
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-resolution in situ photo-irradiation MAS NMR: application to the UV-polymerization of n-butyl acrylate†
Thomas J. N.
Hooper
 *,
Rodrigo
de Oliveira-Silva
and
Dimitrios
Sakellariou
*,
Rodrigo
de Oliveira-Silva
and
Dimitrios
Sakellariou
Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Leuven 3001, Belgium. E-mail: thomas.hooper@kuleuven.be
First published on 2nd December 2024
Abstract
Light plays a pivotal role in many solid-state materials technologies. High-resolution solid-state nuclear magnetic resonance (NMR) is a key tool for characterizing atomic-scale structures and dynamics in solid compounds, but requisite magic angle spinning (MAS) of the sample-containing rotors limits the measurement of these systems in their irradiated states. Here we report on a bespoke methodology for performing solid-state NMR of a sample irradiated with visible/ultraviolet (vis/UV) light, while undergoing MAS at frequencies ≤15 kHz. A fiber optic insert guides vis/UV light to the spinning NMR rotor, where a glass end cap and dispersion rod act to illuminate the sample inside the rotor. This methodology was used to follow the photopolymerization reaction of n-butyl acrylate, providing well-resolved 1H and 13C NMR assignments of the formation of a semi-solid polymer gel.
Introduction
Solid-state photochemistry describes a wide range of photon energy driven reactions of great importance to several industries. UV-cured polymerization has become commonplace in production, used for printing, coating, and additive manufacturing.1 Photodegradation is an obstacle in food science, pharmaceuticals, polymers, solar cells, and space materials.2–5 Optoelectronic semiconductors are finding use as heterogeneous photocatalysts to increase the efficiency of various reactions,6 and have long been used as light emitting diodes and photo-voltaic cells.7–9 Each of these applications is an area of active scientific research as the community searches for greener processes and energy solutions. Photochemistry is also prevalent in biological systems such as photosynthesis, skin damage and vision.10One of the most powerful tools for studying solid materials in general is nuclear magnetic resonance (NMR), which provides atomic-scale structural information of crystalline and amorphous materials alike. In addition, it can probe molecular dynamics over a range of time-scales. Magic angle sample spinning (MAS) is necessary in solid-state NMR to achieve resolution of individual frequencies of different chemical sites. It involves rotating the sample at an angle of 54.7° from the static magnetic field and acts to reduce the NMR line broadening which results from anisotropic interactions. Methodology development towards in situ solid-state NMR harnesses the power of NMR for elucidating complex processes and reactions in solid materials, resulting in interesting bespoke systems tailored for studying specific processes.11–15 Photochemical reactions are often fast and reversible, hence the ability to perform MAS NMR measurements while irradiating the sample with UV/vis light in situ has great utility.
There are existing reports of in situ photo-irradiation MAS NMR. Starting in 1977, D. Raftery, et al. utilized a home-built system to perform in situ photo-irradiation solid-state NMR of various photo-catalytic materials.16–24 Their system utilized a quartz rod to guide light through a 10 mm gap in the RF coil onto the side of a 5 mm glass tube which contained the sample. Hence, a bespoke RF coil was required, and MAS frequencies were limited to <2.7 kHz. In 2004, M. Hunger, et al. developed an impressive system which modified a 7 mm Bruker MAS probe with an injection system allowing in situ continuous flow reaction MAS (3 kHz) NMR measurements.25 The bottom of the rotor was replaced with a quartz glass window, allowing in situ photo irradiation supplied by a fiber optic fed through the bottom of the stator. The photoreaction pathways in retinal-binding photoreceptor proteins have received much attention with nascent photo-irradiation MAS NMR techniques. Over the last 30 years, multiple groups have studied photoreceptor proteins using home-built in situ irradiation setups which guided light through the MAS stator (perpendicular to the rotor) to shine light onto the sides of the rotor walls, achieving sufficient light penetration from thin wall zirconia rotors,26,27 or by using specially designed transparent rotors.28–32 The advent of photo-chemically induced dynamic nuclear polarization (photo-CIDNP) in solid-state, a hyperpolarization technique where nuclear polarization is created by the evolution of spin-correlated radical pairs, has also resulted in multiple groups utilizing photo-irradiation MAS NMR setups.33–38 The majority of these used DNP MAS probes, where light was directed through the existing microwave waveguide onto the sides of clear sapphire rotors spinning at 8 kHz. Bruker states a maximum MAS frequency for its 3.2 mm sapphire rotors of 12 kHz.39 In 2011, A. Naito, et al. developed an in situ irradiation MAS NMR setup which irradiated the sample from inside the rotor. The end cap of a 4 mm Varian pencil rotor was swapped for a tapered glass rod, attached with glue. The narrow end of the glass rod extended into the rotor, allowing dispersal of light into the sample space. The light was supplied by an unattached fiber optic cable directed onto the exposed end of the glass rod. Their system was also used to study photoreceptor proteins, with a maximum MAS frequency of 4 kHz throughout.40–44
Results and discussion
We have developed upon the design of A. Naito, et al., adapting an existing solid state NMR probe and rotors to create a bespoke setup for in situ irradiation MAS NMR. This setup uses low-cost and easy-to-produce parts for adapting existing MAS NMR probes, therefore not requiring coil modification, DNP probes, or specialized rotors. It can achieve stable MAS frequencies of up to 15 kHz, in an arrangement that allows facile rotor (re)packing. In addition, any fiber-coupled light source can be integrated into the setup. The traditional end-cap of a Varian 4 mm pencil rotor is replaced with a tapered glass rod (Fig. 1a), manufactured via the KU Leuven Campus Glasblazerij. Quartz glass cylinders of 3 mm diameter were stretched into shape before cutting and polishing. The widest end of the glass rod fits tightly into the rotor with the long tapered section extending into the rotor sample space. A PTFE insert with a small divot holds the narrow end of the glass rod to ensure stability under spinning conditions. The glass rod is held in place by friction alone, not requiring glue, allowing unhindered repacking of this “windowed” rotor and minimizing 1H rotor background. The tapered glass rod acts to disperse the light from a fiber optic cable into the sample, which is packed around the sides of the light dispersion rod (Fig. 1a). A 4 mm MAS HXY Chemagnetics probe was minimally adapted with a home-built mechanical part to guide the fiber-optic to the rotor, as shown in Fig. 1b and c. The part was 3-D printed in 3DM-ABS resin and attached with BeCu shielding to minimize external RF penetration inside the MAS probe. An utensil holds the end of the fiber optic cable 2 mm from the exposed end of the glass rod within the rotor when placed in the probe stator at the magic angle. To achieve this angle the fiber optic cable is bent out of the top the probe, and then fed back through the length of the probe for connection to a Thorlabs fiber optic coupled LED held at a distance of 2 m from the actively shielded magnet, as demonstrated in Fig. 1c. The fiber optic holding utensil can be partially retracted from the top of the probe, allowing easy rotor removal for sample repacking. The utensil is not in contact with the stator and therefore has no effect on the calibration of the magic angle. The 3DM-ABS utensil has a glass transition temperature of ∼100 °C, limiting the temperature range of this prototype, but 3D printing resins with a higher temperature resistance could be used if necessary.45 Further experimental details can be found in the ESI.†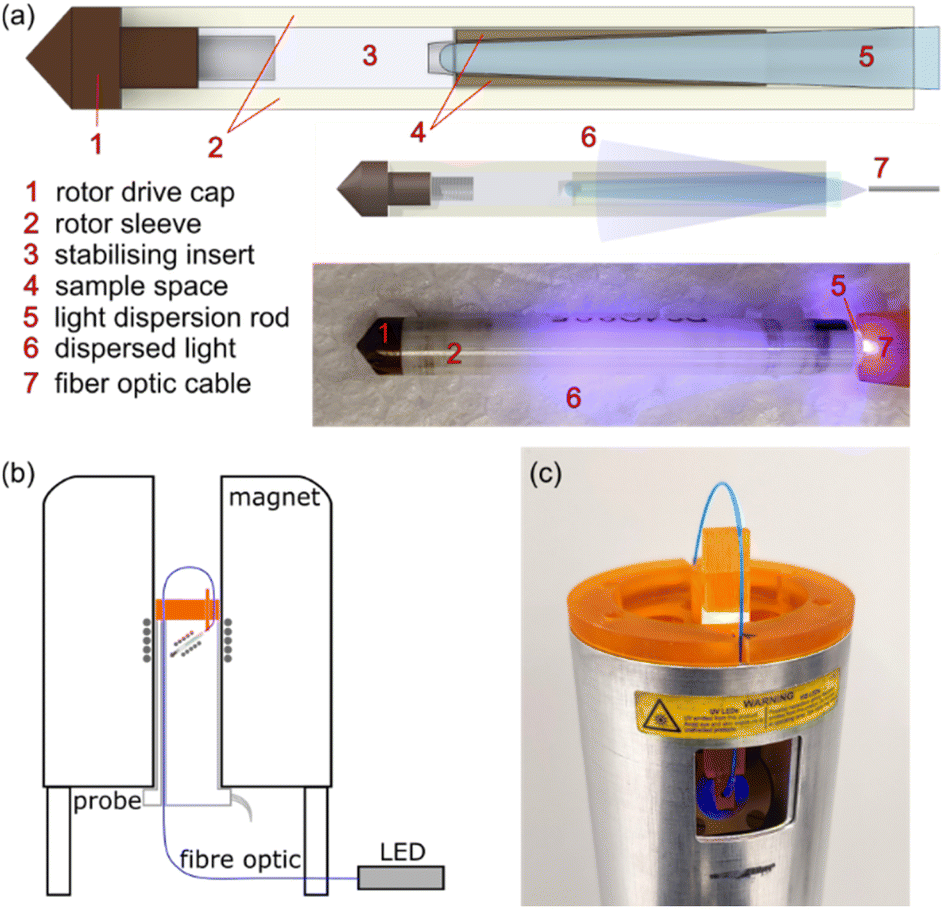 | ||
| Fig. 1 (a) Drawings of the windowed rotor for in situ irradiation and a photo of the adapted rotor while under irradiation with blue light. (b) Schematic of the arrangement of the adapted probe/rotor and the fiber optic cable connected to a light source. (c) Photo of the 3D printed probe-head with fiber optic alignment utensil while the rotor is irradiated with blue light. | ||
To test the in situ photo-irradiation MAS NMR setup, the UV-initiated polymerisation reaction of n-butyl acrylate was chosen. The reaction is demonstrated in Scheme 1. The reactants and products of the reaction are transparent allowing irradiation of a maximally filled sample space and the progress of the reaction can be easily tracked via1H NMR, allowing high NMR sensitivity. Samples were prepared by first passing the liquid n-butyl acrylate (nBA; 98+%) (Alfa Aesar) through an Al2O3 filter, to remove the ∼50 ppm of 4-methoxyphenol which is included to inhibit polymerisation. Then 0.5 wt% of powdered benzoin methyl ether (BME; 97%) was dissolved into the filtered nBA liquid. Irradiation at 365 nm causes photolysis of the BME initiator resulting in two radical products. The radicals open the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the nBA monomer, resulting in another radical, which can react with more monomer. As such, the reaction propagates producing polymer chains, until the reaction terminates via combination with other polymer chains, disproportionation, or, less commonly, reaction with a primary radical.46
C bond of the nBA monomer, resulting in another radical, which can react with more monomer. As such, the reaction propagates producing polymer chains, until the reaction terminates via combination with other polymer chains, disproportionation, or, less commonly, reaction with a primary radical.46
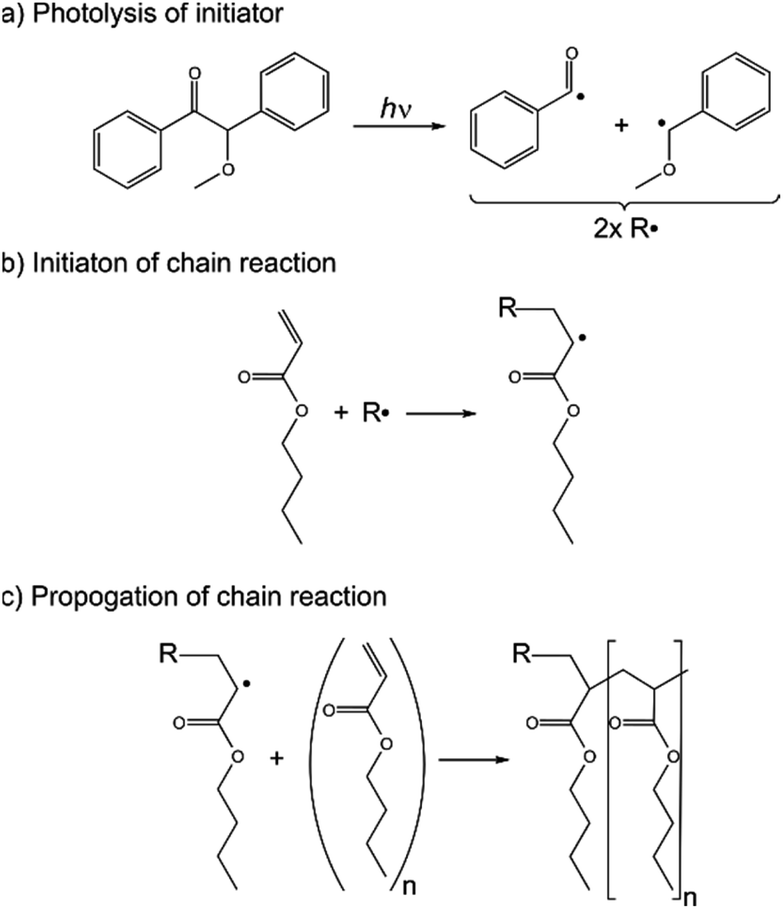 | ||
| Scheme 1 Polymerization reaction of UV-initiator benzoin methyl ether and monomer n-butyl acrylate. | ||
The liquid reactants were packed into the rotor to the sides of the light dispersal rod, which was sealed with the insert and drive cap and then placed in the stator of the probe. The fiber optic position was adjusted for good alignment with the light dispersion rod. A fiber-coupled LED provides UV light with a wavelength of 365 nm with variable radiant power; the radiant power at the fiber optic output ranges from 0–13.3 mW which is linearly dependent on the driving current of the LED.
To test the irradiation efficacy static 1H NMR measurements were acquired at different time points during continuous irradiation of 365 nm UV light with a radiant power of 13.3 mW. The static 1H NMR spectra are shown in Fig. 2a. As the time under UV irradiation increases, the intensity of the acrylate hydrogens resonances (∼6 ppm) is reduced as the polymerization reaction propagates. After sufficient time, there is negligible 1H signal at ∼6 ppm confirming that the whole sample space is effectively irradiated by the light dispersion rod. The resonances also broaden and their T1 relaxation time decreases (Fig. 2b), indicative of the increase in viscosity of the forming polymer. An initial increase in T1 can be attributed to an initial decrease in viscosity due to the exothermic nature of the reaction.
 | ||
| Fig. 2 (a) Static 1H NMR spectra of the nBA polymerization reaction during UV exposure (365 nm at 13.3 mW fiber optic output power). (b) Plot of 1H longitudinal relaxation times (T1) versus monomer conversion (X) correlating to the spectra in (a). (c) 79Br NMR spectra of powdered KBr at different MAS rates. | ||
High-resolution NMR spectra of the polymerization can be achieved via MAS. To demonstrate the stability of the MAS spinning, the windowed rotor was initially packed with powdered KBr. Fig. 2c shows the 79Br NMR spectra of KBr at multiple MAS rates up to 15 kHz. Once the MAS stability of the windowed rotor had been demonstrated the polymerization reaction could be examined under MAS.
However, to avoid spinning liquid material the polymerization reactants were initially exposed to 60 s of UV (365 nm at 13.3 mW) in the windowed rotor under static conditions; this initial polymerization resulted in ∼50% conversion of the monomer providing a sufficient increase in viscosity to avoid possible sample leakage under MAS. Then the rotor was spun to 6 kHz and 1H NMR spectra were acquired at different timepoints while under continuous UV irradiation (365 nm at 13.3 mW). Fig. 3c and d shows two of the 1H NMR spectra taken during the polymerization reaction, where 1H resonances are well resolved for each of the hydrogens in the monomer/polymer (see Fig. 3a and b for assignment). Integration of the well resolved resonances of the remaining monomer acrylate hydrogens (6.8–5.5 ppm) gives the monomer concentration (M) allowing accurate tracking of the progress of the polymerization. The monomer conversion ratio is given by:
 | (1) |
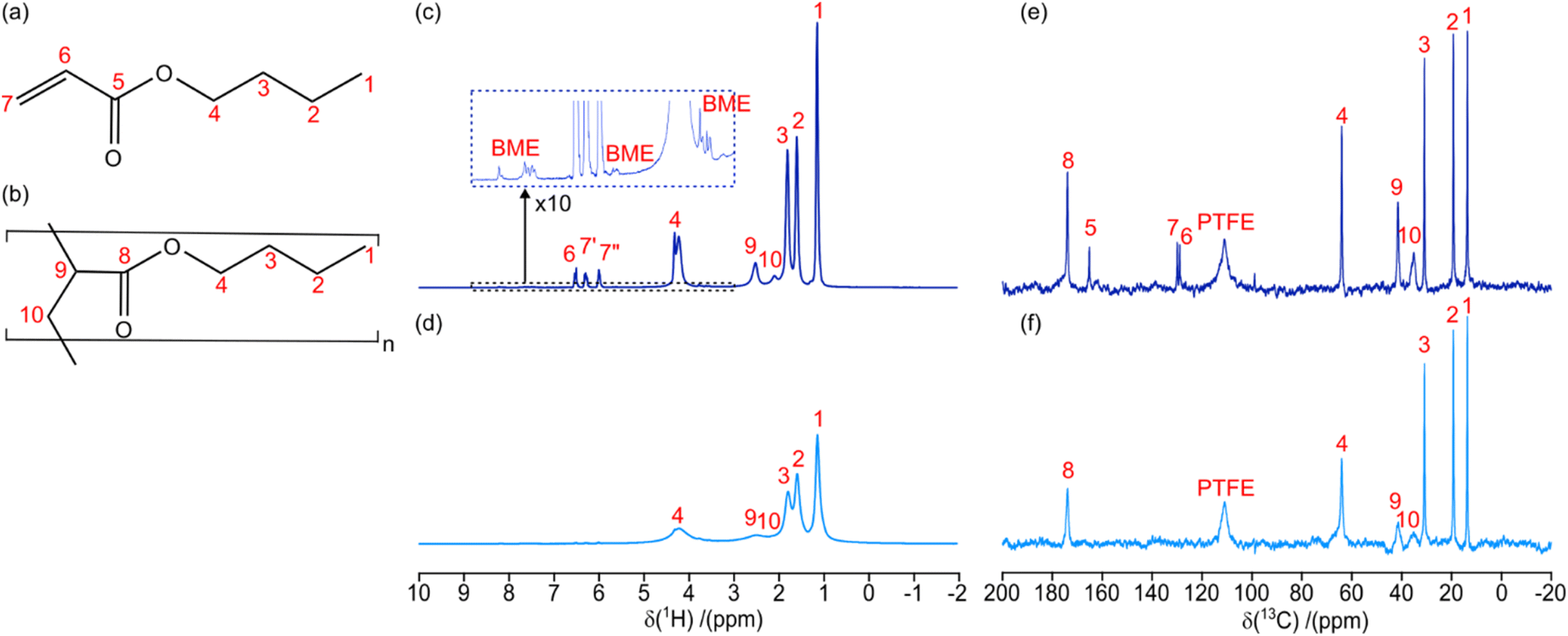 | ||
| Fig. 3 Molecular drawing of (a) nBA monomer and (b) PBA polymer with labelled hydrogen/carbon environments. (c) 1H and (e) 13C MAS (6 kHz) NMR spectra of monomer/polymer mixture (X = 0.84) after 140 s total UV exposure duration. (d) 1H and (f) 13C MAS (6 kHz) NMR spectra of PBA (X = 1.00) after 630 s total UV exposure duration. The resonances in the NMR spectra are labelled with numbers relating to assignments in (a and b), or as relating to the BME initiator or the PTFE insert. The irradiation wavelength was 365 nm with a radiant power of 13.3 mW, throughout. | ||
Hence, the displayed 1H NMR spectra were acquired at 84% (Figure 3c) and 100% (Fig. 3d) monomer conversion. Fig. 3c therefore contains a mixture of both monomer (Fig. 3a) and polymer (Fig. 3b) 1H resonances. On the other hand, Fig. 3d contains only polybutyl acrylate (PBA) resonances and shows significant broadening due to the increased viscosity, as seen previously in the static spectra (Fig. 2a). At various time points the UV irradiation, and hence the polymerization reaction, was stopped to acquire longer 13C NMR spectra (experimental time of 600 s). Fig. 3e and f shows two 13C NMR spectra taken during the polymerization reaction, where 13C resonances are well resolved for each of the carbon sites in the monomer/polymer (see Fig. 3a and b for assignment).
The polymerization reaction was shown to stop rapidly after the UV irradiation was removed, by acquiring static 1H NMR spectra under UV, turning off the UV LED immediately after acquisition, and then re-acquiring in dark conditions (see ESI Fig. S1†). The pre- and post-UV spectra are highly similar throughout the polymerization process demonstrating that the reaction halts quickly. These results also confirm that the paramagnetic radicals created by the UV polymerization process has a negligible effect on the 1H NMR spectra of the bulk materials, due to the low relative concentration of radicals at any one point in time during the UV exposure (<0.6 mol%; i.e. double the initial concentration of the photo-initiator).
Different fiber optic output radiant powers were used to explore the rate of the polymerization reaction under different UV fluxes. As above, each experiment began at ∼50% monomer conversion after 60 s of 365 nm light at 13.3 mW under static conditions to partially solidify the sample. Afterwards, the rotor was spun at 6 kHz and the 1H NMR was recorded every 20 s during continuous exposure to UV irradiation at a set power to track the polymerization reaction. Fig. 4a shows the in situ1H MAS NMR spectra over time during UV irradiation at 6.7 mW. The monomer conversion ratio, determined via1H NMR integration, is shown as a function of continuous UV exposure duration in Fig. 4b. The dataset at 3.3 mW only reaches a maximum conversion of ∼96%, suggesting a small amount of liquid monomer may have leaked beneath the PTFE insert, out of reach of the UV irradiation.
 | ||
| Fig. 4 (a) MAS (6 kHz) 1H NMR spectra of nBA polymerization under continuous UV exposure (365 nm at 6.7 mW fiber optic output power). (b) Plot of monomer conversion X, determined via integration of monomer NMR resonances, versus UV exposure duration at different radiant powers. Exponential polymerisation curves are fitted viaeqn (2) (with an offset where necessary) and reaction rates k are displayed. | ||
At each UV power the reaction begins with an induction period at a slower rate before following an exponential curve fitted with:
| X = 1 − e−kt. | (2) |
The resulting reaction rates constants k, at each power for the exponential section of the reaction are displayed in Fig. 4b. As expected, the rate of polymerization is reduced with reduced UV power, with a non-linear relationship between reaction rate and radiant power. K. V. K. Boodhoo et al., previously studied the polymerization of a thin film of n-butyl acrylate with variable UV intensities via gel permeation chromatography.46 In their work, they mechanistically derived equations describing the polymerization reaction, which, under certain assumptions, provided an analytical solution for the monomer conversion:
 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 L mol−1 s−1; kt = 6 × 107 L mol−1 s−1) the polymerization curves for each radiant power can be simulated;47 these curves are shown as dashed lines in Fig. 5a. The coefficients contain the photo-initiator initial concentration P0, quantum yield of initiation φ, and absorption coefficient α; the average film thickness L and surface area S; and the light wavelength λ. These parameters are provided in the ESI† and were measured directly from our system or have been provided by the literature.48,49 Hence, the results of the in situ NMR experiments can be compared to the predicted polymerization curves of the analytical equation. Solid lines in Fig. 5a show the extrapolated exponential polymerization curves determined in Fig. 4b (shown without induction periods for clarity). As shown, the exponential curves provide a good approximation of the analytically simulated polymerization curves with a similar relationship between reaction rate and radiant power. This corroborates the equation derived by Boodhoo et al., with a larger dataset than provided by their experimentation, giving credence to the proposed values for the polymerization propagation rate kp and termination rate constant kt. The analytically calculated polymerization curves are visibly faster than the experimentally derived polymerization curves. This can be ascribed to the false assumption in the analytical calculation that 100% of the radiant power reaches the sample. In reality, there is light loss due to reflection from the glass rod surface and/or non-ideal alignment. The differences in the curves allow an estimation of the light loss in the in situ NMR setup to be around 5–20%.
300 L mol−1 s−1; kt = 6 × 107 L mol−1 s−1) the polymerization curves for each radiant power can be simulated;47 these curves are shown as dashed lines in Fig. 5a. The coefficients contain the photo-initiator initial concentration P0, quantum yield of initiation φ, and absorption coefficient α; the average film thickness L and surface area S; and the light wavelength λ. These parameters are provided in the ESI† and were measured directly from our system or have been provided by the literature.48,49 Hence, the results of the in situ NMR experiments can be compared to the predicted polymerization curves of the analytical equation. Solid lines in Fig. 5a show the extrapolated exponential polymerization curves determined in Fig. 4b (shown without induction periods for clarity). As shown, the exponential curves provide a good approximation of the analytically simulated polymerization curves with a similar relationship between reaction rate and radiant power. This corroborates the equation derived by Boodhoo et al., with a larger dataset than provided by their experimentation, giving credence to the proposed values for the polymerization propagation rate kp and termination rate constant kt. The analytically calculated polymerization curves are visibly faster than the experimentally derived polymerization curves. This can be ascribed to the false assumption in the analytical calculation that 100% of the radiant power reaches the sample. In reality, there is light loss due to reflection from the glass rod surface and/or non-ideal alignment. The differences in the curves allow an estimation of the light loss in the in situ NMR setup to be around 5–20%. | (4) |
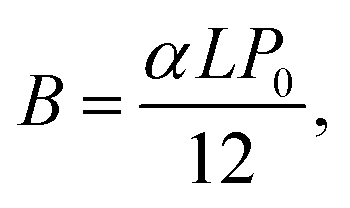 | (5) |
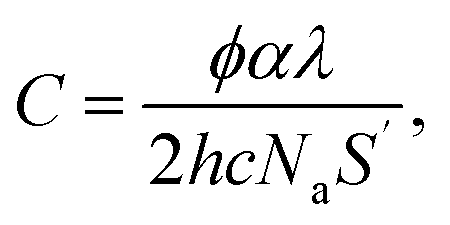 | (6) |
 | ||
| Fig. 5 (a) Comparison of experimental and calculated polymerisation curves at each radiant power. Solid lines represent extrapolated exponential reaction curves (eqn (2)) utilising the reaction rate constants k determined via fitting of the in situ NMR data (Fig. 4b). Dashed lines represent calculated polymerisation curves from the analytical eqn (3) derived by K. V. K. Boodhoo et al.,46 assuming 100% of the radiant power is incident on the sample. (b) Plot of monomer conversion from UV irradiation at 13.3 mW while under MAS (6 kHz) and static conditions. (c) Plot of monomer conversion from UV irradiation at 1.7 mW while under MAS (6 kHz) and static conditions; two static datasets are provided: one with continuous irradiation and one where irradiation was halted at X ∼50% for 10 min before the UV irradiation and 1H NMR acquisition were reinitiated. Exponential polymerisation curves are fitted viaeqn (2) (with an offset where necessary) and reaction rates k are displayed. | ||
The induction period increases in duration with reduced UV power and is most clearly seen at the lower powers of 3.3 and 1.7 mW. To investigate the origin of the induction period, polymerization curves were compared for when the rotor was static versus spinning at 6 kHz. Fig. 5b shows the polymerization curve with strong (13.3 mW) UV power under static and MAS conditions, where the MAS curve begins at ∼50% monomer conversion. Both curves show a small initial induction period from when the UV irradiation begins (or is reinitiated). Beyond the induction period, both line-shapes can be fitted with an exponential curve (eqn (1)), providing similar reaction rates of k ∼0.02 s−1. The static measurement appears to have a slightly faster polymerization rate, but this may encompass systematic errors due to the lower accuracy of measuring the monomer conversion from the broader overlapping resonances in the static 1H spectra. Hence, at relatively high UV power the small induction period can likely be ascribed to chemistry phenomena in the polymerization reaction. Induction periods have been seen previously in photo-initiator reactions and were explained by: effects of exothermic heat on polymer chain dynamics; radical–radical termination reactions being favored over radical–monomer reactions at low radical concentrations; or inhibitory effects of incorporated oxygen.46 However, comparison of the polymerization curves at weak UV power (1.7 mW) shows a stark difference in the data acquired under static and MAS conditions (Fig. 5c). The static polymerization curve begins with a small induction period, similar to the high-power data, before becoming exponential with a slower reaction rate of k = 0.007 s−1. However, the MAS polymerization has a much longer induction period, suggesting that the MAS is hindering the initial polymerisation. To rule out the effect of halting the UV irradiation at 50% monomer conversion to begin MAS spinning, a static polymerisation curve was measured with a 10 minutes halt of the UV irradiation at 50% monomer conversion before restarting the UV LED. As shown in Fig. 5c, the halting of the UV irradiation does not greatly influence the static polymerisation curve at low UV power. Instead, we hypothesize that the MAS acts to centrifuge the monomer/polymer gel mixture. High molecular weight (MW) polymer chains will be removed from low MW monomer units, hampering the polymerisation chain reaction and increasing the termination rate of reaction with other polymer chains. Presumably, the centrifugation effect is lessened once the gel becomes more solid at higher monomer conversion percentages and the reaction curve becomes exponential, as in the static experiments.
Conclusions
In conclusion, an in situ photo-irradiation MAS NMR setup has been demonstrated, which is both easy to produce and use. Results are presented from 1H and 13C MAS NMR tracking of the UV polymerization of nBA. The MAS NMR provides highly resolved spectra proving that the setup can be used to follow light intensity dependent reactions in semi-solids with good accuracy; future mixed-phase investigations could include light induced crystallization reactions50 or surface interactions in heterogeneous photo-catalysts.16,18,20,21,24,25,51 However, care needs to be taken to account for centrifugation of the sample under MAS. Centrifugation would not be an issue in more rigid materials where NMR can provide insights into their atomic-scale structure and dynamic. We hope this will contribute to the characterization of light-dependent solid materials at the forefront of materials research, such as photovoltaic semiconductor layers or photo-stabilized polymers.4,52A Chemagnetics probe and Varian rotor were adapted for use in this work, however MAS NMR equipment from other commercial vendors could be adapted similarly by substituting the MAS sample holder end-cap with a tapered glass rod and feeding a fiber optic cable through the probe. The fiber optic coupled system allows interchange of the light source for observation of photochemical phenomena caused by different wavelengths and intensities. The light source is also controlled via USB and hence could be triggered by the NMR spectrometer. Integration of light pulses into the NMR RF pulse sequences would provide interesting avenues of development in areas such as photo-hyperpolarization NMR techniques.33
Data availability
Raw NMR data for this article (Topspin data file format) are available at KU Leuven Research Data Repository at [https://doi.org/10.48804/VLODK1].Author contributions
Conceptualisation (TJNH), investigation (TJNH), funding acquisition (TJNH, DS), formal analysis (TJNH), methodology (TJNH, ROS), writing – original draft (TJNH), writing – review and editing (ROS, DS), project administration (TJNH, DS).Conflicts of interest
There are no conflicts to declare.Acknowledgements
TJNH thanks the Research Foundation – Flanders (Fonds voor Weten-schappelijk Onderzoek – Vlaanderen) for its support via a senior post-doctoral fellowship (1253824N). DS thanks the KU Leuven grant (STG-18-00289) and Research Foundation – Flanders (Fonds voor Weten-schappelijk Onderzoek – Vlaanderen) PorMedNMR grant (G0D5419N). We are very grateful to the FabLab Leuven and the Campus Glasblazerij for 3-D printing of the bespoke probe-head and manufacturing of the glass light dispersion rods, respectively.Notes and references
- A. Endruweit, M. S. Johnson and A. C. Long, Polym. Compos., 2006, 27, 119–128 CrossRef.
- G. Wypych, Handbook of UV Degradation and Stabilization, Elsevier, 2020 Search PubMed.
- J. Lindroos and H. Savin, Sol. Energy Mater. Sol. Cells, 2016, 147, 115–126 CrossRef.
- S. Kundu and T. L. Kelly, EcoMat, 2020, 2, e12025 CrossRef.
- S. K. R. Miller and B. Banks, MRS Bull., 2010, 35, 20–24 CrossRef.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef PubMed.
- J. Cho, J. H. Park, J. K. Kim and E. F. Schubert, Laser Photonics Rev., 2017, 11, 1600147 CrossRef.
- P. Vashishtha, S. Bishnoi, C.-H. A. Li, M. Jagadeeswararao, T. J. N. Hooper, N. Lohia, S. B. Shivarudraiah, M. S. Ansari, S. N. Sharma and J. E. Halpert, ACS Appl. Electron. Mater., 2020, 2, 3470–3490 CrossRef.
- D.-P. Hader, General Photobiology, Elsevier, 2013 Search PubMed.
- W. Zhang, S. Xu, X. Han and X. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952–1963 CrossRef PubMed.
- R. de Oliveira-Silva, A. Bélime, C. Le Coeur, A. Chennevière, A. Helary, F. Cousin, P. Judeinstein, D. Sakellariou and J.-M. Zanotti, J. Neutron Res., 2019, 21, 155–166 Search PubMed.
- J. Marreiros, R. de Oliveira-Silva, P. Iacomi, P. L. Llewellyn, R. Ameloot and D. Sakellariou, J. Am. Chem. Soc., 2021, 143, 8249–8254 CrossRef PubMed.
- Z. Yu, R. de Oliveira-Silva, Y. Pontikes and D. Sakellariou, Cem. Concr. Res., 2023, 166, 107116 CrossRef.
- S.-J. Hwang, C. Petucci and D. Raftery, J. Am. Chem. Soc., 1997, 119, 7877–7878 CrossRef.
- S.-J. Hwang, C. Petucci and D. Raftery, J. Am. Chem. Soc., 1998, 120, 4388–4397 CrossRef.
- S.-J. Hwang and D. Raftery, Catal. Today, 1999, 49, 353–361 CrossRef.
- C. V. Rice and D. Raftery, Chem. Commun., 1999, 895–896 RSC.
- S. Pilkenton and D. Raftery, Solid State Nucl. Magn. Reson., 2003, 24, 236–253 CrossRef.
- S. Pilkenton, S.-J. Hwang and D. Raftery, J. Phys. Chem. B, 1999, 103, 11152–11160 CrossRef.
- A. R. Pradhan, M. A. Macnaughtan and D. Raftery, J. Am. Chem. Soc., 2000, 122, 404–405 CrossRef.
- W. Xu and D. Raftery, J. Phys. Chem. B, 2001, 105, 4343–4349 Search PubMed.
- J. Borisch, S. Pilkenton, M. L. Miller, D. Raftery and J. S. Francisco, J. Phys. Chem. B, 2004, 108, 5640–5646 Search PubMed.
- M. Hunger and W. Wang, Chem. Commun., 2004, 584–585 Search PubMed.
- M. Concistrè, A. Gansmüller, N. McLean, O. G. Johannessen, I. Marín Montesinos, P. H. M. Bovee-Geurts, P. Verdegem, J. Lugtenburg, R. C. D. Brown, W. J. DeGrip and M. H. Levitt, J. Am. Chem. Soc., 2008, 130, 10490–10491 CrossRef PubMed.
- J. Becker-Baldus, C. Bamann, K. Saxena, H. Gustmann, L. J. Brown, R. C. D. Brown, C. Reiter, E. Bamberg, J. Wachtveitl, H. Schwalbe and C. Glaubitz, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9896–9901 CrossRef PubMed.
- J. G. Hu, B. Q. Sun, M. Bizounok, M. E. Hatcher, J. C. Lansing, J. Raap, P. J. E. Verdegem, J. Lugtenburg, R. G. Griffin and J. Herzfeld, Biochemistry, 1998, 37, 8088–8096 CrossRef PubMed.
- J. G. Hu, B. Q. Sun, A. T. Petkova, R. G. Griffin and J. Herzfeld, Biochemistry, 1997, 36, 9316–9322 CrossRef PubMed.
- A. T. Petkova, M. Hatanaka, C. P. Jaroniec, J. G. Hu, M. Belenky, M. Verhoeven, J. Lugtenburg, R. G. Griffin and J. Herzfeld, Biochemistry, 2002, 41, 2429–2437 CrossRef PubMed.
- M. L. Mak-Jurkauskas, V. S. Bajaj, M. K. Hornstein, M. Belenky, R. G. Griffin and J. Herzfeld, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 883–888 CrossRef CAS PubMed.
- V. S. Bajaj, M. L. Mak-Jurkauskas, M. Belenky, J. Herzfeld and R. G. Griffin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9244–9249 CrossRef CAS.
- J. Matysik, Y. Ding, Y. Kim, P. Kurle, A. Yurkovskaya, K. Ivanov and A. Alia, Appl. Magn. Reson., 2022, 53, 521–537 CrossRef CAS PubMed.
- M. G. Zysmilich and A. McDermott, J. Am. Chem. Soc., 1994, 116, 8362–8363 CrossRef CAS.
- K. B. Sai Sankar Gupta, E. Daviso, G. Jeschke, A. Alia, M. Ernst and J. Matysik, J. Magn. Reson., 2014, 246, 9–17 CrossRef CAS PubMed.
- M. Najdanova, D. Gräsing, A. Alia and J. Matysik, Photochem. Photobiol., 2018, 94, 69–80 CrossRef CAS.
- F. De Biasi, M. A. Hope, Y. Qiu, P. J. Brown, M. Visegrádi, O. Ouari, M. R. Wasielewski and L. Emsley, J. Phys. Chem. Lett., 2024, 15, 5488–5494 CrossRef CAS.
- F. De Biasi, G. Karthikeyan, M. Visegrádi, M. Levien, M. A. Hope, P. J. Brown, M. R. Wasielewski, O. Ouari and L. Emsley, J. Am. Chem. Soc., 2024, 146(29), 19667–19672 CrossRef CAS.
- DNP-NMR|Dynamic Nuclear Polarization|Solid State NMR, https://www.bruker.com/en/products-and-solutions/mr/nmr/dnp-nmr.html, accessed July 18, 2024.
- Y. Tomonaga, T. Hidaka, I. Kawamura, T. Nishio, K. Ohsawa, T. Okitsu, A. Wada, Y. Sudo, N. Kamo, A. Ramamoorthy and A. Naito, Biophys. J., 2011, 101, L50–L52 CrossRef PubMed.
- A. Naito, Y. Makino, A. Shigeta and I. Kawamura, Biophys. Rev., 2019, 11, 167–181 CrossRef PubMed.
- H. Yomoda, Y. Makino, Y. Tomonaga, T. Hidaka, I. Kawamura, T. Okitsu, A. Wada, Y. Sudo and A. Naito, Angew. Chem., Int. Ed., 2014, 53, 6960–6964 CrossRef.
- Y. Makino, I. Kawamura, T. Okitsu, A. Wada, N. Kamo, Y. Sudo, K. Ueda and A. Naito, Biophys. J., 2018, 115, 72–83 CrossRef PubMed.
- I. Kawamura, N. Kihara, M. Ohmine, K. Nishimura, S. Tuzi, H. Saitô and A. Naito, J. Am. Chem. Soc., 2007, 129, 1016–1017 CrossRef PubMed.
- C. E. Wilkes and M. T. Berard, PVC Handbook, Hanser, 2005 Search PubMed.
- K. V. K. Boodhoo, W. a. E. Dunk, M. S. Jassim and R. J. Jachuck, J. Appl. Polym. Sci., 2004, 91, 2079–2095 CrossRef.
- S. Beuermann, D. A. Paquet, J. H. McMinn and R. A. Hutchinson, Macromolecules, 1996, 29, 4206–4215 CrossRef.
- M. D. Goodner and C. N. Bowman, in Solvent-Free Polymerizations and Processes, American Chemical Society, 1999, vol. 713, pp. 220–231 Search PubMed.
- M. G. Joshi, J. Appl. Polym. Sci., 1981, 26, 3945–3946 CrossRef.
- C. E. Hughes, P. A. Williams and K. D. M. Harris, Angew. Chem., Int. Ed. Engl., 2014, 53, 8939 CrossRef.
- M. Hunger and T. Horvath, J. Chem. Soc., Chem. Commun., 1995, 1423–1424 RSC.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Solid-state NMR experimental details, light irradiation equipment, monomer conversion simulation parameters, and additional 1H NMR spectra are provided in pdf format. See DOI: https://doi.org/10.1039/d4ta06729k |
| This journal is © The Royal Society of Chemistry 2025 |