
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Corrosion stability of NASICON-based membranes against aqueous solutions: case study for sodium iodine batteries†
Gerald
Dück
 ab,
Frank
Schäfer
c,
Jürgen Peter
Gross
d,
Subham
Ray
d,
Tarek
Allam
d,
Michael
Holzapfel
c,
Ruth
Schwaiger
d,
Martin
Finsterbusch
*a and
Dina
Fattakhova-Rohlfing
*ab
ab,
Frank
Schäfer
c,
Jürgen Peter
Gross
d,
Subham
Ray
d,
Tarek
Allam
d,
Michael
Holzapfel
c,
Ruth
Schwaiger
d,
Martin
Finsterbusch
*a and
Dina
Fattakhova-Rohlfing
*ab
aForschungszentrum Jülich GmbH, Institute of Energy Materials and Devices, Materials Synthesis and Processing (IMD-2), 52425 Jülich, Germany. E-mail: d.fattakhova@fz-juelich.de; m.finsterbusch@fz-juelich.de
bUniversity Duisburg-Essen, Faculty of Engineering and Center for Nanointegration Duisburg-Essen CENIDE, Lotharstraße 1, 47057 Duisburg, Germany
cFraunhofer-Institute for Chemical Technology (ICT), 76327 Pfinztal, Germany
dForschungszentrum Jülich GmbH, Institute of Energy Materials and Devices, Microstructure and Properties of Materials (IMD-1), 52425 Jülich, Germany
First published on 3rd December 2024
Abstract
Sodium batteries are attractive candidates for medium to large scale stationary energy storage applications. While high-temperature Na–NiCl batteries (Zebra batteries) have been in commercial use for decades, intensive research is being conducted into new cell concepts to mitigate some of the drawbacks, particularly the high operating temperature and the expensive Na-beta-alumina separator. Medium-temperature sodium batteries are promising low-cost alternatives with an operating temperature of only 100 °C (slightly above the melting point of metallic Na), a NaSICON-based separator and a high-capacity aqueous cathode based on NaI/NaI3. Compared to polycrystalline Na-beta-alumina, NaSICONs offer higher Na-ion conductivity and lower manufacturing costs. However, little is known about the stability of this large class of materials under the specific operating conditions in this new cell type. In this study, we systematically investigate the chemical stability of different NaSICONs composition as a function of the state of charge (SOC) of the catholyte. Subsequently, solid separators were prepared and tested in symmetrical NaI/NASICON/NaI3 and full Na/NaSICON/NaI + NaI3 cells to evaluate the degradation under electrochemical cycling. These experiments revealed that the main cause of degradation is Na-proton exchange in the NaSICON and reaction of iodine with secondary phases and that this depends on the SOC. From this fundamental understanding, optimization strategies were derived that led to the development of sub-stoichiometric NaSICON with optimized doping to extend the lifetime of aqueous medium temperature Na batteries. The results obtained here not only enable the use of this exciting new battery technology for medium- to large-scale energy storage, but can also help to increase the lifespan and efficiency of other aqueous-based battery systems, such as seawater-based batteries or membranes for Na extraction from brine solutions.
Introduction
Sodium batteries are a promising alternative to lithium-ion batteries (LIBs), as sodium is much more abundant and less expensive than lithium. The advantages of sodium batteries are particularly evident in the mass market for stationary energy storage, where the cost structure and availability of critical materials play a decisive role. Several sodium battery concepts such as Na–S and Zebra batteries have already been commercialized and used for stationary storage for decades.1–3 These batteries offer competitive energy densities and can be operated for several thousand cycles without degradation due to the use of high energy density sodium metal anodes and molten salt catholytes separated by a ceramic membrane. However, the reliable operation of the known systems requires a relatively high temperature of around 300 °C, which requires expensive housing materials for insulation and corrosion protection as well as complex production processes.The development of transition temperature sodium batteries with sodium metal anodes that can be operated at medium to room temperature is a compelling solution. One of the intensively investigated strategies is the use of aqueous catholytes instead of molten salts, which can be operated at temperatures of about 100 °C and below. Several room temperature batteries with aqueous cathodes have been already introduced, such as Li–Fe,4,5 Li–I2,6,7 Mg–I2,8 or Zn–I2 batteries9 or even sea water batteries,10 which are described in a comprehensive review by Arnold et al.11 In this article, the authors emphasize the importance of the stability of the solid electrolyte in the aqueous medium, with typical cycle numbers stagnating between 20 and 100 cycles. Another promising concept introduced by Zhu and Kee,12 which is discussed in this work, utilizes liquid sodium anodes and aqueous NaI/I2 cathodes operating just above the melting point of sodium at around 100 °C. In this novel approach, iodine and sodium metal are the active electrode materials responsible for the conversion of chemical and electrical energy.
Various room temperature and medium temperature sodium batteries have a potential to achieve competitive energy densities at practical operation temperatures due to the use of sodium metal anodes and aqueous electrolytes. However, their successful operation requires the use of solid Na-ion conducting membrane that is chemically stable to both the liquid sodium anode and the aqueous catholyte.
Solid Na-ion conductors have been studied extensively in the past,13–15 with most research focusing on understanding and improving ionic conductivity. Sodium beta-alumina (Na-β′′-alumina) is often used as ion-conducting separator membrane in sodium batteries. It enables low-cost precursor materials and offers high ionic conductivity and stability to liquid sodium.3,16–18 However, Na-β′′-alumina requires a sintering temperature of about 1600 °C, which increases production cost and negates the benefits of using low-cost precursor materials. In addition, previous studies by Holzapfel et al. have shown that Na-β′′-alumina is not suitable for use in aqueous batteries, as it generally decomposes rapidly in aqueous solutions, including the medium temperature Na/I2 battery described above.19 In contrast, sodium superionic conductors (NaSICON) have shown very promising durability, even in harsh alkaline20 and acidic environment.19
NaSICONs with the chemical formula Na1+xZr2SixP3−xO12 (0 ≤ x ≤ 3) are characterized by their excellent ionic conductivity, a phenomenon first described by Goodenough et al.21 The impressive conductivity, commendable thermal stability and excellent compatibility with sodium metal make NaSICONs excellent solid electrolytes for the development of high energy density sodium-ion batteries (NIBs) and solid-state batteries (SSBs). The superiority of a sodium metal anode lies in its higher theoretical capacity compared to graphite or other insertion materials used in Li-ion or Na-ion batteries.22
Despite the increasing interest to NaSICON materials, the development of NaSICON-based batteries is still in its infancy, as some important issues remain to be answered, especially regarding the chemical stability of NaSICON-based battery components under operation conditions. Little information is available on the chemical stability of NaSICONs in different battery concepts and there is a lack of systematic investigation of chemical and corrosion stability as a function of NaSICON composition. In earlier studies, stability problems with NaSICON in contact with sodium metal were identified.23,24 Later studies proposed innovative ways to circumvent this stability problems and suggested methods such as surface modifications and compositional tuning to improve the stability of NaSICONs, especially when interacting with sodium metal.16,25–28
The question of chemical stability becomes even more complex when considering the interactions of NaSICON with aqueous catholytes. In particular, the exposure of NaSICON materials to an aqueous environment presents an additional challenge. Although some NaSICON compositions exhibit some degree of stability in water,29–32 the overall stability is strongly influenced by the specific exposure conditions. Exposure to water or high humidity can lead to hydrolysis, initiating degradation of the NaSICON structure and potentially compromising ionic conductivity.
These results emphasize that improving the chemical stability of NaSICON materials is a pivotal goal of ongoing research on NaSICON-based aqueous batteries. Understanding the factors that influence the chemical stability of NaSICON materials in different environments and under various conditions forms the basis for further battery development. The knowledge gained will guide the development of strategies to improve the stability of these materials and provide the opportunities to unlock their full potential in energy storage applications.
In this study, a systematic investigation of the stability of different NaSICON materials under the operating conditions relevant for medium temperature batteries was conducted. First, we analyzed the different chemical environments prevailing during the different state of charge (SOC) of the aqueous I−/I2 cathode to identify critical conditions. We then modified the chemical composition of the NaSICON material by doping with different elements and adjusted the Si–P-ratio and sodium content to optimize the chemical and electrochemical performance. Furthermore, we investigated the difference between NaSICON powder and sintered pellets and the effects of different material processing, sinterability and microstructure of the NaSICON membranes. The identification of a suitable membrane material is crucial for the successful commercialization of this promising energy storage technology.
Experimental
Synthesis
The various NaSICON materials (Table 1) were synthesized via a solution-assisted solid-state reaction (SA-SSR) synthesis route.33 Stoichiometric amounts of NaNO3 (ACS, Merck) and ZrO(NO3)2·xH2O (99%, Aldrich) were dissolved in deionized water. Small amounts of HNO3 (69%, ACS, Aldrich) were added to adjust to pH ≈ 3. For the Zr-substituted compounds, appropriate amounts of Mg(NO3)2 (for analysis, Merck), Sc-nitrate and Hf-nitrate were added to the solution, respectively. Afterwards, tetraethyl-orthosilicate (TEOS) (reagent grade, Merck) was added and stirred until fully hydrolyzed. Finally, NH4H2PO4 (ACS, Merck) was added. The gel that was formed was dried at 80 °C and calcined at 800 °C for 4 hours to receive the raw NaSICON powder.| Material | Theoretical composition | T sinter/°C | t dwell/hours | Rel. density/% of sintered pellets | σ total (25 °C)/mS cm−1 | |
|---|---|---|---|---|---|---|
| NZSiP3.4_SSR | Na3.4Zr2Si2.4P0.6O12 | 1260 | 6 | 94.8 ± 0.7 | 3.0 ± 0.2 | |
| Zr-Substitution | NScZSiP3.4 | Na3.4Sc0.4Zr1.6Si2PO12 | 1260 | 6 | 94.2 ± 0.2 | 0.58 ± 0.01 |
| NAlYZSiP3.4 | Na3.4Al0.2Y0.2Zr1.6Si2PO12 | 1150 | 6 | 97.3 ± 0.4 | 0.23 ± 0.01 | |
| NHSiP3.4 | Na3.4Hf2Si2.4P0.6O12 | 1350 | 6 | 96.9 ± 0.3 | 1.1 ± 0.2 | |
| NHSiP3.2 | Na3.2Hf2Si2.2P0.8O12 | 1350 | 6 | Powder only | ||
| NMZSiP3.1 | Na3.1Mg0.05Zr1.95Si2PO12 | 1260 | 6 | Powder only | ||
| Na-Stoichiometry | NZSiP3.0 | Na3Zr2Si2PO12 | 1250 | 5 | Powder only | |
| NZP | NaZr2P3O12 | 1050 | 12 | Powder only | ||
| NZSiP3.0sub | Na3.0Zr2Si2.3P0.7O11.85 | 1260 | 6 | 98.0 ± 0.1 | 2.8 ± 0.2 | |
| NHSiP3.0sub | Na3.0Hf2Si2.3P0.7O11.85 | 1350 | 6 | 98.2 ± 1.5 | 2.9 ± 0.3 | |
| Fabrication | NZSiP3.4 | Na3.4Zr2Si2.4P0.6O12 | 1260 | 6 | 89.8 ± 0.3 | 0.91 ± 0.4 |
The NaSICON materials for the long-term cycling tests were synthesized via a similar SA-SSR route, as described by Holzapfel et al.19 Stoichiometric amounts of basic zirconium carbonate (technical grade, Alfa Aesar) were first dissolved in diluted HNO3. Afterwards a premixed solution of Na2SO3·5H2O (technical grade, Alfa Aesar), TEOS (99%, Sigma-Aldrich) and NH4H2PO4 (technical grade, Alfa Aesar) was added. The formed slurry was dried at 110 °C. The dried powder was first milled in a planetary ball mill in iso-propanol and then calcined at 750 °C for 4 hours.
To prepare a Hf-nitrate solution, HfO2 (99%, Alfa Aesar) was first stirred in five times molar amount of H2SO4 (96%) at 300 °C for 36 hours. The highly viscous solution was mixed into five times the amount of deionized water and brought to pH ≈ 9 with ammonia solution (28%) to precipitate Hf-hydroxide. The mixture was boiled for a few hours to generate crystal growth and then cooled to room temperature. The cold solution was Sc-nitrate solution was prepared by dissolving stoichiometric amounts of Sc2O3 (99.5%, Projector GmbH) in boiling HNO3 solution.
The raw NaSICON powders were ball-milled in ethanol with zirconia milling balls (3 & 5 mm) for around 72 hours using a tumbling mixer. After drying, the final powder was either further heat treated as is or first pressed (PW 10, P-O-Weber) into cylindrical pellets of 13 mm diameter at 100 MPa and then sintered at the same conditions as the respective powder. The sintering conditions for all investigated materials can be taken from Table 1.
Alternatively, one composition (NZSiP3.4_SSR) was also synthesized using a solid-state reaction (SSR). Stoichiometric amounts of Na2CO3, ZrO2, SiO2 and NH4H2PO4 (ACS, Merck) were milled together in a planetary ball mill (PM400, Retsch) with zirconia milling balls (3 mm) for around 1 hour. After drying, the powder was further processed according to the other materials.
Structural and compositional analysis
The powder was characterized by X-ray diffraction (XRD) (D4, Bruker). The lattice parameters and quantities of the crystalline phases were determined by Rietveld refinement of the XRD data. For the refinement the software TOPAS (Version 6, Brucker AXS GmbH, Karlsruhe, Germany) was used. Hydrogen measurements were carried out by Inert Gas Fusion (IGF) technique using a G8 GALILEO ONH analyzer (Bruker). The powder samples were encapsulated in tin capsules and melted at 1600 °C. The released gases from the melted samples were transferred by a nitrogen carrier gas into the detection system. A thermal conductivity detector (TCD) was used to detect the hydrogen. The system was calibrated using calibration gas as well as standard samples prior to the measurements. The chemical composition was determined by ICP-OES analysis (Thermo Scientific iCAP7600 spectrometer with optical scale and CID semi-conductor detector, axial and radial reflection, wavelength 166–847 nm).5 mmol of NaSICON powder were each transferred into 10 mL of 3 M NaI3 solution – which represents 100% state of charge (SOC) – and stored at 100 °C. The 3 M NaI3 solution was prepared by stirring 3 M NaI and 3 M molecular iodine in water at room temperature until completely dissolved. The pH of the 3 M NaI3 solution was 1.3, as determined using a pH meter (pH 20, VWR, Germany). After certain exposure times, the solution was decanted and the powder was washed three times with 50 mL of ethanol until no more discoloration of the washing solution could be observed. Lastly, the powder was dried in a vacuum drying oven at 80 °C and 50 mbar. The aged powder was again investigated by XRD and ICP-OES. Furthermore, NaSICON powder was also stored in saturated NaI solution (≙0% SOC), 3 M NaI solution and 3 M NaI solution with pH adjusted to 1.3, to investigate the influence of salt concentration, acidity and presence of I2 in the catholyte solution on the chemical stability of NaSICON. For the pH-adjusted solution, a few drops of concentrated HCl (ACS reagent, 37%, Sigma-Aldrich) were given to a 3 M NaI-solution until the pH reached the desired value.
To analyze the microstructure of sintered pellets, a thin layer of platinum (Pt) was sputtered onto the samples. The samples were then examined using a scanning electron microscope (SEM) (EVO 15, ZEISS) equipped with both secondary electron (SE) and backscattered electron (BSE) detectors. Additionally, the SEM featured an energy-dispersive X-ray spectroscopy (EDS) detector (ULTIM MAX 100, OXFORD INSTRUMENTS). The electron acceleration voltage was set to 15 kV.
Electrochemical analysis
Sintered pressed pellets were first sanded and polished with SiC sandpaper up to a fine grit of P4000. The samples were characterized by measuring the geometrical density, surface XRD (D4, Bruker) and electrical impedance (VMP-300, Bio-Logic Sciences Instruments Ltd, Claix, France).Sintered pellets were submerged in 3 M NaI3 solution – which represents 100% state of charge (SOC) – and stored at 100 °C. The pellets were removed from solution after specified exposure times, thoroughly rinsed with ethanol and dried overnight at 200 °C to remove all remaining volatile substances. The as-prepared pellets were sputtered with a thin layer of gold (Cressington 108 auto coater, TESCAN GmbH, Dortmund, Germany) on both sides and electrical impedance measurements were performed at 25 °C. Afterwards, some samples were polished with P400 grit sanding paper by hand to carefully remove approx. 10 μm of the surface on each side of the pellet. The pellets where then sputtered with gold on both sides and we measured the electrical impedance again.
Results and discussion
The operation principle of an aqueous sodium iodine battery is illustrated in Fig. 1. The cell contains a dense ceramic NaSICON separator, which is connected to Na metal on the anode side and an aqueous iodine-containing catholyte on the cathode side. During charge and discharge, the catholyte solution cycles between the fully sodiated state (saturated NaI-sol., 0% SOC) and a desodiated state (3 M NaI3-sol., 100% SOC) according to the following equations: | ||
| Fig. 1 Schematic representation of a NaI/I2 battery cell and optimization pathways for NaSICON ceramic electrolytes for corrosion stability at the Na-NaSICON and the NaSICON–cathode interface during charge and discharge. The reaction formulas describe the change in composition of the catholyte at the NaSICON–cathode interface during charge and discharge and are not balanced chemical equations. | ||
Charge (desodiation):
| 9Na+ + 9I− − 6e− (−6Na+ to membrane) ⇌ 3Na+ + 3I3− | (1) |
| I3− ⇌ I2 + I− | (2) |
Discharge (sodiation):
| 3Na+ + 3I3− + 6e− (+6Na+ from membrane) ⇌ 9NaI↓ | (3) |
The change in oxidation state of the iodine cathode active material during charge and discharge leads to a change in the concentration of different iodine species (from molecular iodine to different iodides) and is accompanied by a change in the Na+ ion concentration and the acidity of the catholyte, all of which influence the corrosion stability of the NaSICON separator. Thus, the fully sodiated catholyte (0% SOC) has the highest Na+ ion concentration and therefore the lowest concentration gradient between the Na+ ions in the separator and in the catholyte, which reduces the driving force for ion diffusion. In comparison, the Na+ ion concentration in the desodiated catholyte (3 M NaI3-sol.) is about 3 times lower, which significantly increases the probability of Na+ ion diffusion from the separator. Furthermore, the 3 M NaI3-sol. is inherently acidic – which further increases its corrosivity – whereas the saturated NaI-sol. is neutral.19 The iodine formed during the desodiation mainly forms complexes with the iodide ion to form I3− and to a lesser extent higher iodides, i.e. I5−. To a certain extent, however, the iodine molecules remain dissolved in the catholyte (equilibrium in eqn (2)). Due to the disproportionation equilibria of iodine in water (eqn (4) and (5)), a pH value below 7 develops in the solution as desodiation progresses. Last but not least, the dissolved iodine could also react directly with the ceramic separator.
| I2 + H2O ⇌ HOI + HI | (4) |
| 3I2 + H2O ⇌ HIO3 + 6HI | (5) |
| I2 (aqueous) ⇌ I2 (vapor) | (6) |
In order to understand the effects of these parameters on the corrosion stability of the NaSICON membranes, the experiments were designed in such a way that the individual parameters could be varied. First, the corrosion stability of NaSICON powders in catholytes with different compositions was studied in detail to gain insight into the composition- and catholyte-dependent corrosion stability of NaSICON materials. Subsequently, similar investigations were carried out on polycrystalline NaSICON membranes produced from sintered powders in order to evaluate the influence of the microstructure on the corrosion stability of the membranes.
Chemical stability of NaSICON powders in an aqueous I−/I2 catholyte
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, ICSD no. 202279).34 The unchanged NaSICON phase could be fitted using the NaSICON low structure (space group C12/c1, ICSD no. 38096).35 As can be seen in Fig. 2, the sintered starting powder has a pure NaSICON structure. When exposed to a saturated NaI solution, the first signs (<3%) of a secondary phase are observed after about seven days, which is assigned to proton-exchanged NaSICON phase with an expected composition Na3.4−x (H3O)xZr2Si2.4P0.6O12 (H-NaSICON). When the NaI concentration was reduced to 3 M (as in a catholyte with a 100% SOC), a significant amount of H-NaSICON was already observed after three days (5%). These results indicate a reduced corrosion stability of NZSiP3.4_SSR in solutions with a lower concentration of sodium ions, which is the case in catholytes with a higher SOC. Furthermore, the corrosion rate increases with increasing acidity of the solution, which also occurs at higher SOC. When the pH of a neutral 3 M NaI solution was lowered to about 1.3, which corresponds to the pH of a 3 M NaI3 solution in the fully desodiated catholyte, the amount of the protonated H-NaSICON increased to about 10%. Surprisingly, the presence of molecular iodine further accelerates the corrosion rate, although the pH and Na concentration do not change. After submersion in the 3 M NaI3 solution (pH 1.3), which contained molecular I2 in addition to NaI, the amount of H-NaSICON increased to about 18% within the same exposure time. XRD analysis showed no other secondary phases except for the protonated NaSICON phase. A more detailed analysis of the phase composition as determined by XRD and Rietveld refinement can be found in the ESI (Table S1†). Based on our results, we suggest that the presence of iodine increases the corrosion rate of the NaSICON phase beyond the pH value because the presence of iodine results in faster Na+/H+ exchange than in an iodine-free solution of similar pH. We assume that the higher corrosion activity of the iodine-containing solution is not related to a direct reaction with iodine, but to other processes due to the presence of iodine. One of the most important processes is the reaction of iodine with water, as described by disproportionation equilibria (eqn (2) and (4)), which can lead to the continuous formation of acidic species. The mechanism of this process is, however, still unclear and an additional systematic study is needed to understand it in detail.
c, ICSD no. 202279).34 The unchanged NaSICON phase could be fitted using the NaSICON low structure (space group C12/c1, ICSD no. 38096).35 As can be seen in Fig. 2, the sintered starting powder has a pure NaSICON structure. When exposed to a saturated NaI solution, the first signs (<3%) of a secondary phase are observed after about seven days, which is assigned to proton-exchanged NaSICON phase with an expected composition Na3.4−x (H3O)xZr2Si2.4P0.6O12 (H-NaSICON). When the NaI concentration was reduced to 3 M (as in a catholyte with a 100% SOC), a significant amount of H-NaSICON was already observed after three days (5%). These results indicate a reduced corrosion stability of NZSiP3.4_SSR in solutions with a lower concentration of sodium ions, which is the case in catholytes with a higher SOC. Furthermore, the corrosion rate increases with increasing acidity of the solution, which also occurs at higher SOC. When the pH of a neutral 3 M NaI solution was lowered to about 1.3, which corresponds to the pH of a 3 M NaI3 solution in the fully desodiated catholyte, the amount of the protonated H-NaSICON increased to about 10%. Surprisingly, the presence of molecular iodine further accelerates the corrosion rate, although the pH and Na concentration do not change. After submersion in the 3 M NaI3 solution (pH 1.3), which contained molecular I2 in addition to NaI, the amount of H-NaSICON increased to about 18% within the same exposure time. XRD analysis showed no other secondary phases except for the protonated NaSICON phase. A more detailed analysis of the phase composition as determined by XRD and Rietveld refinement can be found in the ESI (Table S1†). Based on our results, we suggest that the presence of iodine increases the corrosion rate of the NaSICON phase beyond the pH value because the presence of iodine results in faster Na+/H+ exchange than in an iodine-free solution of similar pH. We assume that the higher corrosion activity of the iodine-containing solution is not related to a direct reaction with iodine, but to other processes due to the presence of iodine. One of the most important processes is the reaction of iodine with water, as described by disproportionation equilibria (eqn (2) and (4)), which can lead to the continuous formation of acidic species. The mechanism of this process is, however, still unclear and an additional systematic study is needed to understand it in detail.
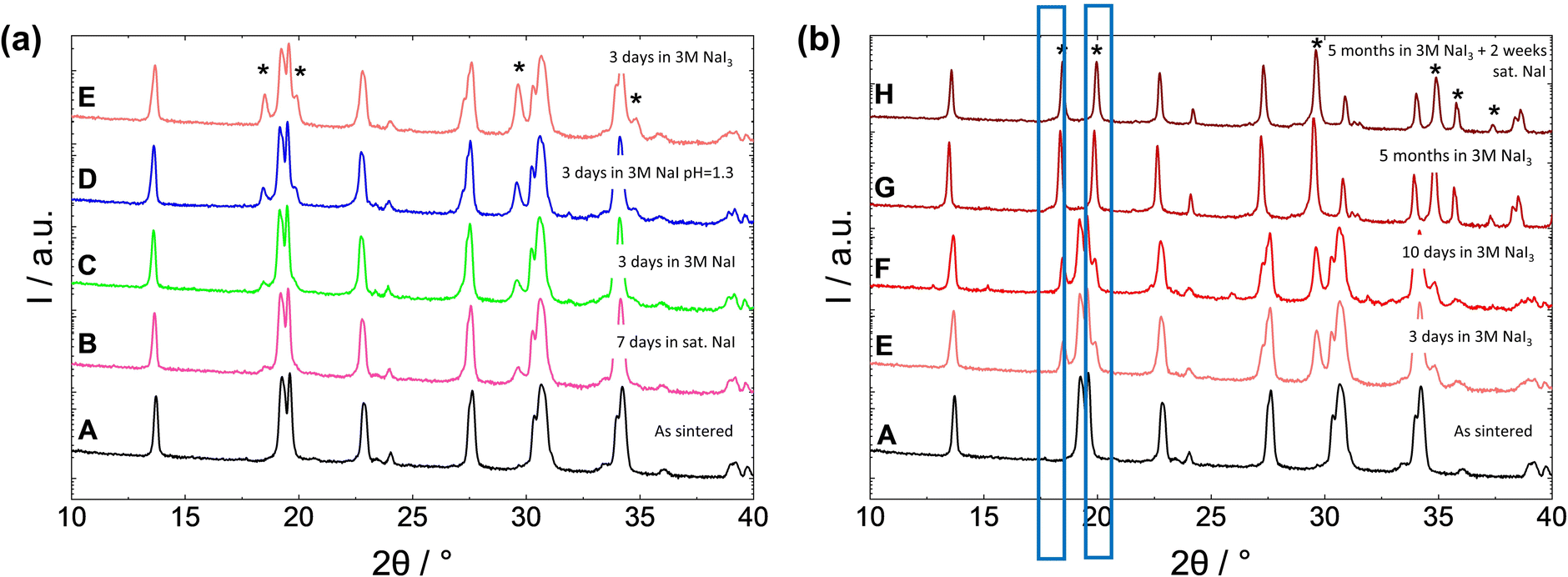 | ||
| Fig. 2 XRD patterns of: (a) NZSiP3.4 powder before (A) and after (E) and (F) exposure to different solutions: saturated NaI for 7 days (B), 3 M NaI for 3 days (C), 3 M NaI adjusted to pH = 1.3 for 3 days (D), 3 M NaI3 for 3 days (E). (b) NZSiP3.4 powder before and after different exposure times to 3 M NaI3 solution: for 10 days (F), for 5 months (G). Sample G was additionally placed in sat. NaI-solution (0% SOC) for 2 weeks (H). Protonated phase formed due to Na+/H+ exchanged NaSICON (H-NaSICON) is labeled with asterisk (*). | ||
The fastest corrosion of NaSICON powders was observed in the 3 M NaI3 solution (100% SOC). The Na+/H+-exchange of the NaSICON phase in this solution progresses steadily until only the fully protonated phase remains (Fig. 2b). This increase in hydrogen in the sample was also confirmed via IGF (ESI Fig. S6†). Interestingly, however, no other side reactions seem to take place as no other secondary phases except H-NaSICON can be observed in the XRD patterns even after 5 months of exposure. To our surprise, the chemical composition of the NaSICON phase, characterized by ICP-OES, remains approximately the same within the error limit even after only the protonated phase remains according to the XRD analysis (see Table 2). This result can be explained by the different distribution of the protonated and non-protonated phases in the particle volume and the different information depths of the characterization methods used. Since the proton exchange proceeds from the surface inwards, XRD detects the phase change quite well up to 2 μm into the particle, but cannot reach the non-protonated particle core. In contrast, ICP-OES measures the entire powder, including the particle core, as well as any amorphous phases that are not detected by XRD. Furthermore, since a detailed investigation of the exchanged proton phase is still pending, it is unclear how much Na is actually removed during the process, and partial proton insertion by interstitial atoms is also still possible. Second, the sensitivity of OES for Na is low and subject to large errors, and despite rinsing the powder, residues of Na from the electrolyte solution cannot be excluded. After an in-depth analysis of the powders with SEM/EDX (see ESI Fig. S7 and S8†), we could identify sodium phosphate particles on the surface of the NaSICON particles. After a prolonged exposure to the 3 M NaI3 solution, these particles had changed from a spherical shape to a needle like structure and also contained iodine. Therefore, we can suggest that although iodine does not directly react with NaSICON, it can nonetheless react with secondary phases which are typically present in sintered NaSICON materials.36–39
| Material composition | |
|---|---|
| After sintering | Na3.12(23)(Zr,Hf)2Si2.58(23)P0.61(05)O12 |
| After 5 months in 3 M NaI3 | Na3.40(13)(Zr,Hf)2Si2.74(12)P0.63(03)O12 |
XRD analysis of as prepared NMZSiP3.1, NSZSiP3.4 and NHSiP3.2 shows high phase purity of these materials with NaSICON structure (Fig. 3). Minor amounts of ZrO2 and HfO2 secondary phases are detected, which is commonly observed in sintered NaSICON materials. After exposing powders of these materials to the 3 M NaI3 solution, all three compounds show the formation of protonated NaSICON phase with a similar degree of protonation. Same as the NZSiP3.4 powder described above, the progressing formation of H-NaSICON with time is observed for all phases, clearly noticeable by the appearance of reflexes around 18.5° and 20°. Due to a handling error, the Sc containing sample test (Fig. 3a) was stopped after three days already. However, we assume a similar development as NMZSiP3.1 (Fig. 3b), which just as NZSiP3.4 developed a purely proton exchanged phase and no other secondary phases as per XRD analysis. In addition to the degradation from H+/Na+-exchange which all compositions showed, the Hf phase NHSiP3.2 also showed another minor decomposition by formation of monoclinic HfO2 after five months in 3 M NaI3 solution at 100 °C (Fig. 3c). Furthermore, in addition to the formation of an unknown NaSICON structure due to proton exchange, we could also observe a continuous change in the lattice parameter of the protonated NaSICON phase for all compositions with prolonged exposure time (Fig. S5†).
 | ||
| Fig. 3 XRD patterns of NSZSiP3.4 (a), NMZSiP3.1 (b) and NHSiP3.2 (c) before (black) and after exposure to 3 M NaI3 solution for 3 days (red) and 5 months (blue), respectively, as well as their respective phase compositions from Rietveld refinement (d–f). Complete diffractograms are shown in ESI (Fig. S1–S3†). | ||
The Na3.4Zr2Si2.4P0.6O12 material discussed above contains 3.4 sodium atoms per structure unit, providing the highest ionic conductivity of this composition series. To investigate the effect of Na stoichiometry on corrosion stability, the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio was set to 0:1 to obtain NaZr2P3O12 composition, which contains only one sodium atom per structure unit (Fig. 4). For the same exposure time in the 3 M NaI3 solution, the NaZr2P3O12 shows a much slower progress of Na+/H+-exchange reaction than the NZSiP3.4 material with higher Na content (Fig. 4A). Even after five months, hardly any Na+/H+-exchange was observed for the NaZr2P3O12 powder, whereas for NZSiP3.4, almost complete protonation with formation of H-NaSICON phase was observed after only three days. Obviously, a reduction in the Na content significantly increases chemical stability of NaSICON phases in an acidic environment. These results experimentally confirm the assumption that a reduction of the Na content could be beneficial for the stability of the NaSICON material.
:P ratio was set to 0:1 to obtain NaZr2P3O12 composition, which contains only one sodium atom per structure unit (Fig. 4). For the same exposure time in the 3 M NaI3 solution, the NaZr2P3O12 shows a much slower progress of Na+/H+-exchange reaction than the NZSiP3.4 material with higher Na content (Fig. 4A). Even after five months, hardly any Na+/H+-exchange was observed for the NaZr2P3O12 powder, whereas for NZSiP3.4, almost complete protonation with formation of H-NaSICON phase was observed after only three days. Obviously, a reduction in the Na content significantly increases chemical stability of NaSICON phases in an acidic environment. These results experimentally confirm the assumption that a reduction of the Na content could be beneficial for the stability of the NaSICON material.
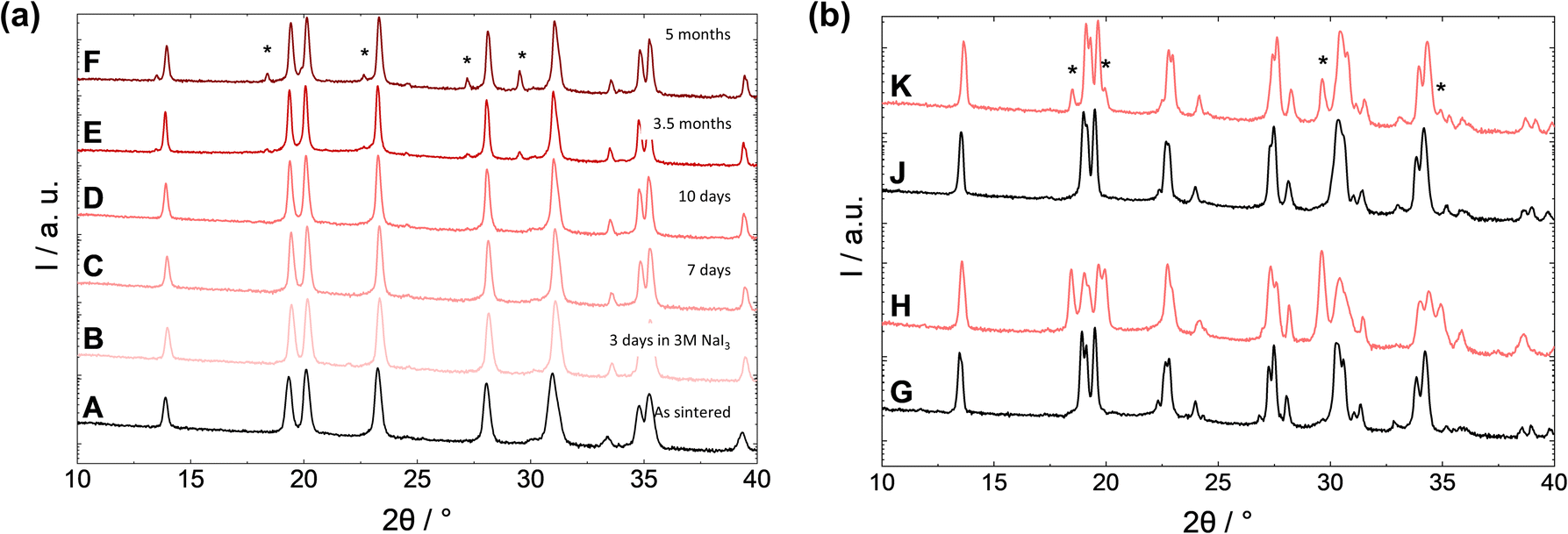 | ||
| Fig. 4 (a): XRD of NZP1.0 before (A) and after exposure to 3 M NaI3 for different times (B–F): 3 days (B), 7 days (C), 10 days (D), 3.5 months (E), 5 months (F). (b): XRD of stoichiometric (G and H) and sub-stoichiometric NZSiP3.0 (I and J) before (G and I) and after exposure to 3 M NaI3 solution (H and J). | ||
Unfortunately, an uncompensated reduction in Na content leads to a reduction in charge carrier concentration and thus to a greatly reduced ionic conductivity,43 so the anionic substitution was further varied to allow a lower Na concentration without significant conductivity loss. As a possible strategy, Holzapfel et al.19 suggested varying the oxygen sub-stoichiometry in a highly ionically conductive Na3.3Zr2Si2.3P0.7O12 phase, following the work of Balagopal & Flinders.44 By reducing the relative O amount, the Na content could be lowered to only 3 Na atoms per structural unit in a sub-stoichiometric Na3.0Zr2Si2.3P0.7O11.85 phase. The sub-stoichiometric composition was compared with a stoichiometric Na3.0Zr2Si2PO12 composition with a similar Na content (Fig. 4b). Both materials show the same monoclinic NaSICON structure with a minor ZrO2 secondary phase. However, after only ten days in 3 M NaI3 solution, about 50% of the stoichiometric NaSICON has transformed into the proton-exchanged phase. In stark contrast, the sub-stoichiometric NaSICON showed a significantly lower amount of the proton-exchanged phase. This means that the further extraction of Na+ ions from the NaSICON lattice is hindered if there is already a sodium deficiency to begin with.
Impact of NaSICON membrane degradation on the electrochemical performance
To evaluate the microstructure effects, sintered dense pellets made of different NaSICON materials were used to represent the dense separators in a possible full cell. The pellets were exposed to the same conditions as the powders (3 M NaI3 solution at 100 °C) to analyze the effects of the observed variations in degradation on the electrochemical performance.
For the powder samples, no significant difference was observed between stoichiometric NaSICON compositions with the same Na content. In contrast to the powders, remarkable differences in performance were observed for the NaSICON pellets. After just one hour in the 3 M NaI3 solution, the pellets discolored from white to brownish/purple for all materials, although to varying degrees depending on the composition (Fig. 5). The discoloration persisted even after thoroughly rinsing the pellets with demineralized water and ethanol to remove NaI/I2. Most striking, however, is the degradation of the Hf-substituted pellets of NHSiP3.4. Within one hour, the samples lost about 1% of their weight and became so brittle that they broke. With the naked eye we could observe the infiltration of the aqueous solution along the grain boundaries. At low pressure, the pellet broke along the visible lines. The degree of discoloration in the other materials correlates with the relative density of the sintered ceramic, which shows more color the lower the density. While most samples show little weight loss, the NZSiP3.4 sample increases in weight after exposure to the solution. Most likely due to the high porosity of the pellet of about 10% (Table 1), the solution can penetrate the sample so deeply that it remains there even after cleaning and drying.
 | ||
| Fig. 5 Nyquist plots of the electrical impedance spectra of NaSICON pellets before and after exposure to 3 M NaI3 solution, as well as after sanding 10 μm off each side of the pellet after exposure: (a) NZSiP3.4_SSR; (b) NZSiP3.0sub. | ||
In addition to the color, the impedance of the pellets also changes after exposure to the catholyte solution. For the as prepared pellets with different composition, the Nyquist plots of the electrical impedance spectra look very similar, showing a semicircle corresponding to the grain boundary resistance (RGB) with an onset at high frequencies corresponding to bulk resistance of the NaSICON phase, and a diffusion part corresponding to the gold blocking electrodes. After exposure to the catholyte, the impedance spectra become much more complex, and a wide suppressed semicircle is observed in the Nyquist plots, consisting of several overlapping semicircles that cannot be fitted with sufficient accuracy. Several phenomena complicate the identification of an appropriate physical model, such as surface degradation, surface contamination and degradation along the grain boundaries that can occur to different extent on the surface and in deeper parts of the pellet depending on how deep the catholyte is able to penetrate due to porosity. The total resistance of the cells, estimated from the intersection of the suppressed semicircle on the real part of the Nyquist plots at low frequencies, increases to different degrees depending on the composition. Among the materials with stoichiometric sodium content, the NZSiP3.4_SSR (Fig. 6), the NScZSiP3.4 (ESI Fig. S9†) and the NAlYZSiP3.4 (ESI Fig. S10†) show the smallest increase in total resistance of around 1000–2000 Ω cm. In addition to the composition, a high relative density of the sintered pellets is important for corrosion stability, because the higher the density, the less surface area is available for the solution to attack. The lower density sample such as NZSiP3.4 (ESI Fig. S11†) shows the greatest change in weight and a much greater increase in total resistance of about 8000 Ω cm. This can be explained by looking at the surface of the pristine and the etched samples in Fig. 7 and 8. Despite thorough rinsing of the sample with ethanol after etching, the entire surface is covered in a layer of sodium iodide (Fig. 8a). Most likely, enough NaI3 solution had penetrated through the porous structure deeper into the pellet, which could not be removed entirely, despite cleaning with ethanol. During the drying of the pellet, the solution must have propagated to the surface, leaving the observed layer after full evaporation of the solvent. After abrading about 10 μm from each side of the etched pellets, the total resistance of most materials decreases significantly. This shows that the increase in resistance is mainly a surface phenomenon. The only sample that did not show the decrease in resistance after polishing is also the only one that showed an increase in mass after etching. An analysis of the fracture surface of such a pellet (Fig. 8b) revealed a nanometer-sized deposit all over the NaSICON grains and a detection of iodine into a sample depth of as much as 30 μm or more. Residual material from the deeper pores must be brought to the surface after polishing and causes additional interlayer resistance. Overall, it can be said that the NaSICON samples with the same Na content degrade at a similar rate and high porosity is detrimental to the performance of the ceramic. Of all the materials tested, the NaSICON pellet made from sub-stoichiometric NaSICON powder NZSiP3_sub, which exhibited high corrosion stability in the powder tests, showed no discoloration, minimal mass loss and a very small increase in total resistance of less than 100 Ω cm (Fig. 6) after exposure to the catholyte.
 | ||
| Fig. 6 Sintered dense NaSICON ceramics after one hour exposure to 3 M NaI3 solution and measured weight gain/loss in percent. | ||
 | ||
| Fig. 7 SEM images of the surfaces of NZSiP3.0sub and NZSiP3.4 before and after etching of the sample in 3 M NaI3 solution at 100 °C for one hour. | ||
 | ||
| Fig. 8 SEM and EDX investigation of NZSiP3.4 pellets after etching in 3 M NaI3 solution at 100 °C for one hour: (a) EDX mapping of the surface; (b) SEM images of the pellet cross-section (fracture surface) (left, center) and EDX spectra (right) taken from spots at different sample depths as indicated in the SEM image (center). | ||
This confirms that the correlation between Na content and chemical stability observed in the powder tests also applies to sintered pellets. Since the sub-stoichiometric NaSICON exhibits a high ionic conductivity of 2.8 mS cm−1 at RT (which is only slightly lower than the highest reported values for NaSICON sodium ion conductors45 and higher than the values reported for the stoichiometric composition with the same Na content, i.e. Na3.0Zr2Si2PO12 (ref. 43)), these materials are promising candidates for corrosion-stable separators for aqueous sodium batteries. Indeed, the anionic substitution with increasing Si content increases the ionic conductivity even if the Na content is not increased, and the chemical stability increases with reduced Na content in the NaSICON structure.
The long-time corrosion stability of the membrane made of sub-stoichiometric NZSiP3.0sub was tested in the 3 M NaI3 solution over a longer period of time. The impedance measurements were carried out after different intervals (Fig. 9). After seven days of etching, the sub-stoichiometric material also showed a clear discoloration. The impedance data show that the bulk ionic conductivity remains about the same, while the grain boundary resistance increases steadily. In the Nyquist plot, further semicircles appear at lower frequencies, which cannot yet be assigned to corresponding processes, as discussed above. It is conceivable that a semicircle at 50 kHz to 50 Hz is related to the formation of a brownish layer on the pellet surface, which can already be observed after seven days of immersion in the catholyte (Fig. 9, inset). The real part of the impedance obtained by semicircle fitting remains approximately constant at around 10000 Ohm.
 | ||
| Fig. 9 Nyquist plot of the electrical impedance spectra of NZSiP3.0sub pellet after different exposure times to 3 M NaI3 solution at 100 °C. Inserts of small impedance magnification and pellet image after 7 days of etching. | ||
From the comparison of the stability of powders versus sintered pellets, which are possible separators in full cells, the strong influence of sintering becomes clear. The grain boundaries and secondary phases in particular are attacked by the electrolyte solution, which leads to failure of the component, especially if the relative density is low and the electrolyte can penetrate into the separator. This clearly shows the importance of testing materials on the component level and not only as powders to fully assess the impact of the microstructure on degradation.
 | ||
| Fig. 10 Polarization curves of NaSICON pellets cycled in symmetric cells with NaI/I2 catholyte solutions (80% SOC) at 5 mA cm−2 (a) and 1 mA cm−2 (b) at 80 °C, and XRD pattern of the surface of NaSICON pellets after cycling (c). | ||
 | ||
| Fig. 11 (a–c) Cycling data of a NaI/I2|NaSICON|NaI/I2 symmetrical cell: (a) cell resistance as a function of cycle number; (b) cell potential as a function of capacity representative for cycle number 100; (c) an image of the cell setup. (d–f) Cycling data of a Na|NaSICON|NaI/I2 full cell: (d) cell potential as a function of time; (e) galvanostatic charge/discharge curves; (f) cell resistance as a function of cycle number. An image of the cell setup is shown in inset. | ||
Similar to the difference between powder and separator testing, these effects demonstrate the significant differences between ex situ and in situ degradation of separators. While “self-healing” was not observed in the ex situ measurements, the interaction between the metal anode, which serves as Na reservoir, and the dynamic changes in I3− concentration during cycling leads to additional reactions in the cracks of the separator that fill them and prevent the cell from failing. Even if the phases formed are not as mechanically stable as the original NaSICON, the separator “heals” to a certain extent and thus extends the life of the cell.
Conclusions
In this study, we analyzed the factors influencing the corrosion stability of NaSICON membranes in aqueous medium-temperature sodium-iodine batteries. Our results provide a comprehensive understanding of the degradation mechanisms and offer strategies for future improvements of NaSICON membrane technology in such cells. The presence of free iodine, the acidity of the aqueous catholyte, the sodium content in the NaSICON structure, the effect of sintering and compositional variations, the balance between ionic conductivity and structural integrity, and the influence of temperature on solubility were identified as key factors for degradation.Free iodine has been pinpointed as one of the major factors in the stability of NaSICON. XRD only points to the formation of protonated NaSICON phase. Therefore, we can suggest the higher corrosion activity of the iodine containing solution is not related to direct reaction of NaSICON with iodine, but should be related to other processes due the presence of iodine. One of the most important processes is the disproportionation of I2 in water. This adds to the acidity of the solution and accelerates the degradation process due to H+/Na+ exchange. More importantly, the sodium content in the NaSICON composition has been shown to play a crucial role. Structures with a lower sodium content exhibit a higher resistance to sodium extraction and consequently a better stability against proton exchange. Furthermore, our research highlights the importance of the sintering process and compositional variations. Although the substitution of elements such as Zr does not directly affect proton exchange in powders, it has a noticeable impact on the chemical stability of sintered pellets, most likely due to its effects on the microstructure (grain boundaries, secondary phases etc.). EDX analysis revealed the presence of iodine in sodium phosphate phase which denotes direct reaction of iodine with phases which are typically present along grain boundaries of sintered polycrystalline NaSICON.
Our results clearly show the importance of the delicate balance between high ionic conductivity and structural integrity. We were able to show that sub-stoichiometric NaSICON exhibits high ionic conductivity and can withstand high current densities over extended periods of time, confirming its potential for practical applications. However, the challenges associated with temperature and solubility, particularly the risk of NaI oversaturation and precipitation, underscore the need for careful temperature management to avoid stress and crack formation within the NaSICON membrane.
Based on these findings, we propose several guidelines for the future development of NaSICON membranes. Optimizing the sodium content to balance conductivity and stability, refining the sintering and surface treatment process to improve pellet quality, controlling the state of charge to manage iodine concentration and acidity, and exploring salt complexing strategies to mitigate iodine-induced degradation are the key factors for optimizing performance. In addition, setting operational limits for current densities and controlling operating temperatures are critical to extending membrane life.
In addition to Na–I batteries, understanding the degradation mechanisms of NaSICON membranes and the strategies identified here to improve their durability and performance can also be useful for other aqueous battery systems, such as seawater-based batteries or membranes for Na extraction from brine solutions. Our research provides a roadmap for the development of more durable and efficient NaSICON membranes for a variety of applications. Overcoming the inherent challenges through a combination of material optimization and operational adaptations paves the way for the next generation of medium to large-scale energy storage solutions using NaSICON-based technologies.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: G. D., M. H. and M. F.; data curation: G. D., F. S. and J. G.; formal analysis: G. D., F. S., J. G., S. R. and T. A.; funding acquisition: M. H., M. F., R. S. and D. F.; investigation: G. D., F. S., J. G., S. R., T. A. and M. H.; methodology: G. D., F. S., J. G., S. R., T. A., M. H. and M. F.; project administration: M. H. and M. F.; resources: M. H., M. F., R. S. and D. F.; supervision: M. H., M. F., T. A., R. S. and D. F.; validation: G. D. and F. S.; writing – original draft: G. D., M. F. and D. F.; writing – review & editing: G. D., F. S., J. G., M. H., M. F., R. S. and D. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr S. W. Scheld for SEM investigations. We gratefully acknowledge the financial support by the Helmholtz Association and the Federal Ministry of Education and Research (BMBF) within the MEET-HiEnD III project (Grant No. 13XP0258B) and the project MiTemp (Grant No. 13XP0183B).Notes and references
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS.
- J. T. Kummer and W. Neill, US Pat., US3413150A, Ford Motor Co., 1966 Search PubMed.
- J. T. Kummer, Prog. Solid State Chem., 1972, 7, 141–175 Search PubMed.
- Y. Lu, J. B. Goodenough and Y. Kim, J. Am. Chem. Soc., 2011, 133, 5756–5759 Search PubMed.
- Y. Wang, Y. Wang and H. Zhou, ChemSusChem, 2011, 4, 1087–1090 Search PubMed.
- Y. Zhao, L. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- Y. Zhao, M. Hong, N. Bonnet Mercier, G. Yu, H. C. Choi and H. R. Byon, Nano Lett., 2014, 14, 1085–1092 CrossRef CAS PubMed.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 Search PubMed.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 Search PubMed.
- C. Lee, T.-U. Wi, W. Go, M. F. Rahman, M. T. McDowell, Y. Kim and H.-W. Lee, J. Mater. Chem. A, 2020, 8, 21804–21811 RSC.
- S. Arnold, L. Wang and V. Presser, Small, 2022, 18, 2107913 Search PubMed.
- H. Zhu and R. J. Kee, Electrochim. Acta, 2016, 219, 70–81 Search PubMed.
- Q. Ma and F. Tietz, ChemElectroChem, 2020, 7, 2693–2713 CrossRef CAS.
- S. Fan, M. Lei, H. Wu, J. Hu, C. Yin, T. Liang and C. Li, Energy Storage Mater., 2020, 31, 87–94 CrossRef.
- Q. Yu, J. Hu, X. Nie, Y. Zeng and C. Li, ACS Nano, 2024, 18, 5790–5804 CAS.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- X. Lu, G. Xia, J. P. Lemmon and Z. Yang, J. Power Sources, 2010, 195, 2431–2442 CrossRef CAS.
- L. Viswanathan and A. V. Virkar, J. Mater. Sci., 1982, 17, 753–759 CrossRef CAS.
- M. Holzapfel, D. Wilde, C. Hupbauer, K. Ahlbrecht and T. Berger, Electrochim. Acta, 2017, 237, 12–21 CrossRef CAS.
- L. J. Small, J. S. Wheeler, J. F. Ihlefeld, P. G. Clem and E. D. Spoerke, J. Mater. Chem. A, 2018, 6, 9691–9698 Search PubMed.
- J. B. Goodenough, H. Y. P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 Search PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 Search PubMed.
- H. Schmid, L. C. De Jonghe and C. Cameron, Solid State Ionics, 1982, 6, 57–63 CrossRef CAS.
- V. Lacivita, Y. Wang, S.-H. Bo and G. Ceder, J. Mater. Chem. A, 2019, 7, 8144–8155 RSC.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS PubMed.
- Y. Zhang, C. Wang, G. Pastel, Y. Kuang, H. Xie, Y. Li, B. Liu, W. Luo, C. Chen and L. Hu, Adv. Energy Mater., 2018, 8, 1800635 CrossRef.
- L. Shen, J. Yang, G. Liu, M. Avdeev and X. Yao, Mater. Today Energy, 2021, 20, 100691 CrossRef CAS.
- Q. Ma, T. Ortmann, A. Yang, D. Sebold, S. Burkhardt, M. Rohnke, F. Tietz, D. Fattakhova-Rohlfing, J. Janek and O. Guillon, Adv. Energy Mater., 2022, 12, 2201680 CrossRef CAS.
- F. Mauvy, E. Siebert and P. Fabry, Talanta, 1999, 48, 293–303 CrossRef CAS PubMed.
- M. Guin, S. Indris, M. Kaus, H. Ehrenberg, F. Tietz and O. Guillon, Solid State Ionics, 2017, 302, 102–106 CrossRef CAS.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y. S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- H. Zhang, S. Jeong, B. Qin, D. Vieira Carvalho, D. Buchholz and S. Passerini, ChemSusChem, 2018, 11, 1382–1389 CrossRef CAS PubMed.
- S. Naqash, Q. Ma, F. Tietz and O. Guillon, Solid State Ionics, 2017, 302, 83–91 CrossRef CAS.
- J. P. Boilot, G. Collin and P. Colomban, Mater. Res. Bull., 1987, 22, 669–676 CrossRef CAS.
- E. A. Cheung, H. Nguyen, M. Avdeev, N. R. de Souza, Y. S. Meng and N. Sharma, Chem. Mater., 2021, 33, 8768–8774 CrossRef CAS.
- R. O. Fuentes, F. Figueiredo, F. M. B. Marques and J. I. Franco, Ionics, 2002, 8, 383–390 CrossRef CAS.
- A. K. Kuriakose, T. A. Wheat, A. Ahmad and J. Dirocco, J. Am. Ceram. Soc., 1984, 67, 179–183 CrossRef CAS.
- Z. Khakpour, Electrochim. Acta, 2016, 196, 337–347 CrossRef CAS.
- J. P. Gross, G. Dück, F. Schäfer, M. Holzapfel, M. Finsterbusch, J. Malzbender and R. Schwaiger, J. Mater. Sci., 2023, 58, 144–156 CrossRef CAS.
- S. Song, H. M. Duong, A. M. Korsunsky, N. Hu and L. Lu, Sci. Rep., 2016, 6, 32330 CrossRef CAS PubMed.
- Q. Ma, M. Guin, S. Naqash, C.-L. Tsai, F. Tietz and O. Guillon, Chem. Mater., 2016, 28, 4821–4828 CrossRef CAS.
- E. M. Vogel, R. J. Cava and E. Rietman, Solid State Ionics, 1984, 14, 1–6 CrossRef CAS.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- S. Balagopal and M. Flinders, US Pat., US8246863B2, Ceramatec, Inc., Salt Lake City, UT (US), 2012 Search PubMed.
- Q. Ma, C.-L. Tsai, X.-K. Wei, M. Heggen, F. Tietz and J. T. S. Irvine, J. Mater. Chem. A, 2019, 7, 7766–7776 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05638h |
| This journal is © The Royal Society of Chemistry 2025 |