
Vinylene-linked donor–acceptor covalent organic polymers with low exciton binding energy for enhanced photocatalytic oxidation of sulfides†
Wenhao
Liu
a,
Yujie
Li
a,
Fang
Duan
 *a,
Haiping
Liu
a,
Yanyan
Ren
a,
Shengrong
Yan
a,
Shuanglong
Lu
a,
Mingliang
Du
a,
Xin
Chen
a and
Jun
Wang
*b
*a,
Haiping
Liu
a,
Yanyan
Ren
a,
Shengrong
Yan
a,
Shuanglong
Lu
a,
Mingliang
Du
a,
Xin
Chen
a and
Jun
Wang
*b
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: duanfang@jiangnan.edu.cn
bJiangsu Key Laboratory of Advanced Food Manufacturing Equipment and Technology, Department of Packaging Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: wangj_1982@jiangnan.edu.cn
First published on 26th November 2024
Abstract
Photocatalytic organic synthesis is a promising technology for green and sustainable organic conversion. Covalent organic polymers (COPs) as an emerging class of porous organic polymers have attracted more and more attention in photocatalytic organic synthesis. However, the relatively high exciton binding energy (Eb) of COPs significantly restricts the exciton dissociation and charge separation in the photocatalytic process, resulting in low photocatalytic efficiency. In this work, a vinylene-linked conjugated COP (Btt-Bdd) with a donor–acceptor (D–A) structure has been prepared through the Knoevenagel condensation reaction. The introduction of a planar π-skeleton donor and vinylene linkage extends the planar π-conjugation of COPs and significantly reduces the Eb to 39.2 meV and improves the charge separation efficiency. Therefore, fully conjugated Btt-Bdd shows excellent photocatalytic performance towards sulfide oxidation with a high conversion and selectivity. The synergistic effect between the D–A structure and vinylene linkage is proved to be an effective way for reducing Eb and thus improving the photocatalytic performance, which provides a good strategy for designing efficient photocatalysts with low Eb for organic synthesis.
Introduction
Organic synthesis, as a branch of chemical synthesis, is an important part of the chemical industry.1–5 Traditional preparation methods often require multiple reaction steps and the use of toxic and harmful reagents, which is harmful to the environment and does not conform to the concept of sustainable development.6,7 Photocatalytic organic synthesis, as a method to convert solar energy into chemical energy, has the advantages of being green and sustainable, mild reaction conditions and fewer side reactions, receiving growing attention.8–11 At present, it has been applied to the aryl boronic acid oxidation reaction, sulfide oxidation reaction, alcohol oxidation reaction, amine oxidation coupling reaction and so on.12–19 However, the low photocatalytic efficiency and the use of precious metals greatly limit the practical application of photocatalysis in organic synthesis.20,21 In order to make full use of light energy and achieve the goal of efficient organic conversion, researchers have been working on the design and synthesis of efficient photocatalysts.22,23 Traditional inorganic semiconductor catalysts, such as TiO2 and ZnO, with inefficient solar energy capture and rapid charge recombination, exhibit low photocatalytic efficiency.24–26 Therefore, it is of great significance to design highly efficient photocatalysts with strong light absorption and fast charge separation for organic conversion.Covalent organic polymers (COPs), as porous organic polymers, have become promising photocatalysts in recent years due to their high porosity, designable structures, and optical properties.27–30 Compared with traditional organic polymers, COPs can be combined in the form of covalent bonds through a variety of structural units to form a periodic network structure, which makes it possible to improve the photocatalytic efficiency for organic synthesis.29,31–34 However, due to the strong excitonic effects of most reported COPs with relatively high exciton binding energy (Eb), it is difficult for them to achieve efficient exciton dissociation and quick charge separation in the photocatalytic process.35–39 Owing to the structural designability of COPs, it is urgent to design and construct COPs with suitable structures to weaken the excitonic effects and reduce the Eb. As known, COPs can be linked by different bonds, such as imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), boron–oxygen (B–O) and vinylene (CC).40–42 Due to the highly reversible nature of the CN bond formed by the Schiff base reaction, COPs connected by imine bonds are easy to obtain with high crystallinity, and imine-linked COPs have been rapidly developed and widely used.43 However, the inherent polarity of imine bonds is not conducive to charge separation for organic synthesis reactions, and the photocatalytic efficiency is therefore limited.44 Vinylene-linked COPs obtained from the Knoevenagel condensation reaction are significantly different for irreversible CC linkage and in-plane π-delocalization on the skeletons, which not only endows polymers with high stability, but also helps reduce the Eb and promote photogenerated charge separation.45,46 Therefore, the construction and development of vinylene-linked COPs are expected to solve the problems of poor stability and low charge separation efficiency for COP photocatalysts. Besides, it has been reported that constructing COPs with a donor–acceptor (D–A) structure is also a good strategy for reducing Eb and thus accelerating photogenerated charge separation.47–49 For example, Lan et al. reported four different linear conjugated polymers with D–A structures to reduce the Eb by modulating the charge transfer pathway, and the Eb value of the optimal sample was 88 meV.48 Liu et al. used a built-in control strategy of D–A interaction to modulate excitonic effects, and the exciton dissociation in Tz-COPs can be greatly facilitated by strengthening the D–A interactions.37 Therefore, combing the advantages of the D–A structure and vinylene linkage, constructing D–A structured COPs with high flatness and extended conjugation of CC linkage can further reduce the Eb and accelerate the charge separation, but this is still a challenge.
N), boron–oxygen (B–O) and vinylene (CC).40–42 Due to the highly reversible nature of the CN bond formed by the Schiff base reaction, COPs connected by imine bonds are easy to obtain with high crystallinity, and imine-linked COPs have been rapidly developed and widely used.43 However, the inherent polarity of imine bonds is not conducive to charge separation for organic synthesis reactions, and the photocatalytic efficiency is therefore limited.44 Vinylene-linked COPs obtained from the Knoevenagel condensation reaction are significantly different for irreversible CC linkage and in-plane π-delocalization on the skeletons, which not only endows polymers with high stability, but also helps reduce the Eb and promote photogenerated charge separation.45,46 Therefore, the construction and development of vinylene-linked COPs are expected to solve the problems of poor stability and low charge separation efficiency for COP photocatalysts. Besides, it has been reported that constructing COPs with a donor–acceptor (D–A) structure is also a good strategy for reducing Eb and thus accelerating photogenerated charge separation.47–49 For example, Lan et al. reported four different linear conjugated polymers with D–A structures to reduce the Eb by modulating the charge transfer pathway, and the Eb value of the optimal sample was 88 meV.48 Liu et al. used a built-in control strategy of D–A interaction to modulate excitonic effects, and the exciton dissociation in Tz-COPs can be greatly facilitated by strengthening the D–A interactions.37 Therefore, combing the advantages of the D–A structure and vinylene linkage, constructing D–A structured COPs with high flatness and extended conjugation of CC linkage can further reduce the Eb and accelerate the charge separation, but this is still a challenge.
Herein, three COPs were fabricated to modulate excitonic effects by introducing a D–A structure and constructing vinylene linkage. As shown in Scheme 1, vinylene-linked Btt-Bdd with a D–A structure was obtained by the Knoevenagel condensation reaction of benzotrithiophene-2,5,8-tricarbaldehyde (Btt) and 2,2′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(4,1-phenylene))diacetonitrile (Bdd). As a planar π-skeleton donor, Btt significantly increased the in-plane π-conjugation of Btt-Bdd. Combined with the linkage of CC double bonds, the in-plane π-conjugation of the COP was further extended, which greatly reduced excitonic effects and facilitated charge separation on the skeleton. Besides, a series of experiments such as temperature-dependent photoluminescence (PL) and photoelectrochemical measurements were conducted to verify this deduction. The results indicated that vinylene-linked Btt-Bdd with higher flatness exhibited a lower Eb (39.2 meV), which effectively showed excellent photocatalytic performance towards sulfide oxidation with a high conversion and selectivity. This work confirms that the Eb value of COPs can be regulated by modulating connection units of the D–A structure and introducing fully conjugated bonds to promote charge separation and meet the needs of photocatalytic organic synthesis.
 | ||
| Scheme 1 The synthetic conditions and chemical structures of Btt-Btd, Btt-Bdd and Btc-Bdd. | ||
Results and discussion
material design, synthesis and characterization
As shown in Scheme 1, three D–A structured COPs (Btt-Btd, Btt-Bdd and Btc-Bdd) with similar frameworks but different connecting units or bonds were prepared through solvothermal methods. More detailed synthesis steps are provided in the ESI.† Imine-linked Btt-Btd with a D–A structure is synthesized through the Schiff base reaction of Btt and Btd. Vinylene-linked Btt-Bdd with a planar π-skeleton donor is fabricated through the Knoevenagel condensation reaction of Btt and Bdd, and vinylene-linked Btc-Bdd with a twisted π-skeleton donor is obtained by the Knoevenagel condensation reaction of Btc and Bdd.The crystal structures of the obtained COPs were investigated by powder X-ray diffraction (PXRD). As shown in Fig. 1a–c, imine-linked Btt-Btd shows higher crystallinity than vinylene-linked Btt-Bdd and Btc-Bdd, which may be related to the irreversible CC double bond linkage. Fourier transform infrared spectroscopy (FT-IR) was used to further verify the successful synthesis of COPs through the Knoevenagel condensation and Schiff base reaction. As shown in Fig. 1d and f, the absorption peaks of the cyano group are observed at 2211 and 2208 cm−1 for Btt-Bdd and Btc-Bdd, respectively, which are about 40 cm−1 lower than that of Bdd. Besides, compared with Btt and Btc, the carbonyl peaks located at 1657 cm−1 and 1654 cm−1 for Btt-Bdd and Btc-Bdd are significantly weakened. Therefore, the vinylene-linked Btt-Bdd and Btc-Bdd are successfully obtained from the Knoevenagel condensation reaction. As shown in Fig. 1e, the absorption peak for the imine bond (CN) in Btt-Btd is observed at 1596 cm−1, and the stretching vibration peak of CO at 1661 cm−1 is significantly weakened compared with that of Btt, indicating that the Schiff base reaction occurred between Btt and Btd. Additionally, the solid-state 13C nuclear magnetic resonance (13C NMR) spectrum was also used to analyze the structures of the obtained COPs. As shown in Fig. 2a–c, a set of characteristic signals at 114 ppm belong to the cyano groups, and the signals at 108 and 147 ppm can be attributed to the vinylene linkages.22 For Btt-Btd, the weak and broad peak that appeared at about 153.2 ppm belongs to the carbon atom of the imine linkage, proving the formation of the imine-linked framework.50
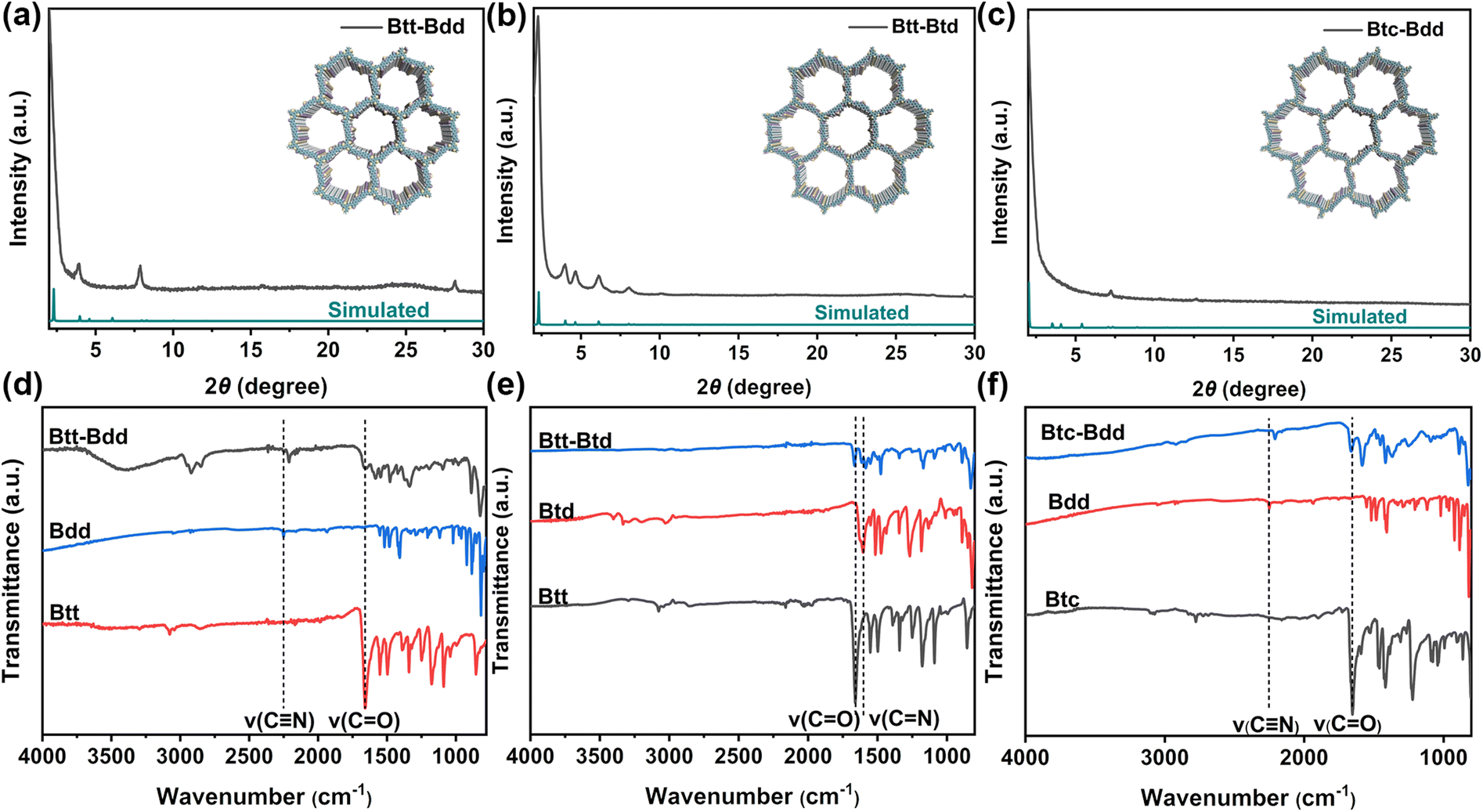 | ||
| Fig. 1 PXRD patterns of (a) Btt-Bdd, (b) Btt-Btd and (c) Btc-Bdd; (d and e) FT-IR spectra of (d) Btt-Bdd, (e) Btt-Btd and (f) Btc-Bdd. | ||
 | ||
| Fig. 2 Solid-state 13C NMR spectra of (a) Btt-Bdd, (b) Btt-Btd and (c) Btc-Bdd; (d) N2 sorption isotherms of the obtained samples. | ||
The specific surface areas and porosities of COPs were tested via the N2 sorption experiments at 77 K. In Fig. 2d, the specific surface areas ofs Btt-Bdd, Btt-Btd and Btc-Bdd are evaluated to be 263, 578 and 170 m2 g−1, respectively, based on the Brunauer–Emmett–Teller (BET) method according to the N2 sorption isotherms. The pore size distributions calculated by the nonlocal density functional theory show that they are all mesoporous polymers with sizes of 2.80, 2.75 and 2.86 nm, respectively (Fig. S1†). Furthermore, X-ray photoelectron spectroscopy (XPS) was used to determine the surface elemental compositions and chemical states of the obtained COPs. From the full spectra in Fig. S2,† the elements of C, N, O and S are presented in the obtained three COPs, and the O element may come from the residual aldehyde groups or surface adsorbed oxygen. As shown in Fig. S3,† the N 1s spectra of Btt-Bdd and Btc-Bdd can both be deconvoluted into two peaks corresponding to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N at 399.2 and CN–S at 400.1 eV.22 The two peaks at 398.5 and 399.5 eV in the N 1s spectra of Btt-Btd are attributed to CN–S in the Btd unit and CN–C, respectively.51 In the S 2p spectrum of Btt-Bdd, the peaks at 165.1 and 164.0 eV are ascribed to S 2p1/2 and S 2p3/2 of C–S–C bonds in the Btt unit, respectively, and the peaks at 166.4 and 165.4 eV belong to S 2p1/2 and S 2p3/2 of CN–S bonds in the Bdd unit.52–54 Similarly, in the S 2p spectra of Btt-Btd and Btc-Bdd, four peaks can also be found and attributed to CN–S and CN–C.50 Moreover, the chemical stability of Btt-Bdd was evaluated by immersion in different solvents such as methanol, acetone, N,N′-dimethylformamide (DMF), tetrahydrofuran (THF), 3 M HCl and 3 M NaOH. The PXRD patterns after treatment are basically consistent with that before, indicating that it has good chemical stability (Fig. S4†). Finally, thermogravimetric analysis (TGA) was conducted to reveal the thermal durability of the three obtained COPs (Fig. S5†). In a nitrogen atmosphere, Btt-Bdd and Btc-Bdd constructed using the conjugated Btt block show no significant mass loss at nearly 400 °C, while Btc-Bdd constructed using the twisted Btc block shows thermal stability below 300 °C, indicating the good thermal durability of the obtained COPs.
N at 399.2 and CN–S at 400.1 eV.22 The two peaks at 398.5 and 399.5 eV in the N 1s spectra of Btt-Btd are attributed to CN–S in the Btd unit and CN–C, respectively.51 In the S 2p spectrum of Btt-Bdd, the peaks at 165.1 and 164.0 eV are ascribed to S 2p1/2 and S 2p3/2 of C–S–C bonds in the Btt unit, respectively, and the peaks at 166.4 and 165.4 eV belong to S 2p1/2 and S 2p3/2 of CN–S bonds in the Bdd unit.52–54 Similarly, in the S 2p spectra of Btt-Btd and Btc-Bdd, four peaks can also be found and attributed to CN–S and CN–C.50 Moreover, the chemical stability of Btt-Bdd was evaluated by immersion in different solvents such as methanol, acetone, N,N′-dimethylformamide (DMF), tetrahydrofuran (THF), 3 M HCl and 3 M NaOH. The PXRD patterns after treatment are basically consistent with that before, indicating that it has good chemical stability (Fig. S4†). Finally, thermogravimetric analysis (TGA) was conducted to reveal the thermal durability of the three obtained COPs (Fig. S5†). In a nitrogen atmosphere, Btt-Bdd and Btc-Bdd constructed using the conjugated Btt block show no significant mass loss at nearly 400 °C, while Btc-Bdd constructed using the twisted Btc block shows thermal stability below 300 °C, indicating the good thermal durability of the obtained COPs.
The morphologies of the three obtained COP samples were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). From the SEM images in Fig. S6a–c,† Btt-Bdd shows irregular particles aggregated by small nanosheets, Btt-Btd exhibits an urchin-like structure assembled by short nanobelts, and Btc-Bdd shows a web-like structure composed of small nanowires. The TEM images shown in Fig. S6d–f† further verify their morphologies and the porous channels can be clearly observed in Btt-Btd. Besides, the EDS elemental mappings in Fig. S7† show that C, N, and S elements are evenly distributed in the obtained samples.
Optical and photoelectrochemical analysis
The optical properties of Btt-Bdd, Btt-Btd, and Btc-Bdd were subsequently investigated using the ultraviolet-visible (UV-vis) absorption spectrum and photoluminescence (PL) spectrum. As depicted in Fig. 3a, Btt-Bdd and Btt-Btd exhibit nearly the same visible light responses in the range of 250 to 650 nm, demonstrating their strong abilities to capture visible light. However, Btc-Bdd shows much lower intensity for visible light absorption, which is attributed to the lower planar π-conjugation of Btc compared to Btt. According to the Tauc plots (Fig. 3b), the optical band gaps (Eg) of Btt-Bdd, Btt-Btd and Btc-Bdd are calculated to be 2.00, 1.98 and 1.85 eV, respectively. | ||
| Fig. 3 (a) UV-vis absorption spectra and (b) Tauc plots of Btt-Bdd, Btt-Btd and Btc-Bdd. (c–e) Integrated PL intensity as a function of temperature (insets: temperature-dependent PL spectra from 80 to 280 K) of Btt-Bdd, Btt-Btd and Btc-Bdd. (f) The fluorescence decay curves of the obtained COPs. | ||
The photoluminescence (PL) spectra were obtained to demonstrate the recombination of electrons and holes for Btt-Bdd, Btt-Btd and Btc-Bdd. As shown in Fig. S8,† Btt-Bdd exhibits obvious lower PL signal intensity than others, which indicates that Btt-Bdd shows lower electron–hole recombination probability and is more favorable for carrier separation. To further investigate the differences in charge separation among the three COPs, the temperature-dependent PL spectra of the as-obtained COPs from 80 to 280 K are recorded and shown in Fig. 3c–e. The integrated PL intensity decreases monotonically with the increase in temperature, and the corresponding Eb values can be calculated using the Arrhenius equation and nonlinear fitting.55 Accordingly, the Eb values of Btt-Bdd, Btt-Btd and Btc-Bdd are calculated to be 39.2, 90.9, and 119.6 meV, respectively. It is worth noting that Btt-Bdd exhibits a much lower Eb value than Btt-Btd and Btc-Bdd. This result indicates that the introduction of conjugated vinylene linkage and a D–A structure with a planar π-skeleton donor significantly facilitates the exciton dissociation and accelerates the charge separation. Moreover, the average fluorescence lifetimes of the three kinds of COPs under the excitation of 420 nm were detected using a transient fluorescence spectrometer. As shown in Fig. 3f, the average fluorescence lifetimes of Btt-Bdd, Btt-Btd and Btc-Bdd are calculated to be 0.65, 0.93 and 1.35 ns, respectively, indicating that the carriers generated by Btt-Bdd can be more rapidly transferred and trapped at the reaction sites, thus greatly inhibiting the recombination of electron–hole pairs.
In addition, in order to demonstrate the difference in the charge separation ability of the three COPs, photoelectrochemical measurements were conducted. From Fig. 4a, transient photocurrent responses of the three COPs during five switching light irradiation cycles show that Btt-Bdd has the highest photocurrent intensity, which suggests the photogenerated charges of Btt-Bdd can be easily separated and transferred. Meanwhile, the smallest radius of Btt-Bdd in the Nyquist plots of electrochemical impedance spectra (Fig. 4b) indicate the lowest interfacial resistance for charge transfer. These results suggest that the vinylene-linked Btt-Bdd with the D–A structure facilitates charge separation and transfer, thus enhancing the photocatalytic activity. To further explore the band structures of Btt-Bdd, Btt-Btd and Btc-Bdd, Mott–Schottky plots were measured at three different frequencies to evaluate the conduction band (CB) positions. The Mott–Schottky plots of all COPs exhibit a positive slope, which is a typical characteristic of N-type semiconductors. For a N-type semiconductor, the CB position is close to its flat band potential. Therefore, the CB positions of Btt-Bdd, Btt-Btd and Btc-Bdd are estimated to be −1.43, −1.25 and −1.10 V vs. Ag/AgCl, and −1.23, −1.05 and −0.90 V vs. NHE, respectively (Fig. 4c–e). According to the formula EVB = ECB + Eg, the valence band (VB) positions of Btt-Bdd, Btt-Btd and Btc-Bdd are calculated to be 0.77 V, 0.93 V and 0.95 V vs. NHE, respectively. The corresponding band structures are schematically shown in Fig. 4f.
 | ||
| Fig. 4 (a) Transient photocurrent responses, (b) EIS Nyquist plots, (c–e) Mott–Schottky plots, and (f) band structure diagram of Btt-Bdd, Btt-Btd and Btc-Bdd. | ||
Photocatalytic performance for the sulfide oxidation reaction
The photocatalytic performances of Btt-Bdd, Btt-Btd and Btc-Bdd were evaluated by selective oxidation of thioanisole to methyl phenyl sulfoxide as a model reaction under white LED light irradiation in the presence of O2 and methanol (Fig. 5). As shown in Fig. 5a, the thioanisole conversion by vinylene-linked Btt-Bdd reaches over 99%, which is much higher than those of Btt-Btd and Btc-Bdd. The kinetics curves of the three COPs are shown in Fig. S9,† from which the conversion rate presents a nearly linear increase over time. Besides, gas chromatography-mass spectrometry (GC-MS) was used to analyze the possible reaction products. As shown in Fig. S10,† it confirms that the main product is methyl phenyl sulfoxide with a high selectivity of 96%. These results prove that the combination of vinylene linkage and a D–A structure with a planar π-skeleton of Btt-Bdd exhibits higher photocatalytic organic conversion efficiency and selectivity. Subsequently, solvent screening experiments were conducted for Btt-Bdd (Table S1†). Among DMF, CH3CN, C2H5OH and CH3OH, the optimum conversion rate and product selectivity could be obtained in CH3OH. Furthermore, we carried out the screening of the optimal volume of methanol, in which 5 ml methanol as the solvent has a better conversion rate and selectivity (Table S2†). In addition, compared with other photocatalysts reported in the literature, Btt-Bdd also showed exceptional selectivity towards sulfide (Table S3†). Moreover, the cyclic experiments using Btt-Bdd were conducted to prove the stability of the obtained COPs in the reaction (Fig. 5b). The experimental results show that the photocatalytic conversion and selectivity remain nearly unchanged during the five cycles, indicating that Btt-Bdd has good stability for photocatalytic performance. In addition, after five cycles of reaction, the recycled photocatalyst is tested by PXRD and FT-IR (Fig. S11†). No significant difference can be found from the spectra before and after reactions, which demonstrates the remarkable stability of vinylene-linked Btt-Bdd. | ||
| Fig. 5 (a) Photocatalytic conversion of sulfide by Btt-Bdd, Btt- Btd and Btc-Bdd. (b) Cyclic experiments of Btt-Bdd. (c) The controlled and quenching experiments for selective aerobic sulfoxidation. Standard reaction conditions: sulfide (0.1 mmol), Btt-Bdd (5 mg), white LED light, methanol (5 mL), quenchers (0.1 equivalent) and O2 (1 atm). (d) EPR spectra of DMPO-O2˙− and TEMPO-1O2 produced by Btt-Bdd under white light irradiation. | ||
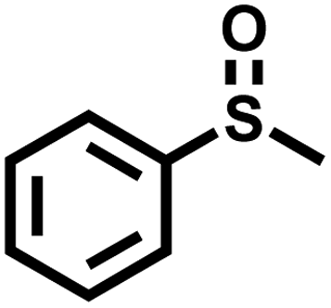
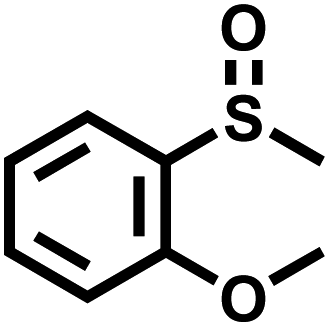
In order to illustrate the general applicability of Btt-Bdd, a series of sulfides with different functional groups and structures were chosen as substrates (Table 1). Most of the substrates can be well oxidized to produce the corresponding sulfoxides. Specifically, the introduction of electron-withdrawing groups such as –F, –Cl, –Br and –I on the benzene ring shows no adverse effect on the photocatalytic reaction and still exhibits a high conversion rate (Table 1, entries 2–5), while introducing methyl as an electron-donating group shows a slight reduction in the conversion (Table 1, entry 9). From the position of substituents, no matter ortho-position, meta-position or para-position on the benzene ring, it shows no obvious effect on the reaction (Table 1, entries 6–8 and 10). From the above analysis, it can be seen that vinylene-linked Btt-Bdd has a wide range of substrate applicability.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) | Sel.b (%) |
| a Standard reaction conditions: 0.1 mmol sulfide, 5 mg Btt-Bdd, 5 mL methanol, white LED irradiation for 8 h, and 1 atm of O2. b Determined by GC-FID using bromobenzene as the internal standard, conversion of sulfides, and selectivity of sulfoxides. | ||||
| 1 |

|
|
99 | 96 |
| 2 |

|

|
98 | 98 |
| 3 |

|

|
99 | 98 |
| 4 |

|

|
99 | 98 |
| 5 |

|

|
99 | 98 |
| 6 |

|
|
99 | 98 |
| 7 |
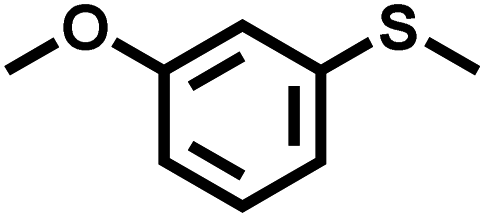
|
|
99 | 96 |
| 8 |

|
|
99 | 95 |
| 9 |

|

|
91 | 96 |
| 10 |

|

|
98 | 96 |

In order to determine the affecting factors of the photocatalytic reaction, we conduct a series of controlled experiments (Fig. 5c). The results show that no products can be formed under the conditions of darkness, no catalyst and an argon atmosphere, respectively. This confirms that the presence of light, photocatalysts and oxygen are the indispensable conditions for the conversion of sulfides to sulfoxides. In order to study the photocatalytic mechanism for the selective oxidation of sulfides to sulfoxides by Btt-Bdd under white light irradiation, different quenchers are added into the reaction to verify the possible reactive oxygen species (ROS).
When p-benzoquinone (P-BQ) as a superoxide (O2˙−) scavenger is added, the conversion is severely decreased. In contrast, when 1,4-diazabicyclo [2.2.2]octane (DABCO) is added as a scavenger of singlet oxygen (1O2), it has almost no effect on the reaction. The results of scavenging experiments show that the major ROS is O2˙− in the oxidation reaction of sulfides to sulfoxides by Btt-Bdd. In addition, when AgNO3 and KI are added as electron (e−) and hole (h+) scavengers, respectively, the conversion rates are only 35% and 26%, indicating that both e− and h+ play a significant role in the oxidation of sulfides to sulfoxides.
To further identify the reactive oxygen species (ROS) involved in the reaction, electron paramagnetic resonance (EPR) tests are conducted and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) is added to detect the generation of O2˙− radicals in the photocatalytic process of sulfides to sulfoxides. As shown in Fig. 5d, under dark conditions, no signal of DMPO-O2˙− can be detected, but after 5 min of illumination, the signal for DMPO-O2˙− is clearly observed, demonstrating the production of O2˙− species after light irradiation. 2,2,6,6-Tetramethylpiperidine-N-oxyl (TEMPO) is used as the 1O2 radical trapping agent; it can be observed that no signal of TEMPO-1O2 is detected under dark or light conditions, indicating that 1O2 is not the active species in this reaction.
According to the above analysis, the possible mechanism for the photocatalytic oxidation of sulfides to sulfoxides by Btt-Bdd is proposed in Fig. 6. Under the irradiation of white light, vinylene-linked Btt-Bdd with planar π-conjugation can be easily excited to generate electrons and holes, and then the photogenerate electrons jump to the LUMO and combine with O2 to produce O2˙−. At the same time, the holes leaving the HOMO react with sulfides to form radical cations, and then the nucleophilic attack of O2˙− can cause the radical cations to further generate peroxy sulfoxide. Finally, it is combined with protons provided by CH3OH to form the methyl phenyl sulfoxide and H2O.
 | ||
| Fig. 6 Possible mechanism for photocatalytic oxidation of sulfides to sulfoxides by Btt-Bdd under white LED irradiation. | ||
Conclusions
In summary, vinylene-linked Btt-Bdd with a D–A structure is successfully obtained through the Knoevenagel condensation reaction. The D–A structure is strengthened by increasing the donor flatness and introducing vinylene linkage, and the in-plane π-conjugation of COPs is extended, which can effectively regulate the excitonic effects and facilitate charge separation and transfer. Therefore, the obtained vinylene-linked Btt-Bdd exhibits excellent photocatalytic performance towards the oxidation of sulfides to sulfoxides with a wide range of substrate applicability. Besides, the mechanism for photocatalytic oxidation of sulfides to sulfoxides by Btt-Bdd is also revealed. This work provides a possible pathway for regulating the excitonic effects of COP materials to enhance their photocatalytic activities.Data availability
The authors confirm that the data supporting this article has been included in the ESI.†Author contributions
Wenhao Liu: designing and performing the experiments and writing this paper. Fang Duan: supervising the project and revising the manuscript. All authors: analyzing and discussing the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2022YFA1203600) and the National Natural Science Foundation of China (No. 52173201). Many thanks to the Central Laboratory, School of Chemical and Material Engineering, Jiangnan University, for their help in the characterization and analysis of the as-obtained samples.Notes and references
- M. A. Emmanuel, S. G. Bender, C. Bilodeau, J. M. Carceller, J. S. DeHovitz, H. Fu, Y. Liu, B. T. Nicholls, Y. Ouyang, C. G. Page, T. Qiao, F. C. Raps, D. R. Sorigué, S.-Z. Sun, J. Turek-Herman, Y. Ye, A. Rivas-Souchet, J. Cao and T. K. Hyster, Chem. Rev., 2023, 123, 5459–5520 CrossRef CAS PubMed.
- S.-Y. Li, K.-F. Huang, Z.-Y. Tang and J.-H. Wang, J. Mater. Chem. A, 2023, 11, 3281–3296 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- X. Sun, X. Zhang and Y. Xie, Matter, 2020, 2, 842–861 CrossRef.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- J.-K. Jin, K. Wu, X.-Y. Liu, G.-Q. Huang, Y.-L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X.-P. Zhou, J. He and D. Li, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- Z. Li, Y. Pi, D. Xu, Y. Li, W. Peng, G. Zhang, F. Zhang and X. Fan, Appl. Catal., B, 2017, 213, 1–8 CrossRef CAS.
- Z. Wang, L. Wang, B. Cheng, H. Yu and J. Yu, Small methods, 2021, 5, 2100979 CrossRef CAS PubMed.
- J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- G. Li, T. Qiu, Q. Wu, Z. Zhao, L. Wang, Y. Li, Y. Geng and H. Tan, Angew. Chem., Int. Ed., 2024, 63, e202405396 CrossRef CAS PubMed.
- M. Li, X. Chi, Z. Zhang, S. Bi, F. Meng, Y. Jiao, K. Mou, Z. Wang, B. Xue, X. Li and F. Zhang, Angew. Chem., Int. Ed., 2024, e202411474 CAS.
- S. Li, L. Hu, Z. Qian, J. Yin, J. Tang, C. Pan, G. Yu and K. A. I. Zhang, ACS Catal., 2023, 13, 12041–12047 CrossRef CAS.
- T.-Y. Qiu, Y.-N. Zhao, W.-S. Tang, H.-Q. Tan, H.-Y. Sun, Z.-H. Kang, X. Zhao and Y.-G. Li, ACS Catal., 2022, 12, 12398–12408 CrossRef CAS.
- S. Suleman, Y. Zhang, Y. Qian, J. Zhang, Z. Lin, O. Metin, Z. Meng and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202314988 CrossRef CAS PubMed.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- Y. Zhao, X. Xu, K. Zhang, Z. Li, H. Wang, Y. Zhao, J. Qiu and J. Wang, ACS Catal., 2024, 14, 3556–3564 CrossRef CAS.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed.
- W. Weng and J. Guo, Nat. Commun., 2022, 13, 5768 CrossRef CAS PubMed.
- L. Dai, A. Dong, X. Meng, H. Liu, Y. Li, P. Li and B. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300224 CrossRef CAS PubMed.
- C. Qin, Y. Yang, X. Wu, L. Chen, Z. Liu, L. Tang, L. Lyu, D. Huang, D. Wang, C. Zhang, X. Yuan, W. Liu and H. Wang, Nat. Commun., 2023, 14, 6740 CrossRef CAS PubMed.
- Z. Sun, M. Wang, J. Fan, R. Feng, Y. Zhou and L. Zhang, Adv. Compos. Hybrid Mater., 2021, 4, 1322–1329 CrossRef CAS.
- Y. Huang, Q. Shang, D. Wang, S. Yang, H. Guan, Q. Lu and S. C. Tsang, Appl. Catal., B, 2016, 187, 59–66 CrossRef CAS.
- H. Zhai, X. Liu, Z. Wang, Y. Liu, Z. Zheng, X. Qin, X. Zhang, P. Wang and B. Huang, Chinese J. Catal., 2020, 41, 1613–1621 CrossRef CAS.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475 CrossRef PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- S. J. Lyle, T. M. Osborn Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- Y. Su, Y. Wan, H. Xu, K.-i. Otake, X. Tang, L. Huang, S. Kitagawa and C. Gu, J. Am. Chem. Soc., 2020, 142, 13316–13321 CrossRef CAS PubMed.
- X. Wang, S. Zhang, X. Li, Z. Zhan, B. Tan, X. Lang and S. Jin, J. Mater. Chem. A, 2021, 9, 16405–16410 RSC.
- L. Hao, R. Shen, S. Chen, W. Bi, L. Wang, G. Liang, P. Zhang and X. Li, J. Mater. Chem. A, 2022, 10, 24064–24072 RSC.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- F. Liu, Y. He, X. Liu, Z. Wang, H.-L. Liu, X. Zhu, C.-C. Hou, Y. Weng, Q. Zhang and Y. Chen, ACS Catal., 2022, 12, 9494–9502 CrossRef CAS.
- J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- W. Zhang, Z. Deng, J. Deng, C.-T. Au, Y. Liao, H. Yang and Q. Liu, J. Mater. Chem. A, 2022, 10, 22419–22427 RSC.
- A. M. Evans, I. Castano, A. Brumberg, L. R. Parent, A. R. Corcos, R. L. Li, N. C. Flanders, D. J. Gosztola, N. C. Gianneschi, R. D. Schaller and W. R. Dichtel, J. Am. Chem. Soc., 2019, 141, 19728–19735 CrossRef CAS PubMed.
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, G. Wu, H. Xia, S. W. Yang and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208919 CrossRef CAS PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Nat. Rev. Chem., 2022, 6, 881–898 CrossRef CAS PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F. J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed.
- S. Bi, F. Meng, D. Wu and F. Zhang, J. Am. Chem. Soc., 2022, 144, 3653–3659 CrossRef CAS PubMed.
- W. Wang, H. Wang, X. Tang, J. Huo, Y. Su, C. Lu, Y. Zhang, H. Xu and C. Gu, Chem. Sci., 2022, 13, 8679–8685 RSC.
- S. Bi, Z.-A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- Z. A. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS PubMed.
- H. Ou, X. Chen, L. Lin, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 8729–8733 CrossRef CAS PubMed.
- J. Li, J. Jia, J. Suo, C. Li, Z. Wang, H. Li, V. Valtchev, S. Qiu, X. Liu and Q. Fang, J. Mater. Chem. A, 2023, 11, 18349–18355 RSC.
- Y. He, G. Liu, Z. Liu, J. Bi, Y. Yu and L. Li, ACS Energy Lett., 2023, 8, 1857–1863 CrossRef CAS.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- G.-B. Wang, Y.-J. Wang, J.-L. Kan, K.-H. Xie, H.-P. Xu, F. Zhao, M.-C. Wang, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 4951–4956 CrossRef CAS PubMed.
- C.-J. Wu, X.-Y. Li, T.-R. Li, M.-Z. Shao, L.-J. Niu, X.-F. Lu, J.-L. Kan, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2022, 144, 18750–18755 CrossRef CAS PubMed.
- X. Gao, J. Yuan, P. Wei, J. Dong, L. Chang, Z. Huang, H. Zheng, J. Liu, J. Jia, T. Luan, B. Zhou, H. Yu and C. Peng, ACS Catal., 2023, 14, 533–546 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06162d |
| This journal is © The Royal Society of Chemistry 2025 |