water molecules at the electrolyte–electrode interface across varying concentrations of KOH electrolytes.
Water electrolysis performances of treated and untreated A100 were first evaluated. Fig. 1a and b show the cathodic HER performances of the treated and untreated A100 under laboratory (1M KOH, ambient temperature) and industrial conditions (6 M KOH, 60 °C), respectively. Obviously, A100-6 exhibits larger current density than other samples at the same potential under both laboratory and industrial conditions. The overpotentials of A100-6 to drive a current density of 500 mA cm−2 are 830 mV and 305 mV under laboratory and industrial conditions, respectively, which are much lower than those of A100 (910 mV and 395 mV) and A100 treated in 1 M (denoted as A100-1, 1000 mV and 350 mV), 3 M (denoted as A100-3, 950 mV and 330 mV) and 8 M KOH (denoted as A100-8, 860 mV and 370 mV). At the same time, both HER and OER performance of A100-6 is higher than that of commercial nickel net (Fig. S13†), indicating a high potential of industrial water splitting application for this cost-effective material derived from industrial steel. Moreover, A100-6 also exhibits a smaller Tafel slope (64 mV dec−1) than A100 (96 mV dec−1), indicating the improvement of reaction kinetics after treating A100 in 6 M KOH (Fig. 1c). Tafel slope values of 120, 40, and 30 mV dec−1 correspond to the Volmer, Heyrovsky, and Tafel reactions of catalysts, respectively.27 Based on the observed Tafel slopes, it can be indicated that the HER process for A100 is primarily controlled by the Volmer step, indicating that water dissociation and electron transfer are the rate-determining steps. In contrast, for A100-6, the HER process appears to be under mixed control involving both the Volmer and Heyrovsky steps, suggesting a balance between water dissociation and hydrogen adsorption–desorption kinetics. Electrochemical impedance spectroscopy (EIS) results further reveal that A100-6 has the smallest charge transfer resistance (Fig. 1g), which explains its best HER performance, and the corresponding equivalent circuit is also shown. The Rs, Rct and CPE represent solution resistance, charge transfer resistance and constant phase element, respectively.28 In addition, the anodic OER performances of the catalysts were also studied. Similar to the HER results, A100-6 exhibits the largest current density among all the samples under both laboratory and industrial conditions, as well as a smaller Tafel slope than A100. The stability of water electrolysis for A100-6 under industrial conditions was further evaluated using a flow cell. As shown in Fig. 1h, after 500 hours of cycling tests at a current density of 400 mA cm−2, the cell potential was decreased from −2.3 V to −2.06 V, demonstrating good stability. The schematic illustration of the in situ treatment of industrial A100 steel and the corresponding cyclic voltammetry (CV) curves are shown in Fig. S1 (ESI).† Scanning electron microscopy (SEM) was utilized to characterize the surface morphology of A100-6. The SEM image in Fig. S2a (ESI)† shows that numerous nanoparticles appear on the surface of A100 following the treatment, which is very different from the smooth surface of A100 (Fig. S3a, ESI†). The corresponding X-ray energy dispersive spectroscopy (EDS) results show that the Fe is dominant in both A100 and A100-6, with small portions of Co, Ni, Cr and Mo (Fig. S2b and S3b, ESI†). The metal content dissolved in electrolyte after the treatment is characterized by inductively coupled plasma mass spectrometry (ICP-MS). The results show an increased dissolution amount for Ni, Co and Fe with the increased KOH treatment concentration (Fig. S4, ESI†). However, decreased dissolution ratios of Cr and Mo can be found following the increased KOH concentration, which is due to their relatively stronger anti-corrosion properties than those of Ni, Co and Fe. Moreover, the X-ray diffraction (XRD) patterns of treated and untreated A100 indicate that α-Fe, characterized by a less compact lattice structure and higher surface activity, dissolves during electrochemical treatment, while γ-Fe, known for its stability,29 is retained in greater amounts (Fig. S5, ESI†).
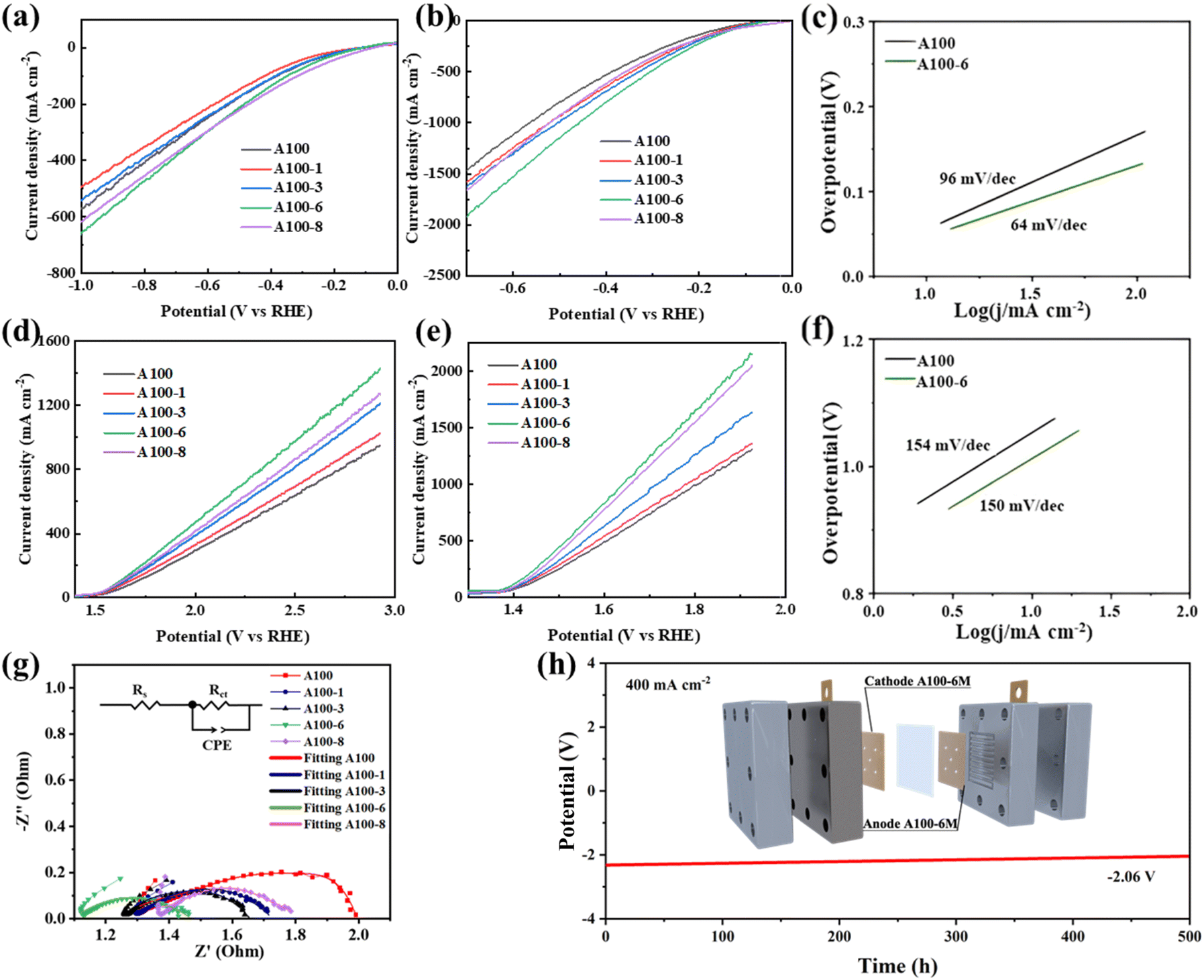 | ||
| Fig. 1 The electrochemical tests of catalysts. The HER polarization curves of treated and untreated A100 under (a) laboratory conditions (1 M KOH and ambient temperature) and (b) industrial conditions (6 M KOH and 60 °C). (c) Tafel slopes of A100 and A100-6 under industrial conditions. The OER polarization curves of treated and untreated A100 under (d) laboratory conditions and (e) industrial conditions. (f) Tafel slopes of A100 and A100-6 under industrial conditions. (g) Nyquist plots and equivalent circuit of treated and untreated A100 under laboratory conditions. (h) Chrono voltage measurement at a current density of 400 mA cm−2 of A100-6 by using a flow cell under industrial conditions. | ||
The surface chemical states of the samples were further analysed by X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of A100, A100-6 and A100-6 after the durability test are shown in Fig. S6 (ESI).† The high-resolution core level XPS spectra show similar chemical states of Fe, Co, Ni and Cr elements for A100-6 and A100 (Fig. S7a–d and S8a–d, ESI†). However, a previously undetectable Mo signal in A100 emerges in the XPS spectra of A100-6 (Fig. S7e and S8e, ESI†). This phenomenon can be attributed to surface dissolution during KOH treatment, exposing the inner Mo content of A100. Moreover, A100-6 exhibits a much higher ratio of M–OH/M–O species (Fig. S7f, ESI†) than that of A100 (Fig. S8f, ESI†), illustrating an enhanced metal hydroxide amount formed on the surface on A100 after treatment. After the durability test, the oxidation states of the metal elements increase (Fig. S9a–e, ESI†), while the M–OH/M–O ratio decreases (Fig. S9f, ESI†), indicating the consumption of surface metal hydroxide during the electrocatalytic process.
The ultraviolet photo-electron spectroscopy (UPS) analysis was conducted to investigate the work functions (WFs) of the samples (Fig. S10a–e, ESI†). The results indicate that the WFs of A100, A100-1, A100-3, A100-6 and A100-8 are 3.86, 3.07, 2.60, 2.23 and 2.26 eV, respectively (Fig. S10f, ESI†). A lower WF value indicates a reduced energy barrier for electron transfer at the material surface.30 This could explain that A100-6 with the lowest WF value exhibits the highest HER performance compared to the other samples.
To systematically investigate the electrode–electrolyte interactions during water electrolysis and elucidate the underlying mechanism, in situ Raman spectroscopy was conducted on A100 and A100-6 in KOH solutions with varying concentrations (1 M, 3 M and 6 M). The in situ Raman spectra of A100 and A100-6 for the HER process are shown in Fig. 2. For A100-6, the bands at ∼473 and ∼680 cm−1 are attributed to the Fe–OH stretching of Fe(OH)2 and Fe(OH)3, which are in accordance with the previously reported results.31 Moreover, an obvious peak appears in the range between 3000 and 3700 cm−1 for both A100-6 and A100, which corresponds to the behaviour of the electrolyte.32 As can be clearly seen, a prominent peak emerges at approximately 3600 cm−1 with the increase in KOH concentration. The peak fitting results (Fig. S11, ESI†) indicate that three peaks at around 3250, 3450 and 3600 cm−1 correspond to the O–H stretching vibration in water molecules, O–H stretching vibration in water molecules with strong hydrogen bonding, and O–H stretching vibration in OH−, respectively.32 The distinct spectral features of these vibration bands render them effective probes for investigating the kinetic behaviour of water molecules.
 | ||
| Fig. 2 In situ Raman spectra (HER process) of A100-6 in (a) 1 M KOH, (b) 3 M KOH and (c) 6 M KOH. In situ Raman spectra (HER process) of A100 in (d) 1 M KOH, (e) 3 M KOH and (f) 6 M KOH solutions at different overpotentials. | ||
In 1 M KOH under the HER process, it is noteworthy that the intensities of water peaks for A100-6 exhibit a decreasing trend (Fig. 2a), while A100 exhibits a seldom changed one (Fig. 2d). In 3 M and 6 M KOH, increasing trends can be found for both A100-6 and A100. Furthermore, for A100-6, the peaks of the O–H stretching vibrations are shifted to lower wave numbers with increasing voltage in both 1 M KOH and 6 M KOH (Fig. S11a and b, ESI†). However, for A100, the position of the O–H stretching peak is essentially unchanged in 1 M KOH and shows a tendency to shift to lower wave numbers in 6 M KOH (Fig. S11c and d, ESI†). Such different trends in the peak intensity and peak position for A100-6 and A100 could be due to the different HBN effects and different interactions between electrode and water, respectively.
To fully understand the HBN effect and the electrode–electrolyte interaction for A100-6 and A100 in different KOH electrolytes, we present a schematic mechanism shown in Fig. 3. As is well established, water molecules usually form a network due to hydrogen bonds (Fig. 3a). However, with the addition of KOH, this network can be disrupted owing to the solvation effects of K+ and OH−. K+ typically binds with 6–8 water molecules through ion–dipole interactions, while OH− usually binds with 3–4 water molecules trough strong hydrogen bonds.27 This suggests that the migration of water molecules is mainly dependent on the movement of OH−. During the water electrolysis process, the electronegative OH− ions migrate towards the anode, while the electropositive K+ ions move towards the cathode. Thus, at a low concentration of KOH (such as 1 M), the HBN will be slightly disrupted (Fig. 3b), but water molecules still face a significant energy barrier from the HBN to reach the electrode surface. Additionally, OH− ions at the electrode surface exert electrostatic repulsion, which carries away some water molecules. This explains the decreased water peak intensity observed in the in situ Raman spectra for A100-6 during the hydrogen evolution reaction (HER) process in 1 M KOH. At high KOH concentrations, the increased OH− ion concentration disrupts the HBN more significantly (Fig. 3c), which is reflected in the increased intensity of the peak at 3600 cm−1 in the Raman spectra for both A100-6 and A100 in 6 M KOH. This higher disruption degree of the HBN facilitates the movement of water molecules, which overpowers the effect from electrostatic repulsion of OH− at the cathode. This explains the continuously increased water peak intensity observed in the in situ Raman spectra for A100-6 during the HER process in 3 M and 6 M KOH.
 | ||
| Fig. 3 Schematic illustration of water migration affected by the HBN on the surface of A100-6. Schematic illustration of the HBN in (a) pure water, (b) 1 M KOH and (c) 6 M KOH. | ||
On the other hand, during the OER process, the anode surface becomes positively charged due to the applied electric field, causing OH− ions to migrate toward the anode under electrostatic force. This movement is opposite to that observed in the HER process. Additionally, owing to the strong hydrogen bonding between OH− and water molecules, some water molecules also migrate toward the anode.33 To further investigate the effect of the HBN on water molecule migration, in situ Raman spectroscopy during the OER process was performed. As shown in Fig. S12 (ESI†), an increase in the Raman peak intensities of water molecules is observed for both A100-6 and A100 during the OER process, regardless of whether the KOH concentration is 1 M, 3 M, or 6 M.
In situ Fourier transform infrared spectroscopy (FTIR) was conducted to further prove the HBN effect. As shown in Fig. 4, four apparent peaks can be found at 3565, 3647, 3743 and 3850 cm−1, which correspond to the O–H stretching of H2O, O–H stretching of OH−, O–H stretching with weak hydrogen bonding of H2O and OH−, and O–H stretching with strong hydrogen bonding of H2O and OH−, respectively.33–37 For A100-6, the intensity of the transmission peaks in 1 M KOH solution shows an overall decreasing trend with increasing voltage (Fig. 4a), while it increases in 3 M and 6 M KOH solutions (Fig. 4b and c). This suggests that the number of water molecules decreases with increasing voltage during the HER in 1 M KOH solution but increases in 3 M and 6 M KOH solutions, which is consistent with the results obtained from the in situ Raman test. For A100, the intensity of the transmission peaks in 1 M, 3 M, and 6 M KOH solutions consistently increases with increasing voltage (Fig. 4d–f). These trends agree with the observations from the in situ Raman tests. Moreover, in the HER process, the intensity of the spectroscopic signals obtained by both in situ Raman and in situ FTIR represents the number of active molecules. The HER processes of A100 and A100-6 in 1 M KOH solution show opposite trends, which suggests that they do not have the same interaction with water molecules, and more specifically this can be attributed to the effect of the electrode surfaces of the two samples on the hydrogen bonding network of water molecules in the solution. In conclusion, the correlation between the in situ Raman and in situ FTIR spectra clearly demonstrates the effect of the HBN on OH−-induced migration of water molecules.
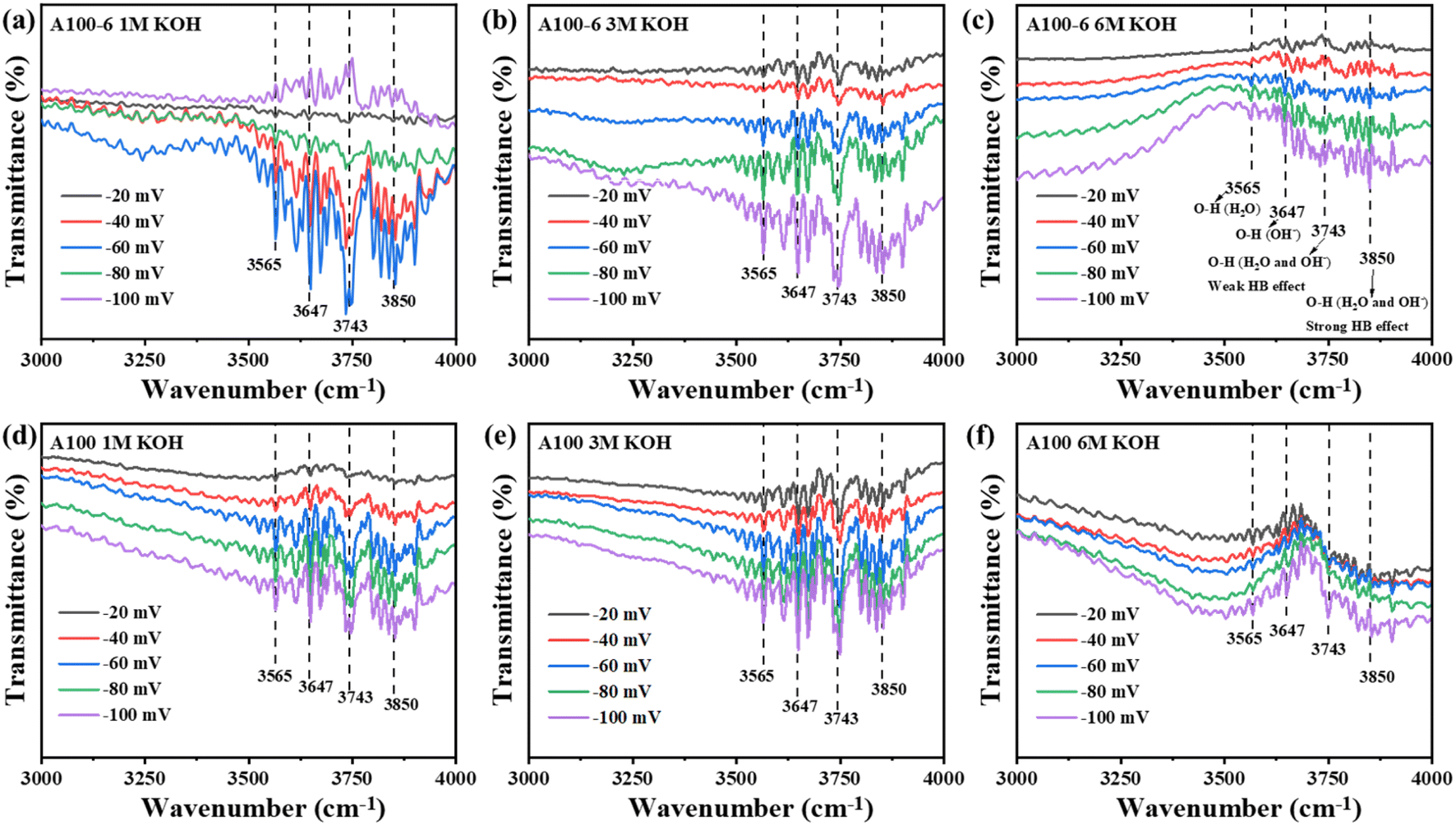 | ||
| Fig. 4 In situ FTIR spectra (HER process) of A100-6 tested in (a) 1 M KOH, (b) 3 M KOH and (c) 6 M KOH, respectively. In situ FTIR spectra of A100 tested in (d) 1 M KOH, (e) 3 M KOH and (f) 6 M KOH at different overpotentials, respectively. | ||
In summary, the effects of the HBN on the migration of water molecules were systematically investigated on the A100 catalyst surface in various KOH electrolytes. A suppressed migration of water molecules as reactants was observed in 1 M KOH solution due to the relatively strong HBN, while it was promoted in 3 M and 6 M KOH solutions. Such phenomena reveal the different HBN effects triggered by the influence of OH− with different concentrations of KOH. This study provides valuable insights into water transport kinetics and offers a pathway for optimizing industrial water electrolysis.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Chengcheng Zhong (conceptualization: lead; formal analysis: lead; investigation: lead; methodology: lead; writing – original draft: lead); Yuxuan Xiao (project administration: equal; writing – review & editing: equal); Jinxian Feng (software: equal; visualization: equal); Chunfa Liu (software: equal; visualization: equal); Lun Li (software: equal; visualization: equal); Weng Fai Ip (funding acquisition: supporting); Shuangpeng Wang (funding acquisition: supporting; resources: supporting); Hui Pan (funding acquisition: lead; project administration: equal; supervision: supporting; writing – review & editing: equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Development Fund (FDCT) from Macau SAR (0050/2023/RIB2, 0023/2023/AFJ, 006/2022/ALC, 0087/2024/AFJ and 0111/2022/A2) and Multi-Year Research Grants (MYRG-GRG2024-00038-IAPME and MYRG-GRG2023-00010-IAPME) from the University of Macau.References
- P. Zhou, P. Niu, J. Liu, N. Zhang, H. Bai, M. Chen, J. Feng, D. Liu, L. Wang and S. Chen, Adv. Funct. Mater., 2022, 32, 2202068 CrossRef CAS.
- N. Farrell, Renewable Sustainable Energy Rev., 2023, 178, 113216 CrossRef CAS.
- F. Osselin, C. Soulaine, C. Fauguerolles, E. C. Gaucher, B. Scaillet and M. Pichavant, Nat. Geosci., 2022, 15, 765–769 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- J.-H. Li, H.-Y. Zhang, Q.-W. Shi, J. Ying and C. Janiak, Prog. Mater. Sci., 2024, 146, 101335 CrossRef CAS.
- J. Ying, M. D. Symes and X.-Y. Yang, Matter, 2023, 6, 674–676 CrossRef CAS.
- J. Ying, Y. Xiao, J. Chen, Z.-Y. Hu, G. Tian, G. V. Tendeloo, Y. Zhang, M. D. Symes, C. Janiak and X.-Y. Yang, Nano Lett., 2023, 23, 7371–7378 CrossRef CAS PubMed.
- Q. Lu, A. L. Wang, Y. Gong, W. Hao, H. Cheng, J. Chen, B. Li, N. Yang, W. Niu, J. Wang, Y. Yu, X. Zhang, Y. Chen, Z. Fan, X. J. Wu, J. Chen, J. Luo, S. Li, L. Gu and H. Zhang, Nat. Chem., 2018, 10, 456–461 CrossRef CAS PubMed.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- T. Chen, C. Qiu, X. Zhang, H. Wang, J. Song, K. Zhang, T. Yang, Y. Zuo, Y. Yang, C. Gao, W. Xiao, Z. Jiang, Y. Wang, Y. Xiang and D. Xia, J. Am. Chem. Soc., 2024, 146, 1174–1184 CrossRef CAS PubMed.
- C. Liu, J. Feng, P. Zhou, D. Liu, L. Qiao, D. Liu, Y. Cao, S.-C. Su, H. Liu and H. Pan, Chem. Eng. J., 2023, 476, 146710 CrossRef CAS.
- Z.-H. Dong, Z. Jiang, T. Tang, Z.-C. Yao, D. Xue, S. Niu, J. Zhang and J.-S. Hu, J. Mater. Chem. A, 2022, 10, 12764–12787 RSC.
- S. Xu, Q. Wu, B.-A. Lu, T. Tang, J.-N. Zhang and J.-S. Hu, Acta Phys.-Chim. Sin., 2023, 39, 2209001 Search PubMed.
- Z.-H. Yin, H. Liu, J.-S. Hu and J.-J. Wang, Natl. Sci. Rev., 2024, 11, nwae362 CrossRef PubMed.
- L. Zhai, T. W. Benedict Lo, Z.-L. Xu, J. Potter, J. Mo, X. Guo, C. C. Tang, S. C. Edman Tsang and S. P. Lau, ACS Energy Lett., 2020, 5, 2483–2491 CrossRef CAS.
- S. Jeong, C. Huang, Z. Levell, R. X. Skalla, W. Hong, N. J. Escorcia, Y. Losovyj, B. Zhu, A. N. Butrum-Griffith, Y. Liu, C. W. Li, D. Reifsnyder Hickey, Y. Liu and X. Ye, J. Am. Chem. Soc., 2024, 146, 4508–4520 CrossRef CAS PubMed.
- J. Ying, Y. Xiao, J. Chen, Z.-Y. Hu, G. Tian, G. V. Tendeloo, Y. Zhang, M. D. Symes, C. Janiak and X.-Y. Yang, Nano Lett., 2023, 23, 7371–7378 CrossRef CAS PubMed.
- J.-B. Chen, J. Ying, Y. Tian, Y.-X. Xiao and X.-Y. Yang, Electrocatalysis under Magnetic Fields, Adv. Funct. Mater., 2024, 24, 2415660 Search PubMed.
- J. Ying, S. Lenaerts, M. D. Symes and X.-Y. Yang, Adv. Sci., 2022, 9, e2106117 CrossRef PubMed.
- L. Su, J. Chen, F. Yang, P. Li, Y. Jin, W. Luo and S. Chen, J. Am. Chem. Soc., 2023, 145, 12051–12058 CrossRef CAS PubMed.
- I. V. Stiopkin, C. WeeRaman, P. A. Pieniazek, F. Y. Shalhout, J. L. Skinner and A. V. Benderskii, Nature, 2011, 474, 192–195 CrossRef CAS PubMed.
- Y. Xu, Y.-B. Ma, F. Gu, S.-S. Yang and C.-S. Tian, Nature, 2023, 621, 506–510 CrossRef CAS PubMed.
- B. Huang, R. R. Rao, S. You, K. Hpone Myint, Y. Song, Y. Wang, W. Ding, L. Giordano, Y. Zhang, T. Wang, S. Muy, Y. Katayama, J. C. Grossman, A. P. Willard, K. Xu, Y. Jiang and Y. Shao-Horn, JACS Au, 2021, 1, 1674–1687 CrossRef CAS PubMed.
- J. Huang, M. Li, M. J. Eslamibidgoli, M. Eikerling and A. Gross, JACS Au, 2021, 1, 1752–1765 CrossRef CAS PubMed.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS.
- O. van der Heijden, S. Park, R. E. Vos, J. J. J. Eggebeen and M. T. M. Koper, ACS Energy Lett., 2024, 9, 1871–1879 CrossRef CAS PubMed.
- S. Niu, T. Tang, Y. Qu, Y. Chen, H. Luo, H. Pan, W.-J. Jiang, J. Zhang and J.-S. Hu, CCS Chem., 2024, 6, 137–148 CrossRef CAS.
- G. P. Pavithra and A. C. Hegde, Appl. Surf. Sci., 2012, 258, 6884–6890 CrossRef CAS.
- Y.-X. Xiao, J. Ying, J.-B. Chen, X. Yang, G. Tian, J.-H. Li, C. Janiak and X.-Y. Yang, Adv. Funct. Mater., 2024, 24, 2418264 CrossRef.
- K. Hedenstedt, J. Bäckström and E. Ahlberg, J. Electrochem. Soc., 2017, 164, H621–H627 CrossRef CAS.
- M. Sbroscia, A. Sodo, F. Bruni, T. Corridoni and M. A. Ricci, J. Phys. Chem. B, 2018, 122, 4077–4082 CrossRef CAS PubMed.
- P.-Y. Wang, J.-F. Zhou, H. Chen, B. Peng and K. Zhang, JACS Au, 2022, 2, 1457–1471 CrossRef CAS PubMed.
- X. Zhang, Y. Xu, Y. Zhou, Y. Gong, Y. Huang and C. Q. Sun, Appl. Surf. Sci., 2017, 422, 475–481 CrossRef CAS.
- N. Agmon, H. J. Bakker, R. K. Campen, R. H. Henchman, P. Pohl, S. Roke, M. Thamer and A. Hassanali, Chem. Rev., 2016, 116, 7642–7672 CrossRef CAS PubMed.
- C. J. Fecko, J. D. Eaves, J. J. Loparo, A. Tokmakoff and P. L. Geissler, Science, 2003, 301, 1698–1702 CrossRef CAS PubMed.
- K. Mizuse, N. Mikami and A. Fujii, Angew. Chem., Int. Ed., 2010, 49, 10119–10122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08772k |
| This journal is © The Royal Society of Chemistry 2025 |