
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the effect of nanocube support morphology on the hydrogenolysis of polypropylene over Ni/CeO2 catalysts†
Donald R.
Inns
 a,
Megan
Carr
b,
Mounib
Bahri
c,
Ajay
Tomer
a,
Troy D.
Manning
a,
Nigel
Browning
c,
Simon A.
Kondrat
d,
John B.
Claridge
a,
Alexandros P.
Katsoulidis
a and
Matthew J.
Rosseinsky
*a
a,
Megan
Carr
b,
Mounib
Bahri
c,
Ajay
Tomer
a,
Troy D.
Manning
a,
Nigel
Browning
c,
Simon A.
Kondrat
d,
John B.
Claridge
a,
Alexandros P.
Katsoulidis
a and
Matthew J.
Rosseinsky
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: m.j.rosseinsky@liverpool.ac.uk
bMaterials Innovation Factory, University of Liverpool, Oxford Street, Liverpool, L7 3NY, UK
cAlbert Crewe Centre for Electron Microscopy, University of Liverpool, Liverpool, L69 3GL, UK
dDepartment of Chemistry, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK
First published on 10th December 2024
Abstract
The catalytic hydrogenolysis process offers the selective production of high-value liquid alkanes from waste polymers. Herein, through normalisation of Ni structure, Ni mass and density, and CeO2 crystallite size, the importance of CeO2 nanocube morphology in the hydrogenolysis of polypropylene (Mw = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1; Mn = 5000 g mol−1) over Ni/CeO2 catalysts was determined. High liquid productivities (65.9–70.9 gliquid gNi−1 h−1) and low methane yields (10%) were achieved over two different Ni/CeO2 catalysts after 16 h reaction due to the high activity and internal scission selectivity of the supported ultrafine Ni particles (<1.3 nm). However, the Ni/CeO2 nanocube catalyst exhibited higher C–C scission rates (838.1 mmol gNi−1 h−1) than a standard benchmark mixed shape Ni/CeO2 catalyst (480.3 mmol gNi−1 h−1) and represents a 75% increase in depolymerisation activity. This led to shorter hydrocarbon chains achieved by the nanocube catalyst (Mw = 2786 g mol−1; Mn = 1442 g mol−1) when compared to the mixed shape catalyst (Mw = 4599 g mol−1; Mn = 2530 g mol−1). The enhanced C–C scission rate of the nanocube catalyst was determined to arise from a combination of improved H-storage and favourable basic properties, with higher weak basic site density key to facilitate a greater degree of hydrocarbon chain adsorption.
000 g mol−1; Mn = 5000 g mol−1) over Ni/CeO2 catalysts was determined. High liquid productivities (65.9–70.9 gliquid gNi−1 h−1) and low methane yields (10%) were achieved over two different Ni/CeO2 catalysts after 16 h reaction due to the high activity and internal scission selectivity of the supported ultrafine Ni particles (<1.3 nm). However, the Ni/CeO2 nanocube catalyst exhibited higher C–C scission rates (838.1 mmol gNi−1 h−1) than a standard benchmark mixed shape Ni/CeO2 catalyst (480.3 mmol gNi−1 h−1) and represents a 75% increase in depolymerisation activity. This led to shorter hydrocarbon chains achieved by the nanocube catalyst (Mw = 2786 g mol−1; Mn = 1442 g mol−1) when compared to the mixed shape catalyst (Mw = 4599 g mol−1; Mn = 2530 g mol−1). The enhanced C–C scission rate of the nanocube catalyst was determined to arise from a combination of improved H-storage and favourable basic properties, with higher weak basic site density key to facilitate a greater degree of hydrocarbon chain adsorption.
Introduction
Polyolefins such as polypropylene (PP) are predominantly used in packaging, construction, and textiles and due to the single use nature of many of these polyolefin products, a large amount of polymer waste is currently being generated.1 Much of this non-degradable waste is placed into landfill, leading to significant environmental damage and water pollution. Other disposal methods include incineration, which generates large amounts of CO2 and little resource gain, and mechanical recycling.2 Mechanical recycling is an important tool in the utilisation of polymer waste, but often leads to lower quality products over several recycle loops, with mixed PP waste streams often problematic and require additional sorting procedures.3 Therefore, chemical recycling methods utilising catalysts have been developed to address the shortfalls of other methods and produce high value products for use in several supply chains.4One such method is the hydrogenolysis process, which is a low-energy valorisation process to generate liquid alkane mixtures.5 The liquid products have the potential to be utilised as lubricant base oils, fuels, and for repolymerisation. The selective scission of the internal C–C backbone of PP chains is key to generating high selectivity towards liquid products, with terminal scission of end chains and methyl side chains leading to the production of methane.6,7 Noble metals such as Ru and Pt have been predominantly researched due to their high activity for C–C scission and H-activation.7–10 However, the high metal loadings required are prohibitively expensive and resource inefficient for scarce elements. Recently, catalysts based on earth-abundant metals such as Ni or Co with SiO2 and Al2O3 have been shown to be effective for the depolymerisation of polyethylene.11–15 Nevertheless, further development is needed to improve the viability of hydrogenolysis.
A strategy for improving C–C scission rates is the engineering of support materials, which facilitate the dispersion of nanoparticles and contribute to the activity of the catalysts.16,17 The mechanism of C–C scission occurs either through hydrogenolysis where bound alkylidyne intermediates on the metal particle site are cleaved with addition of activated hydrogen (H-addition), or hydrocracking, where cleavage occurs from the formation of carbocations and H-addition.18 Significantly, the acid–base properties of the support material define the dominant mechanism, with Brønsted acidic catalysts favouring the hydrocracking mechanism.19 However, both mechanisms have been reported to occur over acidic supported catalysts such as Pt/SiO2–Al2O3 and Ru/TiO2.20,21 Furthermore, the basicity of metal oxide carriers of Ru-supported catalysts was shown to be an important parameter in the selectivity of C–C scission for hydrogenolysis, with weak basic sites improving internal backbone scission through enhanced adsorption of polymer chains.22 Another important factor in polymer hydrogenolysis is utilisation of the H-spillover mechanism, which has been reported to shift the selectivity from terminal to backbone cleavage and improve scission rates.23–25 This mechanism consists of hydrogen being activated on a metal site with migration to a reducible support and reduction of the metal lattice.26 This allows for the storage and increased coverage of activated hydrogen (H-storage and H-coverage) that can be transferred to the metal particle for cleavage of the alkylidyne intermediates in the hydrogenolysis mechanism. Methods for improving H-coverage for polymer hydrogenolysis have been investigated through alterations of metal particle size, promoters, and support polymorph.23–25
A support material that combines favourable weak basic sites for hydrocarbon binding and reducible properties is CeO2, with Ru/CeO2 shown to be effective at hydrogenolysis of various polymer feedstocks with high C–C scission activity.22,23,27–29 Despite Ni/CeO2 catalysts being successfully utilised for lignin and glycerol hydrogenolysis, due to the tailorable surface acid–base and reducible properties of the Ni/CeO2 catalysts, the catalytic system remains underexplored for polymer hydrogenolysis.30,31 The development of highly active Ni catalysts for polymer hydrogenolysis remains challenging, however, utilisation of nanostructured supports is a potential strategy to improve C–C scission rates. CeO2 nanorods and nanocubes have been shown to alter the Ru dispersion and generate highly active catalysts, with the nanocube catalysts being the most effective at depolymerisation.27,28 SrTiO3 nanocubes, another reducible oxide, have also been successfully used to control the geometry and electronic properties of Pt.8,20,32,33 Evidently, the nanocube support morphology has a role in promoting catalyst C–C bond scission activity. However, given the structure sensitive nature of hydrogenolysis, with C–C scission rate and selectivity influenced by metal particle size, the exact promotional role of nanocube supports over standard mixed shape supports is unclear. Herein, we demonstrate active low-loading earth abundant Ni/CeO2 catalysts for PP hydrogenolysis and determine the influence of the nanocube nanostructure when compared to a standard mixed shape catalyst through normalisation of the Ni particle structure and interrogation of the support surface properties.
Results and discussion
Performance assessment of Ni/CeO2 catalysts
The hydrogenolysis of polypropylene was carried out at 280 °C, 30 bar H2, and 500 rpm at different time points. The resulting products (solid, liquid, and gas) were collected at room temperature, with the gas fraction measured from the reactor headspace by gas chromatography equipped with a flame-ionisation detector (GC-FID), and solid and liquid fractions were collected by dissolution of liquid products into CH2Cl2 and separation by filtration. Residual solid and catalyst were dried overnight with no further processing due to difficulties in separation of catalyst from polymer matrix. The separated solid and liquid products were measured gravimetrically after removal of CH2Cl2, and the three product fractions are further classified as: (i) solid products, consisting of unreacted and partially reacted solid polymer that are collected by filtration, with yields calculated by theoretical subtraction of the initial mass of catalyst added; (ii) liquid products, that are liquid or wax alkanes at ambient conditions and are the CH2Cl2 soluble components. The resulting liquid products were analysed by gel permeation chromatography (GPC), to give peak molecular weight (Mp), weight average (Mw), number average (Mn), and dispersity (Đ) values which are used to describe the liquid product distribution, combined with thermogravimetric analysis (TGA) for oxidation profiles and an indication of chain lengths; (iii) gaseous products that are short chain hydrocarbons (C1–C5) in the reactor headspace, with yields to gaseous alkane and alkene products grouped under the appropriate carbon number. Mass balances were assumed to be 100% in all reactions, considering a sealed reactor with no solvents during reaction, with gaseous product mass calculated from the difference between initial polymer used and mass of liquid and solid products.Blank reactions of PP (Mw = 12000 g mol−1, Mn = 5000 g mol−1) and H2 with no catalyst for 24 h show 71% solid recovery (Fig. 1a), with liquid and gas yields of 16% and 13% respectively, indicating partial thermal decomposition of the polymer under the reaction conditions. GPC of the liquid fraction produced at 24 h, with chromatograms given in Fig. S1a,† shows a Mw = 1664 ± 145 g mol−1 and Mn = 819 ± 24 g mol−1, with a dispersity of 2.03, which confirms partial thermal decomposition of the polymer. The gas fraction produced (Fig. 1b) consisted mainly of C3 and C5 products, with some variability in individual C3 and C5 yields but consistent overall product yields, implying thermal decomposition of fragments of hydrocarbon chains. Further blank reactions for 8 and 16 h (Table S1†) confirm the thermal decomposition is relatively consistent over the different time points and suggests the majority of decomposition occurs within the first 8 h, possibly during the initial temperature ramp. Reactions conducted for 24 h with the CeO2 supports (nanocube shaped (NC) and mixed shape commercial (SA)) show no changes in yields or gas distribution, within experimental error, when compared to the blank reactions (Fig. 1). No differences are also noted in the GPC analysis of the liquid fractions (Table S1†) and confirms no catalytic effect of the support materials.
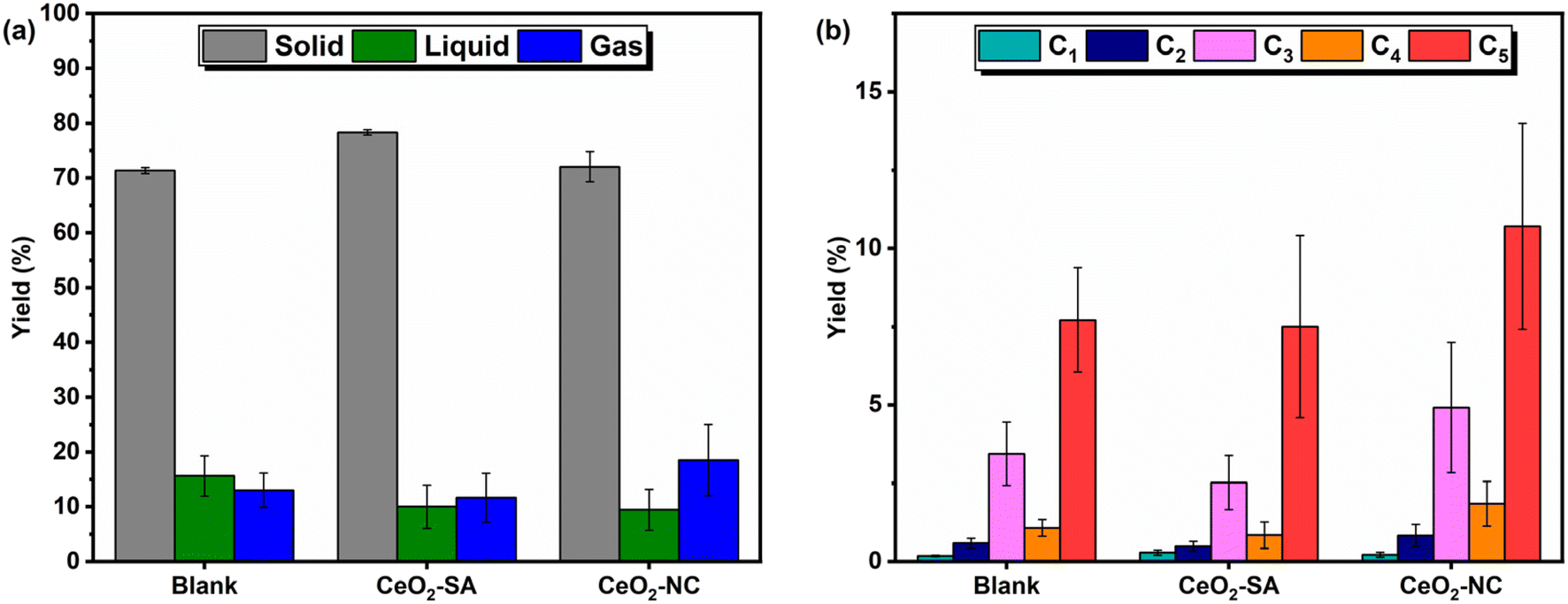 | ||
| Fig. 1 (a) Product distribution and (b) specific gas yields for the hydrogenolysis of PP using no material (blank), CeO2-SA, and CeO2-NC (SA – Sigma Aldrich; NC – nanocube). Hydrogenolysis reaction conditions: PP (2 g), CeO2 (0.2 g), 280 °C, 30 bar H2, 500 rpm, 24 h. Error bars are the standard deviation of multiple reactions. Datasets including full GPC results are reported in Table S1.† | ||
PP hydrogenolysis was conducted over the two Ni/CeO2 catalysts at different time points (Fig. 2a and b and Table S2†). Initial Ni loading of the catalysts during preparation was 1 wt%, however, due to the wash step in the synthesis, some loss of Ni was noted.34 The final Ni loadings on the prepared fresh catalysts, determined by inductively coupled plasma-optical emission spectrometry (ICP-OES), were consistent between batches on a specified support (0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC). Therefore, to eliminate and account for differences in the nominal loading between the catalysts, the Ni mass was fixed (0.00138 g) in the catalytic PP hydrogenolysis reactions.
 | ||
| Fig. 2 Product distribution of the hydrogenolysis of PP over (a) 0.4 wt% Ni/CeO2-SA and (b) 0.7 wt% Ni/CeO2-NC over different time points (SA – Sigma Aldrich; NC – nanocube). (c) Average Mw values of obtained liquids determined by GPC. (d) TGA of the produced liquid samples after 16 h reaction. Specific gas yields for (e) 0.4 wt% Ni/CeO2-SA and (f) 0.7 wt% Ni/CeO2-NC over different time points. Hydrogenolysis reaction conditions: PP (2 g), Ni/CeO2 (fixed Ni content 0.00138 g), 280 °C, 30 bar H2, 500 rpm, 8–24 h. Error bars are the standard deviation of multiple reactions. Datasets including full GPC results are reported in Table S2.† | ||
High liquid yields (78–82%) after 8 h are achieved over both catalysts although a significant fraction of solid remains (9–16%). GPC chromatograms for all the catalytically produced liquids with the Ni/CeO2 catalysts at specified time points are given in Fig. S1b.† GPC analysis of the produced liquids (Fig. 2c) at 8 h shows both catalysts have similar average chain lengths and are within error, with a Mw value of 5381 ± 416 g mol−1 (Mn = 2949 ± 416 g mol−1) and 5741 ± 291 g mol−1 (Mn = 3028 ± 163 g mol−1) for 0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC respectively. This is in agreement with previous literature that the long hydrocarbon chains of the starting polymer are favourably depolymerised before secondary hydrogenolysis reactions occur, due to the higher binding strength of long chain hydrocarbons to the catalyst.6,7,35 The high liquid yields achieved when compared to blank or support reactions confirm the importance of Ni for H-activation and C–C bond scission via hydrogenolysis.
Complete solid deconstruction, within experimental error, is achieved over 0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC in 16 h. High yields of liquid alkanes (73–78%) and liquid productivities (65.9–70.9 gliquid gNi−1 h−1) are achieved over both the catalysts. Comparison of liquid yields, reaction conditions, and productivities with Ni-based catalysts for polymer hydrogenolysis from literature at complete solid deconstruction (Table S3†), shows the Ni/CeO2 catalysts have comparable performance to other Ni-based catalysts for polyethylene (PE) deconstruction.11,12,14 However, it is important to note that comparison between studies should be treated with caution due to various reaction temperatures, large differences between substrates (Mw and side chain), and the description of liquid products.
Despite the similarities in the liquid yields and productivities of the two Ni/CeO2 catalysts, the GPC results of the liquid fraction (Fig. 2c) shows large differences in the Mw at both 16 and 24 h, i.e., post solid deconstruction. The 0.7 wt% Ni/CeO2-NC exhibits Mw values of 2786 ± 219 g mol−1 (Mn = 1442 ± 109 g mol−1) and 2029 ± 23 g mol−1 (Mn = 1226 ± 84 g mol−1) at 16 h and 24 h respectively, whereas the 0.4 wt% Ni/CeO2-SA yields Mw values of 4599 ± 150 g mol−1 (Mn = 2530 ± 117 g mol−1) and 3598 ± 88 g mol−1 (Mn = 1798 ± 75 g mol−1) for 16 and 24 h reactions. Additionally, the 0.7 wt% Ni/CeO2-NC shows a narrowing of the dispersity (Table S2†) throughout the reaction times (1.9 to 1.73), with the 0.4 wt% Ni/CeO2-SA showing broadening of the dispersity (1.82 to 2), indicating different scission selectivity.
To directly compare the differences in catalytic activity between the catalysts, metrics of the average rate of C–C bond scission and average Mw depolymerisation rate were calculated, with the results for 16 h reactions presented in Table 1. The average rate of C–C bond scission normalised per gram of Ni, assuming comparable Ni structure and hence normalised surface Ni atoms (details of Ni structure vide infra), was calculated according to an adjusted method of Lamb et al. (eqn (S7)†) and factors in product yields and Mn, although it is important to note that this calculation also includes gases produced by thermal decomposition.20 However, as the thermal decomposition remains consistent between the catalysts and is a factor in the process, the average C–C scission rates are directly comparable. The average Mw depolymerisation rate (eqn (S9)†) focuses purely on the depolymerisation of solid to liquid and utilises the decrease in Mw of the liquid compared to the starting Mw of the solid normalised by mass of Ni and unit of time. Therefore, it is an indicative measure of internal scission rates due to no consideration of product yields. Both calculated metrics confirm that 0.7 wt% Ni/CeO2-NC show increased depolymerisation when compared to 0.4 wt% Ni/CeO2-SA, with an average C–C scission rate for 0.7 wt% Ni/CeO2-NC (838.1 mmol gNi−1 h−1) showing a 75% improvement in depolymerisation over 0.4 wt% Ni/CeO2-SA (480.3 mmol gNi−1 h−1) at 16 h. Consideration of the average C–C bond scission rates normalised per gram of Ni with only the catalytically produced methane contribution and exclusion of gases from thermal decomposition, further confirm enhanced average C–C scission rates of 0.7 wt% Ni/CeO2-NC when compared to 0.4 wt% Ni/CeO2-SA at both 16 h (Table 1). Furthermore, the average C–C scission rate for 0.7 wt% Ni/CeO2-NC is comparable to that achieved over a reported 0.49 wt% Pt/SrTiO3 in PE hydrogenolysis at 300 °C (2.1 mmol h−1; 525 mmol gPt−1 h−1 at 18 h).20 The enhanced average rate of C–C bond scission with and without gases from thermal decomposition, and average Mw depolymerisation rate for 0.7 wt% Ni/CeO2-NC when compared to 0.4 wt% Ni/CeO2-SA is replicated at 24 h (Table S4†), although reaction rates decrease for both catalysts, possibly due to reduced H2 availability and decreased favourability of shorter chain hydrocarbon binding.
| Catalyst | Liquid productivityb (gliquid gNi−1 h−1) | Average rate of C–C bond scissionc (mmol gNi−1 h−1) | Average rate of C–C bond scission with only C1 gasc (mmol gNi−1 h−1) | Average Mw depolymerisation rated (kg mol−1 gNi−1 h−1) |
|---|---|---|---|---|
| a Ni loading determined by ICP-OES. b Calculated using gravimetric quantities and mass Ni used in the reaction according to eqn (S3). c Calculated according to an adjusted method to Lamb et al. using Mn, gas moles, and mass of Ni (0.00138 g) as given in eqn (S7).20 d Calculated using Mw and mass of Ni as stated in eqn (S9). | ||||
| 0.4 wt% Ni/CeO2-SAa | 70.9 (±6.3) | 480.3 (±20.1) | 353.5 (±36.5) | 335.2 (±9.6) |
| 0.7 wt% Ni/CeO2-NCa | 65.9 (±4.5) | 838.1 (±98.2) | 608.9 (±86.1) | 417.3 (±14.0) |
Further evidence of compositional differences was provided by TGA of the oxidation of the liquid products, with TGA of the liquid products generated after 16 h shown in Fig. 2d. Such oxidation reaction profiling has previously been used to study and classify differences between complex crude or lubricant base oil samples.36–38 The oxidation profiles can be classified into separate regions which consist of multiple and complex reaction pathways and are: (i) low temperature oxidation (LTO; ca. to 350 °C), where volatilisation and partial to full oxidation of short hydrocarbons occurs; (ii) fuel deposition (ca. 350–450 °C), where high molecular weight components are oxidised and cracked to generate coke; and (iii) high temperature oxidation (HTO; ca. 450–800 °C) reactions that convert residual high molecular weight oil and coke into COx.36 Therefore, the oxidation profiles, weight loss in each region, and burnout temperatures are indicative of the composition of the sample. Analysis results are given in Table S5,† with all liquid samples produced from catalytic reactions at different times with both Ni/CeO2 showing a majority of weight loss (95–98%) in the LTO region and a small weight loss in the HTO region (2–5%), confirming a light oil composition for all liquid samples. For reference, the oxidation profile of the solid pristine polypropylene is shown with profiles of the catalytically produced liquid samples. The liquids produced after 8 h reactions (Fig. S2a†) exhibit oxidation profiles that are comparable for the two catalysts. However, after 16 h of reaction (Fig. 2d), the liquid profiles are considerably different, in agreement with the GPC results, with a lower burnout temperature (462 °C) for the 0.7 wt% Ni/CeO2-NC than the 0.4 wt% Ni/CeO2-SA (508 °C). The burnout temperature (462 °C) for the liquid produced at 16 h over the 0.7 wt% Ni/CeO2-NC is comparable to values reported for lubricant base oils (ca. 388–499 °C) and suggests the produced liquids could be used in such applications.39–41 Furthermore, the weight loss is shifted to lower temperatures for the 0.7 wt% Ni/CeO2-NC, indicating the presence of lower molecular weight chains due to higher volatility and lower oxidative stability, when compared to that of the liquid produced by 0.4 wt% Ni/CeO2-SA, with this shift replicated again at 24 h (Fig. S2b†).
The specific gas yields produced over Ni/CeO2 catalysts (Fig. 2e and f) show comparable gas yields, with the catalytically produced gas is methane (C1), from the scission of side or terminal chains, and other gas yields (C2–C5) consistent with blank reactions (Fig. 1b). This suggests catalytic scission by hydrogenolysis from cleavage of alkylidyne intermediates from terminal or side chains, rather than hydrocracking mechanisms which disfavour the formation of methenium cations and therefore generate C3–C5 products, as proposed in mechanistic studies by Lee et al.18 Therefore, it can be hypothesised that the gases produced by partial thermal decomposition of the polymer escape into the reactor headspace and are not in contact with the catalyst surface, with no further scission occurring. The low methane yields achieved (4–11%) compared to liquid yields (73–82%) at all reaction times also verify a high selectivity to internal backbone scission.
The differences in catalytic hydrogenolysis activity and rate of C–C bond scission between the Ni/CeO2 catalysts in this study must be attributable to several factors, namely: (i) differences in active site Ni structure; (ii) differences in Ni loading or Ni density on the supports; (iii) differences in the promotional effects of the support nanostructure. The following discussion will consider each specific factor and the influence that they exhibit on the catalytic activity of the materials.
Ni structure and speciation
The hydrogenolysis mechanism is highly structure sensitive over Pt, Ru, and Ni catalysts due to the changing kinetics and binding energies of each step. Tomer et al. demonstrated experimentally and in silico that large Ru particles, and hence stabilisation of Ru(0001) surfaces, cleave terminal or side chain carbons of PP to generate methane and cleave at lower rates than Ru nanoclusters, with this enhancement trend replicated over Pt catalysts.27,35 Zhao et al. showed that Ni also exhibits structure sensitivity through alteration of particle size, with this further confirmed by Vance et al. over Ni/SiO2 catalysts.11,14 Therefore, to directly compare the differences in hydrogenolysis activity and selectivity, the structure of the Ni active sites should be defined.High angle annular dark field scanning transmission electron microscopy (STEM-HAADF) images (Fig. 3a and b), with further images of different catalyst particles provided in Fig. S3 and S4,† could not be used to identify representative images of Ni species. This is attributed to the probable small size of particles or adatoms, the low loading of Ni, and given the HAADF contrast is proportional to almost square atomic number of elements, coordinatively unsaturated Ni species could not be identified when compared to the high CeO2 contrast. Ni species could also not be identified in STEM-HAADF images of post 16 h reaction catalysts (Fig. S5 and S6†), indicating limited Ni particle growth and high dispersion stability. It is important to note that after 16 h reaction, almost complete solid polymer deconstruction was observed, and no further treatment steps were undertaken on the catalyst after separation from the liquid products. Initial confirmation of Ni speciation was given by X-ray photoelectron spectroscopy (XPS) analysis of the Ni 2p region of the fresh Ni/CeO2 catalysts (Fig. 3c). Peak deconvolution of the Ni 2p region was not undertaken due to the low loading of the catalysts resulting in a low signal to noise ratio, however analysis of peak positions at 885.4–885.8 eV and the associated satellite structures of both fresh and post 16 h reaction catalysts indicate the presence of Ni(II).42,43 As reduction of the Ni(II) species is anticipated under the reaction conditions (280 °C, 30 bar H2), the presence of Ni(II) in the post reaction catalysts suggests re-oxidation after reaction and exposure to air.
 | ||
| Fig. 3 STEM-HAADF images of fresh (a) 0.4 wt% Ni/CeO2-SA and (b) 0.7 wt% Ni/CeO2-NC (SA – Sigma Aldrich; NC – nanocube); (c) XPS analysis of the Ni 2p region of fresh and post 16 h reaction Ni/CeO2 catalysts; (d) nickel K-edge XANES, (e) pre-edge, and (f) white-line energy of fresh and post 16 h reaction Ni/CeO2 catalysts and NiO and Ni standards. | ||
In order to confirm the oxidation state and averaged local coordination environment of the Ni/CeO2 catalysts, ex situ Ni K-edge X-ray absorption spectra (XAS) were taken of the fresh and post-reaction catalysts. Comparison of the X-ray absorption near-edge structure (XANES) spectra of standards, fresh and post 16 h reaction catalysts is shown in Fig. 3d, with linear combination fitting (LCF) of the XANES spectra of the fresh catalysts (Table 2 and Fig. S7†) confirming the presence of NiO, in agreement with the XPS results. However, LCF fitting of the post reaction catalysts (Table 2 and Fig. S8†) shows the partial reduction of the Ni species with the presence of NiO (85–88%) and Ni (12–15%). This is also visualised in the shift in the pre-edge (Fig. 3e) and white-line energy (Fig. 3f) of the catalysts towards pre-edge features and white-line energy associated with Ni(0) foil. Additionally, in both fresh and post-reaction catalysts, the shoulder feature at ∼8655 eV is reduced in intensity when compared to the NiO standard and suggests the presence of small NiO nanostructures due to reduced long-range order, in agreement with Preda et al.44
| Sample | Scattering path | Coordination number | 2σ2 | R | E f | R factor | LCF (wt%) |
|---|---|---|---|---|---|---|---|
| a Fixed parameter: S02 = 0.9334; The fit range for the EXAFS data was 3 < k < 10 A−1, 1.0 < R < 3.3 Å. b fixed CN for Ni–O = 6 and for standard Ni–Ni pathways = 12. c Debye–Waller and amplitude reduction factors fixed. | |||||||
| NiO | Ni–O | 6b | 0.009(3) | 2.075(9) | −2(1) | 0.0093945 | — |
| Ni–Ni(1) | 12b | 0.008(1) | 2.96(1) | ||||
| Ni foil | Ni–Ni(2) | 12b | 0.0076(5) | 2.483(9) | 7(1) | 0.0192813 | — |
| Ni/CeO2-SA fresh | Ni–O | 6b | 0.0059(7) | 2.057(8) | −2.0(8) | 0.0060385 | 100% NiO |
| Ni–Ni(1) | 7(1) | 0.008(2) | 2.957(8) | ||||
| Ni/CeO2-NC fresh | Ni–O | 6b | 0.0094(8) | 2.050(9) | −0.6(9) | 0.0060438 | 100% NiO |
| Ni–Ni(1) | 7(1) | 0.009(2) | 2.967(8) | ||||
| Ni/CeO2-SA post reaction | Ni–O | 6b | 0.009(2) | 2.05(2) | −3(3) | 0.0261900 | 88(1)% NiO |
| Ni–Ni(2) | 2.6(6) | 0.00762c | 2.49(2) | 12(1)% | |||
| Ni–Ni(1) | 1.2(9) | 0.00838c | 2.99(7) | Ni | |||
| Ni/CeO2-NC post reaction | Ni–O | 6b | 0.013(1) | 2.04(2) | −1(2) | 0.0142729 | 85(1)% NiO |
| Ni–Ni(2) | 1.9(4) | 0.00762c | 2.49(2) | 15(1)% | |||
| Ni–Ni(1) | 2.1(6) | 0.00838c | 3.00(3) | Ni | |||
The r-space extended X-ray absorption fine structure (EXAFS) spectra with fitting and the wavelet transformations for the fresh and post reaction Ni/CeO2 are given in Fig. 4, with the fitting parameters and suggested fits given in Table 2. Initial fitting was conducted (fit range: 3 < k < 10 A−1, 1.0 < R < 3.3 Å) on an experimentally measured NiO standard (Fig. S9†) to determine S02 (S02 = 0.9334) with a model of the single scattering Ni–O6 and Ni–Ni(1) paths. It should be noted that the k-space plots (Fig. S9b†) show fitting of the overall profile with the fitting model, however minor features are not captured due to no multiple scattering paths included in the fit. A experimentally measured reference Ni(0) foil (Fig. S10†) was also fit with a single 1st shell Ni–Ni(2) single scattering path with a coordination number (CN) of 12. Confirmation of the validity of the simple models, with only single scattering paths and no multiple scattering paths, was given by the wavelet transformations for the NiO and Ni(0) (Fig. S11 and S12†). The wavelet transformation of NiO shows a feature at ΔR 1–2 Å and Δk 3–6 Å−1 associated with the 1st shell Ni–O6, and a more intense feature at ΔR 2–3 Å and Δk 5–9 Å−1 which is attributed to the 2nd shell single scattering Ni–Ni(1) path and multiple scattering paths. Whereas the wavelet transformation of Ni is dominated by a strong feature at ΔR 1.5–3 Å and Δk 4–10 Å−1, which includes the single scattering Ni–Ni(2) path and multiple scattering paths.
 | ||
| Fig. 4 (a) Ni K-edge EXAFS fitting of k2 weighted FT χR of fresh Ni/CeO2 catalysts. Morlett wavelet transformations of k2 weighted data from Ni k-edge EXAFS spectra of fresh (b) 0.4 wt% Ni/CeO2-SA and (c) 0.7 wt% Ni/CeO2-NC (SA – Sigma Aldrich; NC – nanocube). (d) Ni k-edge EXAFS fitting of k2 weighted FT χR of Ni/CeO2 catalysts post 16 h reaction and exposure to air. Morlett wavelet transformations of k2 weighted data from Ni k-edge EXAFS spectra of post 16 h reaction and air exposed (e) 0.4 wt% Ni/CeO2-SA and (f) 0.7 wt% Ni/CeO2-NC. Fitting plots of NiO, Ni foil and k-space and q-space of k2 weighted data of fresh and post reaction catalysts is given in Fig. S13–S16.† The fit range for the EXAFS data was 3 < k < 10 A−1, 1.0 < R < 3.3 Å. | ||
For the fitting of the catalysts, the 1st shell Ni–O CN was fixed at 6 with floating of the disorder parameter (2σ2), as a large deviation from the ideal CN is not anticipated for the first shell of metal oxides in correspondence with literature.45,46 The Ni–Ni(1) CN was allowed to float to determine the reduction of CN when compared to the bulk NiO standard. The fitting results of both fresh catalysts indicate a comparable averaged structure of the NiO species with a reduced Ni–Ni(1) CN of 7(1) when compared to the NiO standard (Ni–Ni(1) CN = 12). Interestingly, for both catalysts, the path length of 1st shell Ni–O6 is reduced (Ni/CeO2-SA – 2.057(8) Å; Ni/CeO2-NC – 2.050(9) Å) when compared to the bulk NiO (2.075(9) Å). Whereas the path lengths for the 2nd shell Ni–Ni remain relatively consistent between the catalysts and the NiO standard. This observation is in agreement with the structural and molecular dynamic models reported by Anspoks et al. and suggests the formation of Ni vacancies in nanosized NiO species, with averaged NiO particle sizes less than 1.3 nm.47 Furthermore, the wavelet transformation of the catalysts (Fig. 4b and c) shows an increased prominence of the feature associated with Ni–O6 (ΔR 1–2 Å and Δk 3–6 Å−1), which highlights the reduction of intensity of the Ni–Ni(1) feature when compared to the NiO standard. The EXAFS fitting and the wavelet transformations of the fresh catalysts confirm that the globally averaged Ni structure is consistent between the catalysts and direct comparison of the activity of the catalysts can be made.
EXAFS fitting of the catalysts post 16 h of reaction and exposure to air (Fig. 4d and Table 2) show reduced long range order of Ni–Ni(1) paths arising from NiO and the appearance of Ni–Ni(2) paths from Ni(0). This transformation is visualised in the wavelets of the post reaction catalysts (Fig. 4e and f) with features associated with Ni–O6 (ΔR 1–2 Å and Δk 3–6 Å−1), Ni–Ni(2) (ΔR 1.5–3 Å and Δk 4–10 Å−1), and minor contributions of Ni–Ni(1) (ΔR 2–3 Å and Δk 5–9 Å−1) as well as the associated multiple scattering paths. Therefore, it can be postulated that under reaction conditions, the NiO species are reduced to Ni(0) with some degree of redistribution and Ni(0) is the subsequent active species present under reaction conditions. However, upon uncontrolled exposure to air, NiO overlayers of varying degrees are formed, in agreement with the XPS and XANES results. Further evidence of Ni reducibility to Ni(0) under reaction conditions is given by temperature programmed reduction (TPR) of the fresh catalysts (TPR of fresh catalysts vide infra). Importantly, the Ni structures remain comparable and a large growth in Ni structure is not noted after 16 h reaction. Consequently, the differences in activity also cannot be attributed to catalyst deactivation through particle growth nor large differences in metal redistribution.
Extensive advanced characterisation of the Ni speciation and structure of both fresh and post-reaction catalysts confirm that the averaged Ni structure and speciation is consistent between the two catalysts, with particle sizes < 1.3 nm. Therefore, the high selectivity to internal cleavage and comparable liquid yields of the two catalysts are rationalised to be a function of the consistent Ni structures and ultrafine particles. Slight differences in catalytic rates may arise depending on the exact structure of individual adatoms and particles, however, this factor is minimised when considering the averaged structure and cannot wholly explain the large deviations in the C–C scission rate between the catalysts.
Ni loading and density
It is important to consider that despite differences in metal loading, the structure of the Ni determined by EXAFS is consistent for the 0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC catalysts and the same mass of Ni was used in reactions. Thus, differences in the C–C scission rate, giving rise to different liquid alkane compositions despite comparable liquid productivities, do not arise from the Ni structure or loading of Ni. However, differences in Ni density (mol m−2) occur due to the different surface areas (m2 g−1) of the CeO2 supports and must be considered in addition to nominal metal loading.22 Lim et al. studied the hydrogenation of benzaldehyde and showed that the proximity, in effect the density, between PdAu particles significantly affected the selectivity either to benzyl alcohol or the further hydrogenated product toluene.48 For polymer hydrogenolysis with long flexible hydrocarbon carbon chains of polymers as substrates, the multiple adsorption modes within the adsorption layer on the catalyst surface must be considered.49 Therefore, the active site density could be influential due to C–C scission at multiple Ni sites across different points of the same polymer chain. Secondarily for further scission reactions, the diffusion kinetics of hydrocarbon chains to a subsequent Ni active site are altered depending on the distance between Ni sites.The measured BET surface areas of the carriers are 34 and 25 m2 g−1 for the CeO2-SA and CeO2-NC respectively (N2 adsorption isotherms given in Fig. S17†) with a corresponding Ni density of 1.9 and 4.7 × 10−6 mol m−2 for the 0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC respectively, accounting for comparable Ni structures. To evaluate the effects of Ni density, a 1.2 wt% Ni/CeO2-SA was prepared with a Ni density (6.1 × 10−6 mol m−2) comparable to that of the 0.7 wt% Ni/CeO2-NC. Catalytic tests with fixed Ni mass for 16 h showed product yields are comparable to Ni/CeO2-NC within error (Fig. 5a). However, the Mw achieved (Fig. 5b) over the 1.2 wt% Ni/CeO2-SA (Mw = 4115 ± 123 g mol−1; Mn = 2132 ± 55 g mol−1) remains higher than that achieved over 0.7 wt% Ni/CeO2-NC (Mw = 2786 ± 219 g mol−1; Mn = 1442 ± 109 g mol−1), with GPC chromatograms given in Fig. S18.† This is further verified by TGA oxidation of the liquid product (Fig. S19 and Table S5†), with higher oxidation stability and burnout temperature for the 1.2 wt% Ni/CeO2-SA liquid suggesting longer chains present. However, a slightly lower Mw is achieved than 0.4 wt% Ni/CeO2-SA (Mw = 4599 ± 150 g mol−1; Mn = 2530 ± 117 g mol−1) and leads to a slight increase in the average Mw depolymerisation rate and C–C scission rate (Table S6†) for 1.2 wt% Ni/CeO2-SA when compared to 0.4 wt% Ni/CeO2-SA. This shows the activity of the different Ni/CeO2-SA is slightly improved when considering the Ni density. Therefore, Ni density can be considered to have a small role and influence on hydrogenolysis activity, but it cannot rationalise the large differences in rates between Ni/CeO2-SA and Ni/CeO2-NC catalysts.
 | ||
| Fig. 5 (a) Product distribution, (b) Mw values of obtained liquids determined by GPC for the hydrogenolysis of PP over 1.2 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC with comparable Ni densities (SA – Sigma Aldrich; NC – nanocube). Hydrogenolysis reaction conditions: PP (2 g), Ni/CeO2 (fixed Ni content 0.00138 g), 280 °C, 30 bar H2, 500 rpm, 16 h. Error bars are the standard deviation of multiple reactions. Datasets including full GPC results are reported in Table S2.† | ||
Promotional effects of CeO2 support nanostructure
As the differences in C–C scission activity between the catalysts cannot wholly be attributed to factors corresponding to the active Ni metal, loading and density between the catalysts, the support morphology must have a role in the depolymerisation of the polymer chains. Definition of the structural features of the overall catalysts is necessary to conduct comparison of the promotional effects, with confirmation of the mixed morphologies of 0.4 wt% Ni/CeO2-SA (Fig. 6a) and the nanocube morphology in 0.7 wt% Ni/CeO2-NC (Fig. 6b) given by STEM-HAADF. Associated bright field (BF) images and further STEM images (HAADF and BF) of fresh catalysts are given in Fig. S3 and S4.† The exposed surface facet was experimentally confirmed through measurement of the interplanar spacing of the catalysts using STEM-HAADF images. Interplanar spacings of 0.313 nm were measured on fresh 0.4 wt% Ni/CeO2-SA and correspond to enclosure by (111) surface facets (Fig. S20†). Whereas interplanar spacings of 0.270 nm corresponding to the d-spacing of the (200) plane was calculated for fresh 0.7 wt% Ni/CeO2-NC and confirm the exposure of (100) surface facets. Subsequently, in agreement with literature, the facets exposed on the nanocube catalyst are (100), whilst the standard mixed shape catalyst are enclosed by stable (111) facets.50,51 Furthermore, restructuring of the nanocubes was not noted in TEM images after 16 h reaction and exposure to air (Fig. S5 and S6†) with no changes in measured interplanar spacings (Fig. S21†), suggesting stability of the morphology and retention of (100) facets after reaction and exposure to air. However, nanoscale restructuring, disorder, and defects could possibly occur on specific (100) surfaces upon reoxidation after partial reduction.52,53 | ||
| Fig. 6 Characterisation of Ni/CeO2 using STEM-HAADF to determine morphology of (a) Ni/CeO2-SA and (b) Ni/CeO2-NC; (c) synchrotron PXRD of fresh and post 16 h reaction Ni/CeO2 catalysts to confirm CeO2 phases; (d) XPS analysis of Ce 3d region of fresh and post reaction Ni/CeO2 catalysts to confirm cerium surface speciation (SA – Sigma Aldrich; NC – nanocube). Further STEM images (HAADF and BF) of different positions of fresh catalysts to determine morphology are given in Fig. S3 and S4† and STEM images (HAADF and BF) of post-reaction catalysts showing retainment of morphology is given in Fig. S5 and S6.† | ||
Synchrotron PXRD patterns of the Ni/CeO2 catalysts (Fig. 6c) exhibit peaks expected from cubic CeO2 and no peaks associated with NiO or Ni species. This was further confirmed by Pawley fitting of the catalyst patterns using a CeO2 standard collected from the ICSD database, with all peaks related to CeO2 (Fig. S22†). Evaluation of the CeO2 crystallite size (Table 3), calculated using the Scherrer equation, show comparable crystallite sizes between the two fresh catalysts with 29.6 ± 1.2 nm and 23.8 ± 1.6 nm for 0.4 wt% Ni/CeO2-SA and 0.7 wt% Ni/CeO2-NC respectively. Further analysis of the post-reaction catalysts shows limited growth, within fitting error, in the Scherrer crystallite size (Table S7†) or full width half maximum of the characteristic peaks (Table S8†), indicating stability of the catalysts, in correlation with the TEM results.
| Catalyst | CeO2 crystallite sizea (nm) | BET specific surface area (m2 g−1) | CeO2 surface peak temperatureb (°C) | CeO2 surface H2 uptake (μmol g−1) |
|---|---|---|---|---|
| a Calculated using the Scherrer equation, parenthesises show the standard deviation between multiple peaks. b Determined from peak maximum of fitted CeO2 surface reduction peak. | ||||
| 0.4 wt% Ni/CeO2-SA | 29.6 (±1.2) | 34 | 325 | 38 |
| 0.7 wt% Ni/CeO2-NC | 23.8 (±1.6) | 25 | 349 | 240 |
XPS analysis of the Ce 3d region (Fig. 6d) of fresh and post reaction catalysts was undertaken using a complex ten-component peak model adapted from the literature of single crystal and powder CeO2 standards, which accounts for the multiple final-state effects of both Ce(III) and Ce(IV).54–56 For the analysis of fresh 0.7 wt% Ni/CeO2-NC, symmetric components at 880.8, 885.8, 898.8, and 903.2 eV correspond to v0, vI, u0, and uI states of Ce(III) respectively. Further asymmetric-like components at 882.5, 888.8, 898.1, 901.0, 907.5, and 916.4 eV are attributed to the v, vII, vIII and u, uII, uIII states of Ce(IV) respectively. The assignment of components remains the same for the other fresh and post 16 h reaction catalysts, with peak positions and atom percentages listed in Table S9.† Calculated Ce(III) percentages of total Ce 3d show fresh 0.7 wt% Ni/CeO2-NC (11.4%) to contain slightly more defects than 0.4 wt% Ni/CeO2-SA, which could arise from increased hydroxylation of the (100) facet or the higher density of Ni causing disruption of the CeO2 lattice.51 Analysis of the post reaction Ce(III) percentages of total Ce 3d (11% for both catalysts) and atomic percentages of the total elements present in the catalysts (Table S9†) further suggest structural stability of the catalysts with similar defects and surface structure when compared to the fresh catalysts, in agreement with the TEM and PXRD results. Furthermore, only a slight increase in C atom.% (35.6–36.2 at%) is noted in the post 16 h reaction catalysts when compared to the fresh catalysts (29.8–31.6 at%), indicating limited residual polymer or carbon deposition. Comparison of the promotional effects can now be considered within the context of the CeO2 structure and morphology.
An important factor in polymer hydrogenolysis is the availability of activated hydrogen, with high H-coverage reported to shift the selectivity from terminal to backbone cleavage, generating higher liquid yields, and significantly improving scission rates. Chen et al. determined Ru particle size is important on Ru/CeO2 catalysts to improve H-coverage, however in this study, this factor is normalised due to the consistent Ni structure between the catalysts.23 Wang et al. showed promoting catalysts with WOx improved the hydrogen storage capacity via improved H-spillover of Ru/ZrO2 catalysts and subsequently mediated the produced hydrocarbon chain lengths.25 Kim et al. reported the TiO2 rutile polymorph could also improve coverage rates through improved H-spillover.24 Therefore H-spillover can be considered as a key feature in boosting H-storage and subsequent increased activity of the catalysts.26
Temperature programmed reduction (TPR), with a flow of 5% H2/Ar as the reducing gas, is a useful technique to clarify and quantify the reduction behaviour of supports and the effects of H-spillover on supported metal catalysts. Initial TPR of the CeO2 supports (Fig. 7a) show two main regions, which are attributed to surface CeO2 reduction and a higher temperature bulk reduction, as widely accepted in the literature.57 The reduction of CeO2 through interaction with H2 is considered to occur through several steps, as follows: (i) the formation of chemisorbed hydrogen and hydroxyl groups from the dissociation of hydrogen; (ii) reduction of neighbouring Ce(IV) to form oxygen vacancies through electron transfer from surface oxygen; (iii) recombination of hydrogen and hydroxyl to form water and its subsequent desorption; and (iv) oxygen vacancy diffusion into the bulk.51,58 These steps are expressed in the overall equations given in Fig. 7a. The peak temperature of bulk reduction (712–735 °C) is largely invariant when considering the different morphologies.57 However this is not the case when considering the surface reduction peaks, with CeO2-SA exhibiting a smaller surface reduction peak area when compared to CeO2-NC although a higher temperature reduction peak (506 °C) is noted for CeO2-NC when compared to CeO2-SA (415 °C). This is further confirmed by multiple peak fitting of the TPR profiles (Fig. S23 and S24†), with a larger uptake (Table S10†) for surface reduction of CeO2-NC (430 μmol g−1) than CeO2-SA (92 μmol g−1) and a resulting surface uptake ratio (CeO2-NC/CeO2-SA surface uptake) of 5:1. The higher degree of reduction of CeO2-NC and exposed (100) facets over the mixed shape with (111) facets is in agreement with reported experimental and calculated results of CeO2 surface reduction.57,59,60 It is important to note that the activation and consumption of hydrogen by the support materials is above the reaction temperature (280 °C) and therefore activated hydrogen is not available to depolymerise the plastic, which confirms the inactivity of the CeO2 supports in the reaction (Fig. 1) when compared to the blank reactions.
 | ||
| Fig. 7 (a) TPR profile of CeO2 supports; fitted TPR profiles of (b) 0.4 wt% Ni/CeO2-SA and (c) 0.7 wt% Ni/CeO2-NC; equations are given for the reduction of the Ni and CeO2 species through interaction with H2, where Ov is oxygen vacancy. (d) XPS analysis of Ce 3d region of Ni/CeO2 catalysts after subjection to reaction conditions without polymer or stirring (280 °C, 30 bar H2, 8 h) with inert transfer to XPS instrument (SA – Sigma Aldrich; NC – nanocube). | ||
Fitting of the TPR profiles was conducted on 0.4 wt% Ni/CeO2-SA (Fig. 7b) and 0.7 wt% Ni/CeO2-NC (Fig. 7c) catalysts to deconvolute contributions from the reduction of the NiO species, formally NiO + H2 → Ni(0) + H2O, and CeO2 surface and bulk reduction. NiO reduction was fit with two features that occur at 231 °C and 283 °C for 0.4 wt% Ni/CeO2-SA and 228 °C and 310 °C for 0.7 wt% Ni/CeO2-NC.61 Large differences are not noted in Ni reduction peak temperatures or H2 uptake (Table S10†) suggesting similar particle structures and metal–support interactions. Additionally, when considering the globally averaged Ni structure determined by EXAFS is consistent between the catalysts and limited differences in the electronic structure are noted in XANES and XPS, any slight changes in speciation or interaction can be considered to be minimised. It is important to note, that the concentration and pressure of hydrogen affects the reduction temperatures of the TPR profile.62 Given the hydrogenolysis reactions take place under pure H2 at 30 bar and 280 °C, whereas the TPRs are conducted in 5% H2/Ar at atmospheric pressure, the reduction of Ni species and surface CeO2 reduction will shift to lower temperature. Therefore, the active species present under reaction conditions can be considered to be reduced Ni(0) particles.
The surface CeO2 reduction peak of both catalysts, fitted as a single peak, is shifted to lower temperatures (0.4 wt% Ni/CeO2-SA = 325 °C, 0.7 wt% Ni/CeO2-NC = 349 °C) when compared to the support materials (CeO2-SA = 415 °C, CeO2-NC = 506 °C). This shift to lower temperatures and hence enhanced reducibility of the CeO2 surface is a result of the interaction of CeO2 with Ni metal and is evidence of H-spillover effects in both of the catalysts.63 In the catalysts, activated H atoms migrate from the Ni particle to the CeO2, with reduction of the lattice and formation of oxygen vacancies. However, the CeO2 surface H2 uptake (Table 3) for 0.7 wt% Ni/CeO2-NC (240 μmol g−1) is significantly larger than the corresponding peak for 0.4 wt% Ni/CeO2-SA (38 μmol g−1) and implies enhanced degree of reduction and improved H-storage of the 0.7 wt% Ni/CeO2-NC. Importantly, a significant percentage of CeO2 surface reduction is now accessible at hydrogenolysis reaction temperatures (280 °C). Verification of the single CeO2 surface peak validity is given by comparable ratios of surface reduction uptakes between the supports and catalysts (supports = 5:1, catalysts = 6:1), with differences in overall values attributed to simple peak model fitting of the complex H-spillover processes and Ni–CeO2 metal–support interactions. The enhanced degree of reduction also cannot be attributed to large differences in CeO2 crystallite size, due to comparable calculated crystallite sizes, which can strongly affect the catalytic properties as reported for CO oxidation and CO2 hydrogenation.64,65 However, this does correspond to the differences in the CeO2 surface structure due to the morphologies, and correlates well with reported surface reduction energies of (100) facets (0.03 eV), (111) facets (0.65 eV) and (110) facets (1.16 eV).59
To further elucidate the differences in the degree of reduction of the catalysts, catalysts were subjected to reaction conditions (280 °C, 30 bar H2, 8 h) without polymer or stirring. This procedure is advantageous as it allows for study of the catalysts subjected to realistic conditions, with transfer to inert glovebox environments without exposure to air. It is important to consider, that the reactions are conducted under completely static conditions with no substrate and lead to differences in the heating rates, dynamics and kinetics of H-activation and subsequent H-spillover when compared to the catalytic reactions. Consequently, these reactions without polymer should be considered as indicative results to compare the degree of reduction under the reaction conditions rather than an operando study. XPS analysis of the Ce 3d region (Fig. 7d) following inert transfer, confirms reduction of Ce species when compared to the fresh catalysts (Fig. 6d). Furthermore, 0.7 wt% Ni/CeO2-NC (28.6%) has a higher concentration of Ce(III) when compared to 0.4 wt% Ni/CeO2-SA (24.4%), which is consistent with the partial accessibility of CeO2 surface reduction at the reaction temperature. Additionally, reduction in the binding energy by 0.7–1.0 eV of the Ni 2p peak (Table S9†) suggests reduction of the Ni species to Ni(0) under the reaction conditions, in-agreement with the XAS and TPR analysis. Synchrotron PXRD patterns of sealed capillaries following inert transfer confirm retention of cubic CeO2 phase after exposure to reaction conditions (Fig. S25†). Additionally, analysis of the lattice parameters of the reduced catalysts (Table S7†), calculated from Pawley fitting of synchrotron PXRD patterns of sealed capillaries, show a slight expansion of the lattice parameters compared to the fresh catalysts, with a larger expansion for 0.7 wt% Ni/CeO2-NC (5.42385(3) Å to 5.42942(2) Å) when compared to 0.4 wt% Ni/CeO2-SA (5.42384(5) Å to 5.42567(3) Å). This indicates a small degree of subsurface reduction and implies increased Ce(III) concentration in the nanocube catalyst, due to the larger ionic radius of Ce(III) compared to Ce(IV) leading to distortions in the unit cell.66
The Ni/CeO2 nanocube morphology with exposed (100) facets therefore allow for slightly improved H-storage capacity at hydrogenolysis reaction temperatures when compared to the mixed shape catalyst with (111) facets. However, given the degree of reduction is only partially improved under hydrogenolysis reaction conditions, other factors must be considered to rationalise the large differences in activity. In particular, the acid–base properties exhibited by the supports are reported to affect both the mechanism of depolymerisation and the favourability of hydrocarbon binding. Lamb et al. showed the influence of acidic properties on the dominant depolymerisation reaction mechanism of hydrogenolysis or hydrocracking, with acidic sites involved in hydrocracking pathways.20 Jaydev et al. determined that weak basic sites on supports are influential for improving hydrocarbon adsorption and enhancing selectivity to backbone cleavage, and over Pt/C catalysts, oxygen content of the carbon was important for hydrocarbon binding.22,35 Therefore, due to the importance of support acid–base properties in depolymerisation reactions, temperature programmed desorption (TPD) measurements of the supports were performed, using NH3 to probe acid sites and CO2 for basic site analysis.
The acidic properties of CeO2 arise from the Lewis acidity of Ce(IV) and weakly acidic Ce(III) cations, whereas, the basicity originates from Lewis basic surface oxygen and hydroxyl groups. The exposed facet of CeO2 displays different acid–base properties due to variance in the coordination of the surface cations and anions.51,67 Additionally, as CeO2 is a redox active support and is highly dynamic under hydrogenolysis reaction conditions (Ce 3d XPS analysis vide supra), the acid–base properties were analysed on fresh catalysts after a standard He treatment (200 °C for 1 h) and after reduction (550 °C for 1 h) and cooling under 5%H2/Ar flow. Reductions were carried out at 550 °C to reduce the CeO2 surface, as suggested by the TPR profile of the support materials (Fig. 7a), with the TCD profile of the reduction step recorded for each measurement to confirm surface reduction (Fig. S26 and S27†). The exposed CeO2 surface and associated acid–base properties under the reaction conditions can then be considered to transiently evolve between these two states. Blank measurements following pre-treatments showed no peaks and confirm the pre-treatment steps were sufficient to remove any impurities that could be recorded by the thermal conductivity detector.
The NH3-TPD results confirm the low acidity of the materials when normalised by mass and surface area (Table 4), with higher acidity for the fresh CeO2-NC when compared to CeO2-SA. Both supports show decreased acidity after reduction treatment, with a sharp drop in acidity for CeO2-NC, which is associated with the dominant presence of Ce(III) on the material surface. Broad NH3-TPD profiles (Fig. S28†) are exhibited in CeO2 and are representative of the complex nature of multiple binding modes of NH3 to metal oxides.68 The low maximum peak temperatures (Table 4) for the fresh and reduced CeO2 suggest predominantly weak acidity, in agreement with Watanabe et al.69 Given the low acidity exhibited by the support materials and catalytic production of methane in the gas products, it can be concluded that the depolymerisation occurs via a hydrogenolysis mechanism.
| Material | NH3 adsorbed (μmol g−1) | NH3 adsorbed (μmol m−2) | NH3 peak temperaturec (°C) | CO2 adsorbed (μmol g−1) | CO2 adsorbed (μmol m−2) | CO2 peak temperatured (°C) |
|---|---|---|---|---|---|---|
| a Measurements were undertaken with ca. 0.3 g CeO2 to ensure appreciable desorption signals (SA – Sigma Aldrich; NC – nanocube). b Surface reduction of CeO2 was conducted at 550 °C for 1 h under 5% H2/Ar flow as according to CeO2 TPR. c Determined from the peak maximum in the NH3-TPD profile. d Determined from the peak maximum in the CO2-TPD profile. | ||||||
| Fresh CeO2-SA | 420.1 | 12.4 | 178 | 117.1 | 3.5 | 139 |
| Reduced CeO2-SAb | 398.9 | 11.7 | 131 | 132.6 | 3.9 | 94 |
| Fresh CeO2-NC | 509.2 | 20.4 | 136 | 139.4 | 5.6 | 84 |
| Reduced CeO2-NCb | 117.6 | 4.7 | 93 | 155.7 | 6.2 | 72 |
The basicity of the different fresh and reduced CeO2 measured by CO2-TPD and normalised by mass and surface area (Table 4) show higher basicity for the CeO2-NC than CeO2-SA in both instances. In particular, normalisation of the CO2 adsorbed by surface area, or the basicity density, is almost twice as high for the CeO2-NC when compared to the CeO2-SA. A slight increase in amount of basicity for the reduced samples can be attributed to the formation of surface hydroxides. Predominantly weak basic sites are noted over the CO2-TPD profiles (Fig. S29†), with confirmation given by the low maximum temperature peak (Table 4). The maximum peak temperature is lower in the CeO2-NC than CeO2-SA and suggests different weak basic sites. Furthermore, a temperature shift in the peaks to lower temperatures upon reduction suggests alterations in the strength of basicity, which can also be attributed to the formation of surface hydroxides. The differences in acid–base properties between CeO2-SA and CeO2-NC can be rationalised to arise from the different morphologies. The measured results are in agreement with theoretical predictions of acidity and basicity of ideal, or in the case of (100) due to surface polarity, the stable reconstructed O-terminated surfaces (acidity trend: (100) ≈ (110) > (111); basicity trend: (100) > (110) ≈ (111)).50,59,67
Importantly for polymer hydrogenolysis, differences in basicity and oxygen species have been shown to have a profound impact on the favourability of hydrocarbon binding and cleavage, especially when considering the works of Jaydev et al. for Ru-based metal oxide and Pt/C catalysts.22,35 Therefore, to confirm differences in hydrocarbon binding on the different catalyst surfaces, TPDs of 2,2,4-trimethylpentane (2,2,4-TMP), a branched hydrocarbon surrogate for PP, were conducted (Fig. 8). Significant differences are noted in the profiles, peak temperature, and amount adsorbed between the two CeO2 materials. The peak temperature for CeO2-NC (297 °C) confirms the higher binding strength for hydrocarbons when compared to CeO2-SA (142 °C), and suggests that the majority of 2,2,4-TMP can be readily desorbed from the surface of CeO2-SA at hydrogenolysis reaction temperatures (280 °C). CeO2-NC also adsorbs significantly more 2,2,4-TMP (232.3 μmol g−1; 2.7 μmol m−2), when normalised per mass or surface area (Fig. 8b), than CeO2-SA (90.2 μmol g−1; 0.8 μmol m−2). Interestingly, the surface area normalised adsorption correlates to the differences in basicity density of the support materials and further suggests an important role of weak basic sites and their corresponding density. However, the broad desorption profile indicates complex binding modes of 2,2,4-TMP and imply convoluted adsorption–desorption mechanisms involved with the CeO2 surface structure.
 | ||
| Fig. 8 (a) TPD profile of 2,2,4-trimethylpentane (2,2,4-TMP) and (b) quantification of surface adsorption of 2,2,4-TMP with normalisation by mass and surface area. Measurements were undertaken with ca. 0.3 g CeO2 to ensure appreciable desorption signals (SA – Sigma Aldrich; NC – nanocube). Tp is the maximum peak temperature determined from the 2,2,4-TMP-TPD. | ||
Subsequently when considering the basicity and 2,2,4-TMP-TPD measurements and their implications for PP hydrogenolysis, the nanocube with (100) facets improve the binding favourability of hydrocarbon chains onto the CeO2 surface and allows for improved scission of the chains by adjacent Ni particles leading to shorter hydrocarbon chains with a narrowing of the dispersity. Whereas, in the case of CeO2-SA, the decreased favourability of binding when compared to CeO2-NC, results in desorption of longer chains and further cleavage is not necessarily facilitated, with any further cleavage determined by the randomness of Ni-hydrocarbon chain contact and leads to longer chains with a broadening of the dispersity.
The improvement in C–C scission rate for the nanocube catalyst over the mixed shape catalyst is thus two-fold with an interconnected nature between the observed effects. Improvements in the degree of reduction of the CeO2 surface allow for improved H-storage, which partially increases the rate of C–C scission. Over the nanocube catalyst, with increased density of weaker basic sites, the contact between hydrocarbon chains and metal sites is enhanced and gives rise to more scission events. This enhanced contact arising from surface adsorption and weak basicity density is the key parameter to improving the C–C scission activity of the catalyst. Therefore, the morphology of the support structure is determined to be highly influential in directly promoting the activity and selectivity of the catalyst.
Conclusions
The hydrogenolysis of polypropylene was investigated using mixed shape and nanocube Ni/CeO2 catalysts. Both catalysts yielded similar liquid productivities, but with vastly different liquid compositions after solid deconstruction. The high liquid productivities and hence high internal cleavage selectivity of both catalysts arises from the ultrafine Ni particles (<1.3 nm). Analysis of the liquid products showed the Ni/CeO2 nanocube catalyst exhibited a 75% improvement in C–C scission rate over the mixed shape Ni/CeO2 catalyst leading to shorter liquid chains with smaller polydispersity indices. The enhanced C–C scission rate was attributed to improved H-storage and favourable basic properties on the nanocubes due to the exposed (100) facets. The higher weak basic site density of the nanocubes gives rise to higher binding strength and concentration of hydrocarbon chains on the catalyst surface. These effects were considered with normalisation and minimisation of Ni structure, Ni density, and CeO2 particle size, and the catalytic improvement is directly attributed to the promotional effects of the nanocube support morphology over mixed shape materials. The combination of earth abundant metal clusters and support engineering represents a catalyst design strategy to improve C–C scission rates and significantly improves the viability of chemical polymer recycling via hydrogenolysis.Data availability
The data supporting this article have been included as part of the ESI,† with detailed experimental methods including materials synthesis and characterisation methods such as PXRD, ICP-OES, Porosimetry, XPS, XAS, TEM, and chemisorption methods. Methods for reaction testing, liquid and gas analysis procedures (GPC, TGA, and GC-FID), and equations for calculations are included. Additional experimental data and ESI figures† are also available. Metadata (PXRD, XPS, and XAS) for this article is also available via the University of Liverpool data repository at https://doi.org/10.17638/datacat.liverpool.ac.uk/2799.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the UK National Interdisciplinary Centre for Circular Chemical Economy, which is funded by EPSRC (EP/V011863/1). X-ray photoelectron (XPS) data was acquired at the EPSRC National Facility for XPS (“HarwellXPS”, EP/Y023587/1, EP/Y023609/1, EP/Y023536/1, EP/Y023552/1 and EP/Y023544/1). The authors would like to thank Dr Mark Isaacs for support with the XPS and inert transfer XPS measurements. The authors would like to acknowledge the support of the Materials Innovation Factory at the University of Liverpool, created as part of the UK Research Partnership Investment Fund (UKRPIF) initiative, managed by UKRI Research England. The authors acknowledge Professor J. A. Lopez-Sanchez (University of Liverpool) for providing access to the equipment to perform hydrocarbon TPD. XAS measurements were performed at Diamond Light Source on beamline B18 through the UK Catalysis Hub BAG (Proposal SP29271). The authors also thank Diamond Light Source for access to beamline I11 for PXRD measurements (Proposal CY31578).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626(7997), 45–57 CrossRef CAS.
- J.-P. Lange, ACS Sustain. Chem. Eng., 2021, 9(47), 15722–15738 CrossRef CAS.
- A. Piovano and E. Paone, Curr. Res. Green Sustain. Chem, 2022, 5, 100334 CrossRef CAS.
- A. Musa, E. A. Jaseer, S. Barman and N. Garcia, Energy Fuels, 2024, 38(3), 1676–1691 CrossRef CAS.
- L. Chen, Y. Zhu, L. C. Meyer, L. V. Hale, T. T. Le, A. Karkamkar, J. A. Lercher, O. Y. Gutiérrez and J. Szanyi, React. Chem. Eng., 2022, 7(4), 844–854 RSC.
- P. A. Kots, B. C. Vance and D. G. Vlachos, React. Chem. Eng., 2022, 7(1), 41–54 RSC.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5(11), 1795–1803 CrossRef CAS PubMed.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, ACS Catal., 2021, 11(13), 8104–8115 CrossRef CAS.
- B. C. Vance, P. A. Kots, C. Wang, J. E. Granite and D. G. Vlachos, Appl. Catal., B, 2023, 322, 122138 CrossRef CAS.
- B. C. Vance, S. Najmi, P. A. Kots, C. Wang, S. Jeon, E. A. Stach, D. N. Zakharov, N. Marinkovic, S. N. Ehrlich, L. Ma and D. G. Vlachos, JACS Au, 2023, 3(8), 2156–2165 CrossRef CAS PubMed.
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, JACS Au, 2022, 2(10), 2259–2268 CrossRef CAS.
- Z. Zhao, Z. Li, X. Zhang, T. Li, Y. Li, X. Chen and K. Wang, Environ. Pollut., 2022, 313, 120154 CrossRef CAS PubMed.
- M. Chu, X. Wang, X. Wang, P. Xu, L. Zhang, S. Li, K. Feng, J. Zhong, L. Wang, Y. Li, L. He, M. Cao, Q. Zhang, L. Chi and J. Chen, J. Am. Chem. Soc., 2024, 146(15), 10655–10665 CrossRef CAS.
- M. Meirow, A. Tennakoon, X. Wu, J. Willmon, D. Howell, W. Huang, A. D. Sadow and E. Luijten, J. Phys. Chem. C, 2023, 127(49), 23805–23813 CrossRef CAS.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3(11), 893–901 CrossRef CAS.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13(1), 4850 CrossRef CAS.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58(45), 20601–20609 CrossRef CAS.
- J. V. Lamb, Y.-H. Lee, J. Sun, C. Byron, R. Uppuluri, R. M. Kennedy, C. Meng, R. K. Behera, Y.-Y. Wang, L. Qi, A. D. Sadow, W. Huang, M. S. Ferrandon, S. L. Scott, K. R. Poeppelmeier, M. M. Abu-Omar and M. Delferro, ACS Appl. Mater. Interfaces, 2024, 16(9), 11361–11376 CrossRef CAS.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CrossRef CAS.
- S. D. Jaydev, M.-E. Usteri, A. J. Martín and J. Pérez-Ramírez, Chem Catal., 2023, 3(5), 100564 CrossRef CAS.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, ACS Catal., 2022, 12(8), 4618–4627 CrossRef CAS.
- T. Kim, H. Nguyen-Phu, T. Kwon, K. H. Kang and I. Ro, Environ. Pollut., 2023, 331, 121876 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1(9), 1422–1434 CrossRef CAS PubMed.
- M. Li, W. Yin, J. Pan, Y. Zhu, N. Sun, X. Zhang, Y. Wan, Z. Luo, L. Yi and L. Wang, Chem. Eng. J., 2023, 471, 144691 CrossRef CAS.
- A. Tomer, M. M. Islam, M. Bahri, D. R. Inns, T. D. Manning, J. B. Claridge, N. D. Browning, C. R. A. Catlow, A. Roldan, A. P. Katsoulidis and M. J. Rosseinsky, Appl. Catal., A, 2023, 666, 119431 CrossRef CAS.
- S. Lu, Y. Jing, S. Jia, M. Shakouri, Y. Hu, X. Liu, Y. Guo and Y. Wang, ChemCatChem, 2023, 15(3), 1–8 CrossRef.
- H. Ji, X. Wang, X. Wei, Y. Peng, S. Zhang, S. Song and H. Zhang, Small, 2023, 19(35), 2300903 CrossRef CAS.
- C. Jarauta-Córdoba, L. García, J. Ruiz, M. Oliva and J. Arauzo, Molecules, 2024, 29(16), 3797 CrossRef PubMed.
- J. Xie, Y. Xi, W. Gao, H. Zhang, Y. Wu, R. Zhang, H. Yang, Y. Peng, F. Li, Z. Li and C. Li, ACS Catal., 2023, 13(14), 9577–9587 CrossRef CAS.
- K. E. McCullough, I. L. Peczak, R. M. Kennedy, Y.-Y. Wang, J. Lin, X. Wu, A. L. Paterson, F. A. Perras, J. Hall, A. J. Kropf, R. A. Hackler, Y. Shin, J. Niklas, O. G. Poluektov, J. Wen, W. Huang, A. D. Sadow, K. R. Poeppelmeier, M. Delferro and M. S. Ferrandon, J. Mater. Chem. A, 2023, 11(3), 1216–1231 RSC.
- I. L. Peczak, R. M. Kennedy, R. A. Hackler, R. Wang, Y. Shin, M. Delferro and K. R. Poeppelmeier, ACS Appl. Mater. Interfaces, 2021, 13(49), 58691–58700 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17(2), 174–181 CrossRef CAS.
- S. D. Jaydev, A. J. Martín and J. Pérez-Ramírez, ChemSusChem, 2021, 14(23), 5179–5185 CrossRef CAS PubMed.
- Y. Wang, S. Ren, L. Zhang, J. Deng, X. Peng and H. Cheng, Fuel, 2018, 219, 141–150 CrossRef CAS.
- M. V. Kok, Fuel Process. Technol., 2011, 92(5), 1026–1031 CrossRef CAS.
- C. D. Gamlin, N. K. Dutta, N. Roy Choudhury, D. Kehoe and J. Matisons, Thermochim. Acta, 2002, 392–393, 357–369 CrossRef CAS.
- M. K. A. Ali and H. Xianjun, Powder Technol., 2020, 363, 48–58 CrossRef CAS.
- H. Jiang, X. Hou, K. D. Dearn, D. Su and M. Kamal Ahmed Ali, Adv. Powder Technol., 2021, 32(12), 4658–4669 CrossRef CAS.
- Y. Porfiryev, S. Shuvalov, P. Popov, D. Kolybelsky, D. Petrova, E. Ivanov, B. Tonkonogov and V. Vinokurov, ACS Omega, 2020, 5(21), 11946–11954 CrossRef CAS.
- H. Cai, R. Chen, M. Bahri, C. J. Hawkins, M. Sonni, L. M. Daniels, J. Lim, J. A. Evans, M. Zanella, L. A. H. Jones, T. D. Manning, T. D. Veal, L. J. Hardwick, M. S. Dyer, N. D. Browning, J. B. Claridge and M. J. Rosseinsky, ACS Mater. Lett., 2023, 5(2), 527–535 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. St. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600(9), 1771–1779 CrossRef CAS.
- I. Preda, L. Soriano, D. Díaz-Fernández, G. Domínguez-Cañizares, A. Gutiérrez, G. R. Castro and J. Chaboy, J. Synchrotron Radiat., 2013, 20(4), 635–640 CrossRef CAS PubMed.
- D. R. Inns, A. J. Mayer, V. Skukauskas, T. E. Davies, J. Callison and S. A. Kondrat, Top. Catal., 2021, 64(17–20), 992–1009 CrossRef CAS.
- A. Anspoks, A. Kuzmin, A. Kalinko and J. Timoshenko, Solid State Commun., 2010, 150(45–46), 2270–2274 CrossRef CAS.
- A. Anspoks, A. Kalinko, R. Kalendarev and A. Kuzmin, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 86(17), 174114 CrossRef.
- K. R. G. Lim, S. K. Kaiser, H. Wu, S. Garg, M. Perxés Perich, J. E. S. van der Hoeven, M. Aizenberg and J. Aizenberg, Nat. Catal., 2024, 7(2), 172–184 CrossRef CAS.
- M. Zare, P. A. Kots, S. Caratzoulas and D. G. Vlachos, Chem. Sci., 2023, 14(8), 1966–1977 RSC.
- A. Trovarelli and J. Llorca, ACS Catal., 2017, 7(7), 4716–4735 CrossRef CAS.
- L. R. Smith, M. A. Sainna, M. Douthwaite, T. E. Davies, N. F. Dummer, D. J. Willock, D. W. Knight, C. R. A. Catlow, S. H. Taylor and G. J. Hutchings, ACS Catal., 2021, 11(8), 4893–4907 CrossRef CAS.
- H. Nörenberg and J. H. Harding, Surf. Sci., 2001, 477(1), 17–24 CrossRef.
- F. Polo-Garzon, Z. Bao, X. Zhang, W. Huang and Z. Wu, ACS Catal., 2019, 9(6), 5692–5707 CrossRef CAS.
- M. Romeo, K. Bak, J. El Fallah, F. Le Normand and L. Hilaire, Surf. Interface Anal., 1993, 20(6), 508–512 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2023, 55(11), 845–850 CrossRef CAS.
- M. A. Isaacs, C. Drivas, R. Lee, R. Palgrave, C. M. A. Parlett and D. J. Morgan, Appl. Surf. Sci., 2023, 18, 100469 CrossRef.
- T. Désaunay, G. Bonura, V. Chiodo, S. Freni, J.-P. Couzinié, J. Bourgon, A. Ringuedé, F. Labat, C. Adamo and M. Cassir, J. Catal., 2013, 297, 193–201 CrossRef.
- H. Chen, Y. M. Choi, M. Liu and M. C. Lin, ChemPhysChem, 2007, 8(6), 849–855 CrossRef CAS.
- M. Capdevila-Cortada, G. Vilé, D. Teschner, J. Pérez-Ramírez and N. López, Appl. Catal., B, 2016, 197, 299–312 CrossRef CAS.
- O. Matz and M. Calatayud, ACS Omega, 2018, 3(11), 16063–16073 CrossRef CAS PubMed.
- A. Löfberg, J. Guerrero-Caballero, T. Kane, A. Rubbens and L. Jalowiecki-Duhamel, Appl. Catal., B, 2017, 212, 159–174 CrossRef.
- D. Monti and A. Baiker, J. Catal., 1983, 83(2), 323–335 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112(5), 2714–2738 CrossRef CAS PubMed.
- R. Oh, X. Huang, J. Hayward, Y. Zheng, M. Chen, G.-S. Park, G. Hutchings and S. K. Kim, ACS Catal., 2024, 14(2), 897–906 CrossRef CAS.
- V. Muravev, A. Parastaev, Y. van den Bosch, B. Ligt, N. Claes, S. Bals, N. Kosinov and E. J. M. Hensen, Science, 2023, 380(6650), 1174–1179 CrossRef CAS PubMed.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114(30), 12909–12919 CrossRef CAS.
- Z. Wu, A. K. P. Mann, M. Li and S. H. Overbury, J. Phys. Chem. C, 2015, 119(13), 7340–7350 CrossRef CAS.
- C. D. Evans, S. A. Kondrat, P. J. Smith, T. D. Manning, P. J. Miedziak, G. L. Brett, R. D. Armstrong, J. K. Bartley, S. H. Taylor, M. J. Rosseinsky and G. J. Hutchings, Faraday Discuss., 2016, 188, 427–450 RSC.
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113(32), 14249–14257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08111k |
| This journal is © The Royal Society of Chemistry 2025 |