
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the mechanistic role of the catalyst layer microstructure in proton exchange membrane water electrolyzers†
Navneet
Goswami‡
 ,
Abhinand
Ayyaswamy‡
,
Anindya
Nath
,
Bairav S.
Vishnugopi
and
Partha P.
Mukherjee
*
,
Abhinand
Ayyaswamy‡
,
Anindya
Nath
,
Bairav S.
Vishnugopi
and
Partha P.
Mukherjee
*
School of Mechanical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: pmukherjee@purdue.edu
First published on 25th March 2025
Abstract
Proton exchange membrane water electrolyzers (PEMWEs) are prime candidates that can fulfill the target demands of decarbonization through green hydrogen (H2), facilitated by electrochemical water-splitting reactions at low temperatures. In this context, the fast scale-up and cost competitiveness of PEMWE technology necessitate a substantial reduction in expensive catalyst loadings and energy consumption without compromising H2 throughput. We explore avenues to achieve high PEMWE efficacy through our mechanistic modeling framework by evaluating the pertinent electrode architecture and operational metrics. The influence of operating stressors in dictating key performance metrics, such as specific energy consumption, has been highlighted. The microstructural assessment of catalyst layers informs the dominance of associated resistance modes governed by the kinetic transport interplay at the mesoscale level. We identify regimes of limiting PEMWE electrochemical efficacy derived from the conjunction of electrode design and operating conditions, mainly impacted by the variation in ionic, electronic, and species percolation networks. The intrinsic nature of reaction pathways and transport reveals the primary source of electrode-centric limitations affecting the overall performance of PEMWE systems, as analyzed from a resistance evolution perspective. The confluence of PEMWE electrode design and its operational landscape previews a comprehensive insight into the inherent trade-offs controlled by the overall energy consumption and concomitant H2 throughput, steering towards an impactful pathway to decarbonization through water electrolysis.
Introduction
With a rapid rise in the global average temperatures necessitating a change in the existing energy ecosystem, hydrogen (H2) has emerged as the cornerstone of a net zero emission-based infrastructure by 2050. The efforts to usher in a green H2 economy have seen significant strides with the Department of Energy's (DOE) ‘1 1 1’ initiative to reduce the cost of clean H2 by 80% to $1 per kg in a decade.1 This can be primarily attributed to the merits offered by H2 through its carbon-free characteristics and ultra-high energy density (140 MJ kg−1 or equivalently 39200 W h kg−1).2 H2 is already viewed as an important energy carrier mainly produced from fossil fuel reserves.3 Currently, 95% of H2 in the United States is produced via the steam-methane reforming process4 that primarily employs methane as a feedstock.5,6 Low temperature water electrolysis (LTWE) can play a pivotal role in enabling transformative strategies of green H2 production with low carbon intensity. Unsurprisingly, green H2 is touted to decarbonize major high carbon footprint industries7,8 such as cement, steel, fertilizers, and metal refining, in addition to unlocking opportunities in the mobility sector, including heavy-duty transportation and shipping through its deployment as fuel cells.9 The proton exchange membrane water electrolyzer (PEMWE) garners attention through its favorable LTWE capability and its operability with fluctuating renewable energy production.10 However, high capital and operational expenditures pose a significant hindrance to its widespread adoption.11 A myriad of challenges currently plague PEMWE technology, including high iridium (Ir) loadings at the anode catalyst layer (ACL), corrosion propensity of the support structure, mass transport challenges from gas bubble formation, and the lack of novel, inexpensive electrocatalysts with high activity and stability to sustain the slow oxygen evolution reaction (OER).12–14 The escalated price and limited supply of Ir15 employed to catalyze the OER at the ACL serve as a key bottleneck; therefore, concerted efforts are in place to find suitable replacements.16Various physicochemical phenomena control the resistances emanating from the electrode architecture, which eventually dictates the path toward achieving superior electrochemical responses based on the DOE's stipulated target current densities.17 The ionomer present in the ACL serves as a binding agent to the catalyst particles, ensuring structural integrity while providing percolation pathways for the protons through the porous microstructure.18 A judicious amount of the ionomer content is crucial as it affects the key attributes of proton conductivity and pore phase tortuosity. The ionomer content and its interfacial morphology is a net outcome of the electrode processing conditions19 that can influence its associated kinetic and transport processes. Hence, a rational catalyst layer (CL) design based on the ionomer content and morphology can suppress the pertinent overpotential modes. Optimizing the catalyst loading improves catalyst utilization to enhance the reaction efficacy.20–22 Recently, several studies reported the beneficial impact of the porous transport layer (PTL) on CL utilization.23–28 Tailoring the nanostructures of catalyst particles responsible for electrochemical reactions was also decisive in controlling mass transport limitations. Shape engineering at a local particle level29 through morphological modification (spherical vs. needles vs. rods) is a promising avenue toward improving the efficacy of multimodal interactions.
A distinctive signature of PEMWEs is the electro-generation of gas bubbles in the electrodes.30 Typically, the emergence of bubbles progresses through a series of mechanisms such as nucleation, growth, coalescence, and departure.31,32 The presence of bubbles passivates the reaction sites and impacts the local microenvironments, such as the thermal and pH fields.33,34 Some of the earlier works of Vogt35–37 relate the bubble coverage nonlinearly with the current density through empirical relationships. Vogt's equation represents the relationship of bubble coverage with the current density in a stagnant electrolyte and is dependent on several factors, including the wettability of the electrodes, operating conditions, etc. A recent study by Iwata et al.38 used alkaline water splitting as an exemplary electrochemical system to show the relationship among porous electrode wettability, bubble dynamics, and the resulting transport overpotential. The progress in manufacturing techniques during the past decade has resulted in an increasing popularity of surface-engineered micro/nanostructured electrodes with multi-functionalities in water-splitting devices.39 Current research has increasingly focused on realizing the potential of PEMWEs to drive rapid decarbonization by exploring their underlying mechanisms through physics-based modeling. Falcao and Pinto40 provided a detailed review covering state-of-the-art models related to PEMWE technology. Benmehel et al.41 highlighted the integration of approaches among various sub-components of a thermo-electrochemical model and the challenges associated with physics-based modeling of PEMWEs. Recent modeling works have primarily probed the transport phenomena in individual cell components such as PTLs and CLs using pore-scale modeling.42–46 Furthermore, recent models capture degradation events under harsh operating environments, such as chemical degradation of the membrane47 and CL fracture.48 To optimize cell performance, macroscopic models combining heat transfer, charge transport, mass balance, and two-phase flow were employed. García-Salaberri49 presented a one-dimensional, two-phase, non-isothermal model of a PEMWE to delineate the impact of design parameters on the efficacy of electrochemical performance. Comprehensive three-dimensional models developed by Zhou et al.50 and Xu et al.51 reveal the concomitant water and heat distribution characteristics under various operating regimes.
Despite advancements in physics-based analysis, prior modeling efforts still rely on empirical relationships to provide insights into the underlying mechanisms,52–59 that govern PEMWE efficiency. We posit that gaps remain in capturing the pore-scale microstructural attributes of the CLs. Additionally, predicting the electrochemical response and efficacy under the operational landscape of the PEMWE by linking electrode-scale features to the macroscale performance warrants comprehensive investigation. To address these challenges, we have developed a mechanistic modeling framework that unifies microstructure-kinetics-transport interplay to predict PEMWE performance under various operational environments by bridging the implications of electrode recipes and operational metrics under the cognizance of bubble dynamics. We hypothesize that the electrochemical response of a PEMWE is dependent on a host of variables such as operating parameters (current density, temperature, relative humidity, and pressure), microstructural parameters (electrode recipes), and kinetic parameters (material and surface properties). In this work, we reveal the connection between the operational and electrode level descriptors on the overall electrochemical landscape of PEMWEs. Through our mechanistic modeling framework, we decouple the trade-offs between power expenditure and H2 throughput viewed from an electrode engineering lens facilitated through systematic resistance breakdown analysis. We explore the implications of microstructural attributes on kinetic transport interactions through specific energy consumption. By understanding these synergistic electrode-centric mechanisms informed through trade-offs and operational landscape, we reveal the challenges of the limiting electrode in PEMWEs and its pertinent response under various scenarios.
Results and discussion
In this study, we investigated the electrochemical response of a standard PEMWE that constitutes a titanium PTL and Ir oxide CL in the anode, carbon paper as PTL and platinum nanoparticles dispersed on a carbon-supported CL in the cathode, separated by a proton exchange membrane. This representative PEMWE system has various subcomponents of a single cell sandwich that converts input feed water from the anode into a binary mixture of H2 and water (H2O) on the cathode extracted from the gas channels (Fig. 1(a)). | ||
| Fig. 1 (a) Schematic of PEMWE depicting various layers in a single cell sandwich and its constituent phases in stochastically reconstructed microstructures. PTL, CL, PEM and BP indicate the porous transport layer, catalyst layer, proton exchange membrane and bipolar plate, respectively. (b) A representative electrochemical response of the validated PEMWE framework against experimental data from Han et al.59 | ||
Our PEMWE framework (Fig. 1(b)) agrees well with the experimental data of Han et al.59 (see Table S3 for cell parameters and Fig. S7† for polarization under high currents). By incorporating the relevant effects of bubble dynamics (Fig. S1†), this validation process allows our model to explore the various mechanistic interactions in a PEMWE over a wide spectrum of design considerations and operational specifications. A detailed description of the computational framework and the modeling parameters is provided in the ESI (Section S1 and Table S2).† The polarization response shows a non-linear scaling of energy consumption with applied current, implying varying dominance of resistance modes that dictate the rise of overpotential and its associated H2 production. We discern that the polarization signature, H2 throughput, and the overall efficacy informed through the specific energy consumption (SEC) of the PEMWE (Section S5†) are governed by a complex interplay of its operating conditions and the choice of electrode design. Therefore, a comprehensive investigation of the key electrode-centric limitations, operational landscape, and design criterion is critical to enable the long-term viability of green H2 production through electrolysis for decarbonization.
Temperature and relative humidity (RH) of inlet feed water (FW) are crucial in influencing the polarization behavior and H2 throughput from the PEMWE. Details pertaining to the configuration of the PEMWE and its reference operating conditions for all the subsequent analyses conducted in this work are presented in Table S2 of the ESI.† Our computational framework emulates a vapor-fed electrolyzer,60 where RH is defined as the ratio of actual partial pressure of water vapor in the inlet stream to the saturation pressure of water vapor at that FW temperature. Hence, H2 throughput at the CPTL|BP interface is vaporous, presenting various mechanistic implications on the proportion of H2O and H2 at the cathode exit across a spectrum of operating and design conditions. Various mass transport mechanisms including diffusive transport and electro-osmotic drag control the relative concentration of H2O in either electrode. Fig. 2(a) shows the distinct polarization signature influenced by variation in FW temperature. The voltage response of the PEMWE at different current densities is marked by the rise of distinct mechanistic interactions at the mesoscale level. At low current densities, the activation overpotential at the reaction interface significantly contributes to the overpotential; moderate current densities signal the growth of ohmic overpotentials and very high current densities (>2 A cm−2) present the coupled ascension of ohmic and concentration overpotentials resulting from mass-transport limitations.
 | ||
Fig. 2 Implications of operating conditions on the electrochemical performance and efficacy of PEMWE. (a) Influence of feed water temperature on the polarization curve and (b) resistance contributions at 50 °C and 90 °C. (c) Impact of feed water humidity on the polarization curve and (d) resistance contributions at 40% and 100% relative humidity of feed water. The interplay of feed water humidity and temperature on (e) H2 throughput, (f) power consumption, and (g) specific energy consumption (SEC) of a PEMWE stack with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. The PEMWE is operated at a current density of 2 A cm−2 for all cases. 000 cells. The PEMWE is operated at a current density of 2 A cm−2 for all cases. | ||
With increased FW temperatures, the activation overpotential and open-circuit potential decrease due to faster interfacial reaction kinetics stemming from higher exchange current densities in the electrodes. We clearly observe a reduction of 171.3 mV at 0.01 A cm−2 when the water temperature increases from 50 °C to 90 °C (Fig. 2(a)). As the ohmic overpotentials begin to dominate, the rate of increase in voltage is greater, especially at low temperatures, with 1.69 Ω cm2 at 50 °C compared to 1.54 Ω cm2 at 90 °C measured at 1.5 A cm−2. Lower temperatures significantly reduce the protonic conductivity of the system by establishing severe electrolyte gradients in the cell, causing higher ohmic overpotentials (Section S3†). A direct implication of this effect is presented in Fig. 2(b), where we witness a drastic reduction in the ionic resistance with increased temperature (Section S3†). Despite ionic resistance significantly contributing to the overall electrode resistance, there is a reduction in the cathode kinetic resistance and total electronic resistances derived from higher reaction current densities in the CL. A slight increase in kinetic resistance in the ACL is attributed to the influence of decreasing thermodynamic potential (1.21 V at 50 °C vs. 1.17 V at 90 °C) against the evolved overpotential in the electrode.
While mass transport resistances are relatively subdued compared with other modes (ionic, electronic and kinetic) owing to the choice of PEMWE design in this work, understanding the role of operating stressors on major underlying mechanisms like bubble dynamics is critical. Further details on the incorporation of bubble coverage and its physics are described in detail in Section S2 of the ESI.† Under identical bubble formation characteristics, surface attributes (e.g., contact angle) and operating conditions, we observe a distinctive behavior of mass-transport losses with FW temperatures (Fig. S6†). Higher temperatures raise the reaction kinetics, contributing to higher numbers of bubble–surface interactions manifesting as higher bubble coverage on electrochemical active surfaces. An increased bubble radius at elevated temperatures negatively impacts polarization by increasing the mass-transport resistance through increased bubble overpotential. This increased bubble coverage can lower reactive areas and impact the kinetic resistance, despite faster reaction rates.
Interestingly, the H2 throughput inversely scales with temperature since the output is controlled by a binary mixture of permeated H2O and H2 generated from the cathode catalyst layer (CCL). Although H2 production increases in the CL due to faster reaction kinetics at elevated temperatures, the higher concentration of permeated H2O in the cathode results in lower H2 throughput (Fig. S2†). The coupled impact of higher saturation concentrations of FW at the inlet and faster diffusion at higher temperatures creates lower concentration gradients of H2O in the PEMWE while delivering lower mole fractions of H2 during extraction at the CPTL|BP interface. While higher temperatures enhance reaction rates leading to increased H2 concentration at the CCL|CPTL interface, the design of a vapor-fed electrolyzer mandates a binary mixture at the cathode exit. This constraint limits the maximum H2 extraction rate at a given cathode pressure through the CPTL, which is further influenced by the local H2O concentration at the exit.
The H2O concentration directly impacted the overall voltage response in the PEMWE when the RH increased from 40% to 100% at a FW temperature of 70 °C (Fig. 2(c)). The degree of humidification in the cell scales with the RH of input FW; hence, higher H2O concentrations result in greater intrinsic protonic conductivity (15.5 S per m increase from 40% to 100% RH) and lower electrolyte potential gradients throughout the cell sandwich. There is a proportionate rise in total ionic resistances from 100% to 40% RH (Fig. 2(d)), which are a byproduct of enhanced gradients. Since RH significantly affects the electrolyte gradients, we only see a negligible increase in other resistance modes. Fig. 2(e) and (f) show the collective interplay of RH and temperature in influencing the kinetic transport interactions through variation in H2 throughput and total power consumption of the PEMWE. An increase in RH facilitates a greater transport of H2O (electroosmotic drag) across the membrane, thereby affecting the relative proportion of the H2–H2O binary mixture at the CPTL|BP interface. At lower temperatures, changes in RH drastically impact power consumption with minimal influence on H2 production, while elevated temperatures have a consistent variation in both metrics. The coupled effect of slower kinetics and poor protonic conductivity induced from humidification can significantly raise the power consumption (547.4 kW vs. 872.9 kW at 50 °C moving from 100% to 40% RH), although it enables the highest H2 production due to the lower molar composition of H2O in the cathode.
With decreasing temperatures, there was a three-fold increase in H2 throughput at 100% RH as opposed to a 1.5-fold increase when operated at 40% humidity. To understand this joint impact, we probed the SEC (Fig. 2(g)) to demarcate regions of high and low efficacy. A region of moderate to high RH (80–100%) combined with low to intermediate FW temperatures (50–70 °C) facilitates the highest PEMWE efficacy (e.g., 67.74 kW h kgH2−1 at 90% RH and 60 °C). While higher temperatures could lower energy consumption, their equivalent H2 production makes it unfavorable for long-term PEMWE operation (1.2-fold increase at 90% RH and 90 °C). Therefore, understanding the trade-offs resulting from the coupled interdependence of RH and temperature can provide greater opportunities to lower energy expenditure while achieving the target H2 throughput through electrolysis at optimal conditions.
The myriad of physicochemical mechanisms in the PEMWE are regulated by the operating stressors and crucially exhibit varied implications on the overall efficacy and performance through the choice of electrode design. The kinetic and transport descriptors derived from the design of the electrode microstructure control the overall accrued overpotential and H2 production under fixed operating conditions. Fig. 3 and 4 focus our analyses on delineating the microstructural descriptors of the ACL and CCL separately, thus collectively informing the limitations and design space for the practical operability of the PEMWE. More details regarding the generation of synthetic electrode microstructures (Fig. 3(e) and 4(e)) and the evaluation of their intrinsic attributes are described in Section S4 of the ESI.†Fig. 3(a–d) describe the control of local reaction kinetics, proton transport, effective diffusion, and electron percolation pathways in the ACL with variation in catalyst loading and ionomer content. An increase in the catalyst loading from 0.5 to 2 mg cm−2 induces a 3.5-fold increase in the electrochemical active area formed with the ionomer through a three-phase boundary (TPB) (Fig. 3(a)). However, the number of TPB reaction sites saturates once the ionomer fully covers the catalyst, resulting in self-deposition on the ionomer, leading to minimal changes to the reactive area with increasing ionomer content. In contrast, the presence of a high ionomer content creates a stronger ion-transport network in the catalyst layer, facilitating the transport of H+ ions through the membrane (Fig. 3(b)). The protonic conductivity decreases by 94.68% when lowered to a catalyst loading of 0.5 mg cm−2 with the same ionomer content (4.194 S m−1 at 2 mg cm−2 and 30 wt% ionomer). The lack of sufficient catalyst sites prevents the construction of an ionomer bridge across the entire CL, resorting to a gradual buildup onto selective sites, causing greater ion transport tortuosity, and lower effective protonic conductivity.
 | ||
| Fig. 3 Impact of ACL microstructural attributes (catalyst loading and ionomer content) on the (a) electrochemical active area, (b) ratio of effective ionomer conductivity to the bulk ionomer conductivity, (c) pore phase tortuosity, and (d) ratio of effective electronic conductivity to the bulk electronic conductivity. (e) Synthetically generated candidate ACL microstructures illustrating the variation in catalyst loading and ionomer content. | ||
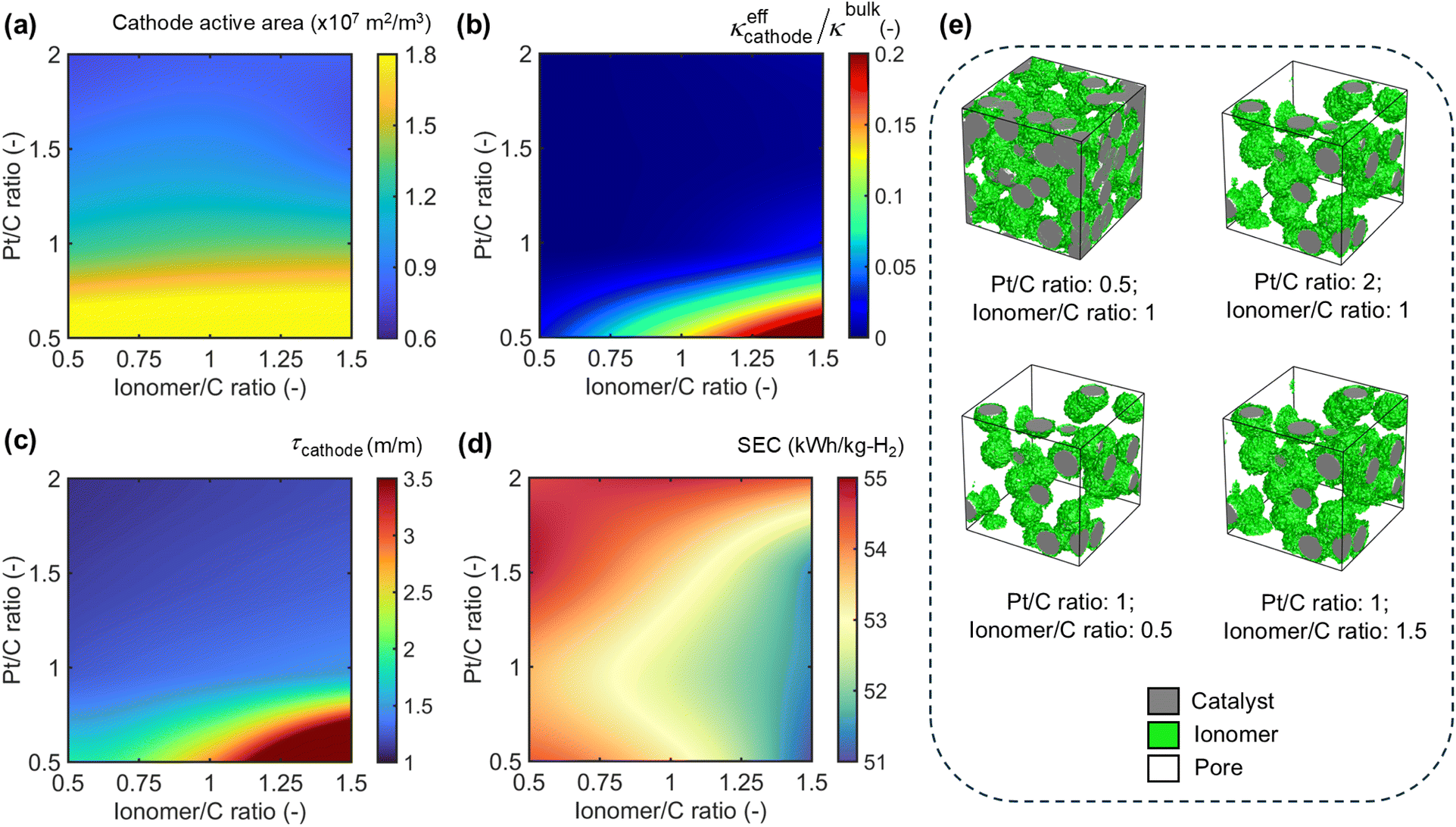 | ||
| Fig. 4 Impact of cathode microstructural attributes (platinum/carbon ratio and ionomer/carbon ratio) on the (a) electrochemical active area, (b) ratio of effective ionomer to bulk ionomer conductivity, (c) pore phase tortuosity and (d) SEC at a current density of 2 A cm−2. (e) Synthetically generated candidate CCL microstructures illustrating variation in the Pt/C ratio and ionomer/C ratio. | ||
With greater occupation sites of the catalyst layer and increased ionomer content, the overall porosity of the CL impacts the diffusive transport of H2O and electro-generated gases (O2 in the ACL). The raised pore-phase tortuosity (Fig. 3(c)) implies large concentration gradients within the CL, thereby affecting the transport of H2O through the membrane. The pore phase tortuosity reaches the highest protonic conductivity of 3.2 at 2 mg cm−2 and 40 wt% ionomer. However, lowering the ionomer wt% by half drastically reduces the tortuosity by 77%, indicating an exponential growth in the pore transport network above a critical threshold of catalyst loading and ionomer content in the ACL. Similarly, we observe a 6.8× increase in the solid-state electronic conductivity when the catalyst loading increases from 0.5 to 2 mg cm−2 (Fig. 3(d)). A similar analysis is conducted to connect the intrinsic electrode design of CCL to its implications on the kinetic and transport resistance of the cell. The cathode electrochemical active area is also a strong function of the catalyst loading, with lower Pt/C ratios translating to higher carbon support structures to foster greater deposition of Pt particles for the same ionomer content (Fig. 4(a)). An increase in the ionomer/C ratio indicates a minimal increase in TPB sites (or electrochemical active area) once sufficient coverage of the catalyst is achieved. Fig. 4(b–c) reveal the implications of higher volumetric proportion in the CL manifesting as greater pore-phase tortuosity derived from low Pt/C and high ionomer/C ratios. Despite increased tortuosity, the denser ionomer bridge allows for a ∼1000× increase in protonic conductivity as opposed to regions of low tortuosity.
We characterize the variation of SEC under an operating current density of 2 A cm−2 and a fixed ACL recipe to examine the collective interplay of diffusive transport, proton conduction pathways, and overall reaction sites (Table S2†). Regions of high Pt/C and low ionomer/C ratios indicate lower interfacial kinetics paired with weaker protonic conduction pathways, resulting in high SEC from increased energy consumption despite possessing lower diffusive transport resistances to assist H2 production (Fig. 4(d)). Conversely, the combined effect of an increased active area and protonic conductivity lowers the overall resistance of the system with higher ionomer content and catalyst loading, delivering lower SEC. Interestingly, the range of SEC over the entire CCL design space is minimal (<5 kW h kgH2−1). We hypothesize a direct correlative trend (or an equivalent strength) between energy consumption and H2 throughput in dictating the overall SEC of the cell under fixed ACL design and operating conditions with variation in the Pt/C and ionomer/C ratios. This ensures that the SEC is effectively invariant with changes to the CCL. While this inference is inherently universal to any CCL of a PEMWE, we can expect a larger variation in the SEC due to CCL design only in a limiting scenario where the anode exchange current density becomes comparable or higher than its cathode counterpart. This microstructure-centric analysis helps illustrate that the overall electrode design variation can instruct contrasting mechanistic implications against operational stressors that impact the overall PEMWE performance.
To further probe the impact of the ACL microstructure on PEMWE performance metrics, we critically examined the polarization and commensurate resistance evolution in the cell. Fig. 5(a) and (c) describe the overpotential rise of the cell due to the growth of microstructure-driven resistance modes (Fig. 5(b) and (d)). Based on the insights from Fig. 3(a), we ascertain that increasing the anode catalyst loading under a fixed ionomer content can drastically reduce the local reaction currents, thus lowering the potential gradients in the cell. Catalyst loading below a specific critical limit dictated by the ionomer content could result in ionomer buildup with no enhancement in the reaction sites, enabling faster growth of polarization, especially at high currents (6.05 V at 0.5 mg cm−2vs. 2.1 V at 2 mg cm−2 when operated at 1 A cm−2). A commensurate resistance response shows a reduction of 4.53 Ω cm2 through the ionic resistance governed by the potential gradients developed within the CL (Fig. 5(b)). Other resistances also decrease with a higher electrochemical active area (such as a 0.26 Ω cm2 reduction in electronic resistance) but present a lower magnitude than ionic resistances. Since mass transport and kinetic resistance are indirectly controlled by the magnitude of overpotentials in the cell, their influence on the overall resistance is minimal.
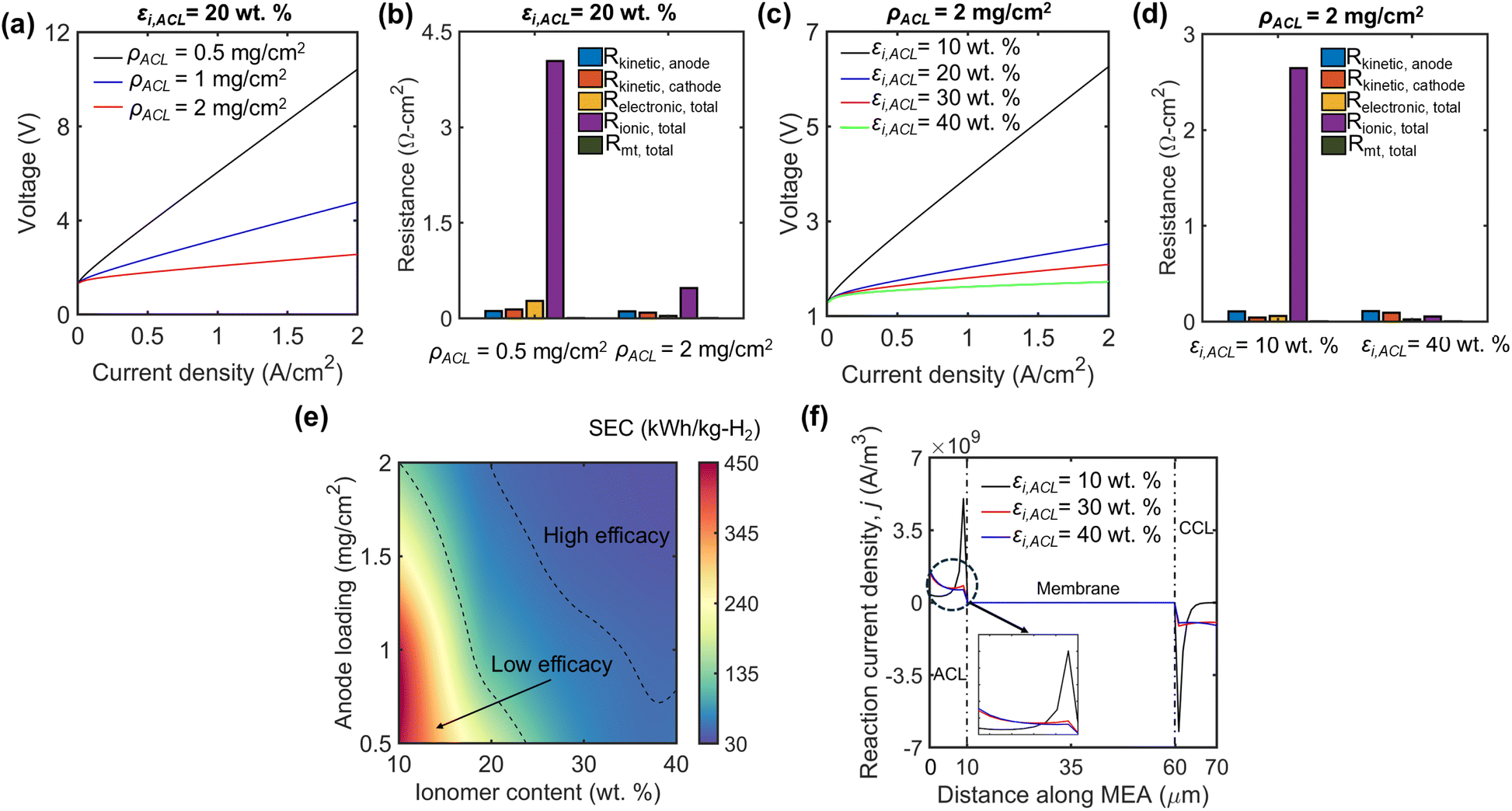 | ||
| Fig. 5 Role of ACL design in dictating the PEMWE electrochemical performance and efficacy. (a) Cell polarization with variable catalyst loading and proportionate (b) resistance contributions at 0.5 and 2 mg cm−2 paired with 20 wt% ionomer content in the ACL. (c) Cell polarization under variable ionomer content and associated (d) resistance contributions at 10 wt% and 40 wt% ionomer content paired with 2 mg cm−2 catalyst loading in the ACL. (e) Efficacy of water electrolysis examined through SEC at a current density of 2 A cm−2. (f) Spatial evolution of reaction current density along the membrane electrode assembly (MEA) under variable ionomer content at a current density of 1 A cm−2. | ||
Similarly, a reduction in ionomer content presents a drastic rise in the polarization response (Fig. 5(c)). The power consumption of the PEMWE cell sandwich significantly rises from 505 kW at 20 wt% to 1253 kW at 10 wt% when operated at a current density of 2 A cm−2. This confirms that an ionomer content below 20 wt% can drastically raise the resistance (Fig. 5(d)) even with sufficient catalyst loading. The spatial evolution of the current density in the membrane electrode assembly (MEA) (Fig. 5(f)) illustrates the heterogeneous nature of local currents at the TPB reaction sites of ACL. At higher ionomer contents, the reaction is equally distributed within the CL, resulting in minimal electrolyte potential gradients in the MEA (Fig. S4†) and CL (minimal Δϕs due to the high electronic conductivity of catalysts). However, the local reaction currents develop a steeper gradient within the CL at very low ionomer contents, with high current densities manifesting at the PEM/CL interface. The low protonic conductivity influenced by ionomer-phase connectivity and a low active area leads to greater overpotentials and resistances within the MEA. Thus, ensuring sufficient ionomer content in the ACL to facilitate enhanced reaction kinetics is vital to regulate energy consumption at acceptable levels.
Fig. 5(e) collectively demarcates regions of acceptable operation by including the proportionate H2 throughput via SEC. Although packing the CL with higher amounts of catalyst and ionomer can raise the pore phase tortuosity and establish higher concentration gradients, its lower energy consumption coupled with higher H2 production stemming from lower H2O concentrations in the cathode (from high tortuosity) creates a domain of high PEMWE efficacy. The ACL microstructural spectrum presents an imbalanced competition between the rise of overpotential and the rate of H2 production within the cell in contrast to Fig. 4(d), where the cathode design space reinforces the balance of energy consumption and H2 throughput. The SEC varies by 22.94 kW h kgH2−1 at 40 wt% ionomer across the range of catalyst loading, whereas it rises to 305 kW h kgH2−1 at 10 wt% ionomer content in the ACL. Similarly, we also explicate a consistent behavior of increasing ionic resistances and polarization with higher ACL thickness (Fig. S3†) due to enhanced transport resistances. This variation in SEC within the ACL design space enables us to mechanistically explain the rationale for identifying the ACL as the limiting electrode in any PEMWE cell.
We probed the variation in the total resistance of the PEMWE cell with electrode design to fully understand the relative impact of electrodes in altering the overall performance and efficacy. Fig. 6(a and b) presents the evolved total resistance of the ACL and CCL under a fixed cathode design (Pt/C = 1 and ionomer/C = 1) at a current density of 2 A cm−2. Contrary to intuition, changes in the ACL design impact the resistance signatures of both electrodes, with CCL having a pronounced significance over the ACL. Our previous analysis indicates that ionic resistances cover a substantial proportion of the total resistance, excluding the membrane resistance under a fixed operating condition (Fig. S5†). The maximum resistance evolved in the ACL at 0.5 mg cm−2 and 10 wt% ionomer is 0.92 Ω cm2, with a range of 0.78 Ω cm2 over the entire design space (Fig. 6(a)). In stark contrast, the highest resistance emanating from the CCL amounts to 2.43 Ω cm2 and a range of 2.34 Ω cm2 under identical conditions. Although the total electrode resistance is substantially controlled by the CCL, its mechanistic interpretation stems from the collective implications directed from the anode side. Under low loading and ionomer content conditions, the potential drop in the ACL is so severe that the electrolyte potential evolution within the MEA is directly influenced by the gradient within the ACL (Fig. S5†). This results in a larger potential drop across the membrane and the CCL, creating extremely high CCL ionic resistances. However, there is only a maximum of 0.25 Ω cm2 in total CCL resistance with changes to cathode design while exhibiting an invariant ACL resistance derived from a fixed ACL recipe (1 mg cm−2 and 20 wt%) (Fig. 6(c)). The electro-osmotic drag of H2O from the anode to the cathode through the PEM, coupled with a 100000× reduction in the OER develops a severe potential gradient that establishes an indirect transfer of raised resistance, even in the cathode. Thus, minute changes in ACL design can inform drastic effects in anode and cathode resistance, due to intrinsic mechanisms controlling the growth of solid and electrolyte phase potentials within the MEA. Changes to cathode design result in identical state variables entering the CCL (Cspecies, ϕs, ϕe), resulting in variation purely from the CCL. A higher volume fraction of catalyst and ionomer could potentially reduce the transport pathways of H2O within the PEMWE, leading to a leaner mole fraction of H2O in the cathode, resulting in higher H2 throughput (Fig. 6(d and e)). A tortuous path to H2O creates large concentration gradients within the CL that potentially offer greater H2 production without compromising the overall energy consumption. A PEMWE cell design cognizant of its intrinsic anode-centric limitations under various operating conditions is pivotal to achieving green H2 targets with favorable SEC limits for large-scale decarbonization.
 | ||
| Fig. 6 Identifying electrode-centric limitations to PEMWE efficacy by characterizing intrinsic resistances under various operational regimes. Influence of ACL design on the concomitant total evolved (a) resistance in the ACL and (b) resistance in the CCL. Influence of cathode electrode design on the total (c) resistance developed in the CCL. All the phase maps correspond to a non-dimensional resistance (R*) with R0 = 1 Ω cm2, operated at a current density of 2 A cm−2 and 80 °C feed water temperature. Spatial evolution of water concentration in the cell-sandwich with (d) ACL design (shown by a ‘star’) paired with a fixed cathode design (Pt/C = 1; ionomer/C = 1) and (e) cathode design (shown by a ‘circle’) paired with a fixed ACL recipe (shown by a ‘triangle’). | ||
Conclusions
The proliferation of H2-based electrochemical energy conversion systems to support the decarbonization of various sectors entails a prudent design of high-performance PEMWEs. This study presents a deep dive into the multi-mechanistic interactions of H2O electrolysis in PEMWEs by developing a microstructure-aware electrochemical model. The coupled interplay of operating stressors (RH and temperature) on the electrochemical signature and efficacy was investigated by incorporating bubble dynamics. Low to intermediate temperatures (50 °C to 70 °C) under moderate to fully humidified conditions (60–100%) supported high operational efficacy with low SEC. The electrode-level resistance breakdown analysis aided an accurate interpretation of the underlying dominant mechanisms during electrolysis at various operating conditions. Specifically, the ionic resistance was dominant purely influenced by thicker membranes, followed by the kinetic, electronic and mass transport resistances in this configuration. A detailed evaluation of microstructure-derived intrinsic kinetic and transport descriptors was conducted to understand the distinctive regions of operation that lead to lower accrued overpotentials. Based on the joint contribution of intrinsic electrode design and operating stressors, we deduced high and low PEMWE efficacy regimes through the juxtaposition of power consumption and concomitant H2 throughput. With ACL loadings >1 mg cm−2 and beyond 20 wt% of ionomer content, we reveal a regime of high PEMWE efficacy (i.e., low SECs) from the coupled impact of anode reaction kinetics and its microstructural implications. The overall electrochemical response, informed partly by cathode resistance, significantly stems from the pertinent mechanistic interactions at the anode, revealing that the PEMWE system is chiefly anode-limited. Avenues for future research include integrating detailed multi-phase physics into the existing reactive transport framework to capture bubble growth dynamics and its interactions with H2O during PEMWE operation. Concerted efforts must be directed toward evaluating the impact of differential pressure and membrane thickness on the kinetic-transport interplay while balancing durability and performance trade-offs to meet stringent DOE targets.17 PEMWE aging models informed by recent advances in fuel cells61–63 and integrated with physics-based predictive models can drive the experimental design of functional materials to reduce SEC, minimize gas crossover through PEMs, and optimize bubble management in PTLs. Lastly, we infer that potential pathways to achieve high throughput green H2 begin with understanding the intrinsic electrode limitations and a knowledge of trade-offs arising from electrode design and PEMWE operating conditions.Data availability
The data supporting this article have been included in the ESI.†Author contributions
Conceptualization: N. G., A. A., and P. P. M.; methodology: N. G., A. A., and A. N.; formal analysis: N. G., A. A, and A. N.; visualization: N. G., A. A., and A. N.; writing – original draft: N. G. and A. A.; writing – review & editing: N. G., A. A., A. N., B. S. V., and P. P. M.; supervision: P. P. M.Conflicts of interest
The authors declare no conflicting interests.Acknowledgements
Financial support from the Alfred P. Sloan Foundation (award no.: G-2022-19482) is gratefully acknowledged.References
- D. N. N. Pivovar, B.S., M. F. Ruth and D. J. Myers, ECS Interface, 2021, 30, 61–65 Search PubMed.
- L. Wan, Z. Xu, Q. Xu, M. Pang, D. Lin, J. Liu and B. Wang, Energy Environ. Sci., 2023, 16, 1384–1430 CAS.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CAS.
- J. Cabana, T. Alaan, G. W. Crabtree, M. C. Hatzell, K. Manthiram, D. A. Steingart, I. Zenyuk, F. Jiao, A. Vojvodic, J. Y. Yang, N. P. Balsara, K. A. Persson, D. J. Siegel, C. L. Haynes, J. Mauzeroll, M. Shen, B. J. Venton, N. Balke, J. Rodríguez-López, D. R. Rolison, R. Shahbazian-Yassar, V. Srinivasan, S. Chaudhuri, A. Couet and J. Hattrick-Simpers, ACS Energy Lett., 2022, 7, 368–374 Search PubMed.
- P. Sun, B. Young, A. Elgowainy, Z. Lu, M. Wang, B. Morelli and T. Hawkins, Environ. Sci. Technol., 2019, 53, 7103–7113 CAS.
- Hydrogen Production, Natural Gas Reforming, https://www.energy.gov/eere/fuelcells/hydrogen-production-natural-gas-reforming, accessed 20 February 2025.
- R. Borup, T. Krause and J. Brouwer, Electrochem. Soc. Interface, 2021, 30, 79–83 CrossRef CAS.
- L. D. Ellis, A. F. Badel, M. L. Chiang, R. J. Y. Park and Y. M. Chiang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12584–12591 CrossRef CAS.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462–474 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- U. Babic, M. Suermann, F. N. Büchi, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, F387–F399 CrossRef CAS.
- S. M. Alia, Curr. Opin. Chem. Eng., 2021, 33, 100703 CrossRef.
- J. Liu, J. Kennedy, A. Marshall, J. Metson and M. P. Taylor, J. Electrochem. Soc., 2022, 169, 114503 CrossRef CAS.
- C. V. Pham, D. Escalera-López, K. Mayrhofer, S. Cherevko and S. Thiele, Adv. Energy Mater., 2021, 11, 2101998 CAS.
- C. Minke, M. Suermann, B. Bensmann and R. Hanke-Rauschenbach, Int. J. Hydrogen Energy, 2021, 46, 23581–23590 CAS.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 33, 2006328 CAS.
- Technical Targets for Proton Exchange Membrane Electrolysis, https://www.energy.gov/eere/fuelcells/technical-targets-proton-exchange-membrane-electrolysis, accessed 20 September 2024.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179–F3189 CAS.
- G. Doo, J. H. Lee, S. Yuk, S. Choi, D. H. Lee, D. W. Lee, H. G. Kim, S. H. Kwon, S. G. Lee and H. T. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 17835–17841 CAS.
- Z. Taie, X. Peng, D. Kulkarni, I. V. Zenyuk, A. Z. Weber, C. Hagen and N. Danilovic, ACS Appl. Mater. Interfaces, 2020, 12, 52701–52712 CAS.
- S. Dong, C. Zhang, Z. Yue, F. Zhang, H. Zhao, Q. Cheng, G. Wang, J. Xu, C. Chen, Z. Zou, Z. Dou and H. Yang, Nano Lett., 2022, 22, 9434–9440 CAS.
- M. A. Hubert, L. A. King and T. F. Jaramillo, ACS Energy Lett., 2022, 7, 17–23 CAS.
- T. Schuler, R. De Bruycker, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2019, 166, F270–F281 CAS.
- T. Schuler, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2019, 166, F555–F565 CAS.
- J. Lopata, Z. Kang, J. Young, G. Bender, J. W. Weidner and S. Shimpalee, J. Electrochem. Soc., 2020, 167, 064507 Search PubMed.
- E. Leonard, A. D. Shum, N. Danilovic, C. Capuano, K. E. Ayers, L. M. Pant, A. Z. Weber, X. Xiao, D. Y. Parkinson and I. V. Zenyuk, Sustainable Energy Fuels, 2020, 4, 921–931 CAS.
- X. Peng, P. Satjaritanun, Z. Taie, L. Wiles, A. Keane, C. Capuano, I. V. Zenyuk and N. Danilovic, Adv. Sci., 2021, 8, 2102950 Search PubMed.
- C. C. Weber, J. A. Wrubel, L. Gubler, G. Bender, S. De Angelis and F. N. Büchi, ACS Appl. Mater. Interfaces, 2023, 15, 34750–34763 CrossRef CAS PubMed.
- T. Burdyny, P. J. Graham, Y. Pang, C. T. Dinh, M. Liu, E. H. Sargent and D. Sinton, ACS Sustain. Chem. Eng., 2017, 5, 4031–4040 CrossRef CAS.
- X. Zhao, H. Ren and L. Luo, Langmuir, 2019, 35, 5392–5408 CrossRef CAS.
- P. Peñas, P. van der Linde, W. Vijselaar, D. van der Meer, D. Lohse, J. Huskens, H. Gardeniers, M. A. Modestino and D. F. Rivas, J. Electrochem. Soc., 2019, 166, H769–H776 Search PubMed.
- B. K. Kim, M. J. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 11940–11947 CrossRef CAS PubMed.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Rivas, Joule, 2020, 4, 555–579 Search PubMed.
- J. K. Lee and A. Bazylak, Joule, 2021, 5, 19–21 Search PubMed.
- H. Vogt and R. J. Balzer, Electrochim. Acta, 2005, 50, 2073–2079 CrossRef CAS.
- H. Vogt, Electrochim. Acta, 2017, 235, 495–499 CrossRef CAS.
- H. Vogt and J. Thonstad, Electrochim. Acta, 2017, 250, 393–398 CrossRef CAS.
- R. Iwata, L. Zhang, K. L. Wilke, S. Gong, M. He, B. M. Gallant and E. N. Wang, Joule, 2021, 5, 887–900 CrossRef CAS.
- M. Li, P. Xie, L. Yu, L. Luo and X. Sun, ACS Nano, 2023, 17, 23299–23316 CAS.
- D. S. Falcão and A. M. F. R. Pinto, J. Cleaner Prod., 2020, 261, 121184 Search PubMed.
- A. Benmehel, S. Chabab, A. L. Do Nascimento Rocha, M. Chepy and T. Kousksou, Energy Convers. Manage.:X, 2024, 24, 100738 CAS.
- T. Seip, N. Shaigan, M. Dinu, K. Fatih and A. Bazylak, J. Power Sources, 2023, 559, 232654 CAS.
- J. Liu, F. Kerner, N. Schlüter and D. Schröder, ACS Appl. Mater. Interfaces, 2023, 15, 54129–54142 CAS.
- J. Liu, M. Li, Y. Yang, N. Schlüter, D. Mimic and D. Schröder, ChemPhysChem, 2023, 24, e202300197 CAS.
- Y. Xu, D. Ye, W. Zhang, Y. Wang, J. Li, L. Zhang, J. Huang, X. Zhu and Q. Liao, Energy Convers. Manage., 2024, 322, 119089 Search PubMed.
- C. Xu, J. Wang, J. Wang, K. Yang, G. Li, W. Gao, H. Wang and S. Zhao, Appl. Energy, 2024, 357, 122541 CAS.
- M. Chandesris, V. Médeau, N. Guillet, S. Chelghoum, D. Thoby and F. Fouda-Onana, Int. J. Hydrogen Energy, 2015, 40, 1353–1366 Search PubMed.
- F. Aldakheel, C. Kandekar, B. Bensmann, H. Dal and R. Hanke-Rauschenbach, Comput. Methods Appl. Mech. Eng., 2022, 400, 115580 Search PubMed.
- P. A. García-Salaberri, J. Power Sources, 2022, 521, 230915 Search PubMed.
- H. Zhou, K. Meng, W. Chen and B. Chen, Int. J. Energy Res., 2022, 46, 17126–17143 CAS.
- G. Xu, X. Du, L. Que, L. Zhang, J. Li, D. Ye, J. Song and J. Gao, Appl. Therm. Eng., 2025, 263, 125384 CAS.
- P. Olivier, C. Bourasseau and P. B. Bouamama, Renewable Sustainable Energy Rev., 2017, 78, 280–300 Search PubMed.
- A. H. Abdol Rahim, A. S. Tijani, S. K. Kamarudin and S. Hanapi, J. Power Sources, 2016, 309, 56–65 CrossRef CAS.
- B. Han, S. M. Steen, J. Mo and F. Y. Zhang, Int. J. Hydrogen Energy, 2015, 40, 7006–7016 CrossRef CAS.
- B. Han, J. Mo, Z. Kang, G. Yang, W. Barnhill and F. Y. Zhang, Int. J. Hydrogen Energy, 2017, 42, 4478–4489 CrossRef CAS.
- F. Marangio, M. Santarelli and M. Calì, Int. J. Hydrogen Energy, 2009, 34, 1143–1158 CrossRef CAS.
- E. T. Ojong, J. T. H. Kwan, A. Nouri-Khorasani, A. Bonakdarpour, D. P. Wilkinson and T. Smolinka, Int. J. Hydrogen Energy, 2017, 42, 25831–25847 CrossRef CAS.
- F. Aubras, J. Deseure, J. J. A. Kadjo, I. Dedigama, J. Majasan, B. Grondin-Perez, J. P. Chabriat and D. J. L. Brett, Int. J. Hydrogen Energy, 2017, 42, 26203–26216 CrossRef CAS.
- B. Han, J. Mo, Z. Kang and F. Y. Zhang, Electrochim. Acta, 2016, 188, 317–326 Search PubMed.
- M. A. Ebbert and S. Litster, Sci. Rep., 2024, 14, 31637 CAS.
- N. Goswami, J. B. Grunewald, T. F. Fuller and P. P. Mukherjee, Electrochim. Acta, 2024, 486, 144143 CAS.
- N. Goswami, J. B. Grunewald, T. F. Fuller and P. P. Mukherjee, J. Mater. Chem. A, 2022, 10, 15101–15115 Search PubMed.
- N. Goswami, K. Chen, X. Wang, J. S. Spendelow, R. L. Borup and P. P. Mukherjee, Chem. Eng. J., 2024, 484, 149672 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08669d |
| ‡ N. G. and A. A. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |