
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles design of a superior electrocatalyst to Pt for hydrogen production in alkaline media†
Haeshik
Lee
a,
Jinwoo
Park
b,
Jae-Min
Myoung
 c and
Byungchan
Han
*a
c and
Byungchan
Han
*a
aDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: bchan@yonsei.ac.kr
bDepartment of Physics, University of Seoul, Seoul 02504, Republic of Korea
cDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Republic of Korea
First published on 12th February 2025
Abstract
Pt electrocatalysts are known to be surprisingly inactive for the hydrogen evolution reaction (HER) in alkaline media due to their high activation barrier for water decomposition. Despite extensive research over several decades, only a few electrocatalysts with exceptional efficiency and stability have been reported, primarily due to an incomplete understanding of the mechanism and catalyst design principles. Therefore, we investigated the underlying mechanism of the proposed catalyst design for Pt monolayer surface alloy (Pt/W(110)) and nanoparticle (W@Pt) structures, focusing on (i) the surface strain effect and (ii) the ligand effect (charge transfer). Using first-principles calculations, we demonstrated that fabricating Pt nanoparticles increases the H2O adsorption energy, with lower dissociation energy barrier expected, only shifts the rate-determining step (RDS) to H2 desorption. When pure Pt structures are modified with W substrates, the dual effects of strain and ligand interactions control the activities of the bulk pseudo-binary Pt/W(110) surface alloy and W@Pt core–shell nanoparticles in the opposite manner. The W@Pt nanoparticles uniformly destabilized all intermediates, increasing the water dissociation activation barrier. In contrast, the Pt/W(110) surface heterogeneously regulated the adsorption intensities of the intermediates, breaking the scaling law of catalysis. We clearly elucidated this heterogeneous behavior in terms of eigenstress-induced surface strain and charge transfer. Our study is expected to provide quantum-mechanical insights into the design of active electrocatalysts by controlling key atomic-level descriptors and addressing the long-standing issue of inactive Pt catalysts in alkaline media.
Introduction
Hydrogen is the most abundant element in the universe, accounting for 90% of all atoms. It is also the lightest gas, with the highest gravimetric energy density of 120 kJ g−1.1 Furthermore, it is also an environmentally friendly and non-toxic gas. These unique properties enable hydrogen gas to play a key role in various chemical reactions from the laboratory to the industrial scale and as a fuel for renewable energy systems and mobile devices. In spite of the attractive aspects, hydrogen production of high purity is difficult in practice because it forms chemically stable mixtures with a wide range of materials (water, hydrides, etc.) or easily smears into bulk crystals.Over the last several decades, extensive efforts have been made to generate hydrogen gas from hydrolysis through thermochemical, electrochemical, and photovoltaic (or photothermal) pathways. Among these, electrochemical hydrogen production via water decomposition is considered a fully green technology. Developed countries are actively working on designing and optimizing water electrolyzers (WEs).2 WEs are largely divided into three different types depending on the working media:3,4 (i) proton exchange membrane (PEM) WEs, (ii) alkaline WEs, and (iii) anion exchange membrane (AEM) WEs. PEMWEs with high activity in the hydrogen evolution reaction (HER) have been studied for a long time to facilitate their commercialization. However, this has not been successful, mainly due to the instability of the catalysts in acidic media, even for Pt metal and novel transition metal oxides.5,6 WEs working in an alkaline medium have recently attracted increasing focus, as they allow the use of cost-effective transition metal catalysts for the HER. However, regardless of the decades of study, the system still faces serious challenges in the identification of highly active and stable catalysts other than Pt and in the reduction of system size. Thus, the AEMWE was suggested to operate in an alkaline medium with a polymer membrane, which was designed by taking the benefits of alkaline and PEM systems. A key requirement for AEMWE systems is improved HER catalytic activity and ionic conductivity for industrial-scale commercialization.
In accordance with previous reports, the sluggish HER in alkaline media is caused by the deficiency of a direct proton source, which leads to a more complicated multistep catalytic mechanism than in acidic media. It is noteworthy that even Pt metal is inactive for the HER in alkaline solution because of the substantially high activation energy required to decompose H2O molecules into H and OH.7 This implies that OH molecules do not adsorb on Pt surfaces as effectively as the H atoms do, and the description of HER activity using a single parameter of H adsorption energy would be insufficient. It is essential to precisely control the adsorption behavior of all intermediates along the entire HER pathway, which is difficult to achieve without understanding the accurate catalytic mechanism of various materials and identifying the principles for its control.
It is widely agreed that there is a clear correlation between the catalytic activity and atomic and electronic structures of catalysts employed for the target reaction. This is called the geometric effect8 or d-band center energy theory.9 Despite well-established theories, few studies have attempted to design highly active HER catalysts as alternatives to Pt metal, and only a few active catalysts with prolonged durability in alkaline solutions have been identified for commercialization. Using first-principles density functional theory (DFT) calculations, we demonstrated the following in this paper: (i) clear elucidation of the underlying mechanism of why nanoparticle construction is not sufficient to increase the HER activity of pure bulk Pt, (ii) identification of two factors (strain and ligand effects) at the atomic and electronic structure levels controlling the HER activity, and (iii) proposal of design principles and selection rules of promising alloying element M with Pt to significantly boost HER activity, surpassing that of Pt bulk and nanoparticles.
Unlike conventional studies, we determined that just the Pt or Pt-M nanoparticles do not actively promote the HER in alkaline media but only shift the RDS of bulk Pt. The two control factors cooperatively worked only for the Pt/W(110) surface, in which a Pt monolayer covered the W(110) plane. To demonstrate the influence of both strain and ligand fields on catalytic activity for the HER, we selected W as the supporting substrate of an active Pt layer. In accordance with the traditional theory, the binding energies of adsorbates with Pt are described by the two factors, which eventually modify the overall catalytic performance. It is difficult, however, to predict the catalytic activity if those variables are coupled with the particle size effect. Therefore, we designed a bulk Pt/W(110) surface and W@Pt nanoparticles to clearly identify a predominant control factor of the catalytic performance and to elucidate its underlying mechanisms. Using DFT calculations, we clearly elucidated the underlying mechanism and proposed unconventional design principles for active electrocatalysts for the HER in alkaline solutions, which can be extended to other targeted catalysts. The rationale of the design is two-fold: (i) decrease Pt loading while maintaining catalytic activity through increased surface area and under-coordinated active sites10,11 and (ii) develop heterogeneous structures by alloying or ligand fields to induce strain.12–14 Through first-principles study, we rigorously characterized the interplay of the two factors (strain and ligand fields) for HER catalysts, which are influential for bulk and nanoparticles in a very dissimilar manner.
Computational details
Spin-polarized DFT calculations were performed as implemented in Vienna ab initio simulation package (VASP)15 for all computations. The interactions between core-valence electrons were described using the projector augmented wave (PAW) pseudopotentials.16,17 A cutoff energy of 450 eV was applied for the plane wave basis set, with a convergence criteria of 10−5 eV and 0.04 eV Å−1 for energy and force, respectively. The cutoff value for force was chosen based on test calculations on adsorption energies (Table S1†). The exchange–correlation energy of the electrons was computed using the generalized gradient approximation (GGA) method18 with Perdew–Burke–Ernzerhof (PBE) functionals.19 Long-range van der Waals interactions were considered using the DFT-D3 method of Grimme.20 DFT calculations for the nanoparticles were carried out using the Γ-point scheme with a vacuum space of 20 Å to prevent interactions between periodic images. Slab calculations were performed with a dense k-point mesh ensuring at least 1000 k-points per atom and a vacuum space of 15 Å.The adsorption energy (Eads) was calculated using eqn (1),
 | (1) |
ΔG = ΔE + ΔZPE − TΔS + kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10)pH, ln(10)pH, | (2) |
ln(10)pH, where kB is the Boltzmann constant. Additionally, dielectric constant correction was applied to the systems with the adsorbate to consider the implicit water environment to simulate the aqueous conditions using VASPsol.22,23 Regarding the electron structure analysis, orbitals were analyzed through the partial density of states (PDOS), and the charge transfer phenomena were examined using the charge density difference plot.
Results and discussion
Model systems
We constructed five model systems (Fig. 1) to study the effects of ligand field and surface strain on the HER catalytic activity by modifying both the bulk and nanoparticle structures. The model systems consisted of two bulk surfaces of the pure Pt(111) surface and Pt/W(110) surface, consisting of a monolayer Pt on the W(110) slab. Additionally, we set up three nanoparticle systems of different sizes and components: pure Pt or W@Pt core–shell configurations. Two differently sized nanoparticles consisting of 55 and 147 atoms were used for the pure Pt system. | ||
| Fig. 1 Model systems of Pt(111), Pt/W(110), Pt55, Pt147, and W@Pt. Pt and W atoms are represented by yellow and blue solid spheres, respectively. | ||
The active sites for the adsorption of each intermediate were determined using DFT calculations, as illustrated in Fig. S1.† The thermodynamically stable structure of each system was determined using DFT calculations. For the bulk Pt/W surface, we identified that the (110) surface of BCC W (Pt/W(110) surface) was the most stable.24 The structures of Pt55 and W@Pt were optimized by comparing the calculated formation and excess energies of similarly sized icosahedron, decahedron, and cuboctahedron structures (Table S2†). Additionally, we calculated energies for BCC-based W@Pt nanoclusters constructed using the Wulff construction method25 to compare the results with the BCC Pt/W(110) surface. Our DFT calculations indicated that icosahedra were the most favorable shapes for both nanoparticles. This is consistent with the fact that the icosahedron exposes only the most stable (111) surface of the FCC crystal structure and that the outermost Pt shell plays a more significant role than the inner W of the BCC structure. The majority of atoms are Pt because the fraction of surface sites is much greater than the inside of the nanoparticles (42 out of 55 in Pt55 and the W@Pt icosahedron, cuboctahedron, and decahedron and 44 out of 59 in the W@Pt BCC-based cluster).26 Based on these results, we also calculated Pt147 of an icosahedron nanoparticle. All the properties of the bulk and nanoparticles were compared to those of a bulk Pt(111) surface, which is the reference model.
HER activity of a pure Pt bulk and nanoparticles
Nanoparticles are the central materials for various electrocatalysts; however, the identification of their ideal structures and compositions for target catalysis is challenging. For electrocatalysis, the conventional Sabatier principle,27 which allows the prediction of catalytic activity using a rather simple descriptor with the scaling law, has been a guideline for searching for optimum materials. However, when catalysts are exposed to aqueous media with non-neutral pH, such as HER catalysts in alkaline or acidic solutions, the catalytic mechanisms are generally much more complicated than the gas-phase reaction under ultra-high vacuum conditions. The scaling law for the adsorption energies of intermediates often does not work, especially in nanoclusters.28,29 The facets of nanoparticles, even those composed of a single component, consist of highly heterogeneous sites.30 Thus, a comprehensive understanding of the guiding principles for electrocatalytic activity in reactive media is crucial, yet remains a significant challenge for both experiments and computations.Fig. 2 illustrates the Gibbs free energy diagram calculated for HER catalysis on the Pt55 icosahedral nanoparticles of approximately 1 nm diameter and on the Pt(111) surface. This indicates that there is a strikingly high activation barrier for the dissociation of H2O molecular bonds into H*–OH* on the Pt(111) surface. This is because the Pt(111) surface adsorbs OH too weakly to cleave the chemical bonds of H2O. However, it shows that the nanoparticles of Pt55 and Pt147 attract all intermediates significantly more strongly (Table S3†) than the Pt(111) surface, as reported previously.28,31 Interestingly, the RDS of the bulk Pt(111) surface, H2O bond dissociation, shifted to H2 adsorbate desorption in Pt55 nanoparticles. The free energy diagram clearly demonstrates that the activation energy for the H2O dissociation on the bulk Pt(111) surface is at least 0.67 eV (∼65 kJ mol−1). In the Pt55 nanoparticle, the RDS activation energy is about 50 kJ mol−1. This scenario is almost the same as that for the larger Pt147 nanoparticles.
 | ||
| Fig. 2 Gibbs free energy diagrams of alkaline HER on the Pt(111) surface and Pt55 nanoparticles. It shows a completely different rate determining step: H2O bond dissociation into H*–OH* on the bulk Pt(111) surface (left inset) and H2 desorption on Pt55 nanoparticles (right inset). | ||
In summary, both Pt55 and Pt147 stabilize all intermediates of the HER excessively, rendering them incapable as active catalysts, and precise but unequal amounts of control of the intermediate adsorption energies for H2O and H2 adsorbates are required to enhance the HER activity in alkaline media.
To control the adsorption energy of individual intermediates, we investigated the surface strain and ligand field effects.32 We achieved it by purposely designing heterogeneous model systems for the bulk Pt/W(110) surface and W@Pt nanoparticles. Both models have larger size element W (1.56 Å, Pt for 1.53 Å)33 to create tensile strain and different work functions to induce charge transfer. In accordance with conventional theory, it is expected that tensile strain should upshift the d-band center energy of Pt, thereby increasing the binding energy of the intermediates. However, the Fermi energy of W (11.56 eV) is higher than that of Pt (11.32 eV), which facilitates the donation of its electron charge to Pt in the shell since the Fermi energy should be equal under the equilibrium conditions. Our electronic structure calculations demonstrated that the transferred electrons from W filled up the antibonding state of the Pt d-orbital and shifted the PDOS downwards. Thus, charge transfer weakens the binding strength of the adsorbates on the W@Pt surface. Likewise, these two factors are expected to influence the catalyst performance in the opposite manner, which implies that they can serve as effective tools for the precise control of the HER catalytic activity through a fine-tuning process to search for optimal conditions.
HER activity of the Pt/W(110) surface
The Pt/W(110) surface consists of a Pt monolayer on a BCC W(110) surface. Our DFT calculations indicated that it was the thermodynamically most stable structure among the FCC(111), BCC(110), and BCC(001) surfaces (Table S4†).24,34,35 Owing to the atomic size difference between Pt and W, the Pt/W(110) surface was under tensile strain. Furthermore, the dissimilar electronic structures induce charge transfer from W to Pt. Both the tensile strain and charge transfer influence the adsorption energy of the adsorbates (H2O, OH, and H) in HER catalysis. It is of interest to characterize which of these is predominant across the varying intermediates. This would lead to the opportunity to fine-tune HER catalysis, and the free energy landscape would be flat (Fig. 3) under optimal conditions. | ||
| Fig. 3 Change in bond lengths on the Pt/W(110) surface by intermediate adsorptions: (a) for H2O, (b) for OH, and (c) for H. (d) Gibbs free energy diagrams of alkaline HER on bulk Pt/W(110) and Pt(111) surfaces. | ||
Khorshidi et al. insisted that the adsorbate should cause eigenstress either in the outward or inward direction by studying CH2 adsorption on a Cu(110) surface.36 When CH2 adsorbs on the bridge site, the Cu atoms adjacent to the adsorbate experience positive eigenstress, whereas the four-fold hollow site-adsorbed CH2 exerts negative eigenstress. Minimization of the potential energy of the system due to stress can be achieved by applying an additional external strain via the formation of a heterostructure similar to the Pt/W(110) surface. As the structure is stabilized, the binding energy is accordingly strengthened and vice versa.
We investigated the HER mechanism on the Pt/W(110) surface under tensile strain. As H2O adsorbed on the Pt/W(110) surface, the surface Pt atoms were pushed outwards by the positive eigenstress. Additionally, the W substrate expanded the top Pt monolayer. Consequently, the positive eigenstress and the external strain were in the same direction and stabilized the potential energy of the structure, eventually leading to enhanced H2O adsorption strength. We found that this rationale can explain the trends of the adsorption energies with varying adsorbates on the active sites, even when the strains are in the same direction. The HER intermediates in alkaline media, H2O, OH, and H, are adsorbed at the atop, long bridge-like hollow sites and atop the Pt surface, respectively. The adsorption energies (Table 1) well agree with the previous report.24 Both the H2O and OH molecules favor adsorption on the Pt/W(110) surface more than on the Pt(111) surface, and it is notable that OH exhibits a stronger preference for the Pt/W(110) surface than for the Pt(111) surface, whereas the binding strength of H is weaker on the Pt/W(110) surface than on the Pt(111) surface (Table 1).
| Pt(111) | Pt/W(110) | |
|---|---|---|
| H2O* | −0.46 eV | −0.48 eV |
| OH* | 0.85 eV | 0.56 eV |
| H* | −0.53 eV | −0.28 eV |
| H*OH* | 0.31 eV | 0.03 eV |
We monitored changes in surface Pt–Pt bond length during adsorption, as shown in Fig. 3. As H2O molecule adsorbs (Fig. 3a), the surface strain is activated by the eigenstress into only one of the directions (+0.20 Å) to slightly stabilize the H2O*–Pt/W(110) surface, thus representing a similar adsorption energy to the H2O*–Pt(111) surface. This implies that the two contradictory factors, tensile strain and charge transfer from W to Pt, offset each other for the H2O adsorption. In contrast, the bond lengths are significantly elongated (+0.35 Å and +0.25 Å) in the two orthogonal directions of the plane during OH adsorption at the long bridge-like hollow site, as shown in Fig. 3b. An extensively increased adsorption energy by 0.29 eV was ascribed to the fact that the local eigenstress in the positive direction was relieved by the tensile strain on the Pt/W(110) surface. By contrast, the H atom adsorbs on the atop site (Fig. 3c) without evident changes in the bond lengths, indicating that atop is less affected by the strain effect37 and the ligand effect mainly governs the binding behavior. Interestingly, the interplay of these two factors improved the overall HER activity, as shown in Fig. 3d. The adsorption energy calculations on the fixed Pt/W(110) surface upon adsorption of the intermediates are provided in Table S5 and Fig. S2.† The comparison enables us to decouple the effect of adsorbate-induced eigenstress. For H2O and OH adsorbates, where strain relieves the stress, the fixed surface inhibits the adsorbate-induced change and binding interaction decreases. In the case of the H atom, no difference in adsorption energy between the fixed and relaxed surfaces upon adsorption is observed. The free energy diagram for the HER on the Pt/W(110) surface demonstrated a flatter landscape than that on the Pt(111) surface. It is clear that the two energy barriers of the Pt/W(110) surface for H2O bond dissociation into H*–OH* and H2* desorption significantly decreased, resulting in higher HER activity.
In short, the heterogeneous atomic layer in Pt/W(110) beneficially modified the intermediate adsorption characteristics of the Pt(111) surface for HER catalysis: stronger adsorption for OH, weaker adsorption of H, and a marginal change for H2O. Consequently, these are characterized by a higher HER activity in the free energy diagram.
HER activity of W@Pt nanoparticles
We calculated the HER mechanisms in the W@Pt nanoparticles to determine whether the two factors interact for high HER activity as on the bulk Pt/W(110) surface. DFT calculations indicated that an average bond length over different sites in W@Pt nanoparticle changes only slightly from that of Pt55: edge–edge (+1.11%) and edge–vertex (−1.05%) (Fig. 4a). The Pt–Pt bond lengths in Pt55 featured two distinct peaks: a shorter one for the edge–vertex and a longer one for the edge–edge sites (Fig. 4b). In contrast, the two bimodal peaks were somewhat obscured in the W@Pt nanoparticles, merging into a smooth peak. This is fairly counterintuitive considering that larger W atoms occupy the inner shells of the W@Pt nanoparticles. We ascribed it to the fact that the attractive interaction of Pt–W atoms was stronger in the W@Pt nanoparticles than that of Pt–Pt, which significantly suppressed the tensile strain in the Pt shell. The stronger core–shell binding interaction in W@Pt is shown by the crystal orbital Hamilton population (COHP) in Fig. 4c, Pt–Pt in Pt55 and W–Pt in W@Pt nanoparticles. This also indicates that the strain effect is marginally influential on the HER catalytic activity, which is consistent with the PDOS and charge density difference plot (Fig. 6b–h). | ||
| Fig. 4 Bond lengths of Pt–Pt atoms on the surfaces of Pt55 and W@Pt nanoparticles in (a); the grey is for edge–vertex and black for edge–edge atoms. In (b) the bond length distribution in W@Pt and Pt55 nanoparticles is shown and (c) illustrates crystal orbital Hamilton populations (COHP) analysis between the core and shell atoms (Pt–Pt for Pt55 and W–Pt for W@Pt) and the negative values of COHP (–COHP) are on the left and integrated (–ICOHP) on the right. | ||
We decoupled the strain effect from the ligand field effect by constructing W@Pt nanoparticles with the same interatomic bond lengths as Pt55 (W@Pt*). Therefore, the difference in the adsorption energies between the W@Pt and W@Pt* nanoparticles should arise only due to the change in the bond length. However, the d-band center energy shift from Pt55 to W@Pt* is solely caused by the ligand effect. Based on the collective effects of the ligand field theory and surface strain, the effect of interatomic distance change seems smaller for all adsorbates, supporting the aforementioned analysis (Fig. 5). The ligand effect estimated using the amount of charge transfer from W to Pt and the degree of Pt antibonding orbital filling, eventually, is more effective in changing the binding strengths of the adsorbates.
 | ||
| Fig. 5 Net change in adsorption energy, a cumulative result of ligand and strain effects for each intermediate adsorbate, (a) H, (b) OH, and (c) H2O. W@Pt* denotes the unstrained surface with its geometry restricted to the coordinates of the relaxed Pt55 system to decouple the ligand effect and strain effect. | ||
Our DFT calculations indicate that both H2O and OH molecules favor the vertices of the Pt55 and W@Pt nanoparticles for chemisorption (Fig. 6a). The component analysis of d-orbitals in the PDOS for the Pt atoms in the vertices of W@Pt (Fig. 6b–g) reveals a downshift of d-band center energies compared to that of Pt55. Three of the five d-orbital components which include the z-direction components (dz2, dxy, and dyz) showed the most significant changes in both the center energy and electron density near the Fermi energy. This implies that the nature of the atomic interactions between the core and shell (vertical direction, i.e., the z-component) substantially influenced the PDOS shift and binding energy. The charge density difference plot in Fig. 6h depicts significant charge transfer from W to Pt. In summary, in the W@Pt nanoparticles, the charge transfer from W to Pt predominantly influences the HER catalytic activity over the surface strain in the outermost shell.
 | ||
| Fig. 6 Vertex Pt atom marked in (a). (b) Total d-orbital and the d-orbital component ((c) dxy, (d) dx2–dy2, (e) dz2, (f) dxz and (g) dyz) partial density of states (PDOS) analyses for the outermost shell Pt atoms located at vertex sites of Pt55 and W@Pt nanoparticles. The charge density difference plot for the W@Pt was in (h) with an isosurface level of 0.01 e Å−3. | ||
The Gibbs free energy diagram along the entire HER catalytic pathway on W@Pt unveils several additional insights (Fig. 7a). The adsorption strengths of all intermediates, H2O, OH, H, and H–OH pairs, became weaker on W@Pt than on Pt55 (Table 2), resulting in an HER activity similar to that of the bulk Pt(111) surface. While the high activation energy problem for H2 desorption in Pt55 was effectively resolved in the W@Pt nanoparticle by the ligand effect, the RDS was still the same as that of the bulk Pt surface (H2O bond dissociation). Notably, the adsorption of the H–OH pair is considerably weak. This was attributed to the differences in the adsorption sites of the Pt55 and W@Pt nanoparticles. In the W@Pt nanoparticles, both H and OH are adsorbed at nearby bridge sites in the edges (Fig. 7b), whereas H is adsorbed on the atop site of the edge and OH on the atop site of the vertex (Fig. 7c) in Pt55. Closer adsorption sites allow the easier formation of hydrogen bonds by H–OH, which in turn weakens the binding between the adsorbates and the catalyst surface. Stronger interactions between the H and OH pairs adsorbed on W@Pt were confirmed by the adsorption energies. When H and OH were placed on different facets at a wider distance (Fig. 7d) while maintaining the edge-bridge adsorption sites for both H and OH just as the coplanar adsorption (Fig. 7e), the binding strength with the catalyst surface was reinforced. However, the corresponding configuration does not occur in the HER catalytic pathway, because the closer adsorption sites of the pair of molecules within the same plane are more energetically feasible.
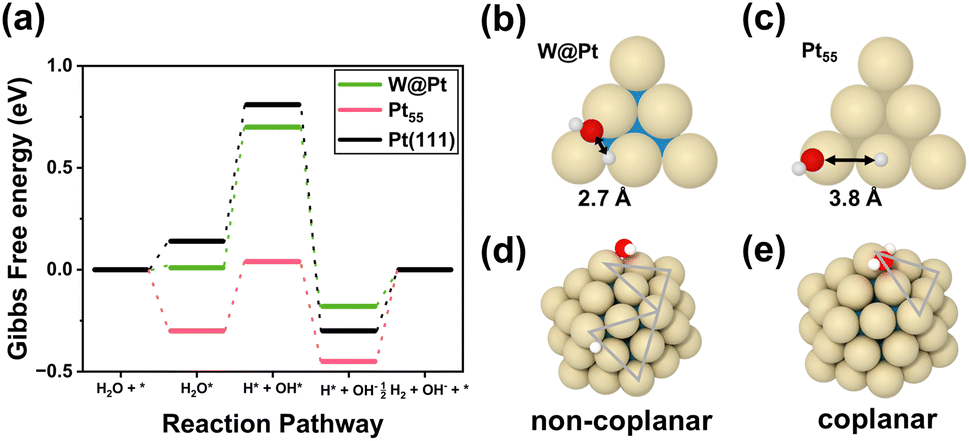 | ||
| Fig. 7 (a) Gibbs free energy diagrams for the alkaline HER on the W@Pt and Pt55 nanoparticles and the bulk Pt(111) surface. The H*–OH* pair adsorption sites were marked in (b) for W@Pt and Pt55 in (c). Non-coplanar (d) and coplanar (e) adsorption of the H*–OH* pair on the bridge sites in the edges of W@Pt nanoparticles. | ||
| Pt55 | W@Pt | |
|---|---|---|
| H2O* | −0.88 eV | −0.55 eV |
| OH* | 0.26 eV | 0.36 eV |
| H* | −0.67 eV | −0.38 eV |
| H*OH* | −0.40 eV | 0.20 eV |
In summary, the surface Pt–Pt bond lengths did not change tangibly in W@Pt, contradicting the conventional idea that the surface strain should play a key role in controlling the adsorption energy. Instead, the ligand effect manifested by charge transfer from W to Pt drives the filling of antibonding orbitals in the Pt d-orbitals, weakening the binding strength of the adsorbates. This means that simply making smaller nanoparticles of either a pure or core–shell alloy does not enhance the HER activity since the adsorption energy is controlled all uniformly, not specifically for the key intermediate in the RDS.
Conclusion
Through first-principles DFT calculations, we identified that the pseudo-binary Pt/W(110) surface is a more active catalyst for the HER in alkaline media than pure bulk Pt and W@Pt core–shell nanoparticles. The Pt/W(110) surface demonstrates a remarkably low activation barrier for H2O bond dissociation, producing a proton intermediate in an alkaline medium. We elucidated the underlying mechanism of the dual-mode interplay in activity, including surface tensile strain from atomic size differences and charge transfer between Pt and W due to their unequal work functions. The two factors influenced uniformly for W@Pt nanoparticles, stabilizing all intermediates and resulting in activity similar to that of the bulk Pt(111) surface. It is also noteworthy that the RDS in Pt nanoparticles is not H2O bond dissociation, but H2 molecule desorption in the final step of the HER. The mechanism on low activity of the W@Pt nanoparticles arises from the strong interaction between Pt atoms in the shell and W atoms in the core regions, which inhibits structural deformation. This interaction, along with the ligand effect, shifts the adsorbate binding properties, reducing HER efficiency. These two factors were optimized on the Pt/W(110) surface, leading to high catalytic activity for the HER and tuning of the adsorption energy of each intermediate. The framework of this study opens new opportunities for developing highly active catalysts for electrochemical reactions in alkaline media.Data availability
The data containing additional calculation results for this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (2022M3H4A04096482) funded by the Ministry of Science, the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (20214000000090, Fostering human resources training in advanced hydrogen energy industry), and the Technology Innovation Program Development Program (“20022479”, “Development of deuterium oxide localization and deuterium benzene synthesis technology to improve OLED lifetime by 25%”) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).References
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- R.-T. Liu, Z.-L. Xu, F.-M. Li, F.-Y. Chen, J.-Y. Yu, Y. Yan, Y. Chen and B. Y. Xia, Chem. Soc. Rev., 2023, 52, 5652–5683 RSC.
- J. Guo, J. Liu, X. Zhang, X. Guan, M. Zeng, J. Shen, J. Zou, Q. Chen, T. Wang and D. Qian, J. Mater. Chem. A, 2022, 10, 13727–13734 RSC.
- T. S. Rodrigues, A. G. M. da Silva and P. H. C. Camargo, J. Mater. Chem. A, 2019, 7, 5857–5874 RSC.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- S. Liu and S. Huang, Int. J. Hydrogen Energy, 2018, 43, 15225–15233 CrossRef CAS.
- Y. Li, W. Pei, J. He, K. Liu, W. Qi, X. Gao, S. Zhou, H. Xie, K. Yin, Y. Gao, J. He, J. Zhao, J. Hu, T.-S. Chan, Z. Li, G. Zhang and M. Liu, ACS Catal., 2019, 9, 10870–10875 CrossRef CAS.
- W. Lee, H. Yun, Y. Kim, S. S. Jeon, H. T. Chung, B. Han and H. Lee, ACS Catal., 2023, 13, 11589–11597 CrossRef CAS.
- X. Zhang, K.-A. Min, W. Zheng, J. Hwang, B. Han and L. Y. S. Lee, Appl. Catal., B, 2020, 273, 118927 CrossRef CAS.
- L. Zhang and G. Henkelman, ACS Catal., 2015, 5, 655–660 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151(23), 234101 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140(8), 084106 CrossRef PubMed.
- J.-H. Meng, C. A. Menning, M. B. Zellner and J. G. Chen, Surf. Sci., 2010, 604, 1845–1853 CrossRef CAS.
- G. Wulff, Z. Kristallogr. Cryst. Mater., 1901, 34, 449–530 CrossRef CAS.
- J.-R. Huo, X.-X. Wang, L. Li, H.-X. Cheng, Y.-J. Su and P. Qian, Sci. Rep., 2016, 6, 35464 CrossRef CAS PubMed.
- J. Tymoczko, F. Calle-Vallejo, W. Schuhmann and A. S. Bandarenka, Nat. Commun., 2016, 7, 10990 CrossRef CAS PubMed.
- J. L. C. Fajín, A. Bruix, M. N. D. S. Cordeiro, J. R. B. Gomes and F. Illas, J. Chem. Phys., 2012, 137(3), 034701 CrossRef PubMed.
- B. C. Han, C. R. Miranda and G. Ceder, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77, 075410 CrossRef.
- B. H. Kim, J. Heo, S. Kim, C. F. Reboul, H. Chun, D. Kang, H. Bae, H. Hyun, J. Lim, H. Lee, B. Han, T. Hyeon, A. P. Alivisatos, P. Ercius, H. Elmlund and J. Park, Science, 2020, 368, 60–67 CrossRef CAS PubMed.
- J. Marchetti, E. Gonzalez, P. Jasen, G. Brizuela and A. Juan, Surf. Rev. Lett., 2008, 15, 319–327 CrossRef CAS.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149(17), 174705 CrossRef PubMed.
- J. M. Wills and W. A. Harrison, Phys. Rev. B:Condens. Matter Mater. Phys., 1983, 28, 4363–4373 CrossRef CAS.
- T. E. Madey, K.-J. Song, C.-Z. Dong and R. A. Demmin, Surf. Sci., 1991, 247, 175–187 CrossRef CAS.
- M. B. Zellner, A. M. Goda, O. Skoplyak, M. A. Barteau and J. G. Chen, Surf. Sci., 2005, 583, 281–296 CrossRef CAS.
- A. Khorshidi, J. Violet, J. Hashemi and A. A. Peterson, Nat. Catal., 2018, 1, 263–268 CrossRef.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08250h |
| This journal is © The Royal Society of Chemistry 2025 |