
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the stability and protonic conductivity in W- and Mo-substituted LaNbO4 under a reducing atmosphere†
Kehan
Huang‡
a,
Yidong
Han‡
a,
Mark A.
Isaacs
 bc and
Stephen J.
Skinner
*ad
bc and
Stephen J.
Skinner
*ad
aDepartment of Materials, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: s.skinner@imperial.ac.uk
bDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
cHarwellXPS, Research Complex at Harwell, Rutherford Appleton Labs, Harwell Campus, Didcot, OX11 0FA, UK
dInternational Institute for Carbon Neutral Energy Research, Kyushu University, Fukuoka, Japan
First published on 23rd December 2024
Abstract
LaNb0.84W0.16O4+δ (LNW16) and LaNb0.84Mo0.16O4+δ (LNM16) have been synthesized via a solid-state reaction route. Both samples underwent 5%-H2 treatments at 800 °C to investigate their stability under a reducing atmosphere. The crystal structures, compositions, and oxidation states were studied before and after treatment showing excellent structural, compositional and redox stability of LaNb0.84W0.16O4+δ, while LaNb0.84Mo0.16O4+δ is reduced significantly. Therefore, the electrochemical performance of LaNb0.84W0.16O4+δ under reducing atmospheres was further investigated. Higher conductivity was observed for the LNW16 samples measured under reducing atmospheres. However, the larger conductivity enhancement in the low temperature range (400 °C to 600 °C) compared with the high temperature range (600 °C to 800 °C) deviated from the conventional electronic conductivity emerging from reduction. DC polarization measurements further proved the predominant protonic conductivity in LaNb0.84W0.16O4+δ under reducing atmospheres.
1 Introduction
LaNbO4-based materials present excellent chemical stability in different atmospheres, especially in CO2-containing atmospheres with appreciable ionic conductivity, which makes this type of material a promising electrolyte for solid oxide fuel cells (SOFCs) and solid oxide electrolysis cells (SOECs).1,2 The recorded protonic conductivity of 0.001 S cm−1 is achieved in 1 at% Ca-substituted LaNbO4 at 950 °C in wet air (pH2O = 0.025 atm).1 However, this conductivity is still not enticing compared to other perovskite-based electrolytes, such as BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb).3 Since these reports, researchers have strived to improve the ionic conductivity by exploring different substitutions such as Sr on the A-site and Ga on the B-site.4,5 Yet, none of them can overcome the low conductivity issue caused by the low substitution solubility.6 Worse, the phase transition nature of LaNbO4-based materials at the device operating temperature tends to introduce a mechanical mismatch between the electrolyte and electrodes further compromising its application.7Recently, new studies have shown that the low solubility and mechanical mismatch issues can be overcome by substituting Nb with hexavalent elements such as W and Mo.8–11 Li et al. studied B-site W-substituted LaNbO4 with different substitution levels and found that the high-temperature tetragonal scheelite-type structure is maintained at room temperature when the substitution level is up to 16 at%.11 Mo substitution exhibits a similar high-temperature phase stabilisation behaviour.9 Compared to other low-solubility, typically lower than 2 at%, substitutions, such as Ca and Sr, the solubility upper limit of W and Mo is about 20 at%, which enables much higher conductivity.9,11,12 While the pure oxide ion conduction and excellent chemical stability in W- and Mo-substituted LaNbO4 were clearly demonstrated by previous works, some authors reported an enhancement of electronic conductivity under reducing atmospheres.8,13 However, the structural and chemical origins of this enhanced conduction under reducing atmospheres remain unknown.
In this work, we aim to understand how reducing atmospheres affect the chemical stability of W- and Mo-substituted LaNbO4 and to explore the change of conductivity under this condition. This work primarily focuses on 16 at% W-substituted LaNbO4, as it shows excellent structural, compositional and redox stabilities under reducing atmospheres. The substitution level of 16 at% was selected to ensure that the tetragonal structure would be maintained through the entire operating temperature range, from 400 °C to 800 °C.11,14 Multiple characterisation techniques, such as X-ray diffraction, X-ray photoelectron spectroscopy and X-ray absorption near edge spectroscopy, are used to probe any possible changes in W- and Mo-substituted LaNbO4 before and after the heat treatment under reducing atmospheres. Electrochemical impedance spectroscopy is used to study the conductivity behaviour of the W-substituted composition.
2 Experimental details
2.1 Sample preparation
La2O3 (99.9%, Alfa Aesar), Nb2O5 (99.99%, Sigma-Aldrich), WO3 (99.8%, Alfa Aesar), and MoO3 (99.95%, Alfa Aesar) were chosen as starting powders. The La2O3 powder was calcined at 1000 °C for 12 hours to ensure dehydration.15 All powders were weighed appropriately and mixed in a rotary ball mill with acetone for 24 h. The mixture was dried at 95 °C for 48 hours to remove the acetone and then calcined at 400 °C for 12 hours to be fully dried. LaNb0.84W0.16O4+δ (denoted as LNW16) and LaNb0.84Mo0.16O4+δ (denoted as LNM16) powder samples were synthesised from these precursors at 1400 °C for 10 hours, followed by manually grinding and passing through a 53 μm sieve. LNW16 and LNM16 pellet samples (Φ = 13 mm) were prepared using a uniaxial press held at 360 MPa for 3 minutes. The sintering condition was the same as the powder samples. The relative densities of LNW16 and LNM16 pellet samples obtained from the Archimedes method were 95.62% and 92.97%, respectively. All the calcination and sintering processes were conducted under ambient atmosphere with a heating and cooling rate of 10 °C min−1.To conduct X-ray photoelectron spectroscopy (XPS) and ultraviolet-visible diffuse reflectance spectroscopy (UV-vis-DRS) measurements, the as-sintered pellets were ground and polished sequentially to 0.25 μm using diamond suspensions (Struers UK Ltd) to minimize the surface roughness effect. After polishing, the samples were ultrasonicated in acetone and deionized water to remove contaminants introduced during the polishing process. These samples were further heat-treated at 400 °C for 4 hours to completely eliminate contaminants.
For 5% H2-treated powders, pristine and polished pellets, the heat treatment was conducted at 800 °C for 2 hours with a heating and cooling rate of 10 °C min−1. The reducing atmosphere was realised by passing forming gas (5% H2/N2) through the tube furnace during the heat treatment, after an initial purge to remove residual air.
2.2 Sample characterisation
The crystal structures of LNW16 and LNM16 powders were determined by X-ray powder diffraction (XRD) using a PANalytical X'Pert PRO diffractometer (Cu Kα radiation, λ = 1.5406 Å). The data were collected in the 2θ range of 20° to 80° with the step size of 0.017° and a holding time of 150 s per step. Rietveld refinement was conducted to determine the lattice parameters of the phases using the GSAS-II software.16 The modulation structure was confirmed with selected area electron diffraction obtained with a JEOL JEM-2100Plus transmission electron microscope (TEM). The d-spacing values and the Miller indices were measured and calculated using GMS 3 software (Gatan Microscopy Suite). The morphology of the pellet samples was investigated with a JEOL JSM 6010 LA SEM. The homogeneity of the phase was confirmed by the backscattered electron (BSE) mode and energy dispersive X-ray spectroscopy (EDS) in mapping mode. The chemical compositions were analysed using EDS (Oxford instrument) implemented in a LEO-1525 FEGSEM (Zeiss). A 15 nm thick chromium layer was sputtered on the top of the samples as a conducting layer. The average grain size was determined using ImageJ.17The oxidation states of each element were investigated using a K-Alpha X-ray photoelectron spectroscopy instrument (Thermo Fisher Scientific). A monochromatic Al Kα (1486.6 eV) X-ray source was used to record the XPS spectra. The spot size of the X-ray beam was 400 μm2. The energy step size was set to 0.1 eV. A dual beam (Ar+ and electron) flood gun was turned on during the measurements to compensate for the charge accumulation. The samples were measured under high vacuum (<5 × 10−7 mbar) conditions. The charge accumulation was corrected by reference to the C 1s signal (284.8 eV). The iteration XPS spectra were collected by setting time-spent of each scan to 30 or 60 minutes, and repeated multiple times. The obtained spectra were analysed and peak fitted using Avantage software (Thermo Fisher Scientific). The Voigt function and Shirley background were used during the peak fitting process.
To further confirm the oxidation state of W and Mo, synchrotron X-ray absorption near edge spectra (XANES) at the W L3-edge and Mo K-edge were collected in transmission mode with the optimized energy step size of 0.35 eV on B18 core beamline at Diamond Light Source (DLS, UK).18 The L3-edge of W metal foil (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 207 eV) and K-edge of Mo metal foil (20000 eV) were used to calibrate the energy shift. Each spectrum was merged from 6 scans and the resulting standard deviation was estimated. The W L3-edge is at an energy that is accessible using bench-top systems, and hence the oxidation state of W can be further investigated using an easyXAFS300 benchtop X-ray absorption spectrometer (EasyXAFS, LLC) in transmission mode located at the Research Complex, Harwell, UK. The energy shift was again calibrated with W metal foil (10207 eV). All samples for XANES measurements were diluted with cellulose powder and pressed into pellets with a diameter of 13 mm and a thickness of approximately 1 mm. The collected XANES data were analysed using the Demeter software package.19
207 eV) and K-edge of Mo metal foil (20000 eV) were used to calibrate the energy shift. Each spectrum was merged from 6 scans and the resulting standard deviation was estimated. The W L3-edge is at an energy that is accessible using bench-top systems, and hence the oxidation state of W can be further investigated using an easyXAFS300 benchtop X-ray absorption spectrometer (EasyXAFS, LLC) in transmission mode located at the Research Complex, Harwell, UK. The energy shift was again calibrated with W metal foil (10207 eV). All samples for XANES measurements were diluted with cellulose powder and pressed into pellets with a diameter of 13 mm and a thickness of approximately 1 mm. The collected XANES data were analysed using the Demeter software package.19
Thermogravimetric analysis (TGA) measurements were conducted using a STA 449 F5 Jupiter simultaneous thermal analyser (Netzsch GmbH, Germany). The heating and cooling rates were 10 °C min−1. The gas flow rate was 50 ml min−1 for the purge gases and 20 ml min−1 for the protective gas (argon). The background, determined by a calibration measurement using Al2O3 powder as a reference, was subtracted and the obtained data were analysed using Proteus software (Netzsch GmbH, Germany). Approximately 30 mg of 5% H2-treated powders were used during each TGA measurement.
To plot the band structure diagrams, UV-vis-DRS and XPS valence band spectroscopy were combined to measure the band gap values as well as the valence band maximum (VBM) positions relative to the Fermi level. The absorption spectra were recorded from 800 nm to 200 nm using a Cary-5000 spectrophotometer (Agilent Technologies, Inc) with the diffuse reflectance sphere accessory and WinUV software. A polytetrafluoroethylene (PTFE) disk was used as the reference to calibrate the absorption baseline. The data interval and the acquisition time per step were set to 0.1 nm and 0.1 s, respectively. An observed data glitch at 350 nm was due to the source changeover. A Tauc plot was calculated and the band gap values were extrapolated by applying the Kubelka–Munk function.20,21 The XPS valence band spectra were measured simultaneously with the oxidation state measurements using the same setup.
To conduct electrochemical impedance spectroscopy (EIS) measurements, platinum paste was brush painted on both sides of the samples and followed by calcining at 900 °C for 1 hour to achieve good adhesion. The platinum electrode area was confined to 0.5 cm2. The data were collected using a Solartron 1260A impedance analyser implemented with a Solartron 1296 dielectric interface with a two-probe configuration. The temperature range was from 400 °C to 800 °C with an interval of 50 °C. The actual temperatures were recorded using a K-type thermocouple placed beside the sample. The frequency range was from 1 MHz to 10 Hz, with 30 sampling points per logarithm decade. An AC potential of 100 mV was applied during the measurements without DC bias. The as-sintered samples underwent heat treatments at 800 °C for 2 hours under different atmospheres before impedance measurements were made in the same atmospheres (O2, compressed air, N2, forming gas (5% H2/N2), BOC UK Ltd). The gases were humidified by pumping the gas through a water bubbler at room temperature (pH2O ≈ 0.025 atm), while the dry atmospheres were obtained by pumping the gases through a commercial gas dryer (Drierite 26800 Drying column, pH2O ≈ 10−5 atm). All obtained data were analysed and fitted using ZView4 software (v4.0h, Scribner Associates).
To conduct DC polarization (Hebb–Wagner) measurements, the ion-blocking polarization cell was synthesized in the configuration of Pt disk/LNW16/porous Pt.22 The platinum paste was brush painted on both sides of the sample as the current collector and followed by the same calcination process mentioned above. The dense platinum disk was fully covered on one side of the sample. This disk works as the ion-blocking electrode and the electrode area was confined to 0.28 cm2. The high-temperature ceramic adhesive (Aremco Ceramabond 885) was used to ensure good sealing between the sample and disk. Then the adhesive was dried at room temperature for 2 hours and then gradually heated up to 800 °C to ensure a gastight seal through the entire operating temperature range. The schematic cell configuration is shown in Fig. S1† which has been widely applied in many works.22–25 A constant DC voltage was applied to the sample by using the electrochemical station (ModuLab XM ECS Electrochemical System, Solartron Analytical Ltd) and the DC current data were collected using the same station. The rest of the setup was the same as that used for the EIS measurements.
3 Results and discussion
3.1 Structural and chemical study
The pristine, undoped LaNbO4 presents a monoclinic phase (I2/a) at low temperature, and transforms to a tetragonal phase (I41/a) at high temperature with the phase transition temperature at approximately 500 °C.26 Given that the tetragonal phase (a = c, β = 90°) has a higher symmetry than the monoclinic phase (a ≠ c, β > 90°), the beta angle (β) was suggested to be used as an indicator of the symmetry of this type of material.9,11,27,28In Fig. 1 and S2,† the XRD patterns of both as-sintered LNW16 and LNM16 show a monoclinic phase (I2/a), while LNW16 shows a slightly higher symmetry (β = 90.6632°) than LNM16 (β = 90.9685°) based on the results of Rietveld refinement. In both cases satellite reflections, attributed to structure modulation, were observed as previously reported.11 This observation contradicts a previous study that suggested that the tetragonal phase could be stabilized at room temperature with 16% of W dopant.11 The relatively small β values obtained highlight the ease with which this monoclinic distortion of the cell can be misinterpreted as a tetragonal cell.7
 | ||
| Fig. 1 XRD patterns of as-sintered LNW16, 5% H2-treated LNW16 and after the 5% H2-annealing for 8, 16 and 24 hours, respectively. The 24 h 5% H2-treated sample was then air-annealed at 800 °C for 4 hours and is labelled as “Air-refired”. The satellite peaks are marked by the arrows. All patterns show a monoclinic phase (I2/a) with a high local coordination symmetry. | ||
After the 5% H2-anneal, the LNW16 phase remained structurally stable showing an almost identical XRD pattern to that obtained after synthesis. The slight sharpening of the main peaks and the merger of peaks at around 48° and 58° two theta can be observed after carefully comparing the patterns of as-sintered LNW16 with the 5% H2-treated one. However, when it came to LNM16 (Fig. S2†), the peaks were more separated with the disappearance of the satellite peaks, which showed that LNM16 cannot maintain its modulation structure in the 5% H2 annealing conditions. After the 5% H2-treatment, this phase became analogous to the undoped LaNbO4 due to the loss of oxygen ions which is further discussed in Section 3.2.8
The lattice parameters of all four samples were determined using Rietveld refinement (Fig. S3†) and the data are listed in Table S1.† Based on the refinements for both compositions, the 5% H2-treatment showed opposite effects on the unit cell volumes: expansion for LNW16 but shrinkage for LNM16. The shrinkage in LNM16 is attributed to local structural changes as the unit cell became more distorted (β = 94.0040°). On the other hand, sharpening of the main reflections, merging of peaks and the volume expansion in LNW16 were due to the relaxation of the monoclinic distortion.7,11 As LNW16 maintained its structural stability under the 5% H2 condition, this treatment could work as an annealing process. To prove that this volume expansion for LNW16 was not due to the 5% H2-atmosphere, the as-sintered LNW16 powder was air-refired using the same temperature program as the 5% H2-treatment. Similar expansion and the relaxation of the monoclinic distortion were suggested by the Rietveld refinements (Table S1†).
The structural stability of LNW16 and LNM16 was further investigated by elongating the 5% H2-treatment time to 8, 16 and 24 hours, respectively. After that, the 24 h-5% H2-treated samples were air-fired at 800 °C for 4 hours. The almost identical XRD patterns shown in Fig. 1 suggest that LNW16 exhibited excellent structural stability under the 5% H2 condition. On the other hand, XRD patterns of LNM16 shown in Fig. S2† suggest that the reduced phase (β = 94.0040°) remained unchanged regardless of the treatment time elongation. However, this reduced phase can be fully re-oxidized after air-refiring. The merger of the main peaks in the range 27° to 30° two theta also suggested the higher symmetry of re-oxidized LNM16 compared with as-sintered LNM16. This was likely due to the similar relaxation of the monoclinic distortion.9,13
The satellite peaks marked by the arrow in Fig. 1 are attributed to the (3 + 2)D incommensurate modulation structure, which is consistent with previous studies.11,13,14 This modulation structure was reported to be highly correlated with the interstitial oxygen defects.29,30 Therefore, it suggests that the interstitial oxygen defects remained in LNW16 but were absent in LNM16 after the 5% H2-treatment. The presence of this modulation structure was further confirmed by TEM and selected area electron diffraction (SAED) for LNW16 (Fig. 2a and b). The calculated d-spacing from TEM is 1.75 nm which refers to the modulation structure indexed by the modulation vectors of q1 ≈ 0.23a + 0.18c and q2 ≈ −0.15a + 0.22c. No modulation along the b axis was observed. The similar satellite peak and the corresponding modulation structure were also found in Mo-substituted LaNbO4 materials and were extensively investigated elsewhere.14
 | ||
| Fig. 2 (a) TEM image of as-sintered LNW16. The calculated d-spacing is 1.75 nm which refers to the modulation structure; (b) SAED pattern of as-sintered LNW16 along the [010] zone axis. A (3 + 2)D modulation structure is clearly confirmed by the presence of satellite reflections (q1 and q2). | ||
Fig. 3a and c show the microstructures of as-sintered and 5% H2-treated LNW16. The extrapolated average grain sizes of both samples were 5.86 ± 1.19 μm. Very few pores were observed suggesting that the sintering conditions were appropriate. The BSE images and EDS maps of both as-sintered and 5% H2-treated LNW16 indicate good phase homogeneity (Fig. 3(b, d) and 4). The measured W contents are presented in Table 1. These values showed good agreement with the theoretical values and suggested that there was no noticeable compositional change after the 5% H2-treatment. The situation was similar in the LNM16 samples (Fig. S4(a and c)†). The extrapolated average grain sizes of both samples were 5.88 ± 0.92 μm. The BSE images and EDS data of as-sintered and 5% H2-treated LNM16 also indicate good phase homogeneity (Fig. S4(b, d) and S5†), and the Mo contents are presented in Table S2.†
 | ||
| Fig. 3 (a and b) SEI and BSE images of as-sintered LNW16; (c and d) SEI and BSE images of 5% H2-treated LNW16. | ||
 | ||
| Fig. 4 (a–c) Corresponding EDS maps of SEM images in Fig. 3(a); (d–f) corresponding EDS maps of SEM images in Fig. 3(c). | ||
| Cation | As-sintered (at%) | 5% H2-treated (at%) | Theoretical (at%) |
|---|---|---|---|
| Nb | 84.7 ± 0.4 | 84.6 ± 0.5 | 84.0 |
| W | 15.3 ± 0.2 | 15.4 ± 0.2 | 16.0 |
3.2 Oxidation state and oxygen stoichiometry study
After the 5% H2-treatment, an obvious color change was observed for both LNW16 and LNM16. The colour of both samples changed from green to black (Fig. S6†). This coloration effect is common with both tungsten and molybdenum oxides as an indicator of the change in the oxidation state of W and Mo as defined by Kröger–Vink defect chemical equations (eqn (1)–(4)).31 | (1) |
 | (2) |
 | (3) |
 | (4) |
To verify these reactions, XPS spectra of all elements were collected for both LNW16 and LNM16 (Fig. 5 and S7†). The La 3d spectra are more complex than other spectra due to the multiplet-splitting, but the magnitude of the multiplet-splitting is a good indicator for chemical state determination.32–34 Therefore, as the magnitudes of each La 3d5/2 peak were approximately 4.6 eV, this clearly showed that the oxidation state of La in all four samples remained stable as La3+ (Fig. 5(a) and S7(a)†). For Nb 3d spectra, all four samples showed well defined doublet peaks suggesting the oxidation state of Nb in all four samples also remained the same, Nb5+ (Fig. 5(b) and S7(b)†).
 | ||
| Fig. 5 Normalized XPS spectra of (a) La 3d, (b) Nb 3d; fitted XPS spectra of W 4f in (c) as-sintered and (e) 5% H2-treated LNW16; fitted XPS spectra of O 1s in (d) as-sintered and (f) 5% H2-treated LNW16. The LF in W 4f spectra represents the loss feature of W6+. | ||
For LNM16, the Mo 3d spectra show a significantly larger ratio of low oxidation states (Mo5+ and Mo4+) with the appearance of shoulders in the 5% H2-treated sample (Fig. S7(c and e)†). These shoulders can be attributed to new oxidation states of Mo which are lower than +4, denoted as δ1+, δ2+ and δ3+. In contrast, for LNW16, the W 4f spectra were almost identical (Fig. 5(c) and (f)). This difference suggested better redox stability of LNW16 than LNM16. The ratio of oxidation states of W and Mo are listed in Tables 2 and S3,† respectively. Note that XPS is not an ideal technique to identify the oxidation state of W and Mo as the flood gun can induce sample reduction to form a continuum of oxidation states.35,36 To verify this phenomenon, the XPS iteration spectra of W 4f and Mo 3d were collected (Fig. S8(a and b)†). The red arrow on each figure suggests the emergence of a feature corresponding to lower oxidation states of W and Mo. It also clearly shows that Mo was easier to reduce than W in this type of material. This analysis highlights that differentiating effects of the H2 treatment from beam effects using XPS is challenging and thus unambiguous determination of W and Mo oxidation states requires an alternative measurement protocol.
| Oxidation state | As-sintered (at%) | 5% H2-treated (at%) |
|---|---|---|
| W6+ | 67.7 (35.8 eV) | 63.0 (35.9 eV) |
| W5+ | 26.2 (34.7 eV) | 29.8 (34.7 eV) |
| W4+ | 6.1 (33.5 eV) | 7.2 (33.5 eV) |
| Oxygen species | As-sintered (at%) | 5% H2-treated (at%) |
|---|---|---|
| [O]lattice | 89.7 (530.5 eV) | 77.8 (530.6 eV) |
| [O]surface | 10.3 (531.5 eV) | 11.1 (531.6 eV) |
| –OH | — | 11.1 (532.9 eV) |
To overcome the challenges of using XPS for oxidation state determination, XANES measurements under ambient atmospheres for W L3-edge and Mo K-edge were conducted (Fig. 6 and S9†). The white line of W L3-edge is correlated with the 2p–5d transitions.37,38 The extrapolated white line energy of both W L3-edge spectra was found to be 10212 eV, which was consistent with the white line energy of W6+ in WO3.39 The standard deviations of both samples were smaller than 0.010 eV. The sharp white line of LNW16 compared to WO3 suggested it maintained an unchanged tetrahedral environment after the 5% H2 treatment.40 The benchtop XANES spectra of W L3-edge were in excellent agreement with the above synchrotron XANES results (Fig. S10†). Therefore, the oxidation state of W in as-sintered and 5% H2-treated LNW16 can be considered as purely W6+, and, in this case, the actual formula of LNW16 can be rewritten as LaNb0.84W0.16O4.08. This result is consistent with Canu's previous work.40
 | ||
| Fig. 6 Normalized synchrotron XANES spectra of W L3-edge measured at ambient condition. The white line of both samples is identical at 10212 eV indicating W6+ oxidation state. | ||
For the Mo K-edge spectra (Fig. S9†), the edge position (E0) of as-sintered LNM16 was the same as MoO3 at 20017 ± 0.005 eV showing purely Mo6+ in this sample. After the 5% H2-treatment, the E0 position shifted to 20015 ± 0.010 eV. Zhou et al. investigated a variety of Mo-containing materials and reported a linear correlation between the E0 position and the oxidation state of Mo, which can be approximated as below:41,42
| ΔE = 3δ − 1 | (5) |
000 eV) and the sample, and δ is the oxidation state of the sample. Then, the oxidation state of Mo in 5% H2-treated LNM16 was extracted to be +5.33. Therefore, the actual formula of as-sintered and 5% H2-treated LNM16 can be rewritten as LaNb0.84Mo0.16O4.08 and LaNb0.84Mo0.16O4.026, respectively. The pre-edge feature which corresponds to the forbidden 1s–4d transition together with the edge feature works as an indicator of the coordination environment of Mo cations.43 The higher intensity of the pre-edge feature and lower intensity of the edge feature in as-sintered LNM16 suggest a more tetrahedral environment, while the opposite suggest a more octahedral environment.43 This result is consistent with the XRD patterns of LNM16 as the higher symmetry phase (β = 90.9685°) presents more four-coordinated B-site cations, while the lower symmetry phase (β = 94.0040°) presents more six-coordinated B-site cations in this type of material.
Based on these XANES results, a conclusion can be made that, despite the multivalent nature of W and Mo, the oxidation state and the coordination environment of W remained unchanged in LNW16 after the 5% H2-treatment, while the oxidation state of Mo in LNM16 was reduced, accompanied by a significant change of its coordination environment after the 5% H2-treatment.
For both LNW16 and LNM16, the as-sintered samples show two O 1s peaks which can be attributed to the oxygen in the lattice and the oxygen in the surface layer (Fig. 5(d) and S7(d)†).44 A small feature at higher binding energy appeared in both 5% H2-treated samples which can be attributed to the hydroxyl species adsorbed on the surface or incorporated in the lattice (Fig. 5(f) and S7(f)†). Tables 2 and S2† list the normalized atomic ratios of the different oxygen species in each sample based on the fitting results of the corresponding XPS spectra. To explain the appearance of these hydroxyl peaks, a protonation mechanism was proposed. This proton incorporation mechanism can be written as eqn (6) and (7).
 | (6) |
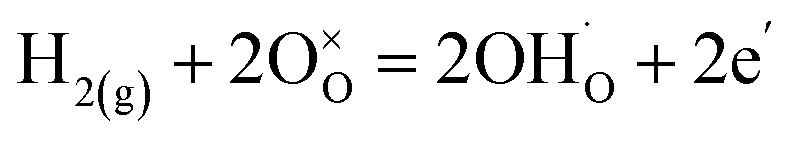 | (7) |
This type of protonation mechanism has been previously reported in other oxides such as ZnO and SmNiO3,45–47 but has never been observed in this type of material. These two possibilities for proton incorporation lead to interstitial or lattice OH− species.
To further study the possible changes of the oxygen species, it is essential to determine the oxygen stoichiometry. Evidently, any change in the oxygen stoichiometry links to the mass change, which makes TGA a widely accepted technique to investigate the change in the oxygen stoichiometry.48 TGA measurements were conducted from RT to 800 °C using compressed air as the purge gas for as-sintered and 5% H2-treated LNW16 (Fig. 7). The mass changes for both samples were subtle and the final mass difference between the two samples was less than 0.1 wt% which was due to the instrumental uncertainty. This was consistent with the fact that the oxidation states of W remained unchanged, as discussed earlier. Besides, as hydrogen is the lightest atom, its incorporation would not cause a significant mass change explaining why no obvious mass change was found in TGA. When it came to LNM16, there was a significant mass increase (0.28 wt%) for the 5% H2-treated LNM16 in air compared to the as-sintered one, which again proved that the Mo species were reduced after the 5% H2-treatment (Fig. S11†). The oxygen stoichiometry of LNM16 after the 5% H2-treatments was reduced by 0.28 wt% giving a nominal composition of LaNb0.84Mo0.16O4.028 if regarding as-sintered LNM16 as LaNb0.84Mo0.16O4.08. This stoichiometric change was again in line with the above XANES studies giving a nominal composition of LaNb0.84Mo0.16O4.026 after the 5% H2-treatment, and within the margins of experimental error for each technique.
 | ||
| Fig. 7 TGA results of as-sintered and 5% H2-treated LNW16 measured from room temperature to 800 °C under flowing air atmosphere. | ||
To further investigate the effects of protonation in LNW16, Tauc plots were calculated from the UV-vis-DRS absorption spectra. The band gap was reduced from 3.49 eV in as-sintered LNW16 to 3.30 eV in the 5% H2-treated sample (Fig. 8(a)). This narrowing was in line with the colouration of LNW16 changing from green to black.49 The tail that appeared before the absorption edge in 5% H2-treated LNW16 is the Urbach tail which is due to the presence of localized defect levels in the band gap resulting from protonation.50 The extrapolated valence band maximum (VBM) of as-sintered and 5% H2-treated LNW16 were the same (Fig. 8(b)). Therefore, the optical band structure diagrams of LNW16 can be plotted (Fig. S6†), and, based on them, protons worked as donors pushing the Fermi level closer to the conduction band minimum (CBM). It is also worth noting that it is common for semiconductors, such as ZnO and WO3, to incorporate protons, resulting in n-type doping.51–54
 | ||
| Fig. 8 (a) Tauc plots of as-sintered LNW16 and 5% H2-treated LNW16; (b) XPS valence spectra of as-sintered LNW16 and 5% H2-treated LNW16. | ||
3.3 Electrochemical study
To understand how the structural and redox stabilities of LNW16 and LNM16 affect their conductivities, electrochemical impedance spectra measurements were conducted to understand the possible effects of protonation on the conductivity. The impedance spectra of LNW16 measured at different temperatures and atmospheres are shown in Fig. S12.† There are three different types of spectra observed through the measurement temperature (400 to 800 °C) and frequency range (1 MHz to 10 Hz). For spectra measured below 570 °C under non-reducing atmospheres, the bulk and grain boundary components are distinguishable, see Fig. S12(a).† However, in the higher temperature range (>570 °C), only the grain boundary component is visible due to the loss of the capacitive component and the transition to an ohmic bulk resistance, see Fig. S12(b).† All spectra measured under the 5% H2 atmosphere at lower temperature are similar to the spectrum shown in Fig. S12(b).† When it comes to the spectra measured under the 5% H2 atmosphere at higher temperature, even the capacitive component of the grain boundary contribution is eliminated resulting in an inductance and an electrode/interfacial component, see Fig. S12(c).† Therefore, for the spectra measured at higher temperature or under the 5% H2 atmosphere, only the total resistances of bulk and grain boundary, Rtot, were extracted from the high frequency intercept of the Z′ axis. Note that although the electrode/interfacial contribution was irrelevant to this work, the fitting was conducted to confirm its nature with a capacitance larger than 10−7 F. The impedance spectra and the fitting processes of LNM16 are similar to those for LNW16.Fig. 9 and S13† show the total conductivities of LNW16 and LNM16 extrapolated from the impedance spectra measured at different atmospheres. For those spectra measured under non-reducing atmospheres, the data points overlapped with each other suggesting a pure oxide ion conduction with negligible protonic and electronic contributions. Besides, a higher conductivity of LNM16 compared with LNW16 under non-reducing atmospheres was observed due to the weaker trapping effect near the Mo6+ cations.9 A significantly enhanced total conductivity in reducing atmospheres can be observed throughout the entire temperature range for both samples. The extrapolated activation energies are summarised in Table 3 and Table S4.† These results were consistent with Canu's and Auckett's works.9,13 For the conductivities of LNW16 measured in reducing atmospheres, the Arrhenius plots for dry and wet conditions deviated from a linear relation at around 570 °C (denoted as Tc). Therefore, these conductivities were divided into two parts, and this division suggested the change of conduction mechanism at Tc. For the conductivities of LNM16 measured in reducing atmospheres, such deviation was absent suggesting a different conduction mechanism in LNW16 and LNM16.
 | ||
| Fig. 9 Total conductivity of LNW16 measured under different atmospheres. The fitting results of the activation energies are presented in Table 2. | ||
| Atmosphere | Humidity | E a (eV) | R 2 |
|---|---|---|---|
| Air | Dry | 1.46 | 0.99 |
| Wet | 1.46 | 0.99 | |
| N2 | Dry | 1.48 | 1.00 |
| Wet | 1.48 | 0.99 | |
| O2 | Dry | 1.46 | 0.99 |
| Wet | 1.47 | 1.00 | |
| 5% H2 | Dry | 0.87 (<Tc) | 0.99 (<Tc) |
| 1.08 (>Tc) | 0.99 (>Tc) | ||
| Wet | 1.14 (<Tc) | 0.99 (<Tc) | |
| 1.32 (>Tc) | 1.00 (>Tc) |
The enhanced conductivity under reducing atmospheres is usually explained by the increase in electronic conductivity which originates from the formation of intrinsic defects and can be summarized using the Brouwer diagram.55,56 For LNM16 under reducing atmospheres, the electronic conduction increased from the reduction of the Mo cations, while the oxygen ion conduction decreased due to the loss of oxygen, as eqn (2) and (3) show. Therefore, at low temperatures, the electronic conduction overshadowed the oxygen ion conduction resulting in more than a three-order of magnitude enhancement of the conductivity. However, at high temperatures, the mobility difference between electrons and oxygen ions was minimized resulting in a smaller difference of total conductivities between LNM16 under non-reducing and reducing conditions. Therefore, LNM16 presented pure oxygen ion conduction in non-reducing atmospheres, while it became a mixed electronic-oxygen ion conductor under reducing atmospheres. No evidence for protonation was observed with the LNM16 composition.
Compared to the LNM16, the conduction behaviour of LNW16 under reducing atmospheres is more complicated. Given the demonstrated redox stability of the W cations, the conduction enhancement cannot be fully electronic i.e. the phase is not reduced and so there is no electronic charge compensation. However, protonic conductivity induced by protonation (eqn (6) and (7)) offers a more reasonable explanation. The enthalpy of the protonation is negative (exothermic) so that it is preferred at low temperatures, 400 °C to 600 °C.57 Therefore, within a low temperature range, the predominant charge carriers are assumed to be electrons and/or protons which reduced the activation energy. When in the high temperature range, 600 °C to 800 °C, the protonation reaction was suppressed and the predominant charge carriers changed to the interstitial oxide ions, which led to the activation energy being similar to the activation energy determined from ambient air measurements. The total conductivity of LNM16 under dry and wet 5% H2 conditions are almost identical, while the total conductivity of LNW16 under the wet 5% H2 condition is over an order smaller than under the dry 5% H2 condition. This is probably attributed to the competition between water molecules and hydroxyl species on the surface suppressing the protonation process. Fully verifying this hypothesis requires further detailed surface studies that are beyond the scope of current work.
From the previous discussion it is clear that the LNW16 composition under reducing conditions exhibits protonic conduction, and therefore determining the transport number of each charge carrier is of importance. In a triple-conducting oxide, the total conductivity is the sum of electronic, protonic and oxide ion partial conductivities, and the transport number of each partial conductivity can be expressed as below:58
| σtot = σe + σO + σp | (8) |
 | (9) |
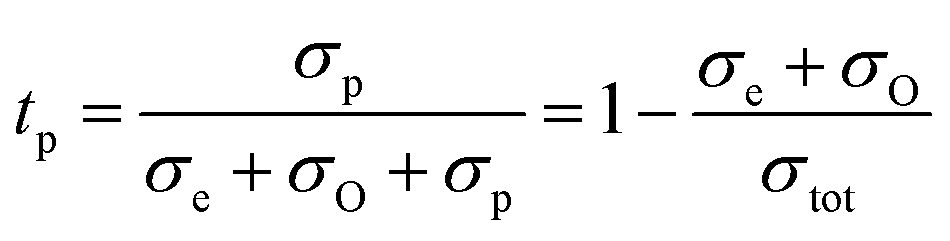 | (10) |
The total conductivity (σtot) can be estimated from EIS results, and the electronic partial conductivity (σe) can be measured by the DC polarization method. Fig. S1† shows that the steadily decreased current relaxation curves to certain values represent the typical relaxation response suggesting a good gas tightness.25 Fig. S1† also suggests higher electronic conductivities of LNW16 under 5% H2 conditions than under O2 conditions. However, the contribution of the electronic conductivity to the total conductivity is still small. Fig. 10(a) shows that the transport number of electrons (telectrons) increased with increasing temperature, but it was lower than 2.0% over the entire temperature range. This result indicated that the ionic conductivity was predominant in LNW16 even under reducing atmospheres. Given the excellent structural and redox stability of LNW16, the intrinsic oxide ion conductivity under reducing atmospheres can be assumed to be analogous to the oxide ion conductivity under ambient atmospheres. Furthermore, LNW16 is proven to be a pure oxide ion conductor under ambient atmospheres both in this work and in a previous study.13 Therefore, the oxide ion partial conductivity (σO) was estimated from the total conductivity measured under ambient atmospheres, and the transport number of protons (tprotons) extracted from eqn (10) (Fig. 10(b)). The results clearly show the predominant protonic conductivity and validates the protonation mechanism proposed in the previous section. The significant decrease in the transport number of protons at high measurement temperatures highlights the exothermic nature of the protonation and is consistent with the change of conduction mechanism at Tc. It also suggests that the electrons were somehow localized. A similar localization phenomenon had been observed in SmNiO3.46
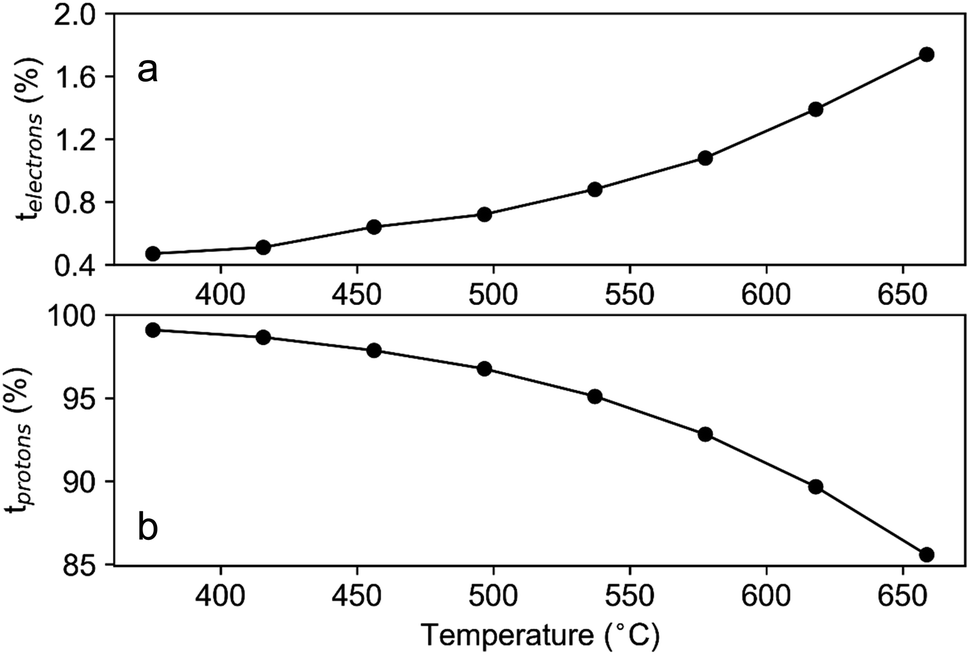 | ||
| Fig. 10 (a) Extracted transport number of electrons in LNW16 under dry 5% H2 atmosphere from DC polarization measurements; (b) estimated transport number of protons in LNW16 under dry 5% H2/N2 atmosphere. | ||
These findings are intriguing as, to the best of our knowledge, this is the first time that protonic conductivity is reported in W-substituted LaNbO4. Previously, this type of material has been proven as a pure oxide ion conductor with negligible protonic conductivity.13 However, this work finds that the proton conductivity can be realised by protonation under hydrogen-containing reducing atmospheres.
4 Conclusion
Samples of LaNb0.84W0.16O4.08 and LaNb0.84Mo0.16O4.08 were successfully prepared using a solid-state reaction method. For the Mo-substituted samples, there was a significant change of structure from higher symmetry to lower symmetry, and both XANES and TGA data showed a change of the oxidation state for Mo in this type of material under a reducing atmosphere. However, compared to the Mo-substituted LaNbO4, the excellent structural, compositional and redox stability of the W-substituted LaNbO4 was clearly exhibited. The tungsten species in LaNb0.84W0.16O4.08 was found to be surprisingly stable as a W6+ ion. The conventional electronic conduction mechanism in solid oxide electrolytes fails to explain the conduction enhancement in LaNb0.84W0.16O4.08 and the protonation mechanism in which hydroxyl species are produced through reaction with hydrogen is proposed to explain the experimental results. The transport number of protons was extracted showing predominant protonic conductivity in LaNb0.84W0.16O4.08 under hydrogen-containing reducing atmospheres. These results demonstrate the potential of this type of material to be developed as proton-conducting electrolytes or hydrogen permeation membranes.Data availability
The research data that supports this article are available on Zenodo (https://zenodo.org/communities/electroceramic-materials).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the beamline scientists Dr Donato Decarolis and Dr Diego Gianolio for their help with the instrument time at the B18 beamline (SP34446) at Diamond Light Source, UK. The authors are grateful for the funding received from UKRI under the following grant numbers that enabled the collection of X-ray absorption spectra (grants EP/Y023587/1, EP/Y023609/1, EP/Y023536/1, EP/Y023552/1 and EP/Y023544/1) at the National Facility for XPS (“HarwellXPS”). SJS also thanks the Royal Academy of Engineering for the award of a Research Chair (RCSRF/2021-1243).References
- R. Haugsrud and T. Norby, J. Am. Ceram. Soc., 2007, 90, 1116 CrossRef CAS.
- R. Haugsrud and T. Norby, Nat. Mater., 2006, 5, 193 CrossRef CAS.
- M. Liu, L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu and Z. Cheng, Science, 2009, 326, 126 CrossRef.
- H. Fjeld, D. M. Kepaptsoglou, R. Haugsrud and T. Norby, Solid State Ionics, 2010, 181, 104 CrossRef CAS.
- A. D. Brandão, J. Gracio, G. C. Mather, V. V. Kharton and D. P. Fagg, J. Solid State Chem., 2011, 184, 863 CrossRef.
- T. Mokkelbost, I. Kaus, R. Haugsrud, T. Norby, T. Grande and M. A. Einarsrud, J. Am. Ceram. Soc., 2008, 91, 879 CrossRef CAS.
- M. Huse, A. W. B. Skilbred, M. Karlsson, S. G. Eriksson, T. Norby, R. Haugsrud and C. S. Knee, J. Solid State Chem., 2012, 187, 27 CrossRef CAS.
- Y. Cao, N. Duan, X. Wang, B. Chi, P. Jian and L. Jian, J. Eur. Ceram. Soc., 2015, 35, 1979 CrossRef CAS.
- J. E. Auckett, L. Lopez-Odriozola, S. J. Clark and I. R. Evans, J. Mater. Chem. A, 2021, 9, 4091 RSC.
- C. Ferrara, A. Mancini, C. Ritter, L. Malavasi and C. Tealdi, J. Mater. Chem. A, 2015, 3, 22258 RSC.
- C. Li, S. S. Pramana, R. D. Bayliss, C. P. Grey, F. Blanc and S. J. Skinner, Chem. Mater., 2020, 32, 2292 CrossRef CAS.
- T. Norby and N. Christiansen, Solid State Ionics, 1995, 77, 240 CrossRef CAS.
- G. Canu, V. Buscaglia, C. Ferrara, P. Mustarelli, S. Gonçalves Patrício, A. I. Batista Rondão, C. Tealdi and F. M. B. Marques, J. Alloys Compd., 2017, 697, 392 CrossRef CAS.
- Y. Han and S. J. Skinner, Mater. Adv., 2023, 4, 3759 RSC.
- A. Neumann and D. Walter, Thermochim. Acta, 2006, 445, 200 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- A. J. Dent, G. Cibin, S. Ramos, A. D. Smith, S. M. Scott, L. Varandas, M. R. Pearson, N. A. Krumpa, C. P. Jones and P. E. Robbins, J. Phys. Conf. Ser., 2009, 190, 012039 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593 Search PubMed.
- J. H. Kim and H. I. Yoo, Solid State Ionics, 2001, 140, 105 CrossRef CAS.
- J. H. Jang and G. M. Choi, Solid State Ionics, 2002, 154–155, 481 CrossRef CAS.
- T. Ishihara, S. Ishikawa, K. Hosoi, H. Nishiguchi and Y. Takita, Solid State Ionics, 2004, 175, 319 CrossRef CAS.
- K. R. Lee, J. H. Lee and H. I. Yoo, Solid State Ionics, 2010, 181, 724 CrossRef CAS.
- L. Jian and C. M. Wayman, J. Am. Ceram. Soc., 1997, 80, 803 CrossRef CAS.
- E. S. Buyanova, Z. A. Mikhailovskaya, Y. V. Emel’yanova, A. A. Levina, M. V. Morozova, S. A. Petrova and N. V. Tarakina, Russ. J. Inorg. Chem., 2017, 62, 211 CrossRef CAS.
- P. Sarin, R. W. Hughes, D. R. Lowry, Z. D. Apostolov and W. M. Kriven, J. Am. Ceram. Soc., 2014, 97, 3307 CrossRef CAS.
- S. S. Pramana, T. Baikie, T. An, M. G. Tucker, J. Wu, M. K. Schreyer, F. Wei, R. D. Bayliss, C. L. Kloc, T. J. White, A. P. Horsfield and S. J. Skinner, J. Am. Ceram. Soc., 2016, 138, 1273 CrossRef CAS.
- J. Li, F. Pan, S. Geng, C. Lin, L. Palatinus, M. Allix, X. Kuang, J. Lin and J. Sun, Nat. Commun., 2020, 11, 1 CrossRef CAS.
- G. Wang, Y. Ling, H. Wang, X. Yang, C. Wang, J. Z. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180 RSC.
- M. F. Sunding, K. Hadidi, S. Diplas, O. M. Løvvik, T. E. Norby and A. E. Gunnæs, J. Electron. Spectrosc. Relat. Phenom., 2011, 184, 399 CrossRef CAS.
- P. A. W. Van Der Heide, Surf. Interface Anal., 2002, 33, 414 CrossRef CAS.
- W. Y. Howng and R. J. Thorn, J. Phys. Chem. Solids, 1980, 41, 75 CrossRef CAS.
- V. Jayaseelan, R. K. Kalaiezhily, N. B. Shinde, K. Kamala Bharathi, M. Navaneethan and S. K. Eswaran, J. Mater. Sci. Mater. Electron., 2022, 33, 8741 CrossRef CAS.
- F. Y. Xie, L. Gong, X. Liu, Y. T. Tao, W. H. Zhang, S. H. Chen, H. Meng and J. Chen, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 112 CrossRef CAS.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2008, 112, 6869 CrossRef CAS.
- H. Asakura, T. Shishido, S. Yamazoe, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2011, 115, 23653 CrossRef CAS.
- M. Nalin, G. Poirier, S. J. L. Ribeiro, Y. Messaddeq and L. Cescato, J. Non-Cryst. Solids, 2007, 353, 1592 CrossRef CAS.
- G. Canu, F. Giannici, A. Chiara, G. Confalonieri, A. Longo, M. T. Buscaglia, M. Dapiaggi, V. Buscaglia and A. Martorana, J. Alloys Compd., 2021, 857, 157532 CrossRef CAS.
- H. Zhou, Z. Chen, A. V. López, E. D. López, E. Lam, A. Tsoukalou, E. Willinger, D. A. Kuznetsov, D. Mance, A. Kierzkowska, F. Donat, P. M. Abdala, A. Comas-Vives, C. Copéret, A. Fedorov and C. R. Müller, Nat. Catal., 2021, 4, 860 CrossRef CAS.
- H. Zhou, Z. Chen, E. Kountoupi, A. Tsoukalou, P. M. Abdala, P. Florian, A. Fedorov and C. R. Müller, Nat. Commun., 2021, 12, 1 CrossRef.
- A. M. Beale and G. Sankar, Chem. Mater., 2003, 15, 146 CrossRef CAS.
- Z. Sha, G. Kerherve, M. A. van Spronsen, G. E. Wilson, J. A. Kilner, G. Held and S. J. Skinner, J. Phys. Chem. C, 2023, 127, 2325 Search PubMed.
- D. G. Thomas and J. J. Lander, J. Chem. Phys., 1956, 25, 1136 CrossRef CAS.
- Y. Zhou, X. Guan, H. Zhou, K. Ramadoss, S. Adam, H. Liu, S. Lee, J. Shi, M. Tsuchiya, D. D. Fong and S. Ramanathan, Nature, 2016, 534, 231 CrossRef CAS PubMed.
- T. Norby and R. Haugsrud, Dense Ceramic Membranes for Hydrogen Separation, in Membranes for Energy Conversion, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, vol. 2, pp 169–216 Search PubMed.
- Y. Yamazaki, F. Blanc, Y. Okuyama, L. Buannic, J. C. Lucio-Vega, C. P. Grey and S. M. Haile, Nat. Mater., 2013, 12, 647 CrossRef CAS.
- B. D. Fahlman, in Materials Chemistry, Springer International Publishing, Cham, 2023, pp. 1–30 Search PubMed.
- B. Choudhury and A. Choudhury, Phys. E, 2014, 56, 364 CrossRef CAS.
- Ç. Kiliç and A. Zunger, Appl. Phys. Lett., 2002, 81, 73 CrossRef.
- C. G. Van de Walle and J. Neugebauer, Nature, 2003, 423, 626 CrossRef CAS PubMed.
- C. G. Van De Walle and J. Neugebauer, Annu. Rev. Mater. Res., 2006, 36, 179 CrossRef CAS.
- M. Vasilopoulou, A. Soultati, D. G. Georgiadou, T. Stergiopoulos, L. C. Palilis, S. Kennou, N. A. Stathopoulos, D. Davazoglou and P. Argitis, J. Mater. Chem. A, 2014, 2, 1738 RSC.
- R. M. C. Marques, F. M. B. Marques and J. R. Frade, Solid State Ionics, 1994, 73, 15 CrossRef CAS.
- K. Singh, J. Nowotny and V. Thangadurai, Chem. Soc. Rev., 2013, 42, 1961 RSC.
- T. Norby, M. Widerøe, R. Glöckner and Y. Larring, Dalton Trans., 2004, 3012 RSC.
- T. Miruszewski, K. Dzierzgowski, P. Winiarz, S. Wachowski, A. Mielewczyk-Gryń and M. Gazda, J. Mater. Chem. A, 2022, 10, 7218 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05501b |
| ‡ Kehan Huang and Yidong Han both contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |