
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning electrochemical performance and interfacial compatibility of oxygen electrodes in proton-conducting solid oxide electrolysis cells†
Saroj
Karki
,
Shuanglin
Zheng
,
Idris Temitope
Bello
,
Allison
Le
and
Hanping
Ding
 *
*
School of Aerospace and Mechanical Engineering, University of Oklahoma, Norman, OK, USA. E-mail: hding@ou.edu
First published on 20th March 2025
Abstract
The growing demand for clean energy and the urgent need to reduce carbon emissions have accelerated the development of alternative energy solutions, with solid oxide electrochemical cells standing out owing to their efficiency in energy conversion between renewable energies and hydrogen. However, the slow reaction kinetics of its oxygen electrode, particularly at intermediate temperatures, impose a significant obstacle in optimizing their performance, reversibility, and durability. To address these challenges, this study introduces a new A-site deficient perovskite oxide as a potential electrode material for reversible protonic ceramic electrochemical cells. The cation deficiency effectively triggered the formation of oxygen vacancies and proton defects after hydration to facilitate multiple charge carrier conduction for enhanced electrode activity. After investigating the effects of cationic deficiency on structure and electrode polarization in a symmetric cell configuration of praseodymium nickel cobaltite perovskite (Pr1−xNi0.7Co0.3O3−δ), the optimal composition was confirmed and used for integration into full cells. The electrochemical performances in fuel cell and electrolysis modes were studied, and reversible operation and short-term stability test were conducted to understand the improved behaviors, providing a pathway to facilitate excessive proton conductivity for enhanced reaction activity in oxygen electrodes.
Introduction
The depletion of fossil fuels and global efforts to reduce carbon emissions have made it essential to seek alternative clean energy technologies to meet future energy needs. Due to the intermittent nature of solar and wind energy, hydrogen is widely considered a key energy carrier with the potential to power the world by generating electricity for driving vehicles, fueling industries, and powering homes, all with net-zero carbon emissions.1 In recent years, reversible protonic ceramic electrochemical cells (R-PCECs) have attracted considerable interest from academia and industry because of their transformative potential.2 R-PCECs are unique in their ability to effortlessly alternate between two operational modes: functioning as a protonic ceramic fuel cell (PCFC) for electricity generation and as a protonic ceramic electrolysis cell (PCEC) for hydrogen production.3 R-PCECs offer impressive advantages over traditional oxide ion-conducting ceramic cells, making them a compelling choice for sustainable energy systems. R-PCECs only require significantly lower activation energy for proton conduction, enabling them to operate more efficiently and produce ultra-pure hydrogen in electrolysis mode, which is crucial for cleaner energy solutions. In fuel cell mode, they promote optimal fuel utilization, thereby maximizing system output.4–6 Moreover, to improve the energy efficiency of R-PCECs, excellent materials systems are required for achieving durable high-performance operation. Oxygen electrodes, where complicated elemental reactions occur, are considered critical components that enable faster water-splitting reactions. However, the improper use of oxygen electrodes could result in slow reaction kinetics and degradation under realistic reversible conditions.7,8An effective oxygen electrode must exhibit high catalytic activity for the oxygen reduction reaction (ORR) in fuel cell mode and for the water oxidation reaction (WOR) in electrolysis mode. It should also have low polarization resistance (Rp) and be highly compatible with the electrolyte, ensuring lower interfacial resistance.9–11 Mixed ion-electron conducting (MIEC) cathode materials have been extensively studied because they can incorporate oxygen ions into the cathode bulk and diffuse them across the entire cathode area.12,13 However, these electrodes are unable to conduct protons, restricting the water formation reaction to the triple-phase boundary (TPB)—the junction where gas, ions, and electrons converge (Fig. 1).14,15 Under polarization, significant accumulation of water at the TPB slows down the rate of subsequent reactions.16 Interestingly, this limitation can be overcome by triggering proton conductivity in oxides to form a triple-conducting oxide (TCO). Theoretically, the use of a TCO electrode would significantly increase the reaction sites because the protons can move freely via surface or bulk.17
 | ||
| Fig. 1 Schematic of an R-PCEC with a triple-conducting oxygen electrode designed to enhance electrochemical reaction kinetics and optimize crystal structure through A-site modifications in perovskite oxides, improving charge carrier conduction properties. (Left) TCO electrode enables efficient transport of protons, oxygen ions, and electrons across the electrode layers, thereby increasing active sites for oxygen reduction and water oxidation reactions. (Right) Creation of oxygen vacancies through A-site deficiency in the crystal structure of the Pr1−xNi0.7Co0.3O3−δ electrode. | ||
In our previous studies, we developed a series of TCO oxygen electrodes based on PrCoO3 for PCECs. Among these, the PrNi0.7Co0.3O3−δ composition emerged as a promising candidate after structural optimization, exhibiting excellent electrochemical performance. Comprehensive characterizations unveiled the underlying mechanisms, revealing that the induced formation of oxygen vacancies facilitated effective hydration, resulting in a reasonable concentration of proton defects. Remarkably, this electrode significantly enhanced activity under dynamic operating conditions. Furthermore, a series of thermal cycling and accelerated stress tests validated its exceptional mechanical stability at the electrode/electrolyte interface, establishing PrNi0.7Co0.3O3−δ as a benchmark oxygen electrode for PCECs.
However, further optimization of this composition was necessary to improve its performance at reduced temperatures (400–500 °C) and enhance system reliability. Lately, it was found that A-site deficiency on the lattice structure of double perovskite (PrBa0.8Ca0.2)0.95Co2O6−δ can trigger more formation of oxygen vacancies and deform the local lattice for better proton diffusion.18 The non-stoichiometry of A-site cations can increase oxygen vacancy concentration due to charge neutrality in the lattice (Fig. 1) and it can transform the chemical and physical properties such as thermal expansion coefficient, electrical conductivity, and polarization resistance.19–23 Contemporary density functional theory studies proved that A-site deficiency reduces vacancy formation energy and promotes charge compensation through the formation of oxygen vacancies. This structural reform also induces a downward shift in the Fermi level, resulting in ligand hole formation that boosts p-type electronic conduction.24 Investigations into the A-site-deficient BaCo0.4Fe0.4Zr0.1Y0.1O3−δ cathode demonstrate that increased A-site deficiency reduces electrical conductivity due to hindered electron and hole hopping caused by elevated oxygen vacancy concentrations. These vacancies play a pivotal role in enhancing oxygen mobility, surface exchange, and bulk diffusion. Remarkably, the area-specific resistance and activation energy progressively decrease with optimized deficiency levels, as evidenced by the superior performance of Ba0.90Co0.4Fe0.4Zr0.1Y0.1O3−δ compared to stoichiometric BaCo0.4Fe0.4Zr0.1Y0.1O3−δ.25 These findings underscore the potential of fine-tuning A-site deficiency to significantly improve cathode catalytic activity and ORR kinetics, offering a promising strategy for advancing electrocatalyst design.
Here, we implemented state-of-the-art A-site deficiency engineering to tailor the defect chemistry and phase stability of Pr1−xNi0.7Co0.3O3−δ (x = 0, 0.1, 0.2, and 0.3) perovskites in PCECs. Our rigorous examination of the structural and electrochemical properties of the material series revealed that Pr0.8Ni0.7Co0.3O3−δ (PNC20) is the optimal composition. PNC20 exhibits a synergistic balance of proton transport, catalytic activity, and long-term durability. It delivers a remarkable peak power density of 0.826 W cm−2 in fuel cell mode and an exceptional current density of 1.07 A cm−2 at 1.3 V in electrolysis mode at 600 °C, significantly outperforming the myriad of conventional MIEC and TCO electrodes. Moreover, transient and long-term durability experiments show that PNC20 is resilient under both fuel cell (FC) and electrochemical cell (EC) modes, with steady performance over 20 cycles of voltage cycling between FC and EC modes and no detectable degradation after 100 h of continuous electrolysis operation at 1.4 V. These findings establish A-site engineering as a powerful strategy for designing next-generation R-PCEC electrodes, positioning PNC20 as a benchmark material for highly efficient and durable protonic ceramic energy conversion technologies.
Experimental
Cathode powder for symmetric and fuel cells
The Pr1−xNi0.7Co0.3O3−δ series electrode powders were created using a wet-chemistry method. Thermo Fisher-sourced Pr(NO3)3·6H2O (99.9% purity), Ni(NO3)2·6H2O (99% purity), and Co(NO3)2·6H2O (99% purity) were dissolved in deionized water. Ethylenediaminetetraacetic acid (EDTA) and citric acid (CA) were utilized as complexing agents and fuels to improve combustion. The molar ratios of EDTA, citric acid, and cations were maintained at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:1. The resultant precursor solution was repeatedly stirred with a magnetic stirrer while heated on a hot plate. The solution became gel-like as the water completely evaporated. After about 3 h of heating at 300 °C, the gel auto-ignited, releasing a considerable volume of powder ash. This ash was then combusted in a muffle furnace at 1000 °C for 5 h, yielding a crystalline perovskite.
:1.5:1. The resultant precursor solution was repeatedly stirred with a magnetic stirrer while heated on a hot plate. The solution became gel-like as the water completely evaporated. After about 3 h of heating at 300 °C, the gel auto-ignited, releasing a considerable volume of powder ash. This ash was then combusted in a muffle furnace at 1000 °C for 5 h, yielding a crystalline perovskite.
Electrolyte powder for fuel cell
BaCe0.7Zr0.1Y0.1Yb0.1O3−δ (BCZYYb7111) electrolyte was synthesized using a sol–gel method with Ba(NO3)2, Ce(NO3)3·6H2O, ZrO(NO3)2·xH2O, Y2O3, and Yb2O3 as precursors, while CA and EDTA were employed as complexing and chelating agents, respectively. First, Y2O3 and Yb2O3 were dissolved in a 20 wt% nitric acid solution at 120 °C, and ZrO(NO3)2·xH2O was dissolved in deionized water to form clear solutions. Concurrently, stoichiometric amounts of Ba(NO3)2, Ce(NO3)3·6H2O, and EDTA were dissolved in deionized water, and the pH was adjusted to 7 using ammonium hydroxide (NH4OH). After achieving a pH of 7, ZrO(NO3)2·xH2O, Y2O3, and Yb2O3 solutions were mixed with the Ba(NO3)2–Ce(NO3)3-EDTA solution and CA, with the pH further adjusted to ∼7 by adding NH4OH. The mixture was stirred for 30 min at room temperature to promote complexation, followed by heating at 250 °C to form a gel that auto-ignited. The resulting powder was calcined at 1000 °C for 5 h in the air to produce the BCZYYb7111 electrolyte.Anode powder for fuel cell
The anode powder consisted of sol–gel-derived BCZYYb7111, NiO, and corn starch, mixed in a weight ratio of 2:3:1.
Anode functional layer power for the fuel cell
The anode functional layer powder was prepared using a sol–gel method similar to that employed for the synthesis of the BCZYYb7111 electrolyte, with the addition of NiO. The BCZYYb7111 and NiO were mixed in a weight ratio of 2:3 to form the final composition.
Electrolyte powder for symmetric cells
The BaCe0.7Zr0.1Y0.1Yb0.1O3−δ electrolyte powder for symmetric cells was produced using solid-state techniques. The precursors BaCO3 (99.8% purity), ZrO2 (99.7% purity), CeO2 (99.5% purity), Y2O3 (99.9% purity), and Yb2O3 (99.9% purity), all obtained from Thermo Fisher Scientific, were thoroughly mixed by ball milling in ethanol at 300 rpm for 24 h. After drying, the mixture was calcined at 1100 °C for 10 h in an air atmosphere. The calcined powder was then ball-milled at 300 rpm in ethanol for another 12 h before being dried and calcined at 1100 °C for 10 h to yield a pure-phase electrolyte powder.Preparation of symmetric cells
To fabricate symmetrical cells, BCZYYb7111 electrolyte pellets were formed by uniaxially pressing the powders (BCZYYb7111 + 2 wt% nickel oxide (NiO)) to obtain green pellets. The pellets were subsequently sintered at 1450 °C for 10 h to densify the product. For PNC electrode slurries, the electrode powders were ball milled with a binder solution (2% ethyl cellulose in terpineol) and ethanol for 10 min. The slurry was mixed and defoamed with a planetary centrifugal mixer (Thinky mixer) until it was thick and sticky. To produce symmetrical cells, an electrode slurry was brush-painted on both sides of the electrolyte pellet and calcined at 920 °C for 5 h to evaporate the organic solvents and binders, yielding full symmetric cells with an active area of 0.178 cm2.Preparation of half-cells
Half-cell pellets were prepared by sequential hydraulic pressing of the NiO-BCZYYb7111-starch anode, functional layer, and electrolyte powder prepared via sol–gel method with a 12 mm die. Half-cell pellets were then sintered at 1425 °C for 10 h to obtain the dense structure. The PNC20s electrode slurry was brush-painted over the electrolyte surface with an active area of 0.178 cm2 and then calcined at 920 °C for 5 h to remove organic solvents and binders from the cathode slurry and generate complete cells.Electrochemical performance measurement
The ASR measurements of Pr1−xNi0.7Co0.3O3−δ series electrodes were conducted using open circuit voltage (OCV) conditions under the supply of wet oxygen (∼3% H2O). The desired amount of humidity was maintained by supplying the gas through the lab-made humidifier. For the PNC20 ASR tests with varying partial pressures of water, the humidifier was maintained at different temperatures to adjust the partial pressure of water. Additionally, ASR measurements were performed under varying oxygen partial pressure by controlling the oxygen and argon (Ar) gas flow rates. The electrochemical stability of the cell was assessed at 600 °C under 3% H2O and a current density of −100 mA cm−2.During full-cell testing, the PNC20 oxygen electrode was exposed to oxygen. The mixed hydrogen (H2, 7 mL min−1) and argon (13 mL min−1) gas was fed to the testing fixture for reducing NiO to metallic nickel. The increase in the OCV was used to track the reduction process. This gas combination was gradually adjusted by increasing the hydrogen flow rate while decreasing the argon flow until the ultimate circumstances were H2 at 20 mL min−1 and no argon flow. When a steady OCV above 1.0 V was achieved, fuel cell mode testing was conducted, and current–voltage (I–V) curves were obtained between 450 °C to 600 °C. Electrochemical impedance spectroscopy (EIS) was measured at OCV, with a frequency range of 106 to 10−1 Hz and an AC signal amplitude of 20 mV.
In electrolysis testing mode, pure H2 was fed to the fuel electrode and humidified oxygen (20% water) was supplied to the air electrode side. The EIS experiments in electrolysis mode were performed at an applied voltage of 1.3 V with a frequency range from 106 to 10−1 Hz and an AC amplitude of 20 mV using VersaStudio software. For a comprehensive analysis of the EIS spectra, the distribution of relaxation times (DRT) was conducted with Gamry Echem analysis software.
Characterizations
The crystalline phase structure of Pr1−xNi0.7Co0.3O3−δ series powders were analyzed using X-ray diffraction (XRD, Rigaku SmartLab) with a Cu rotating anode source. Characterization of the fuel cell was performed using a scanning electron microscope (TFS Quattro S) to analyze the cross-section in secondary electron and backscattered electron modes. Thermogravimetric analysis (TGA, STA 509 Jupiter® Supreme instrument) was conducted to understand the oxygen non-stoichiometry through the study of weight loss by heating the samples in the air from room temperature to 650 °C with a ramping rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) was conducted using a PHI Quantera system to analyze the O 1s spectra of electrodes.Results and discussion
Phase structure
The wet-combustion method was used to synthesize the Pr1−xNi0.7Co0.3O3−δ series (x = 0, 0.1, 0.2, and 0.3, noted as PNC, PNC10, PNC20, and PNC30, respectively). After calcination at 1000 °C, XRD was conducted in these samples at room temperature (Fig. 2a). All the samples exhibit a single pure perovskite orthorhombic structure which is indexed to be a Pbnm space group.26 No discernible impurities were observed, indicating the formation of pure crystal structures. Furthermore, the absence of impurity peaks in the XRD of a homogeneously mixed PNC series oxides and electrolyte powder (Fig. S1†) confirms their excellent chemical compatibility, ensuring a stable electrode–electrolyte interface without the formation of secondary phases or undesired reactions. Additionally, XRD analysis of the post-test PNC20 cathode confirms the stability of the material under reversible operation, with no detectable impurity phase indicating the material's structural integrity. | ||
| Fig. 2 Structural analysis of Pr1−xNi0.7Co0.3O3−δ and its impact on electrode polarization resistance. (a) XRD patterns of Pr1−xNi0.7Co0.3O3−δ (x = 0.1, 0.2, and 0.3), along with post-test PNC20 samples, showing phase purity and structural variations as a function of Pr-site deficiency. (b) Arrhenius plot of electrode polarization resistance from symmetric cells under open-circuit conditions in wet oxygen (∼3% H2O), demonstrating temperature-dependent changes in polarization resistance. | ||
When these electrodes are integrated into the symmetric cells, the electrode polarization resistance can be measured based on the difference between the high and low-frequency intercepts on the real axis to study the catalytic activity towards WOR and ORR.27Fig. 2b depicts the area-specific resistance (ASR) of each composition measured at the OCV condition. As the A-site deficiency increases, ASR continuously drops until x = 0.2 across all intermediate temperature measurements. Beyond this point, an additional deficit causes an increase in ASR. At 600 °C, PNC20 has a Rp of 2.53 Ω cm2, which is 24%, 13.4%, and 9% lower than PNC, PNC10, and PNC30, respectively. Also, at 400 °C, PNC20 stays to outperform the other compositions, with Rp values 16.8%, 6.65%, and 2.7% lower, respectively. This result indicates that introducing Pr deficiency within a definite range (here, x = 0.2) improves catalytic activity by uplifting the formation of oxygen vacancies, improving oxygen dissociation and adsorption on the cathode surface, and facilitating ORR. However, further Pr deficiency can adversely reduce electrode activity. Thus, PNC20 demonstrates the lowest resistance across the entire intermediate temperature range, underscoring its superior electrochemical performance. Furthermore, the activation energies of all the cathode compositions are calculated by using the following equation:26
 | (1) |
The calculated activation energy is 0.427 eV for PNC, 0.411 eV for PNC10, 0.439 eV for PNC20, and 0.414 eV for PNC30. The alterations in activation energies of all the compositions are insignificant with variation of ±0.013. The lower Rp values of PNC20 across the temperature spectrum accentuate its efficient catalytic activity of charge transfer kinetics and reduced operational losses.
Electrochemical performance of the symmetric cell
The electrochemical performance of the PNC20 electrode was evaluated utilizing EIS on symmetric cells. Fig. 3a shows the EIS measurements carried out at different applied voltages, demonstrating a distinct reduction in ASR with increasing voltage. The ASR value was measured to be 0.118 Ω cm2 at an applied voltage of 0.7 V, while it sharply declined to 0.01 Ω cm2 at 1.2 V. This substantial reduction in ASR values shows that the ORR becomes increasingly kinetically favorable at higher potentials, thereby reducing the polarization resistance. Moreover, EIS measurements were also performed under varying water partial pressures (Fig. 3b). At 3%, 10%, 20%, and 30% H2O, the ASR values were 0.47, 0.36, 0.17, and 0.12 Ω cm2, respectively. The dramatic reduction in ASR with increasing water partial pressure indicates that hydration at the cathode in a humidified atmosphere facilitates the formation of protonic defects through the hydration of oxygen vacancies described by the Stotz–Wagner mechanism (eqn (2)), thereby enhancing the proton conduction, improving charge transfer and overall electrochemical performance.28 | (2) |
 | ||
| Fig. 3 Electrochemical performance of symmetric cells at 600 °C under varying operational conditions. (a) EIS measurements under different applied voltages, highlighting voltage-dependent impedance responses. (b) EIS measurements at varying partial pressures of water, showing the effect of water content on cell performance. (c) Distribution of relaxation time analysis at different oxygen partial pressures, revealing oxygen-dependent reaction mechanisms. (d) Polarization resistance stability over time at 3% water content, demonstrating long-term cell durability. (e) Stability of polarization resistance as a function of applied current density (0.1 A cm−2) at 3% water content, showcasing the cell's performance under operational loads. | ||
The increase in oxygen vacancy concentration in A-site deficient PNC20 was further confirmed by XPS analysis. As illustrated in Fig. S2,† the O 1s XPS spectra of the samples were deconvoluted into four distinct sub-peaks located at approximately 528, 529, 531, and 533 eV. These peaks correspond to lattice oxygen (O2−), highly oxidative oxygen (O22−/O−), adsorbed oxygen (OH−/O2), and adsorbed H2O, respectively. The higher percentage of O22−/O− species in PNC20 compared to PNC implies higher oxygen vacancy concentration which results in high proton uptake during hydration reaction when exposed to a wet atmosphere.29–33 The oxygen non-stoichiometry between PNC and PNC20 was also revealed via a thermogravimetric analysis test (TGA). As depicted in Fig. S3,† PNC20 experiences a more significant weight loss than PNC. This pronounced weight reduction directly correlates with the increased concentration of oxygen vacancies in the PNC20 electrode.18,34 This finding corroborates our hypothesis that the introduction of A-site deficiency in PrNi0.7Co0.3O3−δ promotes the formation of oxygen vacancies, which can subsequently facilitate proton conduction and enhance the electrochemical performance of the electrode.
Additionally, EIS measurements were conducted by changing oxygen partial pressures to better distinguish between ORR and the WOR occurring at the PNC20 electrode. DRT analysis was then used to extract particular electrochemical subprocesses. To identify these specific processes, the distinctive frequency range of the EIS spectra (106–10−1 Hz) was used, as illustrated in Fig. 3c. The low-frequency (LF, 10−1–102 Hz) peaks are generally related to gas diffusion or surface adsorption/desorption phenomena; the mid-frequency (MF, 102–104 Hz) peaks are associated with bulk diffusion processes and surface oxygen exchange; and the high-frequency (HF, 104–106 Hz) peaks are associated with charge transfer reactions.34–38 The area under the peaks in each frequency range correlates with the polarization resistance of the respective electrochemical step. It is evident from Fig. 3c that the MF and LF regions contribute the most to the total polarization resistance under different oxygen partial pressures, suggesting that surface exchange, gas diffusion, and bulk diffusion processes dominate the electrode's polarization resistance.39 The MF and LF resistances decrease upon introducing oxygen, indicating improved surface mass transfer and bulk ion transport contributing to the acceleration of the ORR kinetics.40,41
The long-term electrochemical stability of the PNC20 electrode was evaluated by monitoring the Rp over time at a current density of 0.1 A cm−2 and 3% H2O (Fig. 3d and e). Remarkably, no significant decrease was observed in ASR after 82 h of operation in 3% H2O. Furthermore, even after 72 h at a sustained current density of 100 mA cm−2, the ASR exhibited only a modest increase of 12.45%, underscoring the electrode's robust performance stability.
Electrochemical performance of the reversible protonic ceramic cell
The enhanced electrochemical characteristics of PNC20 as a cathode material for reversible protonic ceramic cells were further validated by single-cell performance testing. In fuel cell mode, a cell with a PNC20 cathode and an electrolyte of BCZYYb7111 was assessed at temperatures between 450 °C and 600 °C. Fig. 4a demonstrates the temperature-dependent I–V–P characteristics of the cell in FC mode. The cell achieved a peak power density of 826 mW cm−2 at 600 °C, 522 mW cm−2 at 550 °C, 402 mW cm−2 at 500 °C, and 260 mW cm−2 at 450 °C, surpassing the performance of the same single cell configuration with PNC as air electrode (Fig. S4†) which underscores PNC20's exceptional performance across a range of operating conditions. This further establishes PNC20's enhanced electrocatalytic ORR, and positions it amongst the highest-performing electrodes reported in the literature.42–50 EIS results further support the improved ORR activity of the PNC20 electrode (Fig. 4b). At 600 °C, 550 °C, and 500 °C, the electrolyte resistance (Ro) values were 0.237, 0.313, and 0.4 Ω cm2, respectively, whereas the Rp was 0.144, 0.228, and 0.4 Ω cm2, respectively. It is conspicuous that electrolyte resistance largely controls the total cell resistance across a considerable portion of the intermediate temperature range, which is principally triggered by the electrolyte's conductivity and thickness. | ||
| Fig. 4 Electrochemical electrolysis performance evaluations under different temperatures. (a) I–V–P characteristics in FC mode at 450 °C, 500 °C, 550 °C, and 600 °C, demonstrating the temperature-dependency of power density and current density. (b) Electrochemical impedance spectra measured at different temperatures to study respective contribution from each cell component. (c) Distribution of relaxation time analysis based on EIS data, providing insight into the frequency-dependent polarization resistances and reaction kinetics under FC mode operation. | ||
To gain a more comprehensive understanding of the polarization mechanisms, DRT analysis of EIS was conducted to assess the individual electrochemical subprocesses. As illustrated in Fig. 4c, four distinct peaks, denoted as P1, P2, P2, and P4, emerge when moving from high frequency to low frequency. The LF (<102 Hz) peaks predominantly reflect gas diffusion limitations and surface adsorption/desorption dynamics. MF (102–104 Hz) features are indicative of bulk ion transport and surface oxygen exchange kinetics. Meanwhile, HF (104–106 Hz) peaks capture the rapid charge transfer events at the electrode–electrolyte interface.51 Conspicuously, the peak intensity exhibits a pronounced decline with increasing temperature. Given that the area under these peaks in DRT analysis directly correlates with the polarization resistance of individual electrochemical processes, this trend unequivocally indicates a lower polarization resistance at 600 °C compared to 450 °C, where the peaks are more intense. The high-frequency peak (P1) shifts towards lower frequencies, while the peaks in the MF and LF ranges (P2, P3, and P4) shift towards higher frequencies as the temperature increases. Below 550 °C, the appearance of an additional peak (Padd) in the mid-frequency region, associated with bulk ion transport and surface oxygen exchange, signifies the transition of a previously non-rate-limiting step into a rate-limiting one, introducing an additional energy barrier that must be overcome at this temperature.52 The figure further reveals that the LF peak (P3) and MF peak (P2) dominate as temperature decreases, with P3 being the most pronounced. This suggests that the oxygen adsorption and dissociation processes represent key rate-limiting steps.39 Hence, further optimization of the cathode, potentially through surface modification, could substantially enhance performance and lead to higher power outputs and performances.
Complementing the above discussions, transient voltage experiments on the PNC20 fuel cell were conducted to evaluate its resilience under operating stress. As seen in Fig. 5a, no discernible deterioration of the cell current density was observed over 100 cycles when the applied voltage was swept between 0.5–0.8 V. However, the cell showed a modest decrease in current density under lower voltages, which was probably caused by temporary oxygen starvation.53 Nevertheless, subsequent EIS data (Fig. 5b) demonstrated a reduction in polarization resistance following the transient voltage test, indicating that oxygen starvation did not cause irreversible damage or compromise the long-term stability or functionality of the cell.
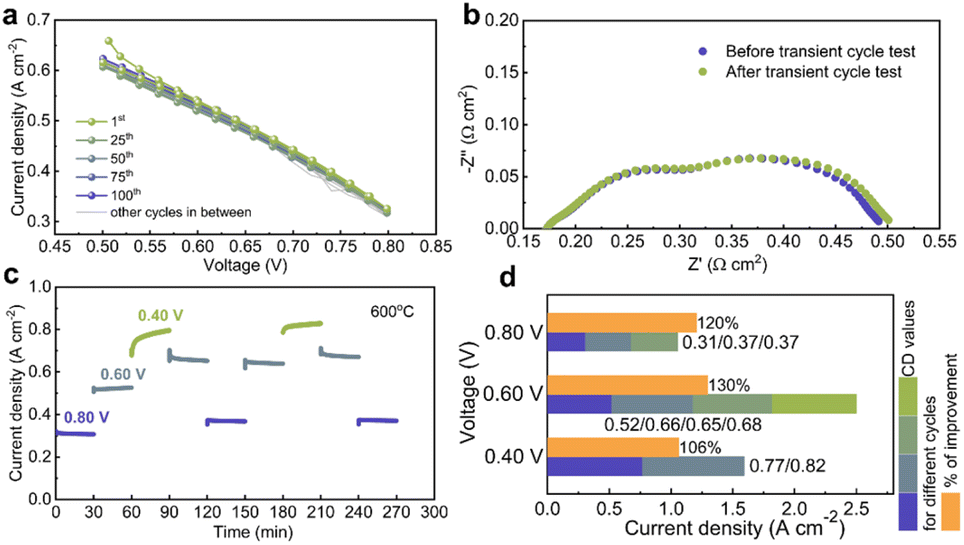 | ||
| Fig. 5 Comprehensive analysis of cell performance under transient voltammetry cycling. (a) Transient voltammetry analysis conducted over 100 cycles, examining the stability and degradation of the cell's electrochemical performance. (b) EIS spectra recorded before and after the transient voltammetry test, highlighting changes in impedance and reaction kinetics. (c) Current density response under stepwise voltage increase and decrease in the number of voltage cycles, assessing the dynamic response of the cell. (d) Bar graph depicting the trend of current density after each cycle, exhibiting the cumulative effect of cycling on electrochemical performance. | ||
Dynamic gains in current density were found by conducting a stepwise voltage test (Fig. 5c), which involved cycling the applied potential from 0.8 V to 0.6 V and then to 0.4 V at 30-minute intervals over two cycles at a fixed temperature of 600 °C. For example, as depicted in Fig. 5d, the current density increased by 20% from 0.31 A cm−2 in the first step to 0.372 A cm−2 in the third step cycle at 0.8 V. Comparable patterns were noted at 0.6 V and 0.4 V, where the current density increased by 6% and 30%, respectively, following two and four voltage steps.
The performance of the R-PCEC was further assessed in the electrolysis mode. The cell's current–voltage (I–V) characteristics, recorded at temperatures between 450 °C and 600 °C, are shown in Fig. 6a where the fuel electrode was exposed to dry hydrogen and the air electrode was supplied with humidified air (20% H2O). At the applied cell voltage of 1.3 V, current densities of 1.07, 0.656, 0.376, and 0.184 A cm−2 were achieved at 600, 550, 500, and 450 °C, respectively. These outcomes pinpoint the substantial activity of the PNC20 cathode toward the oxygen evolution reaction and the WOR in the intermediate temperature range.
 | ||
| Fig. 6 Electrochemical electrolysis performance evaluations under different temperatures. (a) I–V characteristics in EC mode at 450 °C, 500 °C, 550 °C, and 600 °C, demonstrating the temperature-dependency of current density. (b) Electrochemical impedance spectra measured at different temperatures to study the respective contribution from each cell component. | ||
Impedance spectroscopy (Fig. 6b) further revealed that the total cell resistance is predominantly governed by the electrolyte resistance, which is influenced by the electrolyte's thickness and conductivity. The Ro values were determined to be 0.216, 0.297, 0.400, and 0.550 Ω cm2 at 600 °C, 550 °C, 500 °C, and 450 °C, respectively, exhibiting an increasing trend of Ro with decreasing temperature. Alternatively, the Rp, largely contributed by the electrode at a bias of 1.3 V, was determined to be 0.048, 0.1, 0.239, and 0.548 Ω cm2 across the same temperature spectrum. These results prove that Rp is less dominant in the overall resistance, suggesting that reducing electrolyte thickness could significantly lower total resistance, thereby enhancing performance under electrolysis conditions.
The reversible operation of the R-PCEC between electricity generation and hydrogen production was validated through transient testing conducted over a 20-cycle period, with a rapid voltage transition between the electrolysis (1.4 V) and fuel cell (0.7 V) modes. In Fig. 7a, no discernible degradation was seen in the current density during these cycles in both modes, demonstrating the cell's exceptional dynamic response to varying operating conditions. Additionally, the cycling experiment was conducted at 550 °C by alternating between fuel cell mode (0.8 V) and electrolysis mode (1.3 V), with each mode lasting for one hour over a total period of 20 hours (Fig. 7b). The cell exhibited stable performance throughout the test, confirming its high reversibility.
 | ||
| Fig. 7 Reversible operation and stability assessment in reversible operation modes for the as-prepared electrochemical cells. (a–c) Current density responses during reversible operation, illustrating the transient behavior as the working mode alternates between EC and FC modes. (d) Short-term stability evaluation in electrolysis mode upon constant applied voltage of 1.40 V. | ||
Moreover, the cell was tested under cycles of different electrolysis voltages (1.3 V, 1.4 V, and 1.5 V) and fuel cell voltages (0.8 V, 0.7 V, and 0.5 V) for 15 hours as depicted in Fig. 7c. Stable current density was witnessed in fuel cell mode, while an increasing trend in electrolysis mode was perceived. Further, The R-PCEC was measured through 100 h of continuous operation at 1.4 V in electrolysis mode at 550 °C to assess its long-term durability Fig. 7d. It showed sustained performance with only 0.017% degradation per hour, highlighting its robustness and potential for use in reversible systems.
After the electrochemical testing, scanning electron microscopy (SEM) analysis was performed to investigate the interface between the PNC20 electrode and the electrolyte membrane (Fig. 8a). The PNC20 electrode exhibited strong adhesion to the electrolyte and maintained a porous structure, which facilitates efficient gas diffusion across the entire electrode surface. The absence of visible cracks, delamination, or other structural defects highlights the excellent thermal compatibility between PNC20 and the electrolyte, enhancing the mechanical stability and durability of the system.
 | ||
| Fig. 8 Microstructural analysis of the post-test cell after electrochemical measurements using SEM. (a and b) Scanning electron microscopy (SEM) images revealing the microstructural features of the cell after testing, focusing on changes in morphology, surface integrity, and possible degradation of materials resulting from operational stress. | ||
Furthermore, the 8 μm-thick electrolyte membrane exhibits a dense and defect-free morphology, devoid of open pores or pinholes, effectively preventing fuel crossover and ensuring optimal electrochemical performance. As depicted in Fig. 8b, the post-test SEM analysis highlights the microstructural integrity of both the anode functional layer (AFL) and the anode support layer (ASL). Notably, the AFL, measuring approximately 10 μm, undergoes complete reduction to metallic nickel along with ASL, significantly enhances proton transport. The presence of this AFL intermediate layer plays a crucial role in minimizing resistance associated with electrochemical proton incorporation at the anode–electrolyte–gas triple-phase boundaries, thereby facilitating more efficient charge transfer and boosting overall cell performance.54,55 Moreover, the fully reduced nickel phase within the anode is complemented by a well-maintained porosity, ensuring efficient hydrogen diffusion pathways essential for sustained and high-efficiency operation.
Conclusions
The introduction of A-site Pr deficiency in Pr1−xNi0.7Co0.3O3−δ as a TCO electrode offers remarkable improvements for R-PCECs. The PNC20 electrode showed exceptional electrochemical performance in both FC and EC modes, making it an excellent candidate for operation at reduced temperatures. The efficiency of catalyzing ORR and WOR contributes to reduced polarization resistance, leading to higher power densities and long-term durability.The improved electrochemical performances of the PNC20 electrode in R-PCEC are attributed to several key findings. In fuel cell mode, PNC20 achieves a peak power density of 826 mW cm−2 at 600 °C, surpassing many previously reported values in the literature. Similarly, the current density reaches a high current density of 1.07 A cm−2 at 1.3 V in the electrolysis mode. This is because the formation of additional oxygen vacancies induced by Pr deficiency can enhance both surface oxygen exchange and bulk diffusion processes. These oxygen vacancies enable faster ORR kinetics and improve overall charge transfer, making PNC20 the optimal candidate among the tested compositions. DRT analysis of EIS spectra in the intermediate temperature range identifies oxygen adsorption and dissociation as critical rate-limiting processes. This underscores the potential of advanced surface modifications to significantly boost electrochemical performance and achieve greater power output, presenting a promising pathway for further optimization. The long-term durability of PNC20 is validated through transient voltage cycling and reversible operations, where no significant degradation in performance is observed. The electrode's stability across various humidified and oxygen-rich environments emphasizes its robustness for R-PCEC. Additionally, stepwise voltage tests indicate dynamic gains in current density, demonstrating its resilience under fluctuating operational conditions.
In summary, the incorporation of A-site deficiency in Pr1−xNi0.7Co0.3O3−δ (x = 0.2) significantly improves the performance, making PNC20 a promising electrode for the next generation R-PCECs. The material's high activity and excellent stability highlight its potential for advancing this technology.
Data availability
All the relevant data are included in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the U.S. Department of Energy (USDOE), Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office (FCTO) under contract DE-EE0011336. H. D. would like to thank the startup research grant from the University of Oklahoma. Authors thank Dr Yuqing Meng for carrying out TGA measurements.References
- IEA, The Future of Hydrogen, 2019 Search PubMed.
- K. Develos-Bagarinao, T. Ishiyama, H. Kishimoto, H. Shimada and K. Yamaji, Nanoengineering of cathode layers for solid oxide fuel cells to achieve superior power densities, Nat. Commun., 2021, 12, 1–12 CrossRef.
- K. Pei, Y. Zhou, K. Xu, H. Zhang, Y. Ding, B. Zhao, W. Yuan, K. Sasaki, Y. M. Choi, Y. Chen and M. Liu, Surface restructuring of a perovskite-type air electrode for reversible protonic ceramic electrochemical cells, Nat. Commun., 2022, 13(1), 2207 CAS.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, A review of materials and applications for renewable energy conversion and storage, Renew. Sustain. Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- L. Bi, S. Boulfrad and E. Traversa, Steam electrolysis by solid oxide electrolysis cells (SOECs) with proton-conducting oxides, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC.
- W. Wu, H. Ding, Y. Zhang, Y. Ding, P. Katiyar, P. K. Majumdar, T. He and D. Ding, 3D self-architectured steam electrode enabled efficient and durable hydrogen production in a proton-conducting solid oxide electrolysis cell at temperatures lower than 600 °C, Adv. Sci., 2018, 5(11), 1800360 Search PubMed.
- K. Pei, S. Luo, F. He, J. Arbiol, Y. Xu, F. Zhu, Y. Wang and Y. Chen, Constructing an active and stable oxygen electrode surface for reversible protonic ceramic electrochemical cells, Appl. Catal. B., 2023, 330, 1–10 Search PubMed.
- Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao, L. Soule, Y. Niu, Y. Ding, H. Ding, D. Ding and M. Liu, An Active and Robust Air Electrode for Reversible Protonic Ceramic Electrochemical Cells, ACS Energy Lett., 2021, 6, 1511–1520 CAS.
- Y. Gao, M. Zhang, M. Fu, W. Hu, H. Tong and Z. Tao, A comprehensive review of recent progresses in cathode materials for Proton-conducting SOFCs, Energy Rev., 2023, 2(3), 100038 CrossRef.
- M. Papac, V. Stevanović, A. Zakutayev and R. O’Hayre, Triple ionic–electronic conducting oxides for next-generation electrochemical devices, Nat. Mater., 2021, 20, 301–313 CrossRef CAS.
- W. Bian, W. Wu, Y. Gao, J. Y. Gomez, H. Ding, W. Tang, M. Zhou and D. Ding, Regulation of Cathode Mass and Charge Transfer by Structural 3D Engineering for Protonic Ceramic Fuel Cell at 400 °C, Adv. Funct. Mater., 2021, 31, 1–8 Search PubMed.
- A. P. Khandale, B. S. Pahune, S. S. Bhoga, R. V. Kumar and R. Tomov, Development of Pr2−xSrxCuO4±δ mixed ion-electron conducting system as cathode for intermediate temperature solid oxide fuel cell, Int. J. Hydrogen Energy, 2019, 44, 15417–15435 CrossRef CAS.
- S. K. Burnwal, S. Bharadwaj and P. Kistaiah, Review on MIEC Cathode Materials for Solid Oxide Fuel Cells, J. Mol. Eng. Mater., 2016, 04, 1630001 CrossRef CAS.
- M. Wang, C. Su, Z. Zhu, H. Wang and L. Ge, Composite cathodes for protonic ceramic fuel cells: Rationales and materials, Compos. Part B Eng., 2022, 238, 109881 CrossRef CAS.
- Z. Wang, W. Yang, S. P. Shafi, L. Bi, Z. Wang, R. Peng, C. Xia, W. Liu and Y. Lu, A high performance cathode for proton conducting solid oxide fuel cells, J. Mater. Chem. A, 2015, 3, 8405–8412 CAS.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Self-Assembled Triple-Conducting Nanocomposite as a Superior Protonic Ceramic Fuel Cell Cathode, Joule, 2019, 3, 2842–2853 CrossRef CAS.
- W. Zhu, H. Wang, L. Xu, J. Yuan, J. Gong and X. Liu, Pr0.7Ba0.3Co0.8−xFe0.2NixO3−δ perovskite: High activity and durable cathode for intermediate-to-low-temperature proton-conducting solid oxide fuel cells, Int. J. Hydrogen Energy, 2023, 48, 33633–33643 CrossRef CAS.
- W. Tang, H. Ding, W. Bian, W. Wu, W. Li, X. Liu, J. Y. Gomez, C. Y. Regalado Vera, M. Zhou and D. Ding, Understanding of A-site deficiency in layered perovskites: Promotion of dual reaction kinetics for water oxidation and oxygen reduction in protonic ceramic electrochemical cells, J. Mater. Chem. A, 2020, 8, 14600–14608 CAS.
- G. Chen, J. Sunarso, Y. Wang, C. Ge, J. Yang and F. Liang, Evaluation of A-site deficient Sr1−xSc0.175Nb0.025Co0.8O3−δ (x = 0, 0.02, 0.05 and 0.1) perovskite cathodes for intermediate-temperature solid oxide fuel cells, Ceram. Int., 2016, 42, 12894–12900 CAS.
- S. Zhen, W. Sun, G. Tang, D. Rooney, K. Sun and X. Ma, Evaluation of strontium-site-deficient Sr2Fe1.4Co0.1Mo0.5O6−δ-based perovskite oxides as intermediate temperature solid oxide fuel cell cathodes, Int. J. Hydrogen Energy, 2016, 41, 9538–9546 CAS.
- C. Yang, F. Zhao, F. Chen and M. Liu, Investigation of A-site deficient Ba0.9Co0.7Fe0.2Nb0.1O3−δ cathode for proton conducting electrolyte based solid oxide fuel cells, Int. J. Hydrogen Energy, 2014, 39, 8431–8436 CAS.
- F. Meng, T. Xia, J. Wang, Z. Shi and H. Zhao, Praseodymium-deficiency Pr0.94BaCo2O6-δ double perovskite: A promising high performance cathode material for intermediate-temperature solid oxide fuel cells, J. Power Sources, 2015, 293, 741–750 CAS.
- Z. Liu, L. Z. Cheng and M. F. Han, A-site deficient Ba1−xCo0.7Fe0.2Ni0.1O3−δ cathode for intermediate temperature SOFC, J. Power Sources, 2011, 196, 868–871 CrossRef CAS.
- A. Belotti, Y. Wang, A. Curcio, J. Liu, E. Quattrocchi, S. Pepe and F. Ciucci, The influence of A-site deficiency on the electrochemical properties of (Ba0.95La0.05)1−xFeO3−δ as an intermediate temperature solid oxide fuel cell cathode, Int. J. Hydrogen Energy, 2022, 47, 1229–1240 CrossRef CAS.
- W. Wang, X. Zhang, K. Khan, H. Wu, D. Zhang, Y. Yang, Y. Jiang and B. Lin, Enhanced ORR activity of A-site deficiency engineered BaCo0·4Fe0·4Zr0·1Y0·1O3−δ cathode in practical YSZ fuel cells, Int. J. Hydrogen Energy, 2021, 46, 5593–5603 CrossRef CAS.
- W. Tang, H. Ding, W. Bian, C. Y. Regalado Vera, J. Y. Gomez, Y. Dong, J. Li, W. Wu, W. W. Fan, M. Zhou, C. Gore, B. M. Blackburn, H. Luo and D. Ding, An unbalanced battle in excellence: Revealing effect of Ni/Co occupancy on water splitting and oxygen reduction reactions in triple-conducting oxides for protonic ceramic electrochemical cells, Small, 2022, 18(30), 2201953 CrossRef CAS.
- L. Hu, D. Zhou, X. Zhu, N. Wang, J. Bai, H. Gong, Y. Zhang, Y. Chen, W. Yan and Q. Zhu, A high-performance composite cathode based on thermal expansion complementation for SOFC, Fuel, 2024, 362, 130864 CrossRef CAS.
- H. Zheng, M. Riegraf, N. Sata and R. Costa, A double perovskite oxygen electrode in Zr-rich proton conducting ceramic cells for efficient electricity generation and hydrogen production, J. Mater. Chem. A, 2023, 11, 10955–10970 RSC.
- S. Oh, D. Kim, H. J. Ryu and K. T. Lee, A Novel High-Entropy Perovskite Electrolyte with Improved Proton Conductivity and Stability for Reversible Protonic Ceramic Electrochemical Cells, Adv. Funct. Mater., 2024, 34, 1–11 Search PubMed.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, A High-Performance Electrocatalyst for Oxygen Evolution Reaction: LiCo0.8Fe0.2O2, Adv. Mater., 2015, 27, 7150–7155 CrossRef CAS PubMed.
- B. Qian, C. Liu, S. Wang, B. Yin, Y. Zheng, L. Ge, H. Chen and C. Zhang, Ca-doped La0.75Sr0.25Cr0.5Mn0.5O3 cathode with enhanced CO2 electrocatalytic performance for high-temperature solid oxide electrolysis cells, Int. J. Hydrogen Energy, 2021, 46, 33349–33359 CrossRef CAS.
- M. Saqib, I. G. Choi, H. Bae, K. Park, J. S. Shin, Y. D. Kim, J. I. Lee, M. Jo, Y. C. Kim, K. S. Lee, S. J. Song, E. D. Wachsman and J. Y. Park, Transition from perovskite to misfit-layered structure materials: A highly oxygen deficient and stable oxygen electrode catalyst, Energy Environ. Sci., 2021, 14, 2472–2484 CAS.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, Tuning the defects of the triple conducting oxide BaCo0.4Fe0.4Zr0.1Y0.1O3−δ perovskite toward enhanced cathode activity of protonic ceramic fuel cells, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC.
- S. B. Yu, Q. Qi, B. Yang, H. Wang, D. W. Zhang, Y. Liu and Z. T. Li, Enhancing Hydrogen Generation Through Nanoconfinement of Sensitizers and Catalysts in a Homogeneous Supramolecular Organic Framework, Small, 2018, 14, 1801037 CrossRef.
- Y. Bu, S. Joo, Y. Zhang, Y. Wang, D. Meng, X. Ge and G. Kim, A highly efficient composite cathode for proton-conducting solid oxide fuel cells, J. Power Sources, 2020, 451, 227812 CrossRef CAS.
- A. Tahir, A. Belotti, Y. Song, Y. Wang, A. Maradesa, J. Li, Y. Tian and F. Ciucci, Ultrafast Sintered Composite Cathode Incorporating a Negative Thermal Expansion Material for High-Performance Protonic Ceramic Fuel Cells, ACS Appl. Mater. Interfaces, 2024, 16, 44645–44654 CrossRef CAS PubMed.
- M. Ghamarinia, A. Babaei, C. Zamani and H. Aslannejad, Application of the distribution of relaxation time method in electrochemical analysis of the air electrodes in the SOFC/SOEC devices: A review, Chem. Eng. J. Adv., 2023, 15, 100503 CrossRef CAS.
- J. Wang, Z. Li, H. Zang, Y. Sun, Y. Zhao, Z. Wang, Z. Zhu, Z. Wei and Q. Zheng, BaZr0.1Fe0.9−xNixO3−δ cubic perovskite oxides for protonic ceramic fuel cell cathodes, Int. J. Hydrogen Energy, 2022, 47, 9395–9407 CrossRef CAS.
- Y. Zhang, D. Zhou, X. Zhu, N. Wang, J. Bai, L. Hu, H. Gong, B. Zhao and W. Yan, Preparation of Pr, Co co-doped BaFeO3−δ-based nanofiber cathode materials by electrospinning, Int. J. Hydrogen Energy, 2024, 50, 992–1003 CrossRef.
- I. T. Bello, D. Guan, N. Yu, Z. Li, Y. Song, X. Chen, S. Zhao, Q. He, Z. Shao and M. Ni, Revolutionizing material design for protonic ceramic fuel cells: Bridging the limitations of conventional experimental screening and machine learning methods, Chem. Eng. J., 2023, 477, 147098 CrossRef CAS.
- S. Park, S. Lee, H. Baek and D. Shin, Cell performance enhancement facilitated by mixed ionic and electronic conductor fiber for protonic ceramic fuel cells, J. Electroceram., 2020, 45, 1–6 CrossRef CAS.
- D. Wang, Y. Xia, H. Lv, L. Miao, L. Bi and W. Liu, PrBaCo2−xTaxO5+δ based composite materials as cathodes for proton-conducting solid oxide fuel cells with high CO2 resistance, Int. J. Hydrogen Energy, 2020, 45, 31017–31026 CrossRef CAS.
- B. Liu, L. Jia, B. Chi, J. Pu and J. Li, A novel PrBaCo2O5+σ-BaZr0.1Ce0.7Y0.1Yb0.1O3 composite cathode for proton-conducting solid oxide fuel cells, Compos. Part B Eng., 2020, 191, 107936 CrossRef CAS.
- W. Li, B. Guan, L. Ma, S. Hu, N. Zhang and X. Liu, High performing triple-conductive Pr2NiO4+δ anode for proton-conducting steam solid oxide electrolysis cell, J. Mater. Chem. A, 2018, 6, 18057–18066 RSC.
- T. Wu, R. Peng and C. Xia, Sm0.5Sr0.5CoO3−δ–BaCe0.8Sm0.2O3−δ composite cathodes for proton-conducting solid oxide fuel cells, Solid State Ionics, 2008, 179, 1505–1508 CrossRef CAS.
- M. Liu, J. Gao, X. Liu and G. Meng, High performance of anode supported BaZr0.1Ce0.7Y0.2O3−δ(BZCY) electrolyte cell for IT-SOFC, Int. J. Hydrogen Energy, 2011, 36, 13741–13745 CrossRef CAS.
- H. Dai, E. H. Da’as, S. P. Shafi, H. Wang and L. Bi, Tailoring cathode composite boosts the performance of proton-conducting SOFCs fabricated by a one-step co-firing method, J. Eur. Ceram. Soc., 2018, 38, 2903–2908 CAS.
- W. Zhang, X. Zhang, Y. Song and G. Wang, Recent progress on cathode materials for protonic ceramic fuel cells, Next Sustain., 2024, 3, 100028 Search PubMed.
- P. Yao, J. Zhang, Q. Qiu, Y. Zhao, F. Yu and Y. Li, A highly active catalytic cathode La0.8Sr0.2Co0.7Ni0.3O3−δ for protonic ceramic fuel cells: Experimental and computational insights, Int. J. Hydrogen Energy, 2025, 104, 212 CAS.
- I. T. Bello, N. Yu, Y. Song, J. Wang, T. S. Chan, S. Zhao, Z. Li, Y. Dai, J. Yu and M. Ni, Electrokinetic Insights into the Triple Ionic and Electronic Conductivity of a Novel Nanocomposite Functional Material for Protonic Ceramic Fuel Cells, Small, 2022, 18, 1–11 CrossRef PubMed.
- N. Shi, F. Su, D. Huan, Y. Xie, J. Lin, W. Tan, R. Peng, C. Xia, C. Chen and Y. Lu, Performance and DRT analysis of P-SOFCs fabricated using new phase inversion combined tape casting technology, J. Mater. Chem. A, 2017, 5, 19664–19671 RSC.
- S. Zheng, W. Bian and H. Ding, A robust protonic ceramic fuel cell with a triple conducting oxygen electrode under accelerated stress tests, Mater. Adv., 2024, 5, 2296–2305 RSC.
- X. Zhang, Y. Qiu, F. Jin, F. Guo, Y. Song and B. Zhu, A highly active anode functional layer for solid oxide fuel cells based on proton-conducting electrolyte BaZr0.1Ce0.7Y0.2O3−δ, J. Power Sources, 2013, 241, 654–659 CrossRef CAS.
- C. Tang, N. Wang, R. Zhu, S. Kitano, H. Habazaki and Y. Aoki, Design of anode functional layers for protonic solid oxide electrolysis cells, J. Mater. Chem. A, 2022, 10, 15719–15730 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08716j |
| This journal is © The Royal Society of Chemistry 2025 |