
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of Ce co-doping at the A site of Sm0.5−xSr0.5CoO3±δ for a high-performance air electrode for solid oxide reversible cells†
Sovann
Khan
 *a,
Aleksandar
Staykov
a,
Junko
Matsuda
a,
Maksymilian
Kluczny
a,
Kuan-Ting
Wu
a,
Kakeru
Ninomiya
b,
Maiko
Nishibori
b,
Jun Tae
Song
ac,
Motonori
Watanabe
a,
Miki
Inada
ac and
Tatsumi
Ishihara
*ac
*a,
Aleksandar
Staykov
a,
Junko
Matsuda
a,
Maksymilian
Kluczny
a,
Kuan-Ting
Wu
a,
Kakeru
Ninomiya
b,
Maiko
Nishibori
b,
Jun Tae
Song
ac,
Motonori
Watanabe
a,
Miki
Inada
ac and
Tatsumi
Ishihara
*ac
aInternational Institute for Carbon-Neutral Energy Research, (WPI-I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: khan.sovann.455@m.kyushu-u.ac.jp; ishihara@cstf.kyushu-u.ac.jp
bInternational Center for Synchrotron Radiation Innovation Smart, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan
cDepartment of Applied Chemistry Faculty of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
First published on 17th January 2025
Abstract
Oxide perovskites, such as SrCoO3, are considered to be promising air electrode catalysts for solid oxide cells. SrCoO3 is composed of non-precious elements and possesses catalytic activity for various reactions, including an oxygen reduction and an oxygen evolution reaction. However, the catalytic activity of this material is typically limited at reduced temperatures. In this study, the catalytic activity of SrCoO3 was improved by co-doping of cerium (Ce) and samarium (Sm) at the A site (Sr site) of SrCoO3. Although Ce was considered the B-site dopant in terms of ionic size and coordination number, a small amount of Ce was successfully substituted at the Sr site, simultaneously with Sm. Oxygen reduction and evolution activity were significantly increased by substitution of a small amount of cerium (∼2.5 mol%) at the A-site. At 973 K, the maximum power density generated from the LaGaO3-supported cell with Ce–Sm co-doped SrCoO3 as an air electrode was 0.62 W cm−2 in fuel cell mode, and the current density in steam electrolysis mode at 1.5 V was 0.93 A cm−2. The increased air electrode activity could be assigned to the improvement in the surface active sites and electrical conductivity of SrCoO3 by simultaneous substitution of Ce with Sm at the A site.
1. Introduction
The solid oxide fuel cell (SOFC) is considered a green energy conversion device, which enables the production of electricity with high efficiency (>60%).1 Carbon emissions can be suppressed by using SOFCs, particularly when hydrogen is used as fuel. Therefore, using SOFCs as an energy converting technology is highly expected for achieving a carbon-neutral society. Typically, the reversed operation of a fuel cell (FC), turning it into an electrolysis cell (EC), allows the conversion of excess electricity from renewable energy sources into chemicals such as H2via steam electrolysis. This reversible operation gives the solid oxide cell (SOC) more potential as a flexible storage solution for renewable energy. During periods of high energy demand, H2 can be converted to electricity via an FC, and during times of low energy demand, electricity can be utilized to produce H2via an EC. This reversibility is ideal for renewable energy storage in terms of cost-effectiveness and scalability.2,3The development of highly active bifunctional catalysts for the air electrode enabling the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER) is one of the rate-determining steps in the fabrication of SOCs. SOCs have conventionally been operated at high temperatures (>1123 K).4 For cost reasons (using cheaper materials for plant components and metal interconnects) and for durability, efforts to develop SOCs for decreasing the operating temperature (typically at intermediate temperatures of 873–1073 K) have been actively explored.5 However, the large overpotential of the ORR/OER is a major issue for intermediate temperature operation, leading to a drop in the electrochemical performance of cells.4–6 To overcome this issue, the development of highly active oxygen catalysts for the intermediate temperature range is strongly required for realizing the high potential of SOCs with low-cost operation, high efficiency, and long life.
Many methods have been reported to develop highly active cathodes for SOFCs operated at intermediate temperatures, including doping/co-doping, heterojunctions and micro/nanostructures (e.g., core–shell structure, nanofibers, nanoporous structure).7–12 Among these approaches, elemental doping/co-doping shows high potential for tuning the catalytic properties of a wide range of materials. This method has been used to synthesize various structures and compositions of materials, effectively enhancing the ORR activity by improving the surface catalytic activity, electronic and ionic conductivity, structural stability and so on.5
The ABO3 oxide perovskite family, such as SrCoO3, consists of a large cation with a coordination number of 12 at the A site and a small cation with a coordination number of 6 at the B site, and it has been reported as an active catalyst for various reactions, including the ORR and OER.7 Due to the mixed ionic–electronic conductivity of SrCoO3, the reaction can occur on the entire surface of the electrode in addition to the electrode–electrolyte–air three-phase boundary (TPB). This is different from conventional ORR electrodes, where the reaction takes place predominantly at the TPB.13,14 More interestingly, SrCoO3 exhibits diverse physicochemical properties when doped with aliovalent elements. It is well-known that both A- and B-site doping have significant effects on the catalytic activities of oxide perovskites. Generally, the A site can be substituted by large cations (e.g., lanthanides), while the B site can be substituted by small cations (e.g., transition metals, Fe or Mn).5,7 The ionic radii and valence states of dopants are crucial factors affecting the electrochemical properties of materials.
A-site (Sr site) substitution by aliovalent dopants was reported to have beneficial effects on ORR activity by stabilizing the lattice oxygen vacancies, subsequently influencing the electronic and chemical state of the B-site ions due to charge-compensated effects. For example, lanthanide elements such as Sm, La, and Gd have been successfully doped into the Sr site and exhibited high ORR activity for SOFCs.14,15 Besides the defects, doping/co-doping at the A site with redox-active elements has been reported to improve the oxygen exchange capacity of perovskite-based materials.16,17
Among lanthanide elements, cerium (Ce) ion is an interesting dopant due to its different ionic radii and valence states. Several reports have found the positive effects of doping Ce into the A site of perovskites, such as Sm1−xCexFeO3−δ, Sr1−xCexMnO3−δ, and Sr0.9Ce0.1Co0.8Ni0.2O3−δ.18–20 Generally, mixed valence states of Ce3+ and Ce4+ are commonly used as dopants or in alloy/composite materials. The ionic radii of Ce3+ and Ce4+ are significantly different (1.34 Å vs. 1.13 Å).21 Although Ce4+ is the most stable species among Ce valence states, the co-existence of Ce3+ is commonly observed, typically at defect sites.22 The presence of Ce3+ was proved to be crucial for redox cycles (Ce3+ ↔ Ce4+), which improved the oxygen exchange capability of oxide perovskites for thermochemical applications.16,17 However, Ce doping into the A site seems to be limited because of the mismatch of ionic size and it generally forms an impurity phase of CeO2 together with the perovskite phase. The successful incorporation of Ce into a crystal lattice was proposed with an oxygen-deficient lattice.23 Alternatively, the A-site cation deficiency strategy was successfully used to dope Ce ion at the A site of SrTi0.5Mn0.5O3−δ.16
In this study, Ce ions were co-doped with samarium (Sm) into the Sr site of SrCoO3 to form a new compound, CexSm0.5−xSr0.5CoO3−δ (CeSSC), which was used as an air electrode of a La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM)-supported cell operated at temperatures from 973 to 1073 K. With a small amount of Ce co-doping (x = 0.025 or 2.5 mol% of A cations) at the A site, the electrochemical performance as an air electrode was significantly increased. Enhanced surface active sites and electrical conductivity induced by Ce co-doping are considered to play important roles in the improved electrochemical properties of SrCoO3.
2. Experimental
Material synthesis
CexSm0.5−xSr0.5CoO3±δ (CeSSC) was synthesized by using the sol–gel method, as reported in our previous study.15 All metal precursors including 7 − x mmol of Sm(NO)3.6H2O (99.5%, Wako Pure Chemical Industries Co., Ltd, Japan), 7 mmol of Sr(NO)3 (98%, Wako Pure Chemical Industries Co., Ltd, Japan), 14 mmol of Co(NO3)2.6H2O (99.5%, Wako Pure Chemical Industries Co., Ltd, Japan), and x mmol (x = 0–3.5) of Ce(NO2)3.6H2O (98%, Wako Pure Chemical Industries Co., Ltd, Japan) were dissolved in 200 mL of deionized water under magnetic stirring. Then, 56 mmol of citric acid (C6H8O7·H2O, 99.5%, Wako Japan) and 10 mL of polyethylene glycol 400 (PEG400, H(OCH2CH2)nOH, Wako Pure Chemical Industries Co., Ltd, Japan) were subsequently added to prepare the metal precursor solution with a total concentration of 0.14 M. This mixture was heated to 553 K until complete evaporation of water. The solid product was fired at 673 K for 2 h and annealed at 1173 K for 3 h under ambient conditions. Finally, the synthesized powder was ground using a pestle and mortar before use.Other materials, such as La0.9Sr0.1Ga0.8Mg0.2O3−δ (electrolyte powder), NiO–Fe2O3 (anode materials) and TiO2 added Ce0.6La0.4O2−δ (Ti-LDC, used as buffer layer materials), were prepared by solid-state reaction and sol–gel methods, as described in our previous reports.24,25 Detailed processes are described in the ESI.†
Cell fabrication
Electrode slurries of the anode, cathode, and buffer layer were screen-printed on both sides of a 0.3 mm LSGM pellet with an effective area of 0.196 cm−2 for all layers. Ti-LDC was used as the buffer layer between the NiFe anode and the LSGM. The single cell configuration is NiFe|Ti-LDC|LSGM|SSC(CeSSC). After screen-printing, annealing was conducted at 1373 K and 1073 K for 1 h for the anode and cathode sides, respectively. For the symmetric cell, SSC or CeSSC slurry was screen-printed on both sides of a 0.3 mm LSGM pellet and annealed at 1073 K for 1 h. The cell configuration is shown in Fig. S1 in the ESI.†Analysis and characterization
Material characteristics such as morphologies and nanostructures were analyzed using scanning electron microscopy (SEM) (Versa 3D Hivac, FEI, USA) and transmission electron microscopy (TEM) (JEM-ARM200F, JEOL Ltd., Japan). The chemical composition and elemental distribution in each sample were also analyzed by SEM and scanning TEM (STEM) using energy-dispersive X-ray spectroscopy. Elemental mapping and electron energy loss spectroscopy (EELS) were performed using a (S)TEM (Titan ETEM G2, FEI, USA) equipped with a GIF Quantum energy filter (Gatan Inc., USA). The crystal structure was analyzed by X-ray diffraction (XRD) (Cu Kα line, Rigaku Rint 2500, Rigaku Corporation, Japan) with a scanning speed of 5θ degrees per minute and 0.05θ degrees of data acquired step. For Rietveld refinement, selective samples were performed slow-scanned XRD (scanning speed of 2θ degrees per minute and 0.02θ degree of data acquired step) and fitted using the PDXL 2 software package (Rigaku Corporation, Japan). The electronic and oxidation states were determined by X-ray photoelectron spectroscopy (XPS) (KRATOS ULTRA 2, Shimadzu Corporation, Japan). Low energy ion scattering (LEIS) was analyzed for the outermost surface composition (IonToF, Q-tac100).Fuel cell mode
Electrochemical analysis of the cell performance was conducted based on our previous setup.24 Pt meshes were placed on both the anode and cathode of the cell as a current corrector. Pt–lead wires were used to connect the electrodes to the electrochemical equipment. Another Pt wire was connected on the cathode side of the LSGM by Pt paste as the reference electrode. Before electrochemical measurement, the NiFe anode was reduced by humid H2 at 1073 K for 1 hour.For cell performance analysis, 100 mL of humid H2 was supplied to the anode, and 100 mL of dry O2 was supplied to the cathode. Electrochemical analysis of the cells, such as current–voltage–power (I–V–P) measurements, was conducted in the temperature range of 973–1073 K using the four-wire method, with separate current and voltage leads. The current across the cell was controlled using a potentiostat/galvanostat (HAL3001, Hokuto Denko, Japan), and the terminal voltage was measured using a digital multimeter (R6451A, Advantest, Japan). Electrical impedance spectra (EIS) of the electrodes were measured using an impedance/gain-phase analyzer (1260 impedance analyzer and 1287, Solartron, Farnborough, UK) with commercial Z-view software under open-circuit conditions at an AC voltage of 10–25 mV and a frequency range of 105 to 0.1 Hz.
Steam electrolysis cell mode
The cell setup and electrochemical analysis were the same as those used for the fuel cell mode except for the direction of the applied potential. However, the gas composition at the fuel electrode was changed to 5% H2/45% N2/50% steam, while oxygen was used at the air electrode.DFT calculations
Planewave periodic density functional theory (DFT) implemented in the Vienna Ab initio Software Package (VASP) was used.26–28 The simulations were performed using projector-augmented-wave (PAW) pseudopotentials with the Perdew–Burke–Ernzerhof (PBE) functional.29 Calculations were performed with 520 eV cutoff energy and 2 × 4 × 1 k-points sampling for the slabs. Cutoff energies and k-point densities were converged for an orthorhombic unit cell of SrCoO3. DFT + U Hubbart correction was employed to account for the electron correlation at the d-orbitals of Co and the f-orbitals of Ce. The UCo was set to 3.4 eV and the UCe was set to 3.0 eV. Electronic energies were relaxed to 10−6 eV and forces were relaxed to 10−2 eV Å−2. Nudged elastic band calculations (NEB) were used to determine activation energies and were converged to forces of 0.2 eV Å−2.3. Results and discussion
Characterization of materials
The effects of Ce co-doping on the crystal structures of the prepared materials were primarily investigated through XRD analysis. Several studies have already been reported on the use of Sm0.5Sr0.5CoO3−δ (SSC) as an active cathode material for low and intermediate temperature SOFCs.14,15 Therefore, in this study, we mainly focused on the effects of co-doping Ce in the A site of SSC perovskite. From our previous results, 50 mol% Sm doped at the Sr site in Sr(Sm)CoO3 was studied.15 In this experiment, Ce ions are introduced as co-dopants with Sm (CexSm0.5−xSr0.5CoO3±δ, where x = 0, 01, 0.025, 0.05, 0.10, 0.15 and 0.25).Fig. 1 summarizes the effects of different Ce co-doping concentrations on the crystal structures of SSC. A small amount of Ce co-doping (x = 0.01–0.05) maintains the single phase of SrCoO3, which is consistent with the phase observed without Ce co-doping. However, with an increase in the Ce co-doping amount to x = 0.1, a secondary phase is clearly detected (see black arrows in Fig. 1(a)). This secondary phase monotonously increases with the increase of Ce co-doping to x = 0.15 and 0.25. With Ce co-doping of x = 0.25, another impurity phase was clearly observed (see red arrow in Fig. 1(a)). Ce co-doping into the crystal lattice of SSC is further confirmed by magnified XRD peaks at 32–35° (Fig. 1(b)). The shift of the XRD peaks suggests the successful incorporation of Ce ions into the crystal lattice of SrCoO3. At low Ce concentrations (x = 0.01–0.05), a shift of the XRD peak was hardly observed. However, a significant shift to lower angle is observed at high Ce co-doping concentrations (x = 0.10–0.25), which indicates that the lattice constant is increased by the introduction of Ce ions.
 | ||
| Fig. 1 XRD patterns of CeSSC (CexSm0.5−xSr0.5CoO3±δ) with different Ce co-doping contents, x = 0–0.25. | ||
To investigate the detailed crystal structures, Rietveld refinement of the XRD peaks was conducted for samples co-doped with different Ce concentrations (x = 0, 0.025, 0.10, and 0.25), as shown in Fig. 2. The crystal structure information is summarized in Table 1. The XRD pattern of SSC (without Ce co-doping) in Fig. 2(a) is well fitted to the orthorhombic phase according to standard JCPDS#00-053-0112, which agrees with previous reports on the same materials.30–32 Introducing a small amount of Ce co-doping (x = 0.025, Ce0.025Sm0.475Sr0.5CoO3±δ) into the A-site does not change the crystal phase of SSC. XRD peaks in Fig. 2(b) are well matched to the same JCPDS data as the orthorhombic phase. However, when the Ce concentration is increased to x = 0.10, a secondary phase of CeO2 is clearly observed (Fig. 2(c)). The main diffraction peaks are still fitted to the orthorhombic structure. At a Ce concentration of x = 0.25, the Co3O4 phase co-exists with SrCoO3 and CeO2 (Fig. 2(d)). We observed that even at high Ce co-doping concentrations, the orthorhombic phase of SrCoO3 is still maintained, although the CeO2 phase is formed as a secondary phase. This indicates that only a small amount of Ce ions could be possibly doped into the SrCoO3 crystal lattice. Excessive amounts of Ce ions may exceed the solubility limit to form the CeO2 phase.
 | ||
| Fig. 2 XRD patterns and their Rietveld refinements of powder samples (a) without Ce co-doing, x = 0, and (b)–(d) with Ce co-doping at x = 0.025, 0.10 and 0.25 [JCPDS#00-053-0112 of orthorhombic SrCoO3; JCPDS#01-080-5547 of cubic CeO2; JCPDS#00-043-1003 of cubic Co3O4]. | ||
| Ce, x | Phase | Space group | Crystal cell | Reliability factors | ||||
|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | V (Å3) | R WP (%) | x 2 | |||
| 0 | Orth. SrCoO3 | Pnma (62) | 5.376 | 7.580 | 5.350 | 218.013 | 3.03 | 1.0841 |
| 0.025 | Orth. SrCoO3 | Pnma (62) | 5.386 | 7.590 | 5.375 | 219.729 | 2.34 | 1.3020 |
| 0.1 | Orth. SrCoO3 | Pnma (62) | 5.383 | 7.670 | 5.395 | 222.734 | 2.94 | 1.1307 |
| Cubic CeO2 |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) m (225) |
5.387 | 5.387 | 5.387 | 156.329 | |||
| 0.25 | Orth. SrCoO3 | Pnma (62) | 5.460 | 7.620 | 5.980 | 248.799 | 4.71 | 1.0798 |
| Cubic CeO2 |
Fmm (225) |
5.409 | 5.409 | 5.409 | 158.279 | |||
| Cubic Co3O4 |
Fdm (227) |
8.072 | 8.072 | 8.072 | 525.949 | |||
The common co-existence of the multi-valence state Ce3+/4+ ion provides a complex doping behavior into SrCoO3. The ionic sizes of Ce3+, Ce4+, Sr2+ and Sm3+ with coordination number of 12 are 1.34, 1.14, 1.44 and 1.24 Å, respectively. At low Ce concentration (x = 0.025), lattice constants are slightly increased. Because of the low concentration of Ce substituted, a change in the crystal structure is hardly observed. Tilting of the BO6 unit was observed from lanthanum element doping at the A-site of perovskites.33 However, this is not significant in Ce-doped SSC at this low concentration, as shown in Table 1. The cell volume keeps increasing with the increase of Ce co-doping contents, indicating that large-size cations are substituted at the Sm/Sr sites. Ce4+ ion is smaller than Sm3+ and Sr2+. Therefore, the substitution of Ce4+ at Sm/Sr sites should not extend the cell volume. It is likely that the large-sized Ce3+ cation substitutes at the Sm/Sr site in Sm0.5Sr0.5CoO3−δ perovskite.
Ce ions have been used as A- or B-site dopants in perovskites.20,34,35 To determine the doping site of Ce ions, the crystal structures of SSC with Ce co-doped at the B site (Co site) (Sm0.5Sr0.5Co1−xCexO3±δ, SSCCe) were also investigated. The crystal structure significantly changes when Ce ions are introduced into the Co site, even with a small amount (x = 0.025) (Fig. S2†). The size of Ce ions is much larger than that of Co ions. Therefore, the substitution of Ce ions at the Co site causes a large change in the crystal structure of the parent perovskite. More interestingly, when the same amount of Ce ions (x = 0.025) was introduced into the A-site without Sm, a large amount of CeO2 phase was formed (Fig. S3†). Ce doping into the Sr site is very limited without the Sm ion. It is not a surprising result that substitution of Ce ions into the A-site of Sr/LaCoO3 generally formed the impurity phase of CeO2 due to the low solubility of Ce into the A site of the perovskite lattice.16,36 Naik et al. introduced a cation deficiency strategy to improve the solubility of Ce doping at the Sr site of SrTi0.5Mn0.5O3−δ perovskite. They found that mixed valence Ce3+/4+ ions were successfully doped at the Sr site, and performed a redox cycle reaction, resulting in better oxygen exchange capability.16 In our case, it is likely that Sm3+ doped into the Sr2+ site creates lattice defects, which is  , and allows Ce ions to incorporate into the A-site of the perovskite. Therefore, we confirmed that Ce ions were successfully co-doped into the A-site with Sm ions in the CexSm0.5−xSr0.5CoO3±δ sample.
, and allows Ce ions to incorporate into the A-site of the perovskite. Therefore, we confirmed that Ce ions were successfully co-doped into the A-site with Sm ions in the CexSm0.5−xSr0.5CoO3±δ sample.
The morphology and nanostructure with and without Ce co-doping were studied by SEM and TEM/HR-TEM observation. Fig. 3(a)–(d) show SEM images of SSC and CeSSC at Ce doping contents of 0.025, 0.10, and 0.25, respectively. The SEM images indicate that the SSC powder consists of small particles (<500 nm) aggregated together. This is a common structure of sol–gel product particles subjected to high temperature post-annealing. In our case, post-annealing at 1173 K was conducted. Therefore, the aggregated structure of the synthesized particles is unavoidable. With Ce co-doping, no noticeable difference in structure is observed. As expected, Ce co-doped into the SrCoO3 lattice might not significantly affect the particle size/structure. Typically, at this low doping concentration (x < 0.025), the structure of the formed particles is almost identical and it is also expected that the surface area will be similar.
 | ||
| Fig. 3 SEM images of SSC (Sm0.5Sr0.5CoO3−δ) (a) and CeSSC (CexSm0.5−xSr0.5CoO3±δ) at x = 0.025 (b), 0.10 (c), and 0.25 (d). HR-TEM/STEM image and its elemental mapping of CeSSC (x = 0.025) (e) HR-TEM, (f) SAED pattern, (g) STEM and its elemental mapping. | ||
TEM/HR-TEM analysis allows us to investigate the detailed nanostructure and its crystal structure. CeSSC (x = 0.025) was selected as a model sample for HR-TEM analysis. In Fig. 3(e) and (f), the HR-TEM image of CeSSC and its selected area electron diffraction (SAED) pattern show a lattice distance of 0.359 nm, indicating the (101) plane of orthorhombic SrCoO3, according to standard data from JCPDS#00-053-0112, which implies the simple perovskite structure of CeSSC. Elemental mapping in Fig. 3(g) shows the homogeneous distribution of all elements in this sample, allowing us to conclude that Ce–Sm ions are successfully doped into the SrCoO3 lattice.
The elemental compositions of all prepared samples were analyzed with EDS. The EDS elemental compositions are summarized in Table S1.† Three locations from the same sample were analyzed, and average values were summarized. The elemental compositions of all samples are close to the stoichiometric target composition. The oxidation states of each element were analyzed by XPS. Wide-range XPS spectra were used for quick surveys of all elements from samples and main components such as Ce, Sm, Sr, Co, O, and C were detected from the CeSSC sample. No Ce signals were detected from the SSC sample, which is understandable since the SSC sample does not contain Ce element.
High-resolution XPS spectra of each element were intensively analyzed for three samples (Ce, x = 0, 0.025, and 0.10). There was no significant change in the binding energy of Sr2+ and Sm3+ with Ce co-doping (see Fig. S4†). A broad binding energy peak ranging from 875 to 910 eV is identified as the Ce 3d orbital, which comprises both Ce3+ and Ce4+ species (see Fig. 4(a)). The ratio of the area under the curve of each species is summarized in Table S2.† The low oxidation state of Ce3+ slightly decreases at high co-doping concentrations, for which the CeO2 phase was found in the sample by XRD analysis. The presence of Ce3+ should be the reason for the lattice expansion observed with Ce co-doped samples in XRD analysis. The peak at binding energy of 775–800 eV originates from the Co 2p orbital, which is also composed of Co2+ and Co3+ species in all samples (Fig. 4(b)). The shift of the Co 2p peak to lower binding energy with Ce co-doping indicates an increase in the lower valence state of Co. The change in the oxidation state of Co at the B-site might be due to the charge compensation induced by Ce3+/4+ co-doping at the Sr/Sm-site.37
 | ||
| Fig. 4 High-resolution XPS spectra of (a) Ce 3d, (b) Co 2p and (c) O 1s of samples with low and high Ce co-doping contents (x = 0.025 and 0.10). | ||
The clearer evidence can be provided by O 1s spectra in Fig. 4(c) and Table S2.† The broad binding energy from 527 to 536 eV originates from lattice oxygen (OL, 529.1 eV), surface-chemisorbed oxygens including oxygen-defected species (e.g., O−, O22−, O2−) (Odefect, 530.9 eV) and oxygen surface absorption or hydroxyl group (OO–O abs, or OO–OH, 531.7 eV), and absorbed moisture (OH2O abs, 533 eV).38 With Ce co-doping, chemisorbed oxygen increases, which infers that a defect-rich surface is formed with Ce incorporating.39
As a dopant, identification of the Ce species is very important. Previous works have reported that Ce4+ is stable in the Sr site.21,34,40 In this work, we found that mixed valence states of 3+ and 4+ of Ce ions were detected by XPS analysis. To confirm the XPS result, X-ray absorption spectroscopy was used. XAFS spectra of the K and L3 edge of Ce ions in Fig. S5 and S6† confirmed that the Ce specie contained in CeSSC and SSCCe has a lower oxidation state than that in CeO2, which is known for its richness of Ce4+. This indicates that Ce3+ co-exists in these samples. In addition, EELS analysis in Fig. S7† also allows us to conclude that both A and B site co-doped samples contained a mixed valence state of Ce3+ and Ce4+. Ce co-doped at the B-site showed a slightly higher valence state than that of A-site co-doping. By using XPS, XAFS and EELS analysis, it is clearly confirmed that mixed valence states of Ce3+ and Ce4+ were co-doped into the A-site of SrCoO3 perovskite.
Electrochemical performance of CeSSC for the ORR
CeSSC was used as the air electrode in a SOFC to evaluate the ORR activity. A single LSGM-electrolyte-supported cell using CeSSC and NiO–Fe2O3 as the cathode and anode, respectively, was prepared. Ti-LDC was used as the buffer layer to protect against Ni and Fe diffusion. The cell structure and characterization are shown in Fig. S1, S8, and S9.†I–V and I–P curves of both cells were compared at 973, 1023, and 1073 K, as shown in Fig. 5(a) and (b). The maximum power density (Max PD) of the cell using the CeSSC (Ce, x = 0.025) cathode is 0.62, 0.90, and 1.20 W cm−2 at 973, 1023, and 1073 K, respectively. Without Ce co-doping, the power density generated is 0.36 (973 K), 0.53 (1023 K), and 0.77 W cm−2 (1073 K). With a small amount of Ce co-doping (x = 0.025), the power density increased by more than 55% at 1073 K. | ||
| Fig. 5 I–P and I–V curves of cells with (a) SSC and (b) CeSSC cathode at 973, 1023 and 1073 K. (c)–(e) Impedance plots of cells. (f) Estimated polarization resistance as a function of temperature. (g) Long-term stability of FC operation at 973 K. | ||
Fig. 5(c)–(e) show EIS spectra for the whole cell under open-circuit conditions at different temperatures. These spectra consist of ohmic resistance, Rs (intercept on the x-axis), and polarization resistance (Rp), which commonly infers the internal resistance (IR) and overpotential of the cell (also see the equivalent circuit fitted in Fig. S10†). The CeSSC cell generates a smaller IR loss and overpotential than those of the cell made with conventional SSC in all temperature ranges (Fig. 5(c)–(f)). EIS analysis of the cathode half-cell shows a similar trend to the whole cell impedance spectra (Fig. S11†). The CeSSC cathode exhibited smaller Rp and Rs, indicating that the activity of the cathode significantly increased, which is attributed to the reduced overpotential and ohmic loss. The comparison of the estimated polarization resistance and ohmic loss for both cells at 973–1023 K is summarized in Table 2.
| Cathode | Temp. (K) | Max PD (W cm−2) | R s (Ω cm−2) | R p (Ω cm−2) |
|---|---|---|---|---|
| CeSSC | 973 | 0.62 | 0.465 | 0.178 |
| 1023 | 0.90 | 0.331 | 0.108 | |
| 1073 | 1.20 | 0.253 | 0.073 | |
| SSC | 973 | 0.36 | 0.578 | 0.318 |
| 1023 | 0.53 | 0.405 | 0.184 | |
| 1073 | 0.77 | 0.300 | 0.120 |
Further EIS studies were conducted with a symmetric cell, to remove the influences of the fuel electrode, in air and oxygen atmospheres at 773–1073 K. In both air and oxygen atmospheres, the polarization resistance of the CeSSC cell is smaller than that of the SSC cell (see Fig. S12†). The smaller Rp of CeSSC clearly suggests the increased ORR activity, which resulted in increased power density by the Ce co-doped cell.
To demonstrate the stability of Ce at the A site of SSC, a long-term stability test of the CeSSC cell was performed. Fig. 5(g) shows the terminal potential at 0.56 A cm−2 at 973 K as a function of operating time. The potential was decreased by 2.4% after 50 h of operation under O2 at the cathode and humid H2 at the anode site. Since it is well-known that the degradation rate over the initial 50 h is large because of degradation of the Ni base anode, this degradation rate is reasonably low. Impedance analysis of the cell after 50 h is shown in Fig. S13.† The cell polarization was almost the same before and after 50 h of operation, suggesting that the Ce at the A site of SrCoO3 is stable and suitable for air electrodes of SOCs. However, the 50 hours operation was not sufficient to demonstrate practical applicability on an industrial scale. Therefore, further work focusing on longer stability testing will be conducted, and the results will be reported in the future.
The electrochemical analysis shows that the CeSSC air electrode significantly enhanced the cell performance. The reduced IR loss and overpotential of the CeSSC cathode should be explained by the increased conductivity and catalytic activity of the materials. Therefore, the conductivity, Seebeck coefficient and O2-TPD of the SSC and CeSSC samples were further analyzed. The total electrical conductivity of both SSC and CeSSC (Ce, x = 0.025) decreases with the increase of temperatures, indicating the metallic behavior of the materials (Fig. 6(a)). Reduced electrical conductivity should be attributed to the loss of lattice oxygen at high temperature.32,41 The CeSSC sample shows higher total conductivity than that of the SSC sample, typically at low temperature.
 | ||
| Fig. 6 Temperature dependence of (a) electrical conductivity, (b) Seebeck coefficient, and (c) O2-TPD. | ||
The positive values for the Seebeck coefficients further confirmed that both SSC and CeSSC samples are electronic hole conductors (Fig. 6(b)). Compared with SSC, the Seebeck coefficient of CeSSC was smaller, suggesting that the higher charge carriers are induced by Ce co-doping.42,43 The O2-TPD profile in Fig. 6(c) shows the oxygen desorption behavior of SSC and CeSSC from room temperature to 1173 K. Two intense desorption peaks at 573 K to 673 K and >973 K were observed for both samples. At low temperature (573 K to 673 K), α-oxygen is ascribed to surface oxygen desorption or electronic hole formation, while at high temperature (>973 K), β-oxygen is assigned to the lattice oxygen desorption.13,38 The larger amount of oxygen desorption from CeSSC than that from SSC suggests the higher oxygen mobility in bulk, which is effective for expanding the oxygen reaction and diffusion/transport. This result is consistent with previous reports on A-site-doped perovskites, such as La0.8Sr0.2CoO3−δ, PrBa1−xCo2O5+δ and SmxSr1−xCoO3−δ.13,38,41
To propose the reaction mechanism, reactive species on the surface of the catalyst should be identified. LEIS spectroscopy is used to study the outermost surface composition of CeSSC powders, which gives useful insight for understanding the mechanism of increased surface activity for oxygen dissociation.44 The LEIS spectra in Fig. S14† show the strong peaks of Sr for both the SSC and CeSSC samples, which indicates that the surface of both samples was dominated by an SrO layer. Sr surface segregation is commonly observed on doped perovskites.57 By comparing the area under the peak of each element, it shows that a weak peak of co-doped Ce is observed on A-site doped SrCoO3 (CeSSC) since the amount of Ce is just 2.5 mol%. This is another reason explaining the enhanced ORR activity because surface Ce3+ are expected as active sites for the ORR.45
Based on the above results, we propose that the increased ORR activity of SSC by Ce co-doping can be explained by increasing the surface active sites and the feasible redox of Ce3+ ↔ Ce4+. XPS spectra show a large amount of oxygen defect-related species with Ce co-doping, which indicates more active sites for activation of oxygen molecules. LEIS spectra additionally suggest the surface Ce of CeSSC particles, which is commonly known as the active site for the ORR. In addition, the redox cycling reaction of Ce3+ ↔ Ce4+ is expected to increase the conductivity as the acceptor, i.e., the formation of hole and lattice oxygen viaeqn (1) and (2), considering the large amount of oxygen vacancies.
 | (1) |
 | (2) |
The power generation properties of cells using different cathodes with varying Ce co-doping contents were further measured. However, the cathodic performance decreased by increasing the Ce content higher than x = 0.025, indicating that Ce, x = 0.025 was the optimum co-doping amount. This might be related to the solubility of Ce4+/3+ at the A site of SSC, as shown in Fig. 1. Interestingly, the SSCCe cell still exhibits better performance than that of the SSC cell (Fig. S15†). This result indicates that co-doping of Ce ions into the Sr or Co site can enhance the ORR activity of SSC. However, Ce co-doping into the A-site is more effective for increasing cathodic activity due to the outermost surface composition, which largely affects the ORR activity of SSC.
From the experimental results, it was found that Ce co-doping significantly enhances the ORR activity of SSC, demonstrating its potential as a cathode for SOFCs. Surface active sites induced by Ce co-doping are considered an important factor in this enhancement. To confirm this finding, we conduct a theoretical study using density functional theory (DFT) to investigate the surface reaction behavior of Ce doping, particularly Ce3+. Calculations are performed for slabs of eight alternating layers of SrO and CoO2 with total formulae of Sr16Co16O48 and vacuum of 15 Å. The slabs are constructed from a preoptimized orthorhombic unit cell of SrCoO3. In the slabs, all atoms are fully relaxed. Two types of slab are investigated: SrCoO3 and SrCoO3/Ce. In the SrCoO3/Ce slab one Sr atom from the SrO surface is replaced by Ce and one Co atom of the CoO2 surface is replaced by Ce. The formal charges of Sr and Co are 2+ and 4+, respectively, corresponding to 3+ charge for the Ce. Such substitution will improve the electron-donating properties of the SrO/Ce surface as compared to SrO and the hole-donating properties of CoO2/Ce compared to CoO2. The oxygen dissociation reaction is investigated on the SrO and SrO/Ce surfaces.
The results are summarized in Fig. 7, where green color denotes Sr, blue color denotes Co, yellow color denotes Ce, and red color denotes O. The adsorption energy of O2 on the SrCoO3, SrO surface is −0.16 eV. The adsorption energy of O2 on the SrCoO3/Ce, SrO/Ce surface is −1.09 eV. In both cases, the surface adsorption activates the oxygen molecule, and the O–O bond is elongated to 1.26 Å and 1.28 Å, respectively, for the SrO and SrO/Ce surfaces. The oxygen dissociation proceeds on both surfaces to the formation of superoxo (on the SrO surface) or peroxo (on the SrO/Ce surface) species between the dissociated oxygen atoms and the lattice oxygens. The activation barrier on the SrO surface is 2.16 eV, which is comparable to other SrO-terminated perovskites. The activation barrier on the SrO/Ce surface is 0.63 eV. To understand the energy difference, we should look at the products. On the SrO/Ce surface, the transfer of four electrons results in two surface peroxo species. As a result, the oxygen molecule was dissociated and the final total charge of the ions in the system matches the initial as two oxide ions (2− each) are now replaced by two peroxo species (2− each). The reaction is facilitated by the easy electron transfer between Ce(3+) and O2. On the SrO surface, the reaction results in two surface superoxo species. As a result, the oxygen molecule is dissociated and the final total charge of the ions in the system is different from the initial as two oxide ions (2− each) now are replaced by two superoxo species (1− each). As a result, the surface is oxidized. The reaction is facilitated by valence state change in the subsurface Co species. This prediction strongly supports the experimental results, indicating that the surface becomes more active with Ce co-doping.
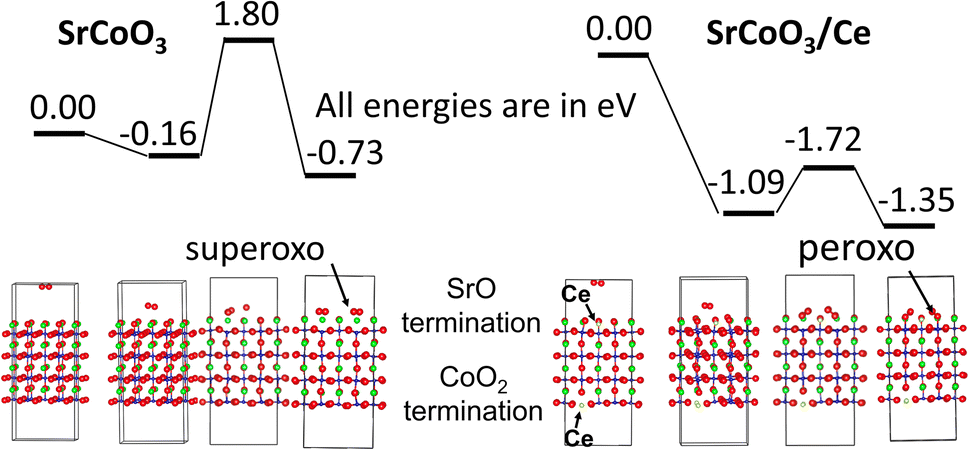 | ||
| Fig. 7 Simulation results for THE ORR on SrCO3 and SrCoO3/Ce. Green color denotes Sr, blue color denotes Co, yellow color denotes Ce, and red color denotes O. | ||
Electrochemical performance of CeSSC for the OER
In the previous section, the ORR activity of SSC was significantly increased with Ce co-doped at the A-site of Sr(Sm)CoO3. In this section, the OER activity of Ce co-doped SSC is studied for the reverse operation of a SOC. Fig. 8 shows the cell performance for steam electrolysis at different temperatures. The current density at 1.5 V is 0.32, 0.58 and 0.93 A cm−2 at 873, 923 and 973 K, respectively, with very small polarization resistance (Fig. 8(a) and (b)). This cell also generated very high efficiencies of hydrogen production, which is close to the theoretical calculation by the Faraday law (Fig. 8(c)). | ||
| Fig. 8 Steam electrolysis cell (EC) operation: (a) temperature dependence of I–V curves of EC operation, (b) impedance plots, (c) hydrogen generation as a function of current density, (d) and (e) comparison of the FC and EC performance of cells made with different cathode materials from previous reports: Pr0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (PBSLCC),46 La0.6Sr0.4Co0.2Fe0.8O3−δ Ce0.9Gd0.1O2−δ (LSCF-GDC (1)),47 SrCo0.5Fe00.45Ti0.05O3−δ (SCFTi),48 La0.1Sr0.8O4−δ (LSC),49 SSC,50 Sr0.9Y0.1CoO3−δ (SYC10)-GDC,51 SSC-SDC and SSC nanofiber,52 LSCF-GDC (2),53 LSCF-SDC,54 Sr2Ti0.8Co0.2FeO6 (STC02F),55 GdBa0.5Sr0.5Co0.9Cu1.1O5+δ (GBSCC),56 Sr2FeCo0.5Mo0.5O6−δ (SFCM).57 | ||
Therefore, Ce co-doping of the perovskite for the air electrode in a solid oxide cell is effective for improving the ORR and OER activity. A cell using CeSSC as an air electrode shows superior performance, which is highly competitive compared to the well-known materials reported earlier as summarized in Fig. 8(d) and (e) (also, see detailed data comparison in Tables S4 and S5†). As a result, Ce co-doping with Sm at the A site of SrCoO3 is highly effective for increasing the surface activity of oxygen dissociation and recombination.
4. Conclusions
Ce–Sm co-doping is highly effective in improving the ORR/OER activity of SrCoO3, making it a promising air electrode material for SOC operated at intermediate temperature. Ce–Sm co-doped SSC increased the surface active sites of SSC, as confirmed by XPS, LEIS analysis and DFT calculation. In addition, the redox reaction of Ce3+ ↔ Ce4+ is also considered to increase the hole concentration resulting in the increased oxide ion diffusion/transport. The formation of electronic holes is another reason for increasing air electrode performance from decreased ohmic loss. The maximum power density achieved on the LSGM-supported cell (NiFe|Ti-LDC|LSGM|CeSSC) is 0.62, 0.90 and 1.20 W cm−2 at 973, 1023 and 1073 K, respectively. Ce–Sm co-doped SSC also exhibits exceptional OER activity in steam electrolysis. The current density at 1.5 V generated from EC operation is 0.32, 0.58 and 0.93 A cm−2 at 873, 923 and 973 K, respectively. This high electrochemical performance of CeSSC is significantly attributed to the surface activity induced by Ce co-doping at the A site, which typically occupies the outermost surface. Therefore, doping Ce ions at the A site of the perovskite is highly effective in enhancing the activity of SrCoO3 as an air electrode.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sovann Khan: conceptualization (lead), data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), validation (lead), visualization (lead), writing–original draft (lead), writing – review & editing (lead). Aleksandar Staykov: data curation (supporting), formal analysis (supporting), investigation (supporting), methodology (supporting), validation (supporting), visualization (supporting), writing – review & editing (supporting). Junko Matsuda, Maksymilian Kluczny, Kuan-Ting Wu, Kakeru Ninomiya, Maiko Nishibori, Jun Tae Song, Motonori Watanabe, Miki Inada: formal analysis (supporting), investigation (supporting), methodology (supporting), writing – review & editing (supporting). Tatsumi Ishihara: conceptualization (lead), data curation (lead), formal analysis (lead), funding acquisition (lead), investigation (lead), methodology (lead), project administration (lead), supervision (lead), validation (lead), visualization (lead), writing – review & editing (lead).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this study was financially supported by the New Energy and Industrial Technology Development Organization (NEDO, JPNP20003), Japan and Grant-in-Aid for Scientific Research(S) (21H05010).Notes and references
- S. Khan and S. Soum, Univers. J. Catal. Sci., 2023, 1, 71–95 CrossRef.
- M. B. Halder, F. Salek, N. Haque, R. Savage, S. Stevanovic, T. A. Bodisco and A. Zare, Int. J. Hydrogen Energy, 2024, 52, 973–1004 CrossRef.
- C. Duan, J. Huang, N. Sullivan and R. O'Hayre, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- J. Zhang, S. Ricote, P. V. Hendriksen and Y. Chen, Adv. Funct. Mater., 2022, 32, 2111205 CrossRef CAS.
- A. Ndubuisi, S. Abouali, K. Singh and V. Thangadurai, J. Mater. Chem. A, 2022, 10, 2196–2227 RSC.
- H. A. Ishfaq, M. Z. Khan, Y. M. Shirke, S. Qamar, A. Hussain, M. T. Mehran, R.-H. Song and M. Saleem, Appl. Catal. B Environ., 2023, 323, 122178 CrossRef CAS.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- Y. Chen, Y. Chen, D. Ding, Y. Ding, Y. Choi, L. Zhang, S. Yoo, D. Chen, B. deGlee, H. Xu, Q. Lu, B. Zhao, G. Vardar, J. Wang, H. Bluhm, E. J. Crumlin, C. Yang, J. Liu, B. Yildiz and M. Liu, Energy Environ. Sci., 2017, 10, 964–971 RSC.
- J. G. Lee, J. H. Park and Y. G. Shul, Nat. Commun., 2014, 5, 4045 CrossRef CAS PubMed.
- P. Qiu, X. Yang, T. Zhu, S. Sun, L. Jia and J. Li, Int. J. Hydrogen Energy, 2020, 45, 23160–23173 CrossRef CAS.
- S. T. Aruna, L. S. Balaji, S. S. Kumar and B. S. Prakash, Renew. Sustain. Energy Rev., 2017, 67, 673–682 CrossRef CAS.
- X. Huang, G. Zhao, G. Wang and J. T. S. Irvine, Chem. Sci., 2018, 9, 3623–3637 RSC.
- F. Dong, M. Ni, Y. Chen, D. Chen, M. O. Tadé and Z. Shao, J. Mater. Chem. A, 2014, 2, 20520–20529 RSC.
- H. Zhang and W. Yang, Chem. Commun., 2007, 2007, 4215–4217 RSC.
- B. S. Kang, J. Matsuda, Y. W. Ju, H. H. Kim and T. Ishihara, Nano Energy, 2019, 56, 382–390 CrossRef CAS.
- J. M. Naik, B. Bulfin, C. A. Triana, D. C. Stoian and G. R. Patzke, ACS Appl. Mater. Interfaces, 2023, 15, 806–817 CrossRef CAS PubMed.
- G. Sai Gautam, E. B. Stechel and E. A. Carter, Chem. Mater., 2020, 32, 9964–9982 CrossRef CAS.
- W. Fan, Z. Sun, J. Wang, J. Zhou, K. Wu and Y. Cheng, J. Power Source, 2016, 312, 223–233 CrossRef CAS.
- H. Gu, H. Chen, L. Gao, Y. Zheng, X. Zhu and L. Guo, Int. J. Hydrogen Energy, 2008, 33, 4681–4688 CrossRef CAS.
- Y. Song, Y. Chen, M. Xu, W. Wang, Y. Zhang, G. Yang, R. Ran, W. Zhou and Z. Shao, Adv. Mater., 2020, 32, 1906979 CrossRef CAS PubMed.
- M. James, K. S. Wallwork, R. L. Withers, D. J. Goossens, K. F. Wilson, J. Horvat, X. L. Wang and M. Colella, Mater. Res. Bull., 2005, 40, 1415–1431 CrossRef CAS.
- K.-W. Park and C. S. Kim, Acta Mater., 2020, 191, 70–80 CrossRef CAS.
- D. J. Cumming, V. V. Kharton, A. A. Yaremchenko, A. V. Kovalevsky and J. A. Kilner, J. Am. Ceram. Soc., 2011, 94, 2993–3000 CrossRef CAS.
- T. Ishihara, N. Jirathiwathanakul and H. Zhong, Energy Environ. Sci., 2010, 3, 665–672 RSC.
- Z. Tan and T. Ishihara, J. Phys. Energy, 2020, 2, 024004 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, Int. J. Quantum Chem., 1985, 28, 497–523 CrossRef.
- J. Chen, X. Yang, D. Wan, B. Li, L. Lei, T. Tian, B. Chi and F. Chen, Electrochim. Acta, 2020, 341, 136031 CrossRef CAS.
- C.-L. Chang, C.-S. Hsu, J.-B. Huang, P.-H. Hsu and B.-H. Hwang, J. Alloys Compd., 2015, 620, 233–239 CrossRef CAS.
- S. Choi, J. Shin, K. M. Ok and G. Kim, Electrochim. Acta, 2012, 81, 217–223 CrossRef CAS.
- D. Muñoz-Gil, M. T. Azcondo, C. Ritter, O. Fabelo, D. Pérez-Coll, G. C. Mather, U. Amador and K. Boulahya, Inorg. Chem., 2020, 59, 12111–12121 CrossRef PubMed.
- W. Yang, T. Hong, S. Li, Z. Ma, C. Sun, C. Xia and L. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 1143–1148 CrossRef CAS PubMed.
- G. Zhang, X. Dong, Z. Liu, W. Zhou, Z. Shao and W. Jin, J. Power Source, 2010, 195, 3386–3393 CrossRef CAS.
- S. A. French, C. R. A. Catlow, R. J. Oldman, S. C. Rogers and S. A. Axon, Chem. Commun., 2002, 2002, 2706–2707 RSC.
- S. Khan, R. J. Oldman, C. R. A. Catlow, S. A. French and S. A. Axon, J. Phys. Chem. C, 2008, 112, 12310–12320 CrossRef CAS.
- J. Yang, S. Hu, Y. Fang, S. Hoang, L. Li, W. Yang, Z. Liang, J. Wu, J. Hu, W. Xiao, C. Pan, Z. Luo, J. Ding, L. Zhang and Y. Guo, ACS Catal., 2019, 9, 9751–9763 CrossRef CAS.
- Y. Song, Q. Zhong and W. Tan, Int. J. Hydrogen Energy, 2014, 39, 13694–13700 CrossRef CAS.
- Q. Wei, R. Guo, F. Wang and H. Li, J. Mater. Sci., 2005, 40, 1317–1319 CrossRef CAS.
- Y. Guo, D. Chen, H. Shi, R. Ran and Z. Shao, Electrochim. Acta, 2011, 56, 2870–2876 CrossRef CAS.
- B. Saha, J. A. Perez-Taborda, J.-H. Bahk, Y. R. Koh, A. Shakouri, M. Martin-Gonzalez and T. D. Sands, Phys. Rev. B, 2018, 97, 085301 CrossRef CAS.
- J. Mizusaki, J. Tabuchi, T. Matsuura, S. Yamauchi and K. Fueki, J. Electrochem. Soc., 1989, 136, 2082 CrossRef CAS.
- J. Druce, T. Ishihara and J. Kilner, Solid State Ionics, 2014, 262, 893–896 CrossRef CAS.
- K. Chen and S. P. Jiang, Electrochem. Energy Rev., 2020, 3, 730–765 CrossRef CAS.
- F. He, F. Zhu, D. Liu, Y. Zhou, K. Sasaki, Y. Choi, M. Liu and Y. Chen, Mater. Today, 2023, 63, 89–98 CrossRef CAS.
- Z. Wang, T. Tan, K. Du and C. Yang, ACS Appl. Energy Mater., 2023, 6, 6781–6789 CrossRef CAS.
- Q. Zhang, H. Qiu, S. Jiang, Y. Liu, J. Xu, W. Wang, D. Chen and C. Su, Energy Fuels, 2023, 37, 6740–6748 CrossRef CAS.
- F. Li, Y. Xu, Q. Wu, D. Zhao and M. Deng, Int. J. Hydrogen Energy, 2023, 48, 3204–3215 CrossRef CAS.
- S. Lee, J. H. Park, K. T. Lee and Y.-W. Ju, J. Alloys Compd., 2021, 875, 159911 CrossRef CAS.
- J. Liu, F. Jin, X. Yang, B. Niu, S. Yu, Y. Li and T. He, Ceram. Int., 2020, 46, 28332–28341 CrossRef CAS.
- S. W. Lee, J.-W. Park, Y.-W. Ju and T. H. Shin, Small Struct., 2024, 5, 2300292 CrossRef CAS.
- K. J. Kim, C. Lim, K. T. Bae, J. J. Lee, M. Y. Oh, H. J. Kim, H. Kim, G. Kim, T. H. Shin, J. W. Han and K. T. Lee, Appl. Catal. B Environ., 2022, 314, 121517 CrossRef CAS.
- Y. Wang, T. Liu, M. Li, C. Xia, B. Zhou and F. Chen, J. Mater. Chem. A, 2016, 4, 14163–14169 RSC.
- B. Niu, C. Lu, W. Yi, S. Luo, X. Li, X. Zhong, X. Zhao and B. Xu, Appl. Catal. B Environ., 2020, 270, 118842 CrossRef CAS.
- K. Li, K. Świerczek, P. Winiarz, A. Brzoza-Kos, A. Stępień, Z. Du, Y. Zhang, K. Zheng, K. Cichy, A. Niemczyk and Y. Naumovich, ACS Appl. Mater. Interfaces, 2023, 15, 39578–39593 CrossRef CAS PubMed.
- J. Zhao, Y. Shen, J. Gu, Y. Wang, Y. Zhao and Y. Bu, J. Power Source, 2024, 592, 233928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08181a |
| This journal is © The Royal Society of Chemistry 2025 |