
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Leveraging self-passivation of quantum dots via nitrogen-doping for multifunctional biopolymer nanocomposites†
Anthony V.
Tuccitto
 ab,
Rafaela
Aguiar
a,
Nello D.
Sansone
a,
Yalda
Chehrehsaz
a,
Boran
Kumral
b,
Peter
Serles
b,
Tobin
Filleter
b,
Chul B.
Park
b and
Patrick C.
Lee
*a
ab,
Rafaela
Aguiar
a,
Nello D.
Sansone
a,
Yalda
Chehrehsaz
a,
Boran
Kumral
b,
Peter
Serles
b,
Tobin
Filleter
b,
Chul B.
Park
b and
Patrick C.
Lee
*a
aMultifunctional Composites Manufacturing Laboratory (MCML), Department of Mechanical and Industrial Engineering, University of Toronto, 5 King's College Road, Toronto, M5S 3G8, Canada. E-mail: patricklee@mie.utoronto.ca
bDepartment of Mechanical and Industrial Engineering, University of Toronto, 5 King's College Road, Toronto, M5S 3G8, Canada
First published on 4th February 2025
Abstract
The plant-based and industrially compostable polymer, poly(L-lactide) (PLLA) is an indispensable substitute for petroleum-based polymers but has limited functionality owing to material and processing-related challenges. To address this, we design graphene oxide-based quantum dots (GO-QDs) with varying degrees of nitrogen-doping from thermal treatment of citric acid and urea, yielding strong and tough nanocomposites with exceptional visible-light transmittance (>70%), selective UV-visible light blocking ability, and fluorescence. For the first time, we clarify that this behaviour arises from the inclusion of carbonyl, pyrrolic, pyridinic, and graphitic-nitrogen in QDs with an increasing degree of nitrogen-doping, each offering distinct light absorption tendencies. The inclusion of just 0.25 wt% of synthesized QDs achieves a 43.4% improvement in toughness with a concomitant increase in stiffness and strength of 13.7% and 13.3%, respectively. This behaviour is attributed to increased craze density confirmed by fractography analysis. Furthermore, GO-QD inclusion can improve the processability of PLLA, increasing its crystallization temperature by over 10 °C, while reducing melt viscosity by up to an order of magnitude owing to size and composition effects, which are valuable in downstream manufacturing processes. This unique combination of properties cannot be achieved with existing micro/nanoparticle UV-absorbers (e.g., zinc oxide, titanium dioxide), reinforcing additives (e.g., cellulose derivatives, polymer nanofibrils), and processing aids (e.g., nucleating agents, plasticizers). This study not only sets a new benchmark for the optical, mechanical and processing-behaviour of PLLA, but also demonstrates the transformative potential of nitrogen-doping in expanding the functional capabilities of green polymer nanocomposites.
1. Introduction
Plastics are ubiquitous materials in modern society; however, the use of most commercially available plastics has been linked to severe environmental consequences at all stages of their life cycles. Specifically, most consumer-destined plastics are sourced from finite petroleum resources and present a variety of disposal and recycling challenges that ultimately result in the accumulation of plastic in landfills and ecosystems worldwide.1 Over recent decades, poly(L-lactide) (PLLA) has been revered as a critical component in efforts to alleviate these concerns, being a bio-based and industrially compostable alternative to petroleum-based polymers. Specifically, PLLA offers increasingly competitive material costs, moderate stiffness (2.5–4 GPa) and strength (60–80 MPa), and melt-processing temperature comparable to petroleum-based polymers like polypropylene. However, its adoption in industrial processes is limited owing to intrinsically slow crystallization kinetics, which limits solidification in molding processes, as well as tuning of mechanical properties for target applications.2 Moreover, from a consumer perspective, PLLA is undesirable owing to its inherently brittle behavior and low glass transition temperature (Tg) compared to other polyesters like polyethylene terephthalate and polybutylene terephthalate. This brittleness gives rise to poor toughness, minimizing energy absorption while under stress.3,4 These limitations severely restrict the applicability of PLA.Beyond its limited mechanical performance and processability, PLLA readily transmits visible-light wavelengths (400–700 nm), making it desirable in applications necessitating good transparency (∼90% visible-light transmittance).5 However, PLLA also readily transmits ultraviolet radiation (UVA: 320–400 nm, UVB: 290–320 nm, UVC: 200–290 nm, respectively), as well as blue-light (∼400–500 nm).6,7 This limits its protective ability for UV- and blue-light sensitive foodstuff, cosmetic products, agricultural products, and personal protective equipment (PPE), thereby rendering it unsuitable for these applications. UV-blocking in polymers typically necessitates the incorporation of organic (typically phenolic derivatives),8 or inorganic UV-blocking additives (e.g., zinc oxide, titanium dioxide).9 While organic UV-blockers are low-cost and permit better absorption wavelength selectivity8 which is necessary in food-related applications (e.g., industrial sterilization processes),10 and agricultural/horticultural crop growth,11 their short working lifespan,9 environmental persistence, as well as toxicity make them unfavorable for use in consumer products.12 In contrast, inorganic UV-blockers generally exhibit UVB/UVC (<350 nm) blocking,8,13 and often necessitate complex compounds of organic and inorganic UV-blockers to achieve absorption targets.8 Beyond this, blue-light blocking is critical for suppressing degradation of food nutrients (e.g., anthocyanins),14 cosmetic products (e.g., L-ascorbic acid),15 as well as in PPE.16 With these considerations in mind, there are limited methods that address each challenge without severely compromising PLLA's sustainable nature. Specifically, inclusion of a secondary component in concentrations greater than 5 wt% can harm PLLA's biodegradation, further limiting composite design alternatives;17 broad range UV- and blue-light blocking may be challenging to achieve with lower filler loadings.18,19
The incorporation of microparticle or nanoparticle fillers is among the most common method of overcoming PLLA's numerous deficiencies. Common micro-fillers include talc,20,21 and micro-fibrillated cellulose.22 Unfortunately, their low specific surface area relative to nanoparticles limits matrix–filler interactions, necessitating beyond 5 wt% for functionality improvements, but at the detriment of transparency, thereby limiting applicability. On the contrary, more pronounced matrix–filler interactions are offered by finer nano-scale particles, which can circumvent this challenge. Common nanoparticle fillers include cellulose nanofibrils and their derivatives,23 synthetic polymer nanofibrils,4,24 and graphene derivatives.25 While cellulose nanofibrils and their derivatives are green additives that can strengthen/toughen PLA, their tendency to undergo self-aggregation owing to strong hydrogen bond interactions can embrittle PLLA and detriment its transparency26,27 unless costly and time-consuming surface treatments or compatibilization strategies are applied. Synthetic polymer nanofibrils have also recently been applied via in situ fibrillation, for toughening PLLA with minimal sacrifice in blend stiffness and strength.4 However, fibrillation approaches are challenging to implement practically owing to material selection constraints (e.g., matrix-dispersed phase viscosity matching, melting temperature difference). Beyond particle and fiber-based fillers, compounding with plasticizers and/or rubbery materials can be an effective method of toughening PLLA, but often at the expense of critical material properties including strength, modulus, and transparency.5,28 Although the use of these additives can target specific deficiencies in PLLA, it is highly desirable to develop a single additive that can be incorporated into PLLA to meet bulk property requirements and provide multi-functionality.
Over the past decade, graphene-oxide based quantum dots (GO-QDs), zero-dimensional graphene derivatives with diameters on the scale of 1–20 nm, have been developed, and offer profuse benefits including strong UV-absorption and photoluminescence arising from quantum confinement effects,29 tunable bandgap, antioxidant effects in biological systems,30 and low cytotoxicity.31,32 These desirable properties make these QDs ideal for applications ranging from anti-counterfeiting to bio-medical imaging.33–36 Additionally, their small size permits retention of transparency in nanocomposites, compared to those containing larger graphene derivatives that typically render nanocomposites black, owing to broad-range light absorption mechanisms.37 It should be noted that while heavy metal-based quantum dots (e.g., cadmium selenide) have found extensive use in literature,38,39 toxicity concerns make them unsuitable for many consumer-destined polymer nanocomposites. On the contrary, bottom-up approaches assembling small biomolecules (e.g., citric acid, glucose, sucrose) into GO-QDs via simple thermal treatments is a greener alternative for fabricating more consumer-friendly QDs, from low-cost and accessible precursors.40–43
The past several years have yielded rich literature on custom design of QDs for target applications based on their size and surface functionalities as typical design criteria. While the size can be challenging to control owing to most synthetic approaches yielding broad particle size distributions, much research has focused on compositional changes in QDs as the primary design input. Typical synthetic and post-synthetic strategies for changing QD composition include passivation control, oxidation, reduction, and heteroatom (e.g., sulfur, boron) doping/functionalization,44 with passivation control and heteroatom doping being the simplest modification to apply in bottom-up synthetic approaches.45 It has been hypothesized that the presence of heteroatoms in QDs can greatly alter UV- and visible-light absorption properties, which can be used to engineer colorful and brighter nanocomposites with good transparency.46,47 Beyond visible and UV-absorption properties, QDs are claimed to offer simultaneous stiffening, strengthening and toughening effects to nanocomposites at relatively low concentrations (e.g., <1 wt%).47 However, despite these synthetic design considerations, the role of green QD chemical composition on the multi-tier properties of biodegradable nanocomposites have yet to be explored.
With these considerations in mind, we design a series of GO-QD derivatives possessing varying compositions, to achieve a unique combination of properties that cannot be achieved with existing micro/nanoparticle UV-absorbers (e.g., zinc oxide, titanium dioxide), reinforcing additives (e.g., cellulose derivatives and polymer nanofibrils), and processing aids (e.g., nucleating agents/plasticizers). Here, three GO-QD derivatives are synthesized via thermal treatment of low-cost organic molecules, namely, citric acid (CA) and urea. Treatment of solely CA yields GO-QDs,40 while nitrogen-doped GO-QDs (NGO-QDs) can be synthesized identically, via substitution of CA with urea as a nitrogen-dopant (N-dopant).48,49 While some N-dopants like ammonia are known to result in larger particle sizes, urea yields QDs with similar size distributions, but different chemical composition.48,50 The prepared GO-QD derivatives are then blended into a PLLA matrix to systematically isolate the role of QD composition on the optical transparency, UV-blocking ability, fluorescence emission, crystallization kinetics, mechanical, and rheological properties of nanocomposites. By engineering QDs with specific proportions of target functional groups, we develop PLLA nanocomposites possessing exceptional visible-light transmittance (>70%), as well as selective UV- and blue-light blocking ability, that open the door for PLLA's use in protective packaging for UV- and blue-light sensitive foodstuff, cosmetic products, agricultural products, and PPE. The nanofiller content used to impart these characteristics is far below standard thresholds that compromise compostability (i.e., 5 wt%),17 a powerful benefit to this technology.
2. Results and discussion
2.1 Chemical and physical characterization of GO-QD derivatives
FTIR analysis of the QD precursors and QDs reveals significant chemical transformations that occur during synthesis (synthesis details found in Materials and methods), in function of precursor composition, as is illustrated in Fig. 1. Specifically, GO-QD is synthesized from CA alone while NGO-QD1 and NGO-QD2 are derived from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:3 precursor molar ratios (CA:urea), respectively. First, the CA possesses bands ranging from 3500 to 3000 cm−1 corresponding to the stretching modes of its four O–H groups. Additionally, the small shoulder-like peak below 3000 cm−1 corresponds to C–H stretching attributed with the methylene groups along the CA. As well, the bands at 1743 and 1692 cm−1 correspond to C
:1 or 1:3 precursor molar ratios (CA:urea), respectively. First, the CA possesses bands ranging from 3500 to 3000 cm−1 corresponding to the stretching modes of its four O–H groups. Additionally, the small shoulder-like peak below 3000 cm−1 corresponds to C–H stretching attributed with the methylene groups along the CA. As well, the bands at 1743 and 1692 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands associated with CA's three carboxylic acid functional groups.51 Furthermore, the series of bands in the range of 1425 to 1340 cm−1 correspond to C–H bending of methylene groups.
O stretching bands associated with CA's three carboxylic acid functional groups.51 Furthermore, the series of bands in the range of 1425 to 1340 cm−1 correspond to C–H bending of methylene groups.
 | ||
| Fig. 1 (A) FTIR spectra of CA and urea QD precursors, as well as each GO-QD derivative. Spectra are shifted along the Y-axis by an arbitrary value for the sake of clarity. Possible chemical structures of (B) GO-QD, (C) NGO-QD1, and (D) NGO-QD2, are provided based on spectroscopic analysis. GO-QD, NGO-QD1, and NGO-QD2 were derived from CA, 3:1, and 1:3 precursor molar ratios (CA:urea), respectively. | ||
In contrast, the urea precursor used for N-doping possesses a high intensity N–H stretching band near 3429 cm−1,52 a band near 1674 cm−1 corresponding to CO stretching, a band near 1590 cm−1 corresponding to N–H bending, and two bands near 1462 and 1149 cm−1 corresponding to C–N bonds.52
Beyond this, the FTIR spectrum of the CA-derived GO-QDs synthesized in this work is consistent with work presented by Dong et al.40 For these GO-QDs, there is a broad peak centered near 3378 cm−1 corresponding to O–H stretching implying the presence of hydroxyl/carboxyl groups. At 2956 cm−1, there is a small shoulder corresponding to C–H stretching in the GO-QD. Beyond this, there is a small shoulder peak at 1700 cm−1 corresponding to CO bond stretching, as well as a larger peak near 1560 cm−1 likely corresponding to CO stretching in carboxylate groups, as were formed during passivation with aqueous NaOH. Others have also attributed this peak to the presence of unoxidized graphitic domains formed during the carbonization process.53 Moreover, the peak present near 1380 cm−1 is characteristic of CA-derived GO-QDs,40 and has been attributed to the presence of C–OH groups,53 resulting from the incomplete carbonization of the CA.
The FTIR spectra of NGO-QDs, in comparison with GO-QDs and CA/urea precursors are also shown in Fig. 1A. The NGO-QD1 sample possesses bands near 3443, 3340, and 3185 cm−1 corresponding to N–H stretching.54 Moreover, these peaks are convoluted with a broad band that is visible near 3585 cm−1, which is not present in the urea precursor, but is present in all QDs fabricated, corresponding to the O–H stretching of the hydroxyl and carboxyl functional groups formed. Akin to GO-QDs, there is a band near 2956 cm−1 corresponding to C–H stretching. As well, the peak near 1700 cm−1 corresponds to either CO stretching, or CN stretching.50 Moreover, the small shoulder peak near 1771 cm−1 likely corresponds to carboxyl groups in the QDs. As well, there is a peak present near 1560 cm−1,53 possibly corresponding to unoxidized graphitic domains. This peak may also be attributed to N–H bending (e.g., pyrrolic-N), similar to observations for the urea precursor. Finally, the peak near 1190 cm−1 likely corresponds to C–N bonds, with one dominant functional group type present.
NGO-QD2 also possesses bands near 3443, 3340, and 3185 cm−1 corresponding to N–H stretching;54 these peak intensities increase compared to NGO-QD1 owing to the greater urea precursor loading used during synthesis, imparting a more abundant source of nitrogen. NGO-QD2 also exhibits a broad O–H and C–H stretching band, like NGO-QD1. Additionally, the peak corresponding to the CO and CN stretching increases in intensity. As such, the NGO-QD2 sample likely possesses more pyridinic and graphitic-N substitution in its structure. Beyond this, two bands arise near 1188 and 1148 cm−1 corresponding to C–N stretching, in contrast to NGO-QD1. This suggests the formation of two distinctly different N-based chemical environments, perhaps pyrrolic and pyridinic-N. It should be noted that an increase in the intensity of bands corresponding to N-containing functional groups is observed with increasing urea precursor loading, suggesting that N-inclusions in the QDs become more abundant with increasing urea precursor loading, as expected. However, this is accompanied by changes in the proportions of N-containing functional groups present in the synthesized QDs. Based on the spectroscopic data provided, the possible chemical structure of a single-layer of each GO-QD derivative is shown in Fig. 1B–D.
XPS was used to quantify the relative proportions of the carbon (C), oxygen (O), and nitrogen (N) containing functional groups present in each GO-QD derivative, as shown in Fig. 2. The relative proportions of these distinct chemical functional groups are of particular importance in engineering QD-containing nanocomposites with tailored properties. For example, certain functional group types may behave as chromophores for selective light absorption. For the GO-QDs, the C1s, sodium (Na) auger, and O1s signals are present at 284, 497, and 530 eV, respectively, as is consistent with literature.55 As well, there was an absence of peaks corresponding to N-containing functional groups, as was expected due to the absence of urea in the precursor. A high-resolution spectrum of the C1s peak was fitted with Gaussian curves to identify the C-based chemical environments present in the prepared QDs, which yielded peaks centered at 284.8 (C–C/CC), 286.3 (C–O), 288.3 (CO), and 290.0 eV (C–(O)C), respectively. These results are consistent with FTIR analysis revealing carboxyl/carboxylate, and hydroxyl groups, along with graphitic carbon. Peak integration reveals that C–C/CC environments are abundantly present (RP: 53.1%), with CO also abundantly present (RP: 38.4%), as is illustrated in Table 1. Other carbon environments are present in low abundance, C–O linkages, and C–(O)C. Moreover, deconvolution of the O1s peak also reveals O-based chemical environments present in the prepared dots, which yielded peaks centered at 530.8 (O–C), 531.7 (OC), and 533.3 eV (OC–O), respectively. Peak integration reveals abundant O–C and OC environments (RP of 47.7 and 43.9%, respectively), with a small fraction of OC–O groups present (RP ∼8.4%). This confirms the presence of abundant O-containing functional groups along the surface of the dots.
 | ||
| Fig. 2 (A) XPS full scan spectra of synthesized GO-QD derivatives, XPS high-resolution spectra illustrating: (B) the C1s peak with peak deconvolutions, (C) the O1s peak with peak deconvolutions, (D) the N1s peak with peak deconvolutions. The arrow shown near 497 eV denotes the Na auger. | ||
| Binding energy (eV) | Functional group | Relative percentage (%) | ||
|---|---|---|---|---|
| GO-QD | NGO-QD1 | NGO-QD2 | ||
| a For the GO-QD and NGO-QD1, we also have a Na auger peak at 497 eV. | ||||
| C1s peak | ||||
| 284.8 | C–C/CC |
53.1 | 54.4 | 50.0 |
| 285.6 | C–N | 0 | 6.7 | 13.1 |
| 286.3 | C–O | 7.0 | 8.1 | 9.8 |
| 288.3 | CO |
38.4 | 29.6 | 26.1 |
| 290.0 | C–(O)C | 1.5 | 1.2 | 1.0 |
|
||||
| O1s peak | ||||
| 530.8 | O–C | 47.7 | 34.0 | 14.2 |
| 531.7 | OC |
43.9 | 60.6 | 54.9 |
| 533.3 | OC–O |
8.4 | 5.4 | 30.9 |
|
||||
| N1s peak | ||||
| 398.9 | Pyridinic-N | 0 | 6.7 | 13.8 |
| 399.8 | Pyrrolic-N | 0 | 86.6 | 75.9 |
| 401.7 | Graphitic-N | 0 | 6.7 | 10.3 |
For NGO-QD1, the C1s, N1s, Na auger, and O1s peaks are present at 284, 399, 497, and 530 eV, respectively. Interestingly, the intensity of the C1s peak increases with the introduction of urea in the precursor, with a concomitant decrease in the O and Na content, owing to the substitution of CA with the urea N-dopant/self-passivator. A high-resolution C1s spectrum is shown in Fig. 2B, with the fitted peaks centered at 284.8 (C–C/CC), 285.6 (C–N), 286.3 (C–O), 288.3 (CO), and 290.0 eV (C–(O)C), respectively. These results are consistent with FTIR analysis revealing carboxyl/carboxylate, and hydroxyl groups, along with graphitic segments. Peak integration reveals that C–C/CC environments are abundantly present (RP: 54.4%), with CO also abundantly present (RP: 29.6%). Other C environments are present in low abundance, including C–O linkages, and C–(O)C. Furthermore, deconvolution of the O1s band shows O-based chemical environments in the NGO-QD1, yielding peaks centered at 530.8 (O–C), 531.7 (OC), and 533.3 eV (OC–O), respectively, akin to GO-QDs. Peak integration of the O1s peak also reveals abundant O–C and OC environments (RP of 34.0 and 60.6%, respectively), with a small fraction of OC–O groups present (RP ∼5.4%). As well, deconvolution of the high-resolution N1s peak reveals three primary N-based chemical environments, namely 398.9 eV (pyridinic-N), 399.8 eV (pyrrolic-N), and 401.7 eV (graphitic-N), which is characteristic of N-doped QDs.55–57 As well, peak integration reveals that pyrrolic-N is most abundant (RP of 86.6%), with both pyridinic-N and graphitic-N present at 6.7% RP, respectively. These findings are consistent with partial passivation of NGO-QD1 with urea.
Finally, the NGO-QD2 possesses C1s, N1s, and O1s peaks akin to NGO-QD1, but with the elimination of the Na auger, owing to the complete self-passivation attributed to the increased urea precursor content. The increased urea content gives rise to a higher N content introduced into the GO-QDs, as illustrated by the increased intensity of the N1s peak. For a quantitative comparison, the degree of N-doping in the carbon framework of NGO-QD1 and NGO-QD2 can be determined using the intensity ratio of the N1s/C1s peaks (RN/C), as has been proposed by Zhu et al.48 The RN/C of NGO-QD1 and NGO-QD2 are 0.20, and 0.32, respectively, confirming the increased degree of N-doping. This is corroborated via elemental analysis shown in Table S1.† Here, pyrrolic-N is most abundant (RP of 75.9%), with pyridinic-N present at 13.8% RP, and graphitic-N present at 10.3% RP. We believe the increase in pyridinic-N and graphitic-N at the expense of pyrrolic-N with increasing urea precursor content yields higher-order N substitutions in the C framework, as opposed to pyrrolic defects. Moreover, the C1s peak shows a high C–C/CC content (RP of 50.0%), with other C environments far less abundant. Finally, the O1s peak still shows the presence of abundant CO groups (RP of 54.9%), confirming the presence of both N- and O-containing functional groups on the surface of this GO-QD derivative.
The zeta potential of the QDs reveals information regarding the electrostatic interactions between nanoparticles, which may affect the self-aggregation tendencies of QDs, thereby affecting nanocomposite bulk properties. The zeta potential of GO-QD, NGO-QD1, and NGO-QD2 are −37 mV, −45 mV, and −56 mV, respectively. The increasing magnitude of zeta potential values with increasing urea precursor loading is likely attributable to the elimination of carboxylate groups with increasing urea passivator content.
Atomic force micrographs reveal the formation of QDs, as shown in Fig. 3A–C. Specifically, all GO-QD derivatives synthesized possessed heights of ∼1–5 nm. These particle sizes are consistent with literature claiming QDs with heights ranging from 0.5–10 nm and diameters ranging from 2–15 nm from similar precursor formulations.40,50,55
 | ||
| Fig. 3 Atomic force micrographs illustrating the prepared (A) GO-QD, (B) NGO-QD1, and (C) NGO-QD2 specimens. | ||
Owing to the successive filtration process to particle sizes <20 nm prior to AFM analysis, lateral dimensions measured beyond this size threshold are a result of agglomeration during drop-casting as has been observed for other QDs.58 This is attributed to the hydrophilic nature of these materials. Additionally, AFM analysis is prone to imaging artifacts and edge effects that are known to overestimate the size of QDs because of sample feature broadening due to tip-sample convolution.59,60 In contrast to larger particle fillers, the nanometric size of prepared QDs gives rise to distinct physical phenomena in polymer matrices that are not otherwise achievable.
2.2 Nanocomposite characterization
O, pyrrolic and pyridinic groups in the QDs.30,55,56,70 Additionally, the absorbance band below 300 nm becomes more apparent likely owing to partial passivation with NaOH increasing the C-content in the dots, making the π → π* transitions of sp2 clusters more obvious. For films containing NGO-QD2, the baseline VLT is slightly reduced (VLT of 72.2 ± 2.0%), likely owing to differences in particle aggregation tendency with compositional changes; this may be responsible for the slight reduction in the baseline light transmittance. As well, the absorption bands previously ascribed to N-containing functional groups increase in intensity, which is consistent with the increasing fraction of N functional groups with increasing degree of N-doping. Interestingly, the absorption band centered at 340 nm attributed to carbonyl, pyrrolic and pyridinic groups of the QDs does not increase dramatically. Rather, the band near 418 nm associated with graphitic-N dramatically increases in intensity, confirming that increasing urea precursor content induces graphitic-N. Here, delocalization of electrons around these graphitic-N substitutions is believed to reduce the energy barrier for this electronic excitation, thereby inducing a red-shift in the absorption relative to pyrrolic and pyridinic functional groups. In fact, Sarkar et al. predicted using density functional theory simulations that graphitic-N is the only N-containing functional group that imparts such a considerable red-shift, owing to a reduced energy gap between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively). This reduction in energy gap facilitates the excitation of electrons into unoccupied π orbitals of the graphitic network with low-energy wavelengths.71 As such, our observations align well with theory. The increased intensity of this absorption band not only improves UVA absorption but imparts selective blue-light absorption with minimal detriment to light transmittance in the remaining visible spectrum. This selective blue-light shielding is highly sought in electronic, optoelectronic, personal-protective equipment, and in food packaging. In particular, blue-light shielding is critical in food and cosmetic packaging.14 It should be noted that while N-doping increases the UV-absorption (UVA: 56.3 ± 1.4%, UVB: 62.1 ± 1.4%, and UVC: 81.2 ± 1.9% light blocking ability), the VLT still remains >70%, even with blue-light absorption becoming more predominant (55.3 ± 1.2% blue-light blocking ability). Unlike larger nanofillers (i.e., >50 nm) that primarily block UV-visible radiation via reflectance, QDs undergo a series of absorptive energy transformations owing to their heteroatom-based surface functional groups, that dissipate the incident radiation.47 This energy dissipation mechanism is confirmed in Fig. 4D, as well as in Fig. S1A and B,† illustrating the fluorescence behavior of the nanocomposites. From these images, the fluorescence emission is clearly intensified with an increased degree of N-doping. It should be noted that this intense fluorescence behavior is not just present with only 0.25 wt% of QDs in PLLA but is present in the absence of external solvents. This is also aligned with Fig. S1C† illustrating the reflectance wavelength of the neat PLLA and QD-containing nanocomposite films between 400–700 nm. Here, the visible light reflectance decreases sharply in the presence of NGO-QD1 and NGO-QD2 between 400 and 500 nm, confirming that changes in the selective light blocking ability of these films is largely attributed to light absorption.
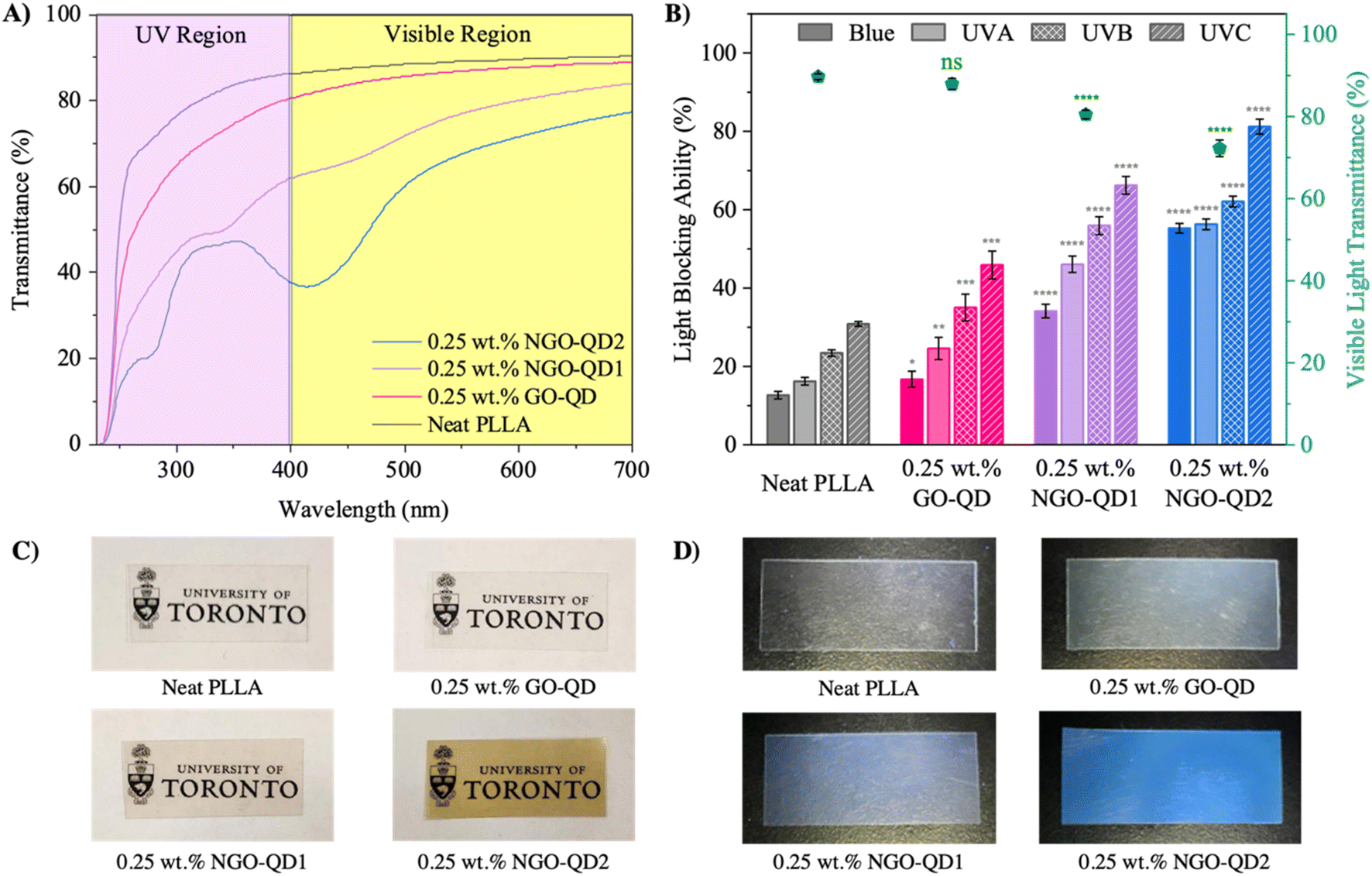 | ||
| Fig. 4 (A) UV-visible transmittance spectra of PLLA and nanocomposite films averaged over 3 replicate measurements, (B) visible-light transmittance and light blocking ability of PLLA and nanocomposite films, (C) images of PLLA and nanocomposite films illustrating their high degree of transparency, (D) images of PLLA and nanocomposite films (150 μm thickness) under exposure to 365 nm blacklight. The thickness of films used in UV-visible spectroscopy measurements is fixed at 150 μm. Blue-light, UVA, UVB, and UVC correspond to wavelength regions of 400–500, 320–400, 290–320, and 260–290 nm, respectively. Statistical analysis was conducted for light blocking ability measurements of QD-containing nanocomposites relative to the control, in each respective wavelength region. | ||
Beyond QD chemical composition, the QD concentration in polymers is known to alter the light absorption properties of QD-containing nanocomposites. As such, we highlight the role of QD concentration on the VLT and light blocking ability in Fig. S1D and E, with discussion in the ESI.†
An important aspect to consider in the VLT of semi-crystalline polymer nanocomposites is the nanocomposite crystallinity. As such, DSC 1st heating thermograms of the fabricated film specimens used for UV-visible spectroscopy analysis are illustrated in Fig. S2.† These reveal a low χc in compression molded films (2.0, 6.3, 6.7, and 0.5% for neat PLLA, and nanocomposite films containing GO-QD, NGO-QD1, and NGO-QD2, respectively), owing to the rapid quenching process used during film preparation.
It is known that solvent- or matrix-QD interactions can greatly affect UV-visible absorption wavelength owing to the sensitivity of electron–hole transitions to the QD surface state.72 Typically, the presence of hydrogen bonds (H-bonds) that reduce the energy barrier to electronic excitation and/or stabilize the excited electronic state may translate to an absorption red-shift.73 As such, FTIR was conducted on neat PLLA and nanocomposite films to probe the intensity of H-bonding interactions in prepared films, with FTIR spectra shown in Fig. S3.† No obvious changes in H-bonding or covalent bonding interactions can be observed in the presence of the QDs, with all spectra appearing like the neat PLLA control. This may be attributed to the lack of mobility of the PLLA chains, being in the glassy state at room temperature, unlike more mobile solvents/matrices that can undergo larger-scale molecular conformational changes under varying chemical environments, that affect a change in the UV-visible spectra of nanocomposite films.
 | ||
| Fig. 5 Calorimetric (A) cooling thermograms, (B) crystallization enthalpies, and (C) heating thermograms obtained after each cooling treatment. Cooling rates are provided in each respective graph. The presented calorimetric data is shifted by an arbitrary value for each respective specimen for easy viewing. | ||
Interestingly, the second heating curves exhibit trends mirroring the cooling curves. The neat PLLA exhibits a broad and prominent cold-crystallization exotherm during heating following cooling at all cooling rates tested. The peak melting temperatures following cooling at rates of 20, 10, 5, and 2 °C min−1 were 167.3, 168.4, 168.9, and 169.4 °C, with all values being rather similar. In contrast, the subsequent reheating curves of the QD-nanocomposites exhibit noticeable cold crystallization peaks after rapid cooling at rates of 20 and 10 °C min−1, with this cold crystallization behaviour eliminated after cooling at rates of 2 and 5 °C min−1. This confirms the strong nucleating effect observed in the presence of the QDs. However, the melting endotherms of the QD-nanocomposites all exhibit consistently reduced melting temperature relative to the neat PLLA, regardless of the previous cooling rate. This suggests either a reduction in crystal perfection or crystal size.76 Moreover, the melting endotherms present in QD-nanocomposites consistently exhibit a bimodal peak distribution, suggesting two distinct crystal populations. Typically, the high-melting temperature endotherm region indicates the melting of PLLA's α crystal polymorph (Tm,α) while the low-melting temperature region indicates the melting of PLLA's disordered α′ crystal polymorph (Tm,α′).77–79 While the α crystal polymorph exists with a pseudo-orthorhombic unit cell (lattice parameters: a = 1.07 nm, b = 0.595 nm, and c = 2.78 nm with a 10/3 helical conformation), the conformationally disordered α′ crystal polymorph typically possesses a slightly looser unit cell (lattice parameters: a = 1.08 nm, b = 0.62 nm, and c = 2.88 nm with a 10/3 helical conformation).80,81 Despite the similar lattice parameters and thus cell volumes, a reduction in the size of ordered domains along the c-axis lessens the thermal stability of α′ crystals relative to α crystals.82,83 Owing to the nucleating effect of the QDs observed under these cooling conditions, it is hypothesized that the presence of QDs can nucleate PLLA crystals, imparting finer and more abundant crystals possessing lower melting temperature.
In general, as the cooling rate decreases, the proportion of the Tm,α′ peak appears to increase, indicating an obstruction of the packing of the α crystals, which yields conformationally disordered α′ crystals. Additionally, the Tm,α appears to decrease suggesting a concomitant decrease in size/order of the α crystal polymorph. As such, the reduction in Tm of both modalities relative to the Tm of neat PLLA suggests obstruction of lamellar packing structure, while the increase in proportion of the conformationally disordered α′ crystals suggest obstruction of the degree of order along the crystal's c-axis. Here, we hypothesize that the reduction in Tm of both crystal modalities is attributed to QDs disrupting the lamellar spacing of the PLLA crystals, which we refer to as inter-lamellar defects. In contrast, we hypothesize that given the size-scale of the dots, which are along the same length-scale of crystal nuclei, it is plausible that some dots may distort the c-axis order of the PLLA crystals, translating to the more dominant proportion of α′ crystals formed at slow cooling rates. Specifically, at slow cooling rates (i.e., 2 °C min−1) crystallization occurs at a lower degree of supercooling (i.e., higher temperature), where nucleation behaviour is dominated by heterogeneous nucleation effects, giving rise to so-called intra-lamellar defects. In fact, the observation of a bimodal melting endotherm in QD-nanocomposites reinforced with our GO-QDs aligns well with observations from Xu et al., who observed similar phenomena. This was attributed to the heterogeneous nucleation effect of their QDs, which induced a higher crystal nucleation density, and thereby translated to more abundant, but less ordered crystals.84
To better illustrate this phenomenon, polarized optical microscopy (POM) was conducted in isothermal mode, with neat PLLA and QD-nanocomposites isothermally crystallized at 130 °C for 30 min (see Fig. 6). It should be noted that non-isothermal crystallization treatment yields crystal sizes that are in fact too fine to clearly illustrate these effects visually. Here, 130 °C was selected to promote the formation of predominantly α crystals,78,85–88 to prove the presence of inter-lamellar defects. After 3 min, neat PLLA exhibits a low nucleation density, in contrast to the GO-QD nanocomposite which exhibits a distinctly higher nucleation density. Moreover, it appears that the role of N-doping on crystal nucleation exhibits a similar trend as is consistent with calorimetric analysis. Interestingly, the QDs impart a birefringence transition from well-ordered, radially oriented crystal lamella in the neat PLLA, to more random birefringence, confirming that the presence of the QDs imparts a greater degree of disorder to crystals. Additionally, the presence of the QDs shrouds the crystal grain boundaries, further confirming that QD inclusion can induce crystallographic disorder.
 | ||
| Fig. 6 Polarized optical microscopy images illustrating isothermal crystallization of (A) neat PLLA, (B) 0.25 wt% GO-QD, (C) 0.25 wt% NGO-QD1, and (D) 0.25 wt% NGO-QD2, at 130 °C. | ||
 | ||
| Fig. 7 (A) Average tensile strength, (B) average tensile modulus, and (C) average tensile toughness of neat PLLA and QD-nanocomposites. | ||
Nanofiller inclusion is known to alter blend mechanical properties relative to non-filled polymer matrices through changes in fracture mechanism, owing to factors like filler size and geometry, concentration, as well as the effect of fillers on matrix crystal size, degree of crystallinity, and interactions with the matrix's amorphous phases. For the case of semi-crystalline polymer matrices, improvements in mechanical properties are generally due to nanofiller-induced crystallization.89–91 As such, we aim to understand the origins of these fracture mechanism changes, by decoupling the effects of crystallization and intrinsic filler inclusion on the mechanical property improvements achieved. Hence, a low mold temperature of 65 °C was selected for injection molding to suppress crystallization. Calorimetric analysis was conducted to confirm if the observed improvements in mechanical properties are attributed to changes in matrix crystallinity/crystal size. The calorimetric 1st heating curves of tensile test specimens are shown in Fig. S4.† There are minimal differences in the glass transition temperature, cold crystallization exotherm, and melting endotherm of the neat PLLA and its nanocomposites with GO-QD derivatives, suggesting that the inclusion of the QDs plays a minimal role in the thermal properties of these polymer nanocomposites under the selected molding conditions. Moreover, the χc remains low, being 4.6, 5.5, 6.4, and 4.8% for the neat PLLA and nanocomposites containing GO-QD, NGO-QD1, and NGO-QD2, respectively. As such, the reinforcing effects induced by QD inclusion are likely owing to other factors unrelated to crystallization. Interestingly, the modulus, strength and toughness of QD-nanocomposites appears sensitive to the degree of N-doping in QDs. Given experimental evidence of increased apparent particle size due to QD–QD interactions with an increasing degree of N-doping, it is possible that increasing apparent particle size may promote craze nucleation, as the craze nucleation ability of reinforcing fillers are a function of particle size.92 This is plausible given the progressive increase in toughness with increasing N-doping degree in QD-containing nanocomposites shown in Fig. 7C.
Fracture surfaces of the neat PLLA and QD-reinforced nanocomposites are shown in Fig. 8 and reveal increased craze density with the presence of NGO-QDs. Crazing is considered a mode of cohesive failure,93 and is characteristic of failure in unmodified, linear PLLA.94 In detail, crazing is a mode of energy absorption in brittle polymer matrices, with craze nucleation, void opening, and local plastic deformation causing the formation of fibrils that bridge micro-cracks, thereby supporting the load applied to the polymer.93,95,96 As such, an increase in craze density typically translates to improvements in strength and toughness owing to local plastic deformation inducing bulk plasticity, as can be observed in the stress–strain behavior of our nanocomposites (see raw data in Fig. S5†). Although reinforcing mechanisms like crack bridging, crack deflection, crack pinning and filler pull-out are possible reinforcing mechanisms in polymer nanocomposites,97–99 it is challenging to discern the extent of these failure modes owing to the fine zero-dimensional nature of the QDs.
 | ||
| Fig. 8 Fracture surface of tensile dogbone specimens corresponding to (A) neat PLLA, (B) 0.25 wt% GO-QD, (C) 0.25 wt% NGO-QD1, and (D) 0.25 wt% NGO-QD2. | ||
While similar improvements in mechanical properties have been reported in PLLA nanocomposites reinforced with coffee ground-derived QDs, both amorphous and crystalline phases were deemed responsible for improvements in mechanical properties, with these dots playing a significant role in altering blend crystal morphology and degree of crystallinity.47 Given the negligible role of the crystalline fraction in our work's nanocomposite mechanical properties (see Table S4†), the reinforcement effect can be linked to the rise in crazing density observed in NGO-QD-reinforced samples when compared to the neat PLLA. This implies that the presence of NGO-QD derivatives contributes to the initiation of crazing nucleation in PLLA. It indicates that the apparent increased particle size of NGO-QD derivatives may function as heterogeneities within the PLLA, resulting in a tougher response by initiating modes of non-uniform deformation in the shape of localized shear bands or dilatation (crazing).100 In fact, Karimi et al. reported similar phenomena with QDs imparting a stiffening, strengthening, and toughening effect in an epoxy-based matrix.101 In this case, improvements in mechanical properties were partially attributed to the interactions of the dots with the resin during curing, but also owing to the fine size of the dots (<5 nm) that fill cavities in the matrix free volume.101
 | ||
| Fig. 9 (A) Complex viscosity of neat PLLA and nanocomposites containing the prepared GO-QD derivatives, measured at 180 °C at 5% strain. (B) Storage (G′) and loss moduli (G′′) obtained from shear oscillatory rheological measurements. Solid symbols denote G′ values, while hollowed symbols denote G′′ values. | ||
For the nanocomposite melts, the observed lubricating effect appears dependent on QD composition. Here, it is hypothesized that while self-passivation improves UV- and select visible-light absorption behaviors, the lack of external passivation agent (i.e., NaOH) makes these dots more prone to self-interactions between nucleophilic N-containing functional groups and electrophilic carbonyl-groups. In contrast, the GO-QD and NGO-QD1 have a lesser tendency to undergo these QD–QD interactions, thereby increasing the interfacial area that contributes to hydrodynamic slip effects. From our spectroscopic analyses, NaOH passivation yields abundant carboxylate groups; these may promote dispersion, minimize agglomeration tendencies, and thus promote hydrodynamic slip. We believe this repulsive behavior translates to further interfacial slip, thereby imparting the most pronounced lubricating effects. Interestingly, this trend is comparable to that observed for nanocomposite crystallization kinetics, and VLT observations, suggesting that the role of passivation is critical in engineering dots with tailored properties.
Additionally, the storage and loss moduli of neat PLLA and QD-containing nanocomposite melts show similar behavior, with the storage (G′) and loss moduli (G′′) of QD-containing nanocomposite melts decreasing analogously to their complex viscosity. To better understand the rheological behavior observed, we estimate the relaxation time (λ) of the melt by the well-accepted relation, λ = 1/ωc, as has been defined previously,107 where ωc is the crossover frequency of the G′ and G′′, as can be found in Fig. 9B. Neat PLLA and NGO-QD2 exhibit relaxation times of 4.9 ms (ωc = 204.5 rad s−1), and 2.2 ms (ωc = 445.8 rad s−1), respectively. In contrast, melts containing GO-QD and NGO-QD1 exhibit no crossover frequency, suggesting that these nanocomposite melts possess predominantly viscous characteristics over their entire frequency range. These findings support the lubricating effect of the QDs that has been previously elucidated. Additionally, these findings suggest that the passivation approach applied to QDs during synthesis has implications on the QD–QD interaction tendency when incorporated into a polymer matrix, with passivation via N-doping imparting a higher QD–QD interaction tendency than partial or complete passivation with NaOH.
3. Conclusion
We designed GO-QD derivatives possessing N-doping degrees of 0, 0.20, and 0.32, to uncover the functional groups needed to fabricate QD-nanocomposites achieving target bulk properties. It was found that an increase in N-containing functional groups increases nanocomposite UV- and blue-light absorption, with both pyrrolic-N and pyridinic-N groups imparting strong UVB/UVA absorption, and red-shifted graphitic-N imparting dominant blue-light absorption, with a minimal reduction in VLT (<20%). This red-shift is attributed to delocalization of the excited electrons in the carbon framework of the QDs. In contrast, the inclusion of solely O-containing functional groups in fact provides only slight UV-absorption in the UVB/UVC region, owing to a higher barrier to less favourable electronic transitions. This selective UV-visible absorption behaviour yields fluorescent nanocomposites that exhibit visible-light emission (e.g., blue, green), with fluorescence emission intensity increasing similarly to UV-visible absorption behaviour. These findings open the door for PLLA's broader use in previously inaccessible applications and inform QD design decisions for similar matrices. As well, we demonstrate the role of GO-QD derivatives in modifying the crystallization behaviour of PLLA. We found that these QDs act as heterogeneous nucleating agents, regardless of their composition. Moreover, the nanometric dimensions of the fabricated GO-QD derivatives may act as inter-lamellar defects in PLLA crystals, disrupting the lamellar spacing as was marked by a reduction in Tm observed by DSC, as well as marked changes in birefringence observed by POM. Additionally, QDs may act as intra-lamellar defects, thereby preferentially promoting the formation of PLLA's conformationally disordered α′ crystals. Beyond this, we demonstrate the role of GO-QD derivatives in modifying the mechanical properties of PLLA, with QDs facilitating crazing in PLLA, which leads to up to a 43% increase in tensile toughness with concomitant increases in stiffness and strength. Finally, we demonstrate that the complex viscosity of PLLA decreases by an order of magnitude in the presence of QDs, a most probable result of hydrodynamic slip related to the passivation applied to each QD formulation. These findings lay the framework for the future design of QDs for nanocomposites with tailored processability and bulk properties.4. Materials and methods
4.1 Materials
A high viscosity grade of PLLA (Mw: 2.1 × 105 g mol−1, Mn: 1.2 × 105 g mol−1, 2% D-isomer content, MFR: 7 g/10 min at 210 °C with 2.16 kg) was kindly supplied from Natureworks LLC, under the trade name of PLLA 4032D.89,102 Anhydrous citric acid (CA, Reagent grade, Mw: 192.13 g mol−1) was purchased from BioShop Canada Inc. As well, urea (BioUltraPure Molecular Biology Grade, purity 99.5%) was obtained from BioShop Canada Inc. Additionally, sodium hydroxide (NaOH) pellets were purchased from Caledon Laboratory Chemicals. Dichloromethane (Certified ACS Stabilized, purity >99.5%) and acetone (Certified ACS, purity >99.5%) were obtained from Fisher Chemical. Anhydrous ethanol was obtained from Commercial Alcohols (Greenfield Global). All materials were used as received without further purification.4.2 Synthesis of graphene oxide-quantum dots
The synthetic procedure used for fabricating the GO-QDs in this work was adapted from Dong et al., using CA as a precursor.40 First, 2 g of CA was added to a 250 mL round bottom flask, which was placed in a silicon oil bath maintained at 200 °C for 30 min, under vigorous stirring. As was reported by Dong et al.,40 the CA melted within 5 min. After this, the liquid turned pale yellow, and then orange. An aqueous solution of NaOH (10 mg mL−1) was then added slowly under vigorous stirring, to passivate and disperse the heat-treated material. Once neutralized to a pH of 7, the solution was transferred to centrifuge tubes and subjected to centrifugation for 10 min at 4000 rpm to allow large sediment particles to settle. The supernatant was recovered and filtered through a 0.22 μm syringe filter, followed by filtration through a 0.02 μm syringe filter to ensure that only particles below 0.02 μm were collected.108 The doubly filtered solution was then stored in a freezer for 24 h, and subject to lyophilization (FreeZone 2.5 Plus from LABCONCO) to isolate the GO-QDs. The isolate powder was dried at 60 °C for 24 h to remove residual moisture and was ground by mortar and pestle to promote dispersion in solution.4.3 Synthesis of N-doped graphene oxide-quantum dots
NGO-QDs were fabricated by slight modification of the GO-QD synthetic approach, substituting CA with urea, a well-reported nitrogen source that can effectively dope CA-based GO-QDs.50,68 Here, molar ratios of 3:1 (CA:urea), and 1:3 (CA:urea) were used to illustrate how differences in composition affect matrix–filler interactions. In short, CA and urea were charged to a round-bottom flask in their desired molar ratios and were subject to incomplete carbonization at 200 °C for 30 min, under vigorous stirring. Within the first minute, the materials were melted, followed by a rapid colour change from clear, to a very dark green. After 30 min, each solution was diluted to passivate and disperse the QDs. Specifically, the QDs prepared from the 3:1 (CA:urea) precursor mixture were passivated by slowly adding an aqueous solution of NaOH (10 mg mL−1) under vigorous stirring to achieve a pH of 7. In contrast, the QDs prepared from the 1:3 molar ratio of CA:urea were self-passivated, with no need for passivation using NaOH; this mixture was diluted with DI water. The QD-containing solutions were then separately transferred to centrifuge tubes and subject to centrifugation for 10 min at 4000 rpm to allow large sediment particles to settle. The supernatant was recovered and subject to identical isolation procedures to the CA-derived dots. QDs prepared with 1:0, 3:1, and 1:3 precursor molar ratios (CA:urea) are denoted as GO-QD, NGO-QD1, and NGO-QD2, respectively. It should be noted that all QDs fabricated likely possess trace amounts of unbound molecular species (e.g., molecular fluorophores) that improve QD optical properties, as discussed elsewhere.57 A detailed rationale for the selection of the QD precursor compositions is provided in the ESI.†
4.4 Solution blending
The synthesized GO-QD derivatives were solution-blended into the PLLA matrix at a concentration of 0.25 wt%. Neat PLLA was prepared analogously, as a control. The detailed solution blending approach can be found in the ESI file.† Nanocomposites containing GO-QD, NGO-QD1, and NGO-QD2 are denoted as 0.25 wt% GO-QD, NGO-QD1, and NGO-QD2, respectively. Details regarding material characterization methods can also be found in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Anthony V. Tuccitto: conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review & editing, Rafaela Aguiar: conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review & editing, Nello D. Sansone: methodology, investigation, writing – review & editing, Yalda Chehrehsaz: formal analysis, Boran Kumral: methodology, writing – review & editing, Peter Serles: methodology, writing – review & editing, Tobin Filleter: writing – review & editing, Chul B. Park: supervision, Patrick C. Lee: conceptualization, writing – review & editing, funding acquisition, project administration, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC) [Discovery Grant RGPIN-2019-05778] for their financial support of this project. We kindly thank Ilya Gourevich for assistance and technical support.References
- M. Ilyas, W. Ahmad, H. Khan, S. Yousaf, K. Khan and S. Nazir, Rev. Environ. Health, 2018, 33, 383–406 CrossRef PubMed.
- L. Wang, R. E. Lee, G. Wang, R. K. M. Chu, J. Zhao and C. B. Park, Chem. Eng. J., 2017, 327, 1151–1162 CrossRef CAS.
- K. Li, Y. Wang, M. Rowe, X. Zhao, T. Li, H. Tekinalp and S. Ozcan, ACS Appl. Polym. Mater., 2020, 2, 411–417 CrossRef CAS.
- A. Anstey, A. Tuccitto, C. Park and P. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 14422–14434 CrossRef CAS.
- A. Reza Monfared, A. V. Tuccitto, H. Omranpour, S. Sakib Rahman, A. Zaoui, A. Salehi, S. Rezaei, R. Rahmati, V. Lotocki, D. S. Seferos and C. B. Park, Chem. Eng. J., 2024, 496, 154181 CrossRef CAS.
- B. L. Diffey, Methods, 2002, 28, 4–13 CrossRef CAS PubMed.
- M. Narayanan, S. Loganathan, R. B. Valapa, S. Thomas and T. O. Varghese, Int. J. Biol. Macromol., 2017, 99, 37–45 CrossRef CAS PubMed.
- B. Mahltig, H. Böttcher, K. Rauch, U. Dieckmann, R. Nitsche and T. Fritz, Thin Solid Films, 2005, 485, 108–114 CrossRef CAS.
- W. Y. Wong, J. K. C. Lam, C. W. Kan and R. Postle, Text. Prog., 2016, 48, 1–54 CrossRef.
- J. W. Ha, K. H. Back, Y. H. Kim and D. H. Kang, Food Microbiol., 2016, 57, 172–177 CrossRef CAS PubMed.
- C. Kittas, M. Tchamitchian, N. Katsoulas, P. Karaiskou and C. Papaioannou, Sci. Hortic., 2006, 110, 30–37 CrossRef.
- Y. Kameda, K. Kimura and M. Miyazaki, Environ. Pollut., 2011, 159, 1570–1576 CrossRef CAS PubMed.
- Y. Man, L. Mu, Y. Wang, S. Lin, G. L. Rempel and Q. Pan, Polym. Polym. Compos., 2015, 36, 1–203 CrossRef.
- S. Thasak, C. A. Arellano, T. Khumsap and L. T. Nguyen, Int. J. Food Eng., 2023, 19, 235–245 CrossRef CAS.
- A. E. Kellie and S. S. Zilva, Biochem. J., 1938, 32, 1561–1565 CrossRef CAS.
- B. T. Smith, S. Belani and A. C. Ho, Int. Ophthalmol. Clin., 2005, 45, 107–115 Search PubMed.
- ASTM D6400 - Standard Specification for Labeling of Plastics Designed to be Aerobically Composted in Municipal or Industrial Facilities, ASTM Int., 2023, vol. 3, DOI:10.1520/D6400-23.
- Y. Kim, J. Suhr, H. W. Seo, H. Sun, S. Kim, I. K. Park, S. H. Kim, Y. Lee, K. J. Kim and J. Do Nam, Sci. Rep., 2017, 7, 1–11 CrossRef.
- K. M. Lim, K. Z. Chiew, S. Y. Pung and W. S. Chow, Mater. Today: Proc., 2019, 17, 853–863 CAS.
- M. Nofar, A. Tabatabaei and C. B. Park, Polymer, 2013, 54, 2382–2391 CrossRef CAS.
- F. Yu, T. Liu, X. Zhao, X. Yu, A. Lu and J. Wang, J. Appl. Polym. Sci., 2012, 125(S2), E99–E109 CrossRef CAS.
- L. Suryanegara, A. N. Nakagaito and H. Yano, Compos. Sci. Technol., 2009, 69, 1187–1192 CrossRef CAS.
- L. Xu, J. Zhao, S. Qian, X. Zhu and J. Takahashi, Compos. Sci. Technol., 2021, 203, 108613 CrossRef CAS.
- A. R. Kakroodi, Y. Kazemi, M. Nofar and C. B. Park, Chem. Eng. J., 2017, 308, 772–782 CrossRef CAS.
- J. M. Campos, A. M. Ferraria, A. M. Botelho Do Rego, M. R. Ribeiro and A. Barros-Timmons, Mater. Chem. Phys., 2015, 166, 122–132 CrossRef CAS.
- N. F. Zaaba, M. Jaafar and H. Ismail, Polym. Eng. Sci., 2021, 61, 22–38 CrossRef CAS.
- A. R. Monfared, H. Omranpour, A. V. Tuccitto, A. Zaoui, S. S. Rahman, M. Kheradmandkeysomi, A. Jalali and C. B. Park, ACS Sustain. Chem. Eng., 2024, 12, 13017–13029 CrossRef CAS.
- M. Baiardo, G. Frisoni, M. Scandola, M. Rimelen, D. Lips, K. Ruffieux and E. Wintermantel, J. Appl. Polym. Sci., 2003, 90, 1731–1738 CrossRef CAS.
- D. Arthisree and G. M. Joshi, J. Mater. Sci.:Mater. Electron., 2017, 28, 10516–10524 CrossRef CAS.
- Z. Ji, Z. Yin, Z. Jia and J. Wei, Langmuir, 2020, 36, 8632–8640 CrossRef CAS PubMed.
- W. Chen, G. Lv, W. Hu, D. Li, S. Chen and Z. Dai, Nanotechnol. Rev., 2018, 7, 157–185 CrossRef CAS.
- Y. Luo, J. Li, Z. Gu and Y. Huang, Sci. Rep., 2023, 13, 1–9 CrossRef.
- Y. Chen, Y. Liu, J. Li, C. Zhao, Y. Yang, L. He, M. Wu, Q. Wang, Y. Yang, C. Wang, D. Zhou and J. Qiu, Chem. Eng. J., 2024, 481, 148600 CrossRef CAS.
- F. Chen, Q. Li, M. Li, F. Huang, H. Zhang, J. Kang and P. Wang, Chem. Eng. J., 2021, 411, 128350 CrossRef CAS.
- M. K. Kumawat, M. Thakur, R. B. Gurung and R. Srivastava, Sci. Rep., 2017, 7, 1–16 CrossRef CAS.
- H. Liu, X. Lv, J. Qian, H. Li, Y. Qian, X. Wang, X. Meng, W. Lin and H. Wang, ACS Nano, 2020, 14, 13304–13315 CrossRef CAS PubMed.
- C. Wang, X. Wang, J. Yu and B. Ding, ACS Nano, 2023, 17, 10888–10897 CrossRef CAS.
- V. Biju, T. Itoh, A. Anas, A. Sujith and M. Ishikawa, Anal. Bioanal. Chem., 2008, 391, 2469–2495 CrossRef CAS PubMed.
- L. C. Spangler, L. Lu, C. J. Kiely, B. W. Berger and S. McIntosh, J. Mater. Chem. A, 2016, 4, 6107–6115 RSC.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- S. Cailotto, E. Amadio, M. Facchin, M. Selva, E. Pontoglio, F. Rizzolio, P. Riello, G. Toffoli, A. Benedetti and A. Perosa, ACS Med. Chem. Lett., 2018, 9, 832–837 CrossRef CAS PubMed.
- T. C. Wareing, P. Gentile and A. N. Phan, ACS Nano, 2021, 15, 15471–15501 CrossRef CAS PubMed.
- V. B. Kumar, S. K. Mirsky, N. T. Shaked and E. Gazit, ACS Nano, 2024, 18, 2421–2433 CrossRef CAS PubMed.
- S. Agnoli and M. Favaro, J. Mater. Chem. A, 2016, 4, 5002–5025 RSC.
- K. Bin Cai, H. Y. Huang, M. L. Hsieh, P. W. Chen, S. E. Chiang, S. H. Chang, J. L. Shen, W. R. Liu and C. T. Yuan, ACS Nano, 2022, 16, 3994–4003 CrossRef.
- N. Gobi, D. Vijayakumar, O. Keles and F. Erogbogbo, ACS Omega, 2017, 2, 4356–4362 CrossRef CAS PubMed.
- H. Xu, L. Xie, J. Li and M. Hakkarainen, ACS Appl. Mater. Interfaces, 2017, 9, 27972–27983 CrossRef CAS.
- Y. Zhu, L. Yan, M. Xu, Y. Li, X. Song and L. Yin, Colloids Surf., A, 2021, 610, 125703 CrossRef CAS.
- C. Wang, Y. He, Y. Xu, L. Sui, T. Jiang, G. Ran and Q. Song, J. Mater. Chem. A, 2022, 10, 2085–2095 RSC.
- S. Gu, C. Te Hsieh, C. Y. Yuan, Y. A. Gandomi, J. K. Chang, C. C. Fu, J. W. Yang and R. S. Juang, J. Lumin., 2020, 217, 116774 CrossRef CAS.
- F. Soheilmoghaddam, G. Sharifzadeh, H. Adelnia and M. U. Wahit, J. Polym. Environ., 2022, 30, 613–621 CrossRef CAS.
- Y. Monroy, D. M. Cabezas, S. Rivero and M. A. García, Int. J. Adhes. Adhes., 2023, 126, 103470 CrossRef CAS.
- J. Chen, Z. Long, S. Wang, Y. Meng, G. Zhang and S. Nie, Int. J. Biol. Macromol., 2019, 139, 367–376 CrossRef CAS PubMed.
- Y. N. Hao, H. L. Guo, L. Tian and X. Kang, RSC Adv., 2015, 5, 43750–43755 RSC.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Sci. Rep., 2014, 4, 1–11 Search PubMed.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272–12277 RSC.
- J. D. Stachowska, A. Murphy, C. Mellor, D. Fernandes, E. N. Gibbons, M. J. Krysmann, A. Kelarakis, E. Burgaz, J. Moore and S. G. Yeates, Sci. Rep., 2021, 11, 1–14 CrossRef.
- J. R. Prekodravac, M. D. Budimir, D. N. Kleut, B. R. Vasiljević, V. B. Rajić, G. Ciasca and B. M. Todorović, Diamond Relat. Mater., 2022, 129, 109366 CrossRef CAS.
- A. Kühle, A. H. Sørensen, J. B. Zandbergen and J. Bohr, Appl. Phys. A:Mater. Sci. Process., 1998, 66, 329–332 CrossRef.
- F. Gołek, P. Mazur, Z. Ryszka and S. Zuber, Appl. Surf. Sci., 2014, 304, 11–19 CrossRef.
- C. Liu, Z. Li, T. J. Hajagos, D. Kishpaugh, D. Y. Chen and Q. Pei, ACS Nano, 2017, 11, 6422–6430 CrossRef CAS PubMed.
- J. T. Seo, J. Han, T. Lim, K. H. Lee, J. Hwang, H. Yang and S. Ju, ACS Nano, 2014, 8, 12476–12482 CrossRef CAS PubMed.
- C. Yoon, K. P. Yang, J. Kim, K. Shin and K. Lee, Chem. Eng. J., 2020, 382, 122792 CrossRef CAS.
- W. J. Peveler, A. Roldan, N. Hollingsworth, M. J. Porter and I. P. Parkin, ACS Nano, 2016, 10, 1139–1146 CrossRef CAS PubMed.
- K. Ming, J. Kim, M. J. Biondi, A. Syed, K. Chen, A. Lam, M. Ostrowski, A. Rebbapragada, J. J. Feld and W. C. W. Chan, ACS Nano, 2015, 9, 3060–3074 CrossRef CAS PubMed.
- G. Wang, Y. Leng, H. Dou, L. Wang, W. Li, X. Wang, K. Sun, L. Shen, X. Yuan, J. Li, K. Sun, J. Han, H. Xiao and Y. Li, ACS Nano, 2013, 7, 471–481 CrossRef CAS PubMed.
- Y. Dong, H. Pang, H. Bin Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS.
- T. Ogi, H. Iwasaki, K. Aishima, F. Iskandar, W. N. Wang, K. Takimiya and K. Okuyama, RSC Adv., 2014, 4, 55709–55715 RSC.
- S. Gu, C. Te Hsieh, Y. Ashraf Gandomi, J. K. Chang, J. Li, J. Li, H. Zhang, Q. Guo, K. C. Lau and R. Pandey, J. Mater. Chem. C, 2019, 7, 5468–5476 RSC.
- B. Vercelli, R. Donnini, F. Ghezzi, A. Sansonetti, U. Giovanella and B. La Ferla, Electrochim. Acta, 2021, 387, 138557 CrossRef CAS.
- S. Sarkar, M. Sudolská, M. Dubecký, C. J. Reckmeier, A. L. Rogach, R. Zbořil and M. Otyepka, J. Phys. Chem. C, 2016, 120, 1303–1308 CrossRef CAS.
- X. Sun, X. Chen, X. Li, J. Xie, X. Lin, Q. Shen, L. Wu and S. Chen, ACS Nano, 2024, 18, 5898–5906 CAS.
- H. Wang, T. Zhang, J. Zhu, Y. Zhai, H. Wang, X. Bai, B. Dong and H. Song, Nanoscale, 2017, 9, 13042–13051 RSC.
- B. Yang, D. Wang, F. Chen, L. F. Su, J. Bin Miao, P. Chen, J. S. Qian, R. Xia and J. W. Liu, J. Macromol. Sci., Part B:Phys., 2019, 58, 290–304 CrossRef CAS.
- D. V. Guseva, A. A. Lazutin and V. V. Vasilevskaya, Polymer, 2021, 221, 123577 CrossRef CAS.
- J. M. Pin, A. V. Tuccitto, M. E. Shivokhin and P. C. Lee, Polymer, 2021, 212, 123123 CrossRef CAS.
- Z. Refaa, M. Boutaous, S. Xin and D. A. Siginer, J. Therm. Anal. Calorim., 2017, 128, 687–698 CrossRef CAS.
- T. Tábi, S. Hajba and J. G. Kovács, Eur. Polym. J., 2016, 82, 232–243 CrossRef.
- M. C. Righetti, M. Gazzano, M. L. Di Lorenzo and R. Androsch, Eur. Polym. J., 2015, 70, 215–220 CrossRef CAS.
- T. Okihara, M. Tsuji, A. Kawaguchi, K. I. Katayama, H. Tsuji, S. H. Hyon and Y. Ikada, J. Macromol. Sci., Part B:Phys., 1991, 30, 119–140 CrossRef CAS.
- H. Wang, J. Zhang and K. Tashiro, Macromolecules, 2017, 50, 3285–3300 CrossRef CAS.
- K. Wasanasuk and K. Tashiro, Polymer, 2011, 52, 6097–6109 CrossRef CAS.
- M. L. Di Lorenzo and R. Androsch, Synthesis, Structure and Properties of Poly(lactic Acid), Springer, 2017, vol. 279 Search PubMed.
- H. Xu, K. H. Adolfsson, L. Xie, S. Hassanzadeh, T. Pettersson and M. Hakkarainen, ACS Sustain. Chem. Eng., 2016, 4, 5618–5631 CrossRef CAS.
- H. Simmons, P. Tiwary, J. E. Colwell and M. Kontopoulou, Polym. Degrad. Stab., 2019, 166, 248–257 CrossRef CAS.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS.
- J. Zhang, Y. Duan, H. Sato, H. Tsuji, I. Noda, S. Yan and Y. Ozaki, Macromolecules, 2005, 38, 8012–8021 CrossRef CAS.
- P. Pan, B. Zhu, W. Kai, T. Dong and Y. Inoue, J. Appl. Polym. Sci., 2008, 107, 54–62 CrossRef CAS.
- A. V Tuccitto, A. Anstey, N. D. Sansone, C. B. Park and P. C. Lee, Int. J. Biol. Macromol., 2022, 218, 22–32 CrossRef PubMed.
- N. D. Sansone, Z. Razzaz, M. Salari, A. V. Tuccitto, R. Aguiar, M. Leroux and P. C. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 40232–40246 CrossRef CAS PubMed.
- A. R. Kakroodi, Y. Kazemi, W. D. Ding, A. Ameli and C. B. Park, Biomacromolecules, 2015, 16, 3925–3935 CrossRef CAS PubMed.
- A. M. Donald and E. J. Kramer, J. Appl. Polym. Sci., 1982, 27, 3729–3741 CrossRef CAS.
- S. S. Rahman, M. B. Mahmud, A. R. Monfared, P. C. Lee and C. B. Park, Compos. Sci. Technol., 2023, 236, 109994 CrossRef CAS.
- M. Razavi, S. Cheng, D. Huang, S. Zhang and S. Q. Wang, Polymer, 2020, 197, 122445 CrossRef CAS.
- T. H. Yu, Y. H. Su, H. H. Huang, H. J. Tsai and W. K. Hsu, Polym. Test., 2020, 91, 106731 CrossRef CAS.
- S. S. Rahman, M. B. Mahmud, A. Salehi, A. R. Monfared, M. A. Islam, T. Filleter, P. C. Lee and C. B. Park, Chem. Eng. J., 2024, 480, 148311 CrossRef CAS.
- C. Shuai, W. Guo, P. Wu, W. Yang, S. Hu, Y. Xia and P. Feng, Chem. Eng. J., 2018, 347, 322–333 CrossRef CAS.
- P. Feng, S. Shen, L. Yang, Y. Kong, S. Yang and C. Shuai, Virtual Phys. Prototyp., 2022, 18(1), e2115384 CrossRef.
- C. Shuai, B. Peng, P. Feng, L. Yu, R. Lai and A. Min, J. Adv. Res., 2022, 35, 13–24 CrossRef CAS PubMed.
- A. S. Argon, Pure Appl. Chem., 1975, 43, 247–272 CrossRef CAS.
- B. Karimi and B. Ramezanzadeh, J. Colloid Interface Sci., 2017, 493, 62–76 CrossRef CAS PubMed.
- X. F. Wei, R. Y. Bao, Z. Q. Cao, W. Yang, B. H. Xie and M. B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- R. Kotsilkova, S. Tabakova and R. Ivanova, Mech. Time-Depend. Mater., 2022, 26, 611–632 CrossRef CAS.
- H. Haridas and M. Kontopoulou, J. Rheol., 2023, 67, 601–619 CrossRef CAS.
- E. H. C. Ferreira, R. J. E. Andrade and G. J. M. Fechine, Macromolecules, 2019, 52, 9620–9631 CrossRef CAS.
- C. Kamal, S. Gravelle and L. Botto, Nat. Commun., 2020, 11, 1–10 CrossRef.
- F. J. Stadler, C. Piel, J. Kaschta, S. Rulhoff, W. Kaminsky and H. Münstedt, Rheol. Acta, 2006, 45, 755–764 CrossRef CAS.
- É. C. Duarte and R. L. Oréfice, J. Appl. Polym. Sci., 2021, 138, 1–14 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and methods, elemental analysis obtained from XPS, photographs of nanocomposites under blacklight excitation, visible light reflectance measurements, UV-visible spectroscopy measurements, calorimetric heating thermograms of nanocomposite films, FTIR spectra of nanocomposite films, thermal properties obtained from non-isothermal calorimetric heating and cooling, calorimetric heating thermograms of nanocomposite dogbone specimens, summary of thermal properties obtained from calorimetric analysis, stress–strain curves obtained for nanocomposite dogbones, statistical analysis. See DOI: https://doi.org/10.1039/d4ta07202b |
| This journal is © The Royal Society of Chemistry 2025 |