
Hierarchical porous Co-rich PtCo thin films for alkaline seawater hydrogen evolution with chlorine corrosion resistance†
Meilin
Zhang
 a,
Hanzhong
Cui
bc,
Zhou
Yang
bc,
Jie
Yan
bc,
Jin
Zhang
*bc,
Huan
Ma
bc and
Renguo
Guan
*bc
a,
Hanzhong
Cui
bc,
Zhou
Yang
bc,
Jie
Yan
bc,
Jin
Zhang
*bc,
Huan
Ma
bc and
Renguo
Guan
*bc
aSchool of Materials Science and Engineering, Northeastern University, Shenyang, 110819, China
bKey Laboratory of Near-Net Forming of Light Metals of Liaoning Province, Dalian Jiaotong University, Dalian, 116028, China. E-mail: jinzhang@djtu.edu.cn; guanrenguo@sina.cn
cEngineering Research Center of Continuous Extrusion, Ministry of Education, Dalian Jiaotong University, Dalian, 116028, China
First published on 17th February 2025
Abstract
The creation of cost-effective and efficient catalysts for hydrogen evolution in seawater is highly desirable, yet it still poses considerable challenges. In this work, hierarchical porous Co-rich PtCo thin films were successfully prepared by one-step electrodeposition with nonionic surfactant F127 as a soft template to introduce mesoporosity. The effect of varying deposition potentials on the composition, morphology, structure and porosity of the films was systematically investigated. The composition can be adjusted with the deposition potential to achieve a Co content within 50–84 at%. XRD, HRTEM, and SAED analyses show that the films are single-phase nanocrystalline fcc PtCo. The hydrogen evolution reaction (HER) activity and stability of these films were assessed under multiple conditions: 1 M KOH, 1 M KOH with 3.5 wt% NaCl, and 1 M KOH with natural seawater. For the Pt/Co ratio of 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :66, the overpotential is as low as 30 mV at a current density of −10 mA cm−2 in alkaline natural seawater. Furthermore, it demonstrated enhanced stability, showing negligible performance degradation even after continuous 24 h at 10 mA cm−2. The excellent seawater HER performance of Pt34Co66 can be attributed to its hierarchical porous structure with mesopores and electron accumulation on Pt. The results presented herein have the potential to promote the large-scale development of seawater hydrogen production.
:66, the overpotential is as low as 30 mV at a current density of −10 mA cm−2 in alkaline natural seawater. Furthermore, it demonstrated enhanced stability, showing negligible performance degradation even after continuous 24 h at 10 mA cm−2. The excellent seawater HER performance of Pt34Co66 can be attributed to its hierarchical porous structure with mesopores and electron accumulation on Pt. The results presented herein have the potential to promote the large-scale development of seawater hydrogen production.
1. Introduction
Environmental pollution and the energy crisis have collectively spurred the development and utilization of hydrogen energy,1–3 which has emerged as a promising alternative energy source. Currently, water electrolysis has gained recognition as an environmentally friendly and efficient method for hydrogen production.4,5 However, a significant challenge lies in the fact that freshwater resources, which are essential for such electrolysis processes, are limited on our planet. Conversely, seawater resources are abundant, covering 70% of the Earth's surface, offering vast potential to produce H2 on a large scale.6,7However, despite the vast potential of seawater electrolysis, it remains a huge challenge. The high concentrations of chloride ions in seawater pose significant obstacles, as these ions can either corrode catalysts and substrates or trigger competing reactions, such as the chlorine evolution reaction, thereby decreasing the hydrogen evolution efficiency and potentially damaging the electrodes.8–10 In attempts to overcome this challenge, researchers have explored various strategies. For example, Zhang et al. successfully integrated Pt with Ni for the seawater hydrogen evolution reaction (HER), demonstrating a notable enhancement in the corrosion resistance of the catalyst to chlorides.11 The incorporation of Ni facilitates electron accumulation on Pt, thus creating a competitive environment that inhibits the direct contact of Cl− with the catalyst surface. This approach was further confirmed by Su et al., who also demonstrated that the introduction of non-noble metals can protect the Pt species while maintaining an efficient hydrogen evolution rate.12 Chen and coworkers carried out first-principles calculations, revealing that Co doping in electrocatalysts can optimize the hydrogen adsorption free energy for the seawater HER, while simultaneously increasing the adsorption energy of Cl−.13 These studies inspired the fabrication of high-efficiency, low-cost, corrosion-resistant Pt-based seawater electrocatalysts. By strategically introducing non-noble metals, it is possible to construct protective barriers that prevent Cl− penetration into the catalyst layer, thereby preserving the integrity and performance of the catalyst.
Moreover, another issue arises in the HER from seawater, where metal cations such as Mg2+ and Ca2+ can form insoluble precipitates that block the active sites of the catalyst.14–16 Therefore, it is necessary to construct efficient and stable electrocatalysts capable of exposing more active sites. In this context, many studies have shown that porous, especially mesoporous structures can endow catalysts with this capability.17–19 For example, Huang et al. pointed out that the mesopores provide abundant active sites and accessibility in the mesoporous FeP electrode, yielding superior HER activity and rapid kinetics.20 Pellicer et al. showed that Fe-rich Pt–Fe catalysts exhibited excellent HER performance and stability over 50 cycles, due to the inherent mesoporous nature.21 Our previous work also demonstrated that mesoporous Pt-rich PtCo films exhibited excellent HER performance in acidic aqueous solution.22 Nonetheless, PtCo with higher Co content has yet to be explored in terms of its affordability, accessibility and underlying catalytic mechanism in seawater hydrogen evolution.
In this work, we employed a one-step electrodeposition technique to fabricate hierarchical porous Co-rich PtCo thin films, using nonionic surfactant F127 as a soft template to induce mesoporosity. The effects of deposition potential on the Pt:Co ratios, morphology, structure, and porosity of the deposits were studied. The HER activity and stability of the prepared deposits were investigated in 1 M KOH, 1 M KOH + 3.5 wt% NaCl, and 1 M KOH + natural seawater, respectively. The results showed that the as-prepared hierarchical porous Co-rich PtCo thin films exhibited superior seawater HER activity and chorine corrosion resistance, outperforming the commercial Pt/C. The results of this study will promote the development of large-scale seawater hydrogen production, especially in the preparation of chlorine-resistant seawater hydrogen evolution catalysts.
2. Experimental
2.1 Chemicals
Cobalt(II) chloride (CoCl2), polyethylene-polypropylene glycol F127 (H(OCH2CH2)x(OCH2CHCH3)y(OCH2CH2)zOH), potassium chloride (KCl), sodium chloride (NaCl), 20 wt% platinum on carbon (Pt/C), and Nafion perfluorinated resin were purchased from Macklin. Sodium hexachloroplatinate(IV) (Na2PtCl6) was purchased from Rhawn. Hydrochloric acid (HCl) and acetone were purchased from Kermel. Potassium hydroxide (KOH) was purchased from Guanghua Sci-Tech. Ammonium chloride (NH4Cl) and boric acid (H3BO3) were purchased from Damao. Isopropyl alcohol and ethanol were purchased from Tianjinkemao. All chemicals were of analytical grade and used without further purification. Natural seawater was collected from the Yellow Sea (E121.58°, N38.88°, Dalian, China). Ultrapure Millipore water (18.2 mΩ cm) was used throughout the experiments.2.2 Electrodeposition
The deposition of PtCo thin films was carried out with a three-electrode system using a CHI760E electrochemical workstation. Au(125 nm)/Ti(25 nm)/Si sheets, a Pt sheet and a double junction Ag|AgCl 3 M KCl electrode were used as the working electrode, counter electrode and reference electrode, respectively. The effective working area of the working electrode (0.5 × 1 cm−2) was 0.5 × 0.5 cm−2. The electrodeposition was carried out using 100 mL of electrolyte with the following components: 2 mM Na2PtCl6, 200 mM CoCl2, 0.1 M NH4Cl, 0.2 M H3BO3, and 40 g L−1 F127. PtCo films were deposited under the same conditions at various deposition potentials: −0.58 V, −0.60 V, −0.62 V, and −0.64 V vs. Ag|AgCl for 1200 s. The pH of the bath was adjusted to 2.2 using 3 M HCl. And the deposition process was carried out in a double-layer electrolytic cell at 45 ± 0.05 °C with 600 rpm stirring. Moreover, the nonionic surfactant F127 forms micelles above its micelle concentration (cmc, 0.7 wt%) in the bath to obtain deposits with mesopores.23,24 For F127 aqueous solution above the critical micelle concentration (cmc, 0.7 wt%), the hydrophobic PPO occupies the core of micelles and hydrated PEO binding with metal cations is distributed on the outer shell. Upon the application of external potential, the metal cations are thought to move toward the cathode together with the F127 micelles, which act as a soft template. Finally, the deposits were immersed in isopropanol for 24 hours in order to remove F127, and then sequentially rinsed with acetone, ethanol and deionized water. Additionally, H3BO3 and NH4Cl were used as the pH buffer and complexing agent, respectively.2.3 Characterization
For characterization of the electrolyte, a three-electrode set-up was used, with an Ag|AgCl 3 M KCl electrode, a platinum sheet and a vitreous carbon electrode as the reference electrode, counter electrode and working electrode, respectively. Cyclic voltammetry (CV) was conducted in the range of −1 to 0.6 V vs. Ag|AgCl at a scan rate of 50 mV s−1. CV was scanned at first toward negative potential to reduce metal ions in solution at the working electrode and then toward positive potential to oxidize the deposit.The surface morphology of deposits was observed using a Zeiss Supra55 field emission scanning electron microscope (FESEM) at 5 kV in InLens mode. The particle size distribution was statistically analyzed using Image Pro Plus 6.0 (IPP) software. The Pt:Co ratios of deposits were obtained by PerkinElmer NexION 300X inductively coupled plasma mass spectrometry (ICP-MS). The crystallographic information of the PtCo thin films was obtained by Bruker D8 Advance X-ray diffraction (GIXRD) at a grazing incidence angle of 0.3° and a scanning rate of 3° min−1 in the range of 2θ = 32.5°–50°. The surface chemical composition and electronic state of the PtCo thin films were detected by Thermofisher Nexsa X-ray photoelectron spectroscopy (XPS) using an Al-Kα monochromator to provide incident photon energy. And the binding energy was corrected with reference to the C 1s signal (284.8 eV). The PtCo thin films were further characterized using an FEI Talos F200X G2 transmission electron microscope (TEM) equipped with an energy dispersive X-ray detector (EDX) at a voltage of 200 kV. The high-resolution transmission electron microscopy (HRTEM) images and selected area electron diffraction (SAED) information were recorded.
2.4 Electrochemical performance measurements
All electrochemical measurements were performed on a CHI760E electrochemical workstation using a three-electrode system (25 °C). The deposited PtCo thin films and a glass-carbon disk electrode (RDE, 5 mm in diameter) drop-casted with Pt/C were used as the working electrodes. A graphite rod and a double junction Ag|AgCl 3 M KCl electrode served as the counter electrode and reference electrode, respectively. The electrocatalytic hydrogen evolution performance was tested in three solutions: 1 M KOH (alkaline aqueous solution), 1 M KOH + 3.5 wt% NaCl (alkaline simulated seawater), and 1 M KOH + natural seawater (alkaline natural seawater), respectively. The HER polarization curves of the working electrode were recorded by linear sweep voltammetry (LSV) with 100% iR compensation at a scan rate of 2 mV s−1. Additionally, the Tafel slopes were derived from the LSV curves. CV curves were recorded at open circuit potential ±50 mV (the non-Faraday range) with a scan rate from 10 to 200 mV s−1, from which the Cdl values, electrochemical active surface area (ECSA), and roughness factor (RF) were calculated. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 100 kHz to 0.1 Hz at an overpotential of −10 mA cm−2. The stability of the catalysts was assessed through 24 h continuous electrolysis at a constant current density of −10 mA cm−2 using chronoamperometry. The related calculation process and the preparation of the Pt/C electrode are given in the ESI.†The measured potentials (vs. Ag|AgCl) were converted to those against the reversible hydrogen electrode (RHE) by using the standard Nernst function, eqn (1):25
| ERHE = EAg|AgCl + 0.059 pH + E0Ag|AgCl | (1) |
3. Results and discussion
3.1 Chemical composition, morphology and structure
By altering the deposition potential, the Pt/Co ratio within the films can be regulated. The ICP-MS results, as displayed in Fig. 1a, reveal that the Pt:Co ratios in the as-prepared PtCo films are 50:50, 34:66, 21:79 and 16:84, corresponding to deposition potentials of −0.58 V, −0.60 V, −0.62 V and −0.64 V, respectively. It can be found that the increased negative deposition potential facilitates the incorporation of additional Co into PtCo films. This is because the more negative deposition potential assists the reduction of Co(II) in the electrolyte.25,26 As shown in Fig. S1,† three reduction peaks were observed during the negative potential scanning process, located near −0.2, −0.5 and −0.8 V, corresponding to the reduction of Pt, PtCo and Co, respectively, with reference to Eiler’s work.25 Based on the ICP results, the PtCo thin films are designated as PtxCoy, where the Pt:Co ratio is represented by x:y (the sum of x and y is 100 in atomic percent). Furthermore, the top-view SEM images of Pt50Co50, Pt34Co66, Pt21Co79 and Pt16Co84 are shown in Fig. 1b–e. The surface morphology of all the samples generally consists of aggregates of nanonodules (as indicated by the yellow dotted circle), forming a hierarchical porous architecture. For Pt50Co50, these nanonodules are densely packed and thus have a lower degree of porosity (Fig. 1b). However, for higher Co content, the PtCo film exhibits a homogeneous microstructure, with a rather distinguished nanoporosity (Fig. 1c–e). As the applied potential becomes more negative, Pt34Co66, Pt21Co79 and Pt16Co84 display a loosely packed arrangement. Thus, it is evident that the transformation from dense to loose packing in the mesoporous PtCo films is attributable to the applied potential. The magnified SEM image of Pt34Co66 is shown in Fig. 1f, in which the size of interstices left by the nanonodule aggregates varies from about 10 to nearly 225 nm. And the magnified SEM images of Pt50Co50, Pt21Co79, and Pt16Co84 are shown in Fig. S2.† Moreover, as displayed in Fig. S3,† the average diameters of the nanonodule aggregates of Pt50Co50, Pt34Co66, Pt21Co79, and Pt16Co84 are 65.25 nm, 85.51 nm, 87.86 nm, and 97.02 nm, respectively.
 | ||
| Fig. 1 (a) ICP-MS results and (b)–(e) top-view SEM images of PtCo films deposited at (b) −0.58 V, (c)−0.60 V, (d) −0.62 V, and (e) −0.64 V. (f) Magnified SEM image of Pt34Co66. | ||
Fig. 2a and b present the Pt 4f and Co 2p signals of the PtCo films obtained from XPS measurements, with the results summarized in Table S1.† Analysis of the Pt 4f7/2 signals indicates that both Pt34Co66 and Pt16Co84 exhibit a binding energy of 71.48 eV, which is 0.1 eV lower than that of Pt50Co50 and Pt21Co79 (both at 71.58 eV). This suggests that optimal electron aggregation occurs around Pt sites (Table S1†).11 Furthermore, the Co 2p3/2 signals in the Co 2p region reveal that Pt34Co66 has the lowest binding energy (780.38 eV), which is 0.5 eV lower than that of Pt21Co79 (780.88 eV), implying that this sample has the largest electron accumulation around Pt (Fig. 2b and Table S1†). In particular, as indicated by the green dotted line in Fig. 2b, Pt34Co66 displays distinct peaks in the Co 2p spectrum, with Co 2p3/2 and Co 2p1/2 signals at 778.38 eV and 793.08 eV, respectively. This phenomenon is attributed to the presence of metallic cobalt on the surface, which will be elaborated in the deconvolution part of the XPS results. The deconvolution results of XPS spectra are displayed in Fig. S4† and 8, and the separate fitting results are summarized in Table S2.†
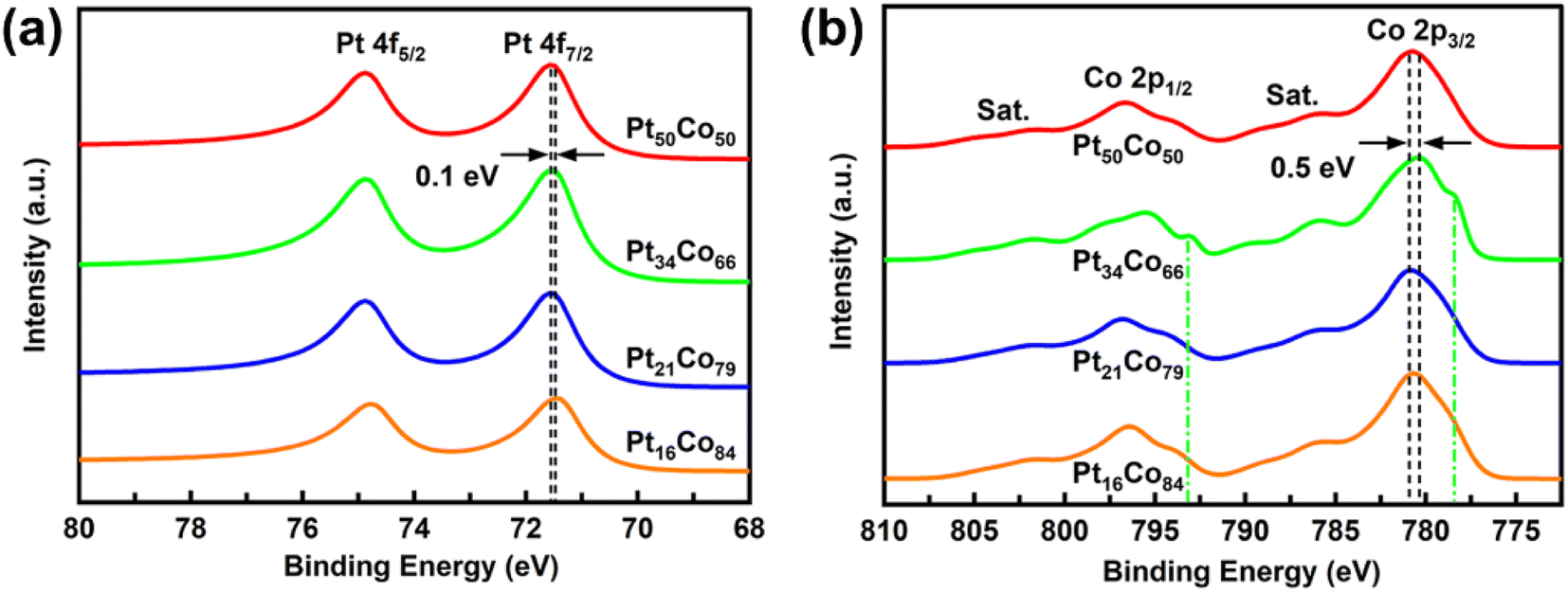 | ||
| Fig. 2 XPS analysis of as-deposited PtCo films: (a) Pt 4f and (b) Co 2p signals. | ||
The cross-sectional characteristics of the Pt34Co66 film were examined by TEM, as shown in Fig. 3. Fig. 3a shows that Pt34Co66 is composed of nano-sized nodules, which leave in between a high density of nanopores with sizes ranging from 3 to 20 nm. The size of the nanonodules varies between 2 and 8 nm, as indicated in Fig. 3b. Furthermore, Fig. 3b1 shows a HRTEM image of the area enclosed in the yellow dotted box in Fig. 3b. The lattice fringes with interplanar distances of 0.221 nm and 0.192 nm can be assigned to the (111) and (002) planes of the fcc PtCo alloy (reference PDF#98-010-2621). Moreover, the corresponding electron diffraction patterns resulting from the fast Fourier transform (FFT) process are shown in Fig. 3d2, exhibiting the (111), (200), and (022) crystal planes of the fcc PtCo alloy. Additionally, the diffraction peaks near 2θ = 40.72° and 47.37° correspond to the (111) and (200) crystal planes of the fcc PtCo alloy and can also be observed in other deposits, as shown in Fig. 3c (reference PDF#98-010-2621). Notably, in the XRD pattern, the diffraction intensities observed near 2θ = 38.26° and 44.47° come from the Au substrate, as referenced from PDF#98-016-3723. Moreover, the SAED result shown in Fig. 1d displays in detail the diffraction rings of Pt34Co66, corresponding to the (111), (200), (022), (113), and (024) planes of the fcc PtCo alloy, respectively (reference PDF#98-010-2621). The EDX elemental distribution mapping shows that the Pt and Co elements are homogeneously distributed throughout the selected area (Fig. 3e1–e3). These results confirm that the deposited films are single-phase polycrystalline fcc PtCo.
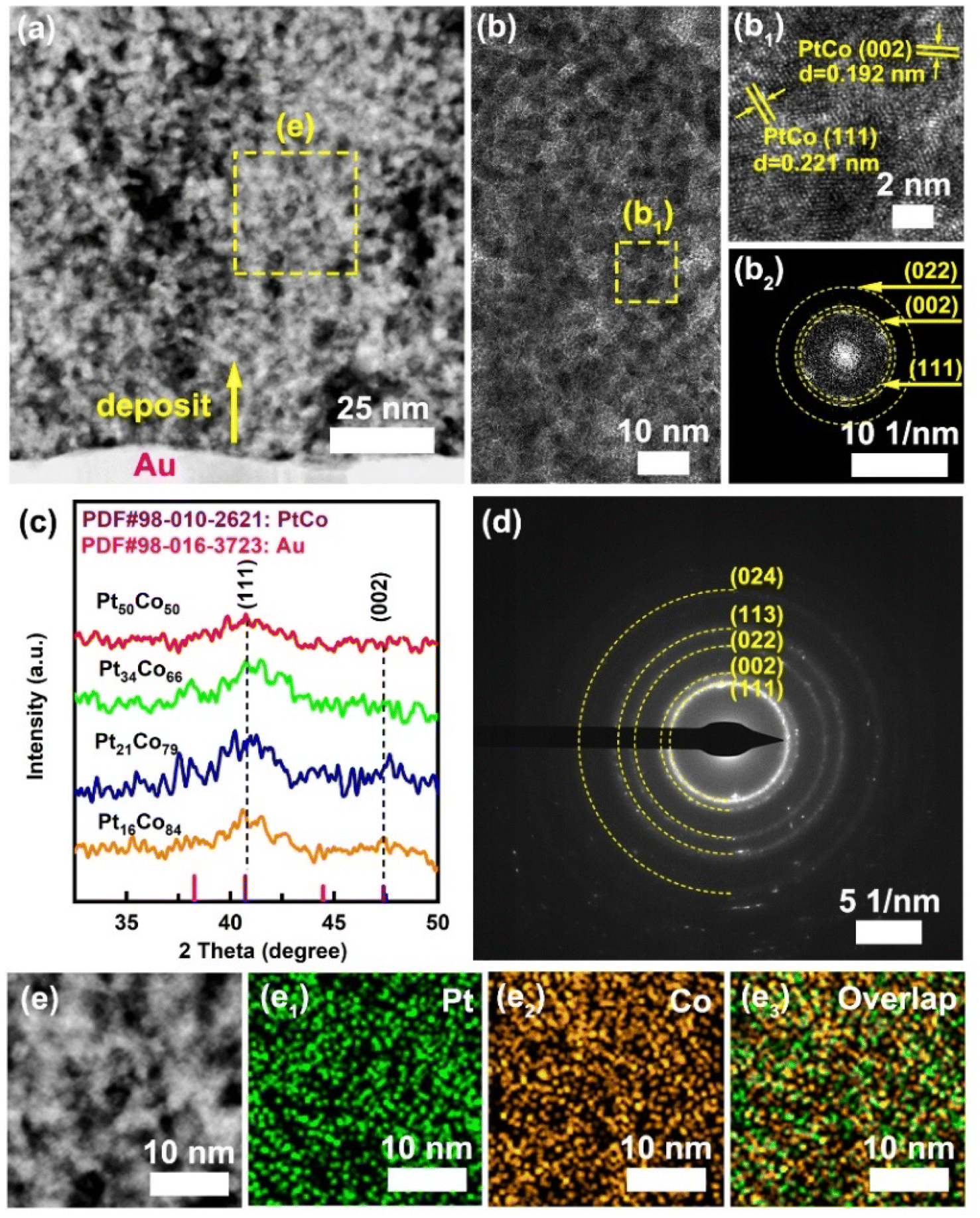 | ||
| Fig. 3 (a) TEM and (b) HRTEM images of the cross-section of the Pt34Co66 film, (b1) the enlarged image and (b2) FFT of the region (b1) in (b). (c) XRD patterns of Pt50Co50, Pt34Co66, Pt21Co79, and Pt16Co84 films. (d) SAED pattern of the Pt34Co66 film. TEM image (e) and the corresponding EDX mapping results (e1–e3). | ||
3.2 Electrochemical performances
The HER polarization curves and Tafel slopes of the deposited PtCo films are shown in Fig. 4. And Fig. S5† displays the LSV curves of the deposited Pt film. These tests were conducted in three different electrolytes: 1 M KOH, 1 M KOH + 3.5 wt% NaCl, and 1 M KOH + natural seawater. The electrocatalytic hydrogen evolution activities of commercial Pt/C and the bare substrate (the Au(125 nm)/Ti(25 nm)/Si sheet) were also assessed as a reference. The Au substrate shows weak HER activity, and its effect on the performance of the catalyst can be ignored. The overall HER performance is summarized in Table S3.† In the three electrolytes, the five catalysts exhibit very similar onset potentials, indicating that the initial electrochemical activity required to initiate the HER is comparable. However, difference emerges as the HER process progresses. Pt34Co66 shows the lowest overpotential compared to other catalysts at a current density of −10 mA cm−2, with a value of 35 mV in 1 M KOH, 33 mV in 1 M KOH + 3.5 wt% NaCl and 30 mV in 1 M KOH + natural seawater. At −50 mA cm−2 and −70 mA cm−2, Pt34Co66 continues to maintain the lowest overpotential in alkaline aqueous solution and alkaline simulated seawater. However, in alkaline natural seawater, the overpotential increases with the cobalt content, indicating that Pt50Co50 exhibits superior HER performance, with respective overpotentials of 71 mV at −50 mA cm−2 and 83 mV at −70 mA cm−2. This performance can be attributed to the presence of various corrosive ions in seawater, which can degrade the catalyst, particularly at high current densities. Notably, Pt50Co50, with the lowest surface area (as shown in Fig. 1b), may demonstrate better stability and corrosion resistance compared to the other samples. Moreover, the overpotentials at −10 mA cm−2 of the four PtCo films fall in the order of alkaline nature seawater < alkaline simulated seawater < alkaline aqueous solution. The presence and interaction of various ions in alkaline seawater can enhance ionic conductivity, provide buffering effects, facilitate favorable surface interactions, and improve catalyst stability, all of which contribute to better HER performance compared to pure alkaline conditions. The corresponding Tafel slopes are shown in Fig. 4b–d. In the three electrolytes, Pt34Co66 exhibits the smallest Tafel slope, which is 24 mV dec−1 in 1 M KOH, 22 mV dec−1 in 1 M KOH + 3.5 wt% NaCl, and 35 mV dec−1 in 1 M KOH + natural seawater, indicating that it has faster hydrogen evolution kinetics. Moreover, in both alkaline aqueous solution and alkaline simulated seawater, the rate-determining step for Pt34Co66 catalyzing the HER is the Heyrovsky step, while in alkaline natural seawater, it is the Volmer step. Interestingly, the overpotentials and Tafel slopes of the four deposits in alkaline natural seawater are significantly smaller than those of commercial Pt/C, indicating that the synergistic effect of Pt and Co species enhances the seawater hydrogen evolution performance. | ||
| Fig. 4 (a) LSV curves and (b) Tafel slopes of PtCo films in 1 M KOH. (c) LSV curves and (d) Tafel slopes of PtCo films in 1 M KOH + 3.5 wt% NaCl. (e) LSV curves and (f) Tafel slopes of PtCo films in 1 M KOH + natural seawater. | ||
Furthermore, in order to better understand the intrinsic electron transfer kinetics at the electrode/electrolyte interface, the EIS spectra of the deposited PtCo films and commercial Pt/C were measured. The corresponding fitting data are listed in Table S4.†Fig. 5d is the corresponding equivalent circuit diagram of Fig. 5a–c, where Rs represents the environmental internal resistance composed of the electrolyte and the test system, Rct represents the charge transfer resistance on the catalyst surface, CPE represents the constant phase element, and W represents the diffusion Warburg impedance. As illustrated in Fig. 5a–c, the Nyquist plots of the five catalysts in the three electrolytes are all composed of a small semicircle in the high frequency region and an inclined line in the low frequency region, corresponding to Rct and W, respectively. And the Rct values for Pt50Co50, Pt34Co66, Pt21Co79 and Pt16Co84 are quite small and similar to that of Pt/C, indicating that the deposited PtCo films possess good conductivity (as shown in the insets of Fig. 5a–c and details in Table S4†). This is mainly because the interaction between Pt and Co elements enhances electron transfer.27 However, the slope values of the lines corresponding to the Warburg impedance signal vary significantly. A larger slope value indicates a smaller W value, suggesting a faster reactant diffusion rate on the catalyst surface.28,29 In alkaline aqueous solution, the W value of Pt34Co66 is 1.52 Ω, which is smaller than that of Pt50Co50 (1.72 Ω), Pt21Co79 (2.43 Ω), Pt16Co84 (3.08 Ω), and Pt/C (11.50 Ω) (Fig. 5a). For alkaline simulated seawater, the W values of Pt50Co50 (1.38 Ω), Pt34Co66 (1.47 Ω), Pt21Co79 (1.95 Ω) and Pt16Co84 (1.43 Ω) are similar to each other, and all are significantly smaller than that of Pt/C (11.85 Ω). This trend is consistent with the observations in alkaline seawater (Fig. 5b and c). These results indicate that the deposited PtCo films possess faster electron transfer and reactant diffusion rates during the HER process than commercial Pt/C, which is due to the porosity.
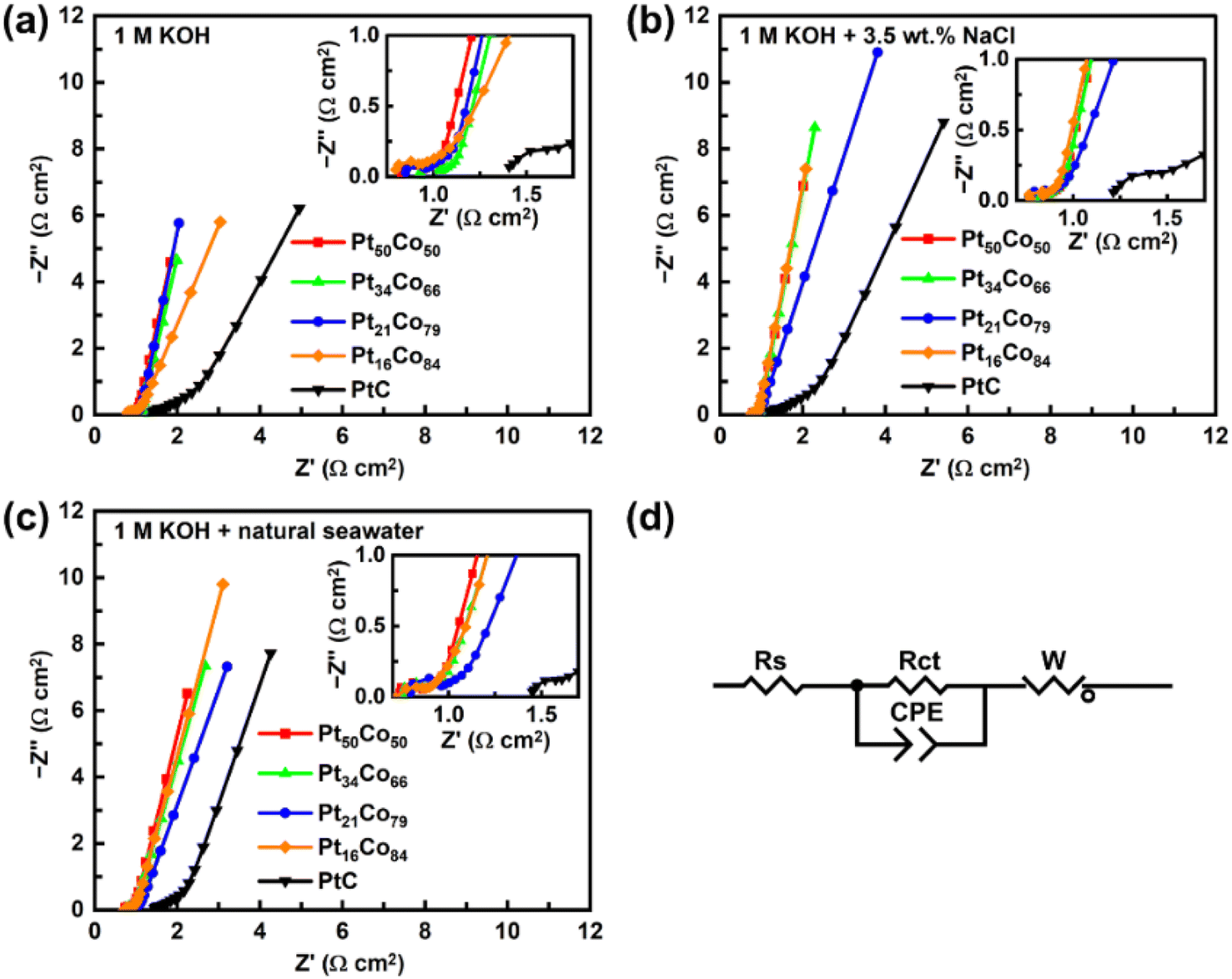 | ||
| Fig. 5 Nyquist plots of PtCo films in (a) 1 M KOH, (b) 1 M KOH + 3.5 wt% NaCl, and (c) 1 M KOH + natural seawater, respectively (inset: Nyquist diagram of the high-frequency region). (d) An equivalent circuit proposed for EIS measurements. | ||
In order to verify the above speculation, CV tests were performed on the catalysts in alkaline simulated seawater, and the results are shown in Fig. 6. From the CV curves, the deposited PtCo films exhibit better rectangularity than commercial Pt/C, indicating that these deposits facilitate the diffusion of electrolytes during the HER process, which is consistent with the previous results. In addition, the calculated Cdl value of Pt34Co66 is 3.48 mF, which is larger than that of Pt50Co50 (1.89 mF), Pt21Co79 (2.53 mF), Pt16Co84 (2.45 mF), and Pt/C (1.88 mF) (Fig. 6f). The largest Cdl value of Pt34Co66 may be due to the optimal electron accumulation around Pt (Fig. 2c and d). The calculated ECSA and RF values are shown in Table S5,† indicating that Pt34Co66 surpasses other catalysts in both aspects, with values of 86.94 cm2 and 347.75, respectively. The substantial RF provides further evidence for the large surface area of the Pt34Co66 film. In other words, the largest ECSA and RF of Pt34Co66 can be attributed to its unique hierarchical porous structure, consisting of aggregates of nanonodules. This structure enhances electrolyte transport efficiency, and thus promotes fast HER kinetics (Fig. 4a, c and f). In addition, after ECSA normalization, the HER activities of PtCo films are positively correlated with the content of Pt, indicating that Pt species is still the main HER active site in the catalyst (Fig. S6†).
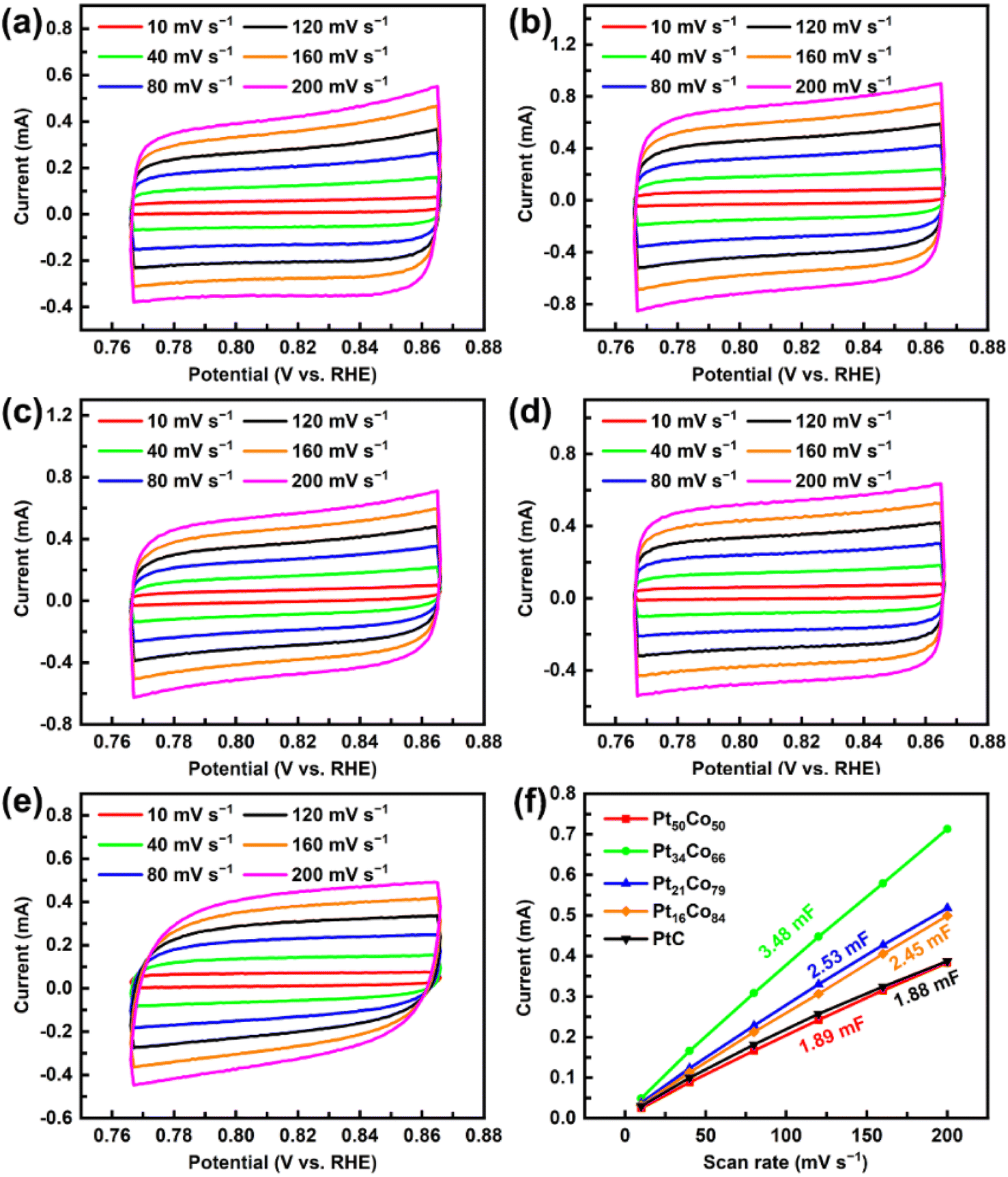 | ||
| Fig. 6 (a)–(e) CV and (f) Cdl curves of PtCo films in 1 M KOH + 3.5 wt% NaCl. | ||
3.3 Stability and durability
Fig. 7 presents the stability and durability results of Pt34Co66 and commercial Pt/C during continuous hydrogen evolution in the three electrolytes. In alkaline aqueous solution, both Pt34Co66 and Pt/C exhibit high durability, with only a minor drop in overpotential (Fig. 7a). However, in alkaline simulated seawater and alkaline natural seawater, Pt34Co66 maintains its high durability, whereas the overpotential of Pt/C decreases significantly (Fig. 7b and c). This degradation in Pt/C is attributed to the corrosion of Pt by chloride ions, resulting in a sharp decline in its stability. As displayed in Table S6,† after the long-term aging stability test in 1 M KOH + natural seawater, the species containing Mg and Ca elements were formed on the surface of the catalyst. These products may be insoluble precipitates formed by metal cations such as Mg2+ and Ca2+, which will cause blockage of active sites.30–32 Moreover, the overpotential of Pt/C decreases more sharply in alkaline seawater, which is because the insoluble precipitates block the active sites, further aggravating the decrease in durability. The excellent HER stability and durability of Pt34Co66 are attributed to its unique hierarchical porous structure, composed of aggregates of nanonodules. On the one hand, the increased number of active sites could compensate for the decrease in catalytic activity caused by impurity blockage. On the other hand, the accumulation of electrons on the more electronegative Pt repels Cl−, thereby reducing the corrosion by chlorides in the process of seawater hydrogen evolution (Fig. 7d and formulae (S11)–(S13)†). | ||
| Fig. 7 Chronoamperometric curves of the Pt34Co66 film and commercial Pt/C in (a) 1 M KOH, (b) 1 M KOH + 3.5 wt% NaCl, and (c) 1 M KOH + natural seawater. (d) Schematic showing electron accumulation brought about by Pt34Co66 composed of aggregates of nanonodules that facilitates resistance to chloride corrosion. | ||
XPS analyses of Pt 4f and Co 2p of the Pt34Co66 film in the as-deposited state and after the long-term stability test were conducted to further evaluate the stability. As shown in Fig. 8a, the Pt 4f peak of the as-prepared Pt34Co66 exhibits two peaks at 71.48 eV and 74.88 eV after deconvolution, corresponding to Pt 4f7/2 and Pt 4f5/2, respectively. This indicates the presence of metallic platinum. It is worth mentioning that compared with pure Pt (71.2 and 74.55 eV), the Pt 4f peaks of Pt34Co66 are shifted to higher energies upon alloy formation with Co, which is in line with the reported theory.33,34 Meanwhile, the upward shift of the core level of the PtCo alloy is due to the charge transfer from Co to Pt atoms.34,35 Moreover, the Pt 4f signals of as-deposited Pt appear as two peaks at 71.6 eV and 74.9 eV after deconvolution, which also correspond to Pt 4f7/2 and Pt 4f5/2 of metallic platinum, respectively (Fig. S7†). However, Co 2p can be deconvoluted into 10 peaks (Fig. 8b). First, two peaks at 778.28 eV and 792.98 eV are attributed to Co (0) 2p3/2 and Co (0) 2p1/2, respectively. Peaks located at 779.98 e V and 795.28 e V correspond to Co(III) 2p3/2 and Co(III) 2p1/2. Additionally, two peaks at 781.18 eV and 797.88 eV are designated to Co(II) 2p3/2 and Co(II) 2p1/2. And the remaining four peaks at 786.08 eV, 789.78 eV, 801.88 eV and 805.08 eV are satellite peaks of Co(II) and Co(III). These results indicate that both metallic cobalt and oxidized cobalt (Co3O4 and/or CoO) exist on the surface of the as-prepared Pt34Co66 film. This is likely due to the reaction of Co metal with oxygen, leading to surface passivation. Notably, metal oxides can serve as effective additional adsorption sites that promote water splitting.36–38 After the 24 h stability test, as displayed in Fig. 8c, Pt species is still present in the metallic state, with no significant changes in the XPS spectrum. As for Co 2p, two peaks at 778.08 eV and 792.78 eV can be assigned to Co (0) 2p3/2 and Co (0) 2p1/2, indicating that metallic cobalt remains on the surface. However, the binding energies of trivalent and divalent cobalt become higher. And four new peaks at 780.78 eV, 782.78 eV, 795.98 eV, and 798.58 eV correspond to Co(III) 2p3/2, Co(II) 2p3/2, Co(III) 2p1/2 and Co(II) 2p1/2, indicate the formation of CoOOH and Co(OH)2. It is worth noting that CoOOH and Co(OH)2 can significantly increase the active sites of the catalyst and help reduce the water dissociation energy barrier during the alkaline HER process.39,40 These results demonstrate that the introduction of Co species in the PtCo films can protect Pt while enhancing HER performance.41,42 Unfortunately, as displayed in Table S6,† the content of Pt and Co is both 0.01 at% in the electrolyte, indicating that the precipitation of Pt and Co in the PtCo film occurs during the long-term continuous HER process.
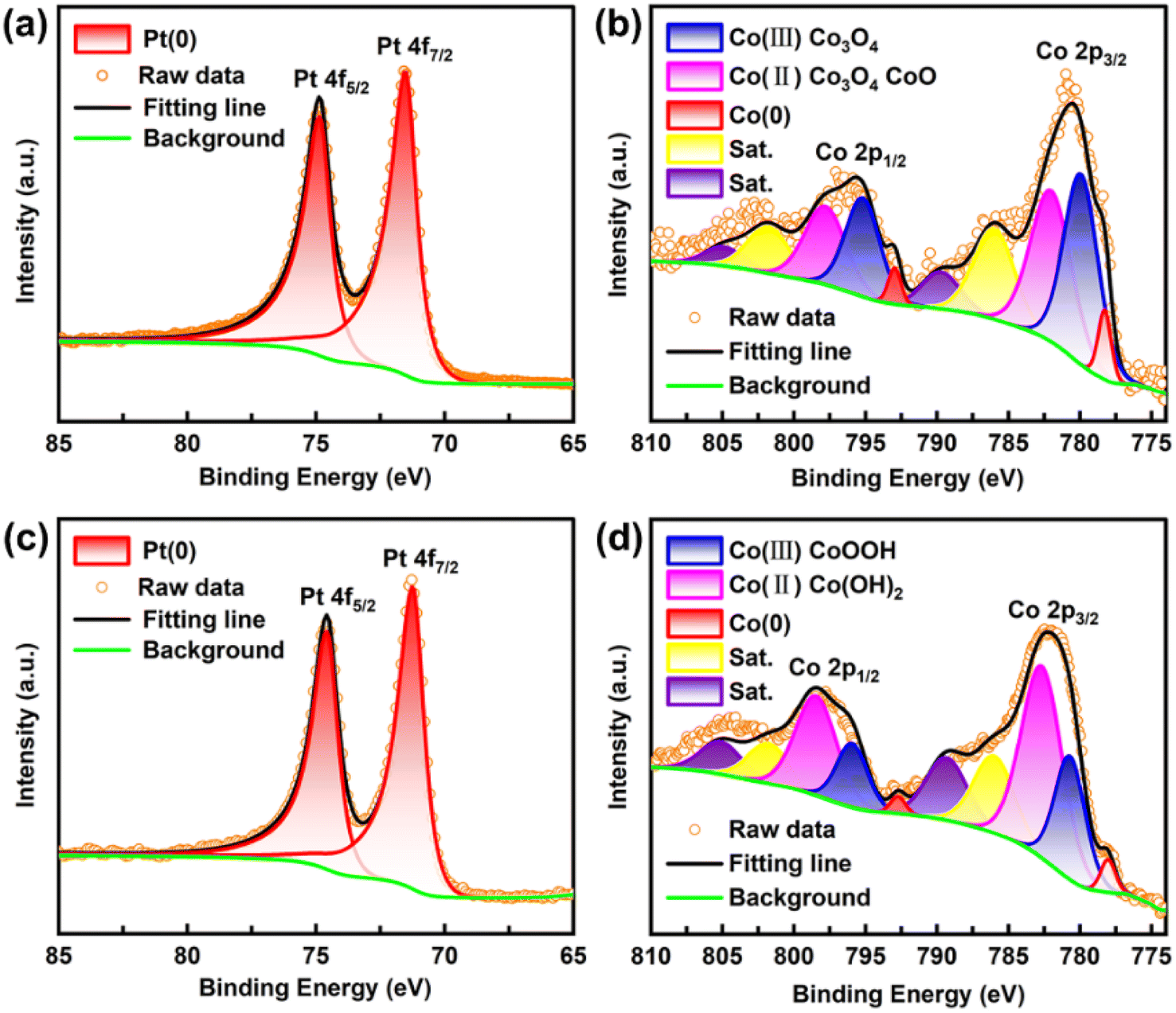 | ||
| Fig. 8 High resolution XPS spectra of the Pt34Co66 film: (a) Pt 4f and (b) Co 2p of the as-prepared state, (c) Pt 4f and (d) Co 2p after the long-term aging stability test in 1 M KOH + natural seawater. | ||
In summary, for the Pt34Co66 film, the incorporation of a certain proportion of Co into Pt changes the electronic structure of Pt and improves the electrocatalytic behavior in alkaline seawater.43 The unique hierarchical porous structure enriches the active sites and enhances the intrinsic activity of the catalyst. Additionally, some Co species convert into CoOOH and Co(OH)2 during the alkaline HER process, increasing the adsorption sites of water molecules and reducing the water dissociation energy barrier.39,40,44
4. Conclusions
Hierarchical porous Co-rich PtCo thin films have been successfully prepared by one-step F127 assisted electrodeposition. The applied potential has been varied to adjust the composition and morphology of the films. Notably, PtCo films with Co content ranging from 50 at% to 84 at% and tunable porosity have been obtained. SEM and TEM images demonstrate the occurrence of a hierarchical porous structure, composed of aggregates of nanonodules. XRD, HRTEM, and SAED analyses indicate the successful construction of an fcc PtCo alloy. The prepared hierarchical porous PtCo films exhibit enhanced HER activity over Pt/C in alkaline aqueous solution, alkaline simulated seawater and alkaline natural seawater. In particular, Pt34Co66 shows the highest specific activity and long-term stability. ECSA and RF reveal that the unique hierarchical porous structure increases the active sites and enhances electrolyte transport efficiency, resulting in faster hydrogen evolution kinetics. XPS analysis revealed that the Pt34Co66 component offers the largest electron accumulation and lowest binding energy around Pt, contributing to its superior stability and durability in natural seawater electrolysis. And the Co element can protect Pt species in the long-term continuous hydrogen evolution process. The results presented herein have the potential to promote the large-scale development of seawater hydrogen production.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. Z.: design of experiments, data analysis, writing – original draft. H. C.: investigation, visualization. Z. Y. and J. Y.: methodology, visualization. J. Z.: writing – review and editing, conceptualization, supervision, funding acquisition. H. M.: writing – review and editing, conceptualization. R. G.: supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Liaoning Province “Xingliao Talent Plan” (XLYC2002070), Young Elite Scientists Sponsorship Program by CAST (2022QNRC001), and Dalian High-level Talents Innovation Support Program (2021RD06) is acknowledged.References
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33(31), 2007100 CrossRef CAS PubMed.
- X. F. Zhang, W. H. Huang, L. Yu, M. G. Melchor, D. S. Wang, L. J. Zhi and H. B. Zhang, Carbon Energy, 2024, 6, 3 CAS.
- X. M. Xu, Z. P. Shao and S. P. Jiang, Energy Technol., 2022, 10(11), 2200573 CrossRef CAS.
- S. X. Zhang, W. W. Xu, H. C. Chen, Q. H. Yang, H. Liu, S. J. Bao, Z. Q. Tian, E. Slavcheva and Z. Y. Lu, Adv. Mater., 2024, 36, 37 Search PubMed.
- S. W. Xu, J. Y. Li, N. Zhang, W. Shen, Y. Zheng and P. X. Xi, Chem. Commun., 2023, 59(65), 9792–9802 RSC.
- Q. Liu, S. J. Sun, L. C. Zhang, Y. S. Luo, Q. Yang, K. Dong, X. D. Fang, D. D. Zheng, A. A. Alshehri and X. P. Sun, Nano Res., 2022, 15(10), 8922–8927 CrossRef CAS.
- H. Y. Jin, X. Liu, A. Vasileff, Y. Jiao, Y. Q. Zhao, Y. Zheng and S. Z. Qiao, ACS Nano, 2018, 12(12), 12761–12769 CrossRef CAS PubMed.
- L. Yu, Q. Zhu, S. W. Song, B. McElhenny, D. Z. Wang, C. Z. Wu, Z. J. Qin, J. M. Bao, Y. Yu, S. Chen and Z. F. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- W. J. Zang, T. Sun, T. Yang, S. B. Xi, M. Waqar, Z. K. Kou, Z. Y. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33(8), 2003846 CrossRef CAS PubMed.
- Y. Kuang, M. J. Kenney, Y. T. Meng, W. H. Hung, Y. J. Liu, J. E. Huang, R. Prasanna, P. S. Li, Y. P. Li, L. Wang, M. C. Lin, M. D. McGehee, X. M. Sun and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(14), 6624–6629 CrossRef CAS PubMed.
- X. Q. Zhang, Y. X. Xiao, G. Tian, X. Yang, Y. Dong, F. Zhang and X. Y. Yang, Chemistry, 2023, 29(5), e202202811 CrossRef CAS PubMed.
- J. Su, Q. Wang, M. Fang, Y. Wang, J. Ke, Q. Shao and J. Lu, ACS Appl. Mater. Interfaces, 2023, 15(44), 51160–51169 CrossRef CAS PubMed.
- X. Chen, Y. R. Duan, H. L. Dou, W. S. Zhang and X. M. Wang, Chem. Eng. J., 2024, 499, 155925 CrossRef CAS.
- R. Andaveh, A. S. Rouhaghdam, J. P. Ai, M. Maleki, K. Wang, A. Seif, G. B. Darband and J. Y. Li, Appl. Catal., B, 2023, 325(14), 122355 CrossRef CAS.
- J. Liang, Z. X. Li, X. He, Y. S. Luo, D. D. Zheng, Y. Wang, T. S. Li, B. W. Ying, S. J. Sun, Z. W. Cai, Q. Liu, B. Tang and X. P. Sun, Mater. Today, 2023, 69, 193–235 CrossRef CAS.
- J. Y. Liu, S. Duan, H. Shi, T. Y. Wang, X. X. Yang, Y. H. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, 45 Search PubMed.
- A. S. Nugraha, J. Na, M. S. A. Hossain, J. J. Lin, Y. V. Kaneti, M. Iqbal, B. Jiang, Y. Bando, T. Asahi and Y. Yamauchi, Appl. Mater. Today, 2020, 18, 100526 CrossRef.
- C. Zhang, M. S. Xia, Z. P. Liu, G. Q. Huang, S. S. Yuan, J. Ai, N. Li and X. T. Li, ChemCatChem, 2020, 12(9), 2589–2594 CrossRef CAS.
- C. Fu, L. Feng, H. Yin, Y. Li, Y. Xie, Y. Feng, Y. Zhao, L. Cao, J. Huang and Y. Liu, Nanoscale, 2022, 14(39), 14779–14788 RSC.
- G. Huang, C. Zhang, Z. Liu, S. Yuan, G. Yang, K. Wang, X. Li, N. Li and S. Jing, ChemElectroChem, 2020, 7(24), 4943–4948 CrossRef CAS.
- E. Isarain-Chávez, M. D. Baró, E. Pellicer and J. Sort, Nanoscale, 2017, 9(45), 18081–18093 RSC.
- M. L. Zhang, Y. H. Shi, X. C. Li, J. Zhang, X. L. Chen, Z. H. Zhou and R. G. Guan, Fuel, 2025, 381, 133288 CrossRef CAS.
- H. J. Wang, L. Wang, T. Sato, Y. Sakamoto, S. Tominaka, K. Miyasaka, N. Miyamoto, Y. Nemoto, O. Terasaki and Y. Yamauchi, Chem. Mater., 2012, 24(9), 1591–1598 CrossRef CAS.
- T. Sakai and P. Alexandridis, J. Phys. Chem. B, 2005, 109(16), 7766–7777 CrossRef CAS PubMed.
- K. Eiler, S. Suriñach, J. Sort and E. Pellicer, Appl. Catal., B, 2020, 265, 118597 CrossRef CAS.
- K. Eiler, J. Fornell, C. Navarro-Senent, E. Pellicer and J. Sort, Nanoscale, 2020, 12(14), 7749–7758 RSC.
- Y. Liu, Y. Wang, P. Yang and G. Gao, Int. J. Hydrogen Energy, 2021, 46(75), 37311–37320 CrossRef CAS.
- M. P. Dabir, S. M. Masoudpanah and M. Mamizadeh, J. Energy Storage, 2023, 70, 107951 CrossRef.
- N. Chauhan, H. W. Choi, M. Kumar and D. H. Yoon, Electrochim. Acta, 2023, 460, 142634 CrossRef CAS.
- T. Van Tam, K. C. Bhamu, M. J. Kim, S. G. Kang, J. S. Chung, S. H. Hur and W. M. Choi, Chem. Eng. J., 2024, 480, 148190 CrossRef CAS.
- L. B. Wu, L. Yu, B. McElhenny, X. X. Xing, D. Luo, F. H. Zhang, J. M. Bao, S. Chen and Z. F. Ren, Appl. Catal., B, 2021, 294, 120256 CrossRef CAS.
- H. Shi, T. Y. Wang, J. Y. Liu, W. W. Chen, S. Z. Li, J. S. Liang, S. X. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. H. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14(1), 3934 CrossRef CAS PubMed.
- Y. Vasquez, A. K. Sra and R. E. Schaak, J. Am. Chem. Soc., 2005, 127, 12504–12505 CrossRef CAS PubMed.
- T. T. Yang, H. Zhu, M. Wan, L. Dong, M. Zhang and M. L. Du, Chem. Commun., 2016, 52, 990–993 RSC.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, J. Sort, E. Pellicer and R. Guan, Int. J. Hydrogen Energy, 2024, 59, 625–634 CrossRef CAS.
- F. Zhang, R. J. Ji, Y. H. Liu, Y. Pan, B. P. Cai, Z. J. Li, Z. Liu, S. C. Lu, Y. T. Wang, H. Jin, C. Ma and X. L. Wu, Appl. Catal., B, 2020, 276, 119141 CrossRef CAS.
- M. Lee, G. Yoon, M. K. Kim, J. Hong, S. Lee and G. H. Ryu, J. Alloys Compd., 2024, 976, 173282 CrossRef CAS.
- X. R. Kong, T. H. Hao, C. C. Lv, Z. Y. Li, F. D. Zhao, S. B. Ning and J. H. Ye, Appl. Surf. Sci., 2024, 664, 160261 CrossRef CAS.
- N. Mahmood, Y. Yao, J. W. Zhang, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2018, 5(2), 1700464 CrossRef PubMed.
- F. R. Nikkuni, L. Dubau, E. A. Ticianelli and M. Chatenet, Appl. Catal., B, 2015, 176, 486–499 CrossRef.
- F. L. Jiang, F. J. Zhu, F. Yang, X. H. Yan, A. M. Wu, L. X. Luo, X. L. Li and J. L. Zhang, ACS Catal., 2020, 10(1), 604–612 CrossRef CAS.
- L. Y. Jiang, X. X. Lin, A. J. Wang, J. H. Yuan, J. J. Feng and X. S. Li, Electrochim. Acta, 2017, 225, 525–532 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation process of the Pt/C electrode, Tafel slope and the HER mechanism, calculation formulae of ECSA and RF, CV of the electrolyte on a vitreous carbon electrode under stagnant conditions, magnified SEM images of Pt50Co50, Pt21Co79, and Pt16Co84, particle size distribution statistics of PtCo films, binding energies for Pt 4f and Co 2p signals of as-deposited PtCo thin films, high resolution XPS spectra of as-deposited Pt50Co50, Pt21Co79, and Pt16Co84 films, separate fitting results of the XPS peaks of as-deposited PtCo films, LSV curves and SEM image of as-deposited Pt films, HER performance of the deposited PtCo thin films, Pt, and commercial Pt/C in different electrolytes, EIS spectrum fitting data of the deposited PtCo thin films and commercial Pt/C in different electrolytes, Cdl values, ECSA, and RF of the deposited PtCo thin films and commercial Pt/C obtained in 1 M KOH + 3.5 wt% NaCl, LSV curves normalized by ECSA, composition of Pt34Co66 (the as-prepared state and after the long-term aging stability test in 1 M KOH + natural seawater) and electrolyte (1 M KOH + natural seawater after the long-term aging stability test) determined by ICP-MS, chlorine corrosion mechanism of the seawater HER, and high resolution XPS spectra of Pt 4f peaks of Pt deposited. See DOI: https://doi.org/10.1039/d4ta08678c |
| This journal is © The Royal Society of Chemistry 2025 |