
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Print-light-synthesis of electrocatalytically active gas diffusion electrodes for fuel cell applications†
Wanderson O.
Silva
 *a,
Alexandre
Mabillard
a,
Mathieu
Soutrenon
a,
Grégoire
Gschwend
b,
Yorick
Ligen
c,
Steve
Joris
a,
Luc
Bondaz
d,
Kumar Varoon
Agrawal
d and
Hubert H.
Girault
ef
*a,
Alexandre
Mabillard
a,
Mathieu
Soutrenon
a,
Grégoire
Gschwend
b,
Yorick
Ligen
c,
Steve
Joris
a,
Luc
Bondaz
d,
Kumar Varoon
Agrawal
d and
Hubert H.
Girault
ef
aInstitute of Systems Engineering, HES-SO Valais- Wallis, CH-1950 Sion, Switzerland. E-mail: wanderson.oliveiradasilva@hevs.ch
bCentre Suisse d'Électronique et de Microtechnique, Battery Innovation Hub, Rue de Monruz 17, CH-2002 Neuchâtel, Switzerland
cGREENGT SA, Rue de Lausanne 57, CH-1110 Morges, Switzerland
dLaboratory of Advanced Separations, Ecole Polytechnique Fédérale de Lausanne (EPFL), Valais Wallis, Rue de l'Industrie 17, CH-1950 Sion, Switzerland
eInstitut des Sciences et Ingénierie Chimiques (ISIC), École Polytechnique Fédérale de Lausanne (EPFL) Station 6, CH-1015 Lausanne, Switzerland
fMaterial Science, Energy and Nanoengineering (MSN) Department, University Mohammed VI Polytechnic, 43150 Ben Guerir, Morocco
First published on 28th January 2025
Abstract
The present work reports a simple approach to manufacture electrocatalytically active gas diffusion electrodes (GDEs) in two steps: (i) inkjet printing and (ii) flash light irradiation from a xenon flash lamp, a process called Print-light-synthesis (PLS). Pt/C PLS GDEs were manufactured from a Pt precursor ink printed directly over a carbon paper gas diffusion layer (GDL) with a microporous layer (MPL) of carbon with a metal precursor loading of 0.5 mgPt−1 cm−2, the precursor film was then exposed to flash light irradiation at 450 V-pulse for 100 ms. SEM images showed a uniform and thin Pt catalyst layer deposited on top of the GDL. XRD and XPS spectra evidenced metallic Pt with face-centered cubic crystalline structures. TEM analysis provided an average particle size of 5.0 ± 0.3 nm with uniform particle distribution over the MPL. Electrochemical characterization was performed on half-cell and fuel cell setups showing electrocatalytic performances comparable to that of a reference GDE Pt/C. Pt/C PLS shows even better fuel cell performance per gram of Pt catalyst compared to the reference Pt/C. This work shows that PLS is a very simple approach, e.g. to manufacture GDEs on a roll-to-roll basis for applications in energy conversion devices such as fuel cells, batteries, electrolyzers, etc.
Introduction
Proton-exchange membrane fuel cells (PEMFCs) are power sources with zero-emission of greenhouse gases used in portable, transport, and stationary power applications.1–5 However, their relative high cost, low durability and limited power density are still limiting factors for their large-scale commercialization. Platinum is widely applied as a catalyst in PEMFCs for the hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR), in the anode and cathode, respectively. However, it corresponds to 40% of the membrane electrode assembly (MEA) cost, primarily due to the high price of this noble metal.6 To achieve large scale PEMFC commercialization, a key challenge is to reduce the amount of platinum from 0.2–0.5 g per kW to lower than 0.1 g per kW.7,8 Several studies have already reported the performances of PEMFCs with such lower Pt loading, however there are still significant performance losses, in particular at higher current densities (1.5 A cm−2).9,10 Aiming to address those limitations and make PEMFCs more economically viable for large-scale implementation in the future, some strategies have been proposed: (i) optimization of the catalyst composition and structure;11–14 (ii) improvement of the catalyst support,7,10,15 (iii) ionomer and electrolyte optimization with an improved triple-phase boundary (TPB),16–19 (iv) advanced catalyst deposition techniques,20–23 (v) optimization of the operational fuel cell conditions,24–29 (vi) integration of new materials and components,6,30–35etc. In addition, the PEMFC stack system itself has also several levers for improvements, which include cathode and anode catalyst layers, gas diffusion layers, flow fields, end plates, etc.36–38To achieve optimal platinum loading, a crucial parameter for practical applications is to find a balance between maximizing fuel cell power density and minimizing overall material costs. This balance ensures robust fuel cell performance, durability, and efficiency throughout the practical PEMFC operation.39,40 Advanced catalyst designs, along with improved electrode structures and novel materials, are continuously explored to enhance catalytic efficiency and decrease platinum usage, aiming to meet the practical demands of commercial-scale PEMFC applications.20,34 The interactions between the Nafion ionomer and Pt/C catalyst play a pivotal role in determining the structure and performance of the catalyst layers in PEMFCs. Nafion ionomers can influence few catalyst layer properties such as porosity, ionomer distribution, and proton conductivity, which are crucial factors that affect the TPB and the overall electrochemical activity and fuel cell performance.19,20 As already highlighted, Pt loading plays an important role in the number of active sites and catalytic activity, which influence directly the reaction kinetics for the HOR and ORR and efficiency.41 In addition, the carbon support where the Pt nanoparticles (Pt-NPs) are supported also affects the catalyst's stability, dispersion, and electron transfer properties.15 Even the typical dispersing solvents such as water and isopropanol used in the catalyst ink formulations can have an impact on the morphology and structure of the catalyst layer, affecting catalyst distribution and PEMFC performance.42 Therefore, optimizing all those parameters is key to achieve an ideal catalyst layer structure, maximizing active sites, proton conductivity, and mass transport, which will ultimately impact the practical performance of the fuel cell in terms of power density, durability, and efficiency.
The MEA preparation with the optimal Pt/C catalyst layer involves several steps to maximize the triple-phase boundary and one of those steps involves the catalyst layer fabrication. For the PEMFC, this step is typically done by one of these three main approaches: Catalyst Coated Substrate (CCS), Catalyst Coated Membrane (CCM), and Decal Transfer Method (DTM).22 Based on this, several ink deposition methods can be applied to create efficient and uniform catalyst layers such as: blade coating, brush printing, ultrasonic spraying, rolling, screen printing, inkjet printing, electrospinning, slot die and electrospraying.22 Here, particular attention will be given to inkjet printing since it offers several advantages compared to the other deposition methods such as: (i) a very precise control of the catalyst deposition, (ii) versatility allowing controlled deposition of catalyst layers with different loadings, surface areas, designs, catalyst layers or gradients with high deposition resolution and accuracy, (iii) low material cost since it significantly reduces the material waste by precisely placing droplets only where needed, optimizing the catalyst utilization, etc.43–46 Furthermore, the non-contact nature of inkjet printing minimizes any possible damage of the substrate and/or catalyst layer. These advantages make inkjet printing a suitable approach for catalyst deposition compared to conventional methods for GDE fabrication in PEMFCs.
The combination of inkjet printing, to deposit the catalyst precursor, with a post-printing process carried out through flash light irradiation from a xenon lamp introduces a new method called Print-light-synthesis (PLS).47,48 The key advantage of PLS is the ability to precisely control the catalyst precursor deposition while harnessing light as a trigger for the in situ catalyst synthesis. This approach also presents great flexibility, allowing for tailored catalyst patterns and fine-tuning of catalyst characteristics such as size, shape, and crystallinity.48,49 In addition, the process is expected to be faster and more eco-friendly compared to conventional high-temperature synthesis methods, reducing energy consumption and waste generation. Therefore, PLS emerges as a promising approach for efficient and customizable catalyst fabrication in PEMFCs, offering advantages in terms of control, speed, and environmental impact over traditional methods.
In this work, PLS was applied to manufacture electroactive gas diffusion electrodes, containing small Pt nanoparticles well dispersed over the carbon support. This is a very simple approach where a Pt precursor salt and citric acid as a reducing agent are initially dissolved in ethanol and then deposited by inkjet printing directly over a GDL containing a microporous layer (MPL). After drying, the precursor film was exposed to high intensity flash light irradiation from a xenon lamp. With the flash light exposure, the carbon black present in the GDL acts as light-to-heat absorber and then it heats up in a very short time (milliseconds). The citric acid is decomposed thermally and photochemically and acts as a sacrificial electron donor, which subsequently promotes the Pt salt conversion into the metallic nanoparticles. The Pt catalyst synthesized by PLS was later characterized by Inductively coupled plasma mass spectrometry (ICP-MS), X-ray diffraction (XRD), scanning electron microscopy (SEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), X-ray photoelectron spectroscopy (XPS), electrochemical measurements in a conventional and half-cell setup to analyze the oxygen reduction reaction and polarization curves in a PEM fuel cell H2/air.
Experimental
Materials
Hexachloroplatinic acid hexahydrate (H2PtCl6·6H2O), citric acid (C6H8O7), perchloric acid (HClO4) and ethanol were purchased from Sigma-Aldrich. Aqueous solutions were prepared from ultrapure water (Millipore Milli-Q, specific resistivity 18.2 MΩ cm−1). Nafion membrane NR211 (thickness: 25 μm) and ionomer solutions (5 wt%) were provided by DuPont. A carbon paper gas diffusion layer (GDL) with a Microporous Layer (MPL) H23C2 with 255 μm and 10 mΩ cm2 (Freudenberg, article number: 11924) containing a porous carbon was used as a GDL during the PLS process to manufacture Pt/C PLS. A benchmark Pt/C catalyst from H23C6 with 0.5 mgPt−1 cm2 and 270 μm of thickness was used as the reference (Freudenberg, reference number: 12038).Methods
Pt-NPs were synthesized directly over the GDL by print-light-synthesis from a Pt precursor ink as shown in Fig. 1. The precursor ink formulated from ethanol, H2PtCl6·6H2O and citric acid (Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid, 1:2) was printed using a Fujifilm Dimatix DMP-2850 printer and disposable Dimatix cartridge DMCLCP-11610. The viscosity and surface tension of the ink were measured with a SV-10A viscometer (A & D Instruments Limited) and drop shape analyzer DSA30 (Krüss), respectively. Some printing parameters such as the number of printing nozzles, voltage pulse, jetting frequency, printhead–substrate distance, cleaning cycles, etc. were carefully adjusted to get a high printing resolution (Fig. S1†). The dried Pt precursor film was exposed to flash light irradiation using a xenon flash lamp (PulseForge 1300, Novacentrix, USA), under a controlled argon atmosphere in an environmental stainless-steel chamber sealed with a 3 mm thickness quartz glass window (Fig. S2†). The flash light irradiation process was carried out at 450 V-pulse with 25 consecutive single flash light shots, providing a total irradiation time of 100 ms, each single pulse had an energy density of 6.7 J cm−2 (Fig. S3†). The distance between the GDL + Pt precursor film and the xenon flash lamp window was 9 mm. After the printing, drying and flash light irradiation steps, the GDEs were washed 5 times with Milli-Q water and ethanol to remove the remaining Pt salt and afterwards the electrodes were dried at room temperature under a stream of air. More details of the flash light synthesis process can be found in previous publications.47–49
:citric acid, 1:2) was printed using a Fujifilm Dimatix DMP-2850 printer and disposable Dimatix cartridge DMCLCP-11610. The viscosity and surface tension of the ink were measured with a SV-10A viscometer (A & D Instruments Limited) and drop shape analyzer DSA30 (Krüss), respectively. Some printing parameters such as the number of printing nozzles, voltage pulse, jetting frequency, printhead–substrate distance, cleaning cycles, etc. were carefully adjusted to get a high printing resolution (Fig. S1†). The dried Pt precursor film was exposed to flash light irradiation using a xenon flash lamp (PulseForge 1300, Novacentrix, USA), under a controlled argon atmosphere in an environmental stainless-steel chamber sealed with a 3 mm thickness quartz glass window (Fig. S2†). The flash light irradiation process was carried out at 450 V-pulse with 25 consecutive single flash light shots, providing a total irradiation time of 100 ms, each single pulse had an energy density of 6.7 J cm−2 (Fig. S3†). The distance between the GDL + Pt precursor film and the xenon flash lamp window was 9 mm. After the printing, drying and flash light irradiation steps, the GDEs were washed 5 times with Milli-Q water and ethanol to remove the remaining Pt salt and afterwards the electrodes were dried at room temperature under a stream of air. More details of the flash light synthesis process can be found in previous publications.47–49
 | ||
| Fig. 1 Scheme of the overall print-light-synthesis process to fabricate gas diffusion electrodes. | ||
Characterization
Scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra were obtained using a ZEISS GeminiSEM 460 with an EHT voltage of 20 kV, and a probe current of 10 nA at 200× magnification. X-ray diffraction (XRD) analyses were carried out using a Bruker D8 Discover diffractometer equipped with a Cu Kα radiation source and a VANTEC-1 detector. Diffraction patterns were recorded in the 2θ range of 10–90°. Inductively coupled plasma mass spectrometry (ICP-MS) was employed to estimate the metal loading after the PLS process. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Physical Electronics Versa Probe II X-ray photoelectron spectrometer. Transmission electron microscopy (TEM) images were acquired from a FEI Tecnai Spirit instrument operated at 120 kV. The Scanning Transmission Electron Microscopy (STEM) images were acquired under the high-angle annular dark-field (HAADF-STEM) condition using a probe-corrected Thermo Fisher Scientific microscope 200 S/TEM operated at 200 kV with a beam current of 150 pA. This microscope is equipped with an ultra-high-brightness cold field emission gun (X-CFEG), a Super-X EDS system comprising four silicon drift detectors, and Velox acquisition software. Energy-dispersive X-ray spectroscopy (EDS) data were collected as spectrum images, in which a focused electron probe was scanned in raster across a region of interest in the STEM mode. For TEM and HAADF-STEM analyses a piece of 0.28 cm2 of the GDE Pt/C PLS was first immersed in isopropanol and then sonicated for 10 min, afterwards 30 μL of the resulting ink was deposited by drop casting over a standard Cu TEM grid with an ultrathin carbon film. Electrochemical characterization was performed in a conventional and half-cell setup (Fig. S4†) at room temperature using a MultiEmStat-8CH potentiostat (PalmSens). Cyclic voltammetry measurements were performed in a standard 3-electrode electrochemical cell in 2 mol L−1 HClO4 electrolyte free of O2. In this setup, the working electrode (GDE) was assembled to a standard glassy carbon support with an electrical contact by using a small lid, made from polyester ether ketone (PEEK). In this WE setup, the glassy carbon was pressed against the gas diffusion electrode and fixed with this small lid to provide an electrical contact with a small gas diffusion electrode area (0.0314 cm2). Linear sweep voltammetry (LSV) measurements for the oxygen reduction reaction (ORR) were performed in a home-made half-cell50 configuration using a 1.5 cm2 GDE with a working electrode area of 0.0314 cm2, and the electrode + Nafion membrane assembly was placed in contact with 2 mol L−1 HClO4 electrolyte and air as presented in Fig. S4.† A platinum mesh and silver|silver chloride (Ag|AgCl) electrodes were employed as counter and reference electrodes, respectively. PEM fuel cell tests were performed from membrane electrode assemblies, containing Pt/C PLS and Pt/C reference catalysts, prepared from 25 cm2 GDEs, Nafion® ionomer 5 wt% and Nafion® membrane 211 (DuPont) hot pressed at 130 °C for 3 min under a compression weight of 350 kg (1.37 MPa) with a Carver 3891 benchtop press, and pieces of soft papers were placed between the aluminum plates and the MEA to protect it from possible damage. A Nafion® ionomer film was deposited by spray coating using a Preeflow eco-SPRAY, a dosing control unit eco-Control EC200-K and a control unit eco-control SC 1200 from Viscotec. The spray coating was performed with an automated and programmable system (robotic arm UR3, Universal Robots) to move the spray unit over the substrate, using a hot plate at 65 °C and a nozzle diameter of 0.5 mm, and the dried Nafion ionomer loading was controlled using an analytical balance by adjusting the carbon/ionomer ratio at 0.35. The prepared MEA was assembled into a single cell test unit from Baltic FuelCells as presented in Fig. S5† and tested at 80 °C, hydrogen and air with flow rates of 300 and 600 mL min−1, respectively, back pressure of 2 bar and 100% of relative humidity.Results and discussion
The print-light-synthesis of Pt/C was performed in two simple steps as presented in Fig. 1: (I) inkjet printing and (II) flash light irradiation. First, the Pt precursor salt was deposited by inkjet printing on the gas diffusion layer (GDL), in this step an appropriate Pt precursor loading was diluted in a citric acid + ethanol solution, providing a final ink loading of 75 mgPt−1 mL−1. The printability parameter (Z) of the ink was calculated by Z = (γρα)0.5η−1, where γ, ρ and η correspond to the surface tension, viscosity, and density, respectively, and η represents the characteristic length, which is related to the nozzle diameter of 21.5 μm. A Z value of 2.2 was estimated and it is in the printable range of 1 < Z < 10.51 The ink was stable providing droplets with 9.5 pL as presented in Fig. S1.† The GDEs were prepared with a precursor metal loading of 0.5 mgPt−1 cm2. After the flash light irradiation process, carried out at 450 V-pulse and 100 ms, the carbon paper was washed and the solution was analyzed by ICP-MS, providing a conversion ratio into metallic Pt nanoparticles of 50%. This is a promising result and demonstrates that Pt-NPs can be synthesized directly over a GDL without pre- and post-treatments processes, within a shorter time (few minutes for 25 cm2), significantly reducing the processing time compared to the conventional manufacturing processes that are typically performed in 1–4 days52 with several toxic chemicals and steps. The present process can be scaled up for industrial applications on a roll-to-roll production line and it is expected that the conversion efficiency can be improved by tuning the printing and flash light irradiation parameters.SEM images in Fig. 2 show very uniform Pt nanoparticle deposition over a microporous layer, which evidences inkjet printing as a suitable deposition method that minimizes the precursor salt waste once the platinum content stay on the GDL surface, and the flash light irradiation process promotes the fast synthesis of Pt nanoparticles. Thin and uniform catalyst deposition is one of the key factors to improve PEMFC performance. Therefore, the proposed protocol can potentially maximize the triple phase boundary with the possibility to manufacture GDEs with less Pt loading keeping the same catalytic performance and then making the technology cheaper and economically viable for large-scale commercialization.
 | ||
| Fig. 2 SEM-EDX images of the GDE cross-section of the GDL and Pt catalyst layer synthesized by print-light-synthesis. | ||
Fig. 3 presents the XRD diffraction patterns of the Pt/C synthesized by PLS in comparison with the bare GDL that contains only carbon black and PTFE, and commercial Pt/C. The XRD spectrum for Pt/C PLS presents characteristic peaks of the face-centered cubic crystalline structure which corroborate with the diffraction peaks of the Pt/C ref.53 All three diffraction patterns show a broad diffraction peak around 25° 2θ, corresponding to the (002) plane of carbon graphite. The diffraction peaks placed at 39.8, 46.1, 67.6 and 81.4° are ascribed to the Pt (111), (200), (220) and (311) planes,53 respectively. The Pt (111) plane is one of the most common crystallographic planes reported for Pt catalysts, and it presents a high atomic density, which is typically associated with high reactivity in catalysis for example the hydrogen oxidation reaction and oxygen reduction reaction. Therefore, the Pt (111) plane was used as a reference to estimate the crystallite size by using the Sherrer's equation: D = Kλ/βcosθ. A crystallite size of 3.0 nm was estimated for Pt/C PLS and it is in the range comparable to the particle size by TEM.
 | ||
| Fig. 3 X-ray diffractograms of the synthesized Pt/C PLS compared with the bare GDL and commercial Pt/C. | ||
TEM images of the Pt/C PLS and its corresponding histogram from the average nanoparticle size distribution are shown in Fig. 4. The acquired TEM images show uniform nanoparticle distribution over the carbon support with sphere shape and average nanoparticle size mostly in the range of 2–6 nm. The TEM images were recorded from different areas as illustrated in Fig. S6† and evidence the absence of large nanoparticle agglomerations; therefore the present PLS method provides a satisfactory nanoparticle distribution and homogeneity. Based on these TEM images, 600 nanoparticles were selected randomly and measured individually by using ImageJ software and all those values were used to construct a histogram providing an average nanoparticle size of 5.0 ± 0.3 nm. In addition, the histogram shows that 0.5% of the Pt nanoparticles are smaller than 2 nm, and 79.2% of the nanoparticles have an average size in the range of 4–6 nm, followed by 16.8% in the range of 8–10 nm, and 2.3% in the range of 12–14 nm and only 1.2% of those nanoparticles have a particle size larger than 14 nm. Therefore, the controlled Pt salt deposition and distribution performed by inkjet printing, followed by the fast nanoparticle synthesis process carried out in 100 ms using a high-power light source delivered by a xenon lamp, not only generate small nanoparticles but also minimize the nanoparticle agglomeration, which could be attributed to the control of the particle nucleation and growth from the fast-heating and cooling process.54
 | ||
| Fig. 4 Transmission electron microscopy images and average particle size distribution histogram for Pt/C PLS (Pt loading: 0.25 mgPt−1 cm−2). | ||
Chemical synthesis is one of the main protocols to synthesize Pt nanoparticles.55 This method normally uses a water-soluble Pt cation as a precursor, a reducing agent and caping agent, the last one is used to stop the nanoparticle growth and agglomeration, both agents act to control the particle size and shape, for example some colloidal methods can generate Pt particles with different shapes such as sphere, cube, cuboctahedra, octahedra, triangular plates, hexagonal plates, nanobars, etc.11,56 However, several caping agents have been applied to minimize the particles aggregation, such as poly-vinylpyrrolidone (PVP), poly vinyl alcohol (PVA), cetyltrimethylammonium bromide (CTAB), dodecylammonium bromide (DTAB), etc.56 However, on the other hand those caping agents also take place blocking active catalytic sites which limits the catalyst performance. Therefore, several post-treatment steps and processes are required to remove those caping agents, which is high time consuming and costly. Therefore, the PLS protocol proposed in this study can also bring new insights and advantages as a free-capping agent protocol to synthesize small Pt nanoparticles with uniform dispersion.
X-ray photoelectron spectra (XPS) of Pt from Pt/C PLS and Pt/C ref catalysts are illustrated in Fig. 5. Both Pt 4f spectra were calibrated using the adventitious C 1s peak at 284.8 eV. The high-resolution spectra related to the Pt 4f region can be deconvoluted into 2 doublets, with components Pt 4f7/2 and Pt 4f5/2 separated by 3.35 eV. The main deconvoluted peaks for both catalysts centered at 71.3 and 74.6 for Pt 4f7/2 and Pt 4f5/2, respectively, can be attributed to the metallic platinum Pt0, which corresponds to 92.7% for Pt/C ref and 82.0% for Pt/C PLS. Overall, both catalysts present similar chemical states. The deconvoluted peaks placed at 72.7 and 76.0 for Pt 4f7/2 and Pt 4f5/2, respectively, are ascribed to the chemical state +2 which is related to the PtO or Pt(OH)2 species. Therefore, Pt/C synthesized by PLS provided 18% of platinum oxides, which is almost three times higher compared to the Pt/C ref that presents only 6.3%. It is well known that a higher Pt0 fraction and lower Pt oxygenate species present a weaker oxyphilicity and then higher electrocatalytic performance for the oxygen reduction reaction, which can explain the slight difference in the electrochemical performance as will be discussed later.57 In addition, it is important to point out the absence of the Cl 2p peak (Fig. S7†) from metal chlorine for the Pt/C PLS catalyst, typically detected at 198.5–199 eV, which evidences a satisfactory Pt salt conversion into Pt metallic, and the remaining salt was then removed by washing several times with water and isopropanol.
 | ||
| Fig. 5 High-resolution XPS spectra of Pt 4f for (a) Pt/C PLS and (b) Pt/C ref. | ||
Therefore, the Pt 4f spectrum for Pt/C PLS is related to the synthesized amount of Pt provided by flash light irradiation. The higher Pt oxide content was also observed by Cha et al.54 who synthesized high-entropy alloys by flash-thermal shock (FTS) also using a xenon lamp source, this similar approach was followed for carbon nanofiber (CNF) membranes impregnated with different metal precursors (Pt, Ir, Fe, Ni, Co, La, Ce, In, and Sr), which were exposed to flashlight irradiation for 20 ms and an energy density of 4.9 J cm−2. A similar approach was then applied in this work, however with the advantages to control the metal precursor loading deposition and layer thickness by inkjet printing. This evidences PLS as a suitable strategy to synthesize GDEs on a large scale for several energy storage applications such as fuel cells, batteries, electrolyzers, etc.
Catalyst activity of the Pt/C PLS was initially investigated by cyclic voltammetry (CV) from a standard three electrode electrochemical cell in HClO4 2 M electrolyte saturated with argon and a scan rate of 100 mV s−1 as shown in Fig. 6a. The CV profile presents three characteristic peaks related to the hydrogen adsorption–desorption region from 0.05 to 0.35 V (vs. RHE), double-layer region 0.35 to 0.8 V (vs. RHE) and Pt-oxide formation/reduction from 0.8 to 1.2 V (vs. RHE).11 The CV from Pt/C PLS presents a wider double layer region compared to Pt/C ref, which can be ascribed to the higher capacitive behavior, probably related to the higher surface area provided by the higher carbon support content.
 | ||
| Fig. 6 (a) Cyclic voltammograms of Pt/C PLS and Pt/C ref catalysts in HClO4 2 M with O2-free at 100 mV s−1 and their respective (b) linear sweep voltammetry curves for the oxygen reduction reaction compared to the bare GDL recorded in HClO4 2 M/air at 5 mV s−1. | ||
The electrochemically active surface areas (ECSA) for both catalysts were measured by CO stripping following the equation:
| ECSA (m2 gPt−1) = QCO/QML × mPt |
STEM-HAADF images presented in Fig. 7 show Pt nanoparticles dispersed on the carbon support (Fig. 7a), which is confirmed by STEM-EDXS Pt elemental mapping as illustrated in Fig. 7b. In addition, Pt single atoms dispersed on the carbon support are clearly observed in Fig. 7e and f. Therefore, this evidences Print-light-synthesis as a promising approach to successfully convert the Pt precursor into small nanoparticles and single dispersed atoms from two single steps: (I) inkjet printing and (ii) flash light irradiation without complex and timing consuming steps. In addition, the presence of single Pt single atoms could be indirectly correlated with the higher electrochemically active surface area for Pt/C synthesized by PLS compared to the reference Pt/C.
 | ||
| Fig. 7 (a and c–f) HAADF-STEM images of Pt single atoms supported on carbon (from the GDL) synthesized by print-light-synthesis and (b) corresponding EDS elemental mapping for platinum from inset (a). | ||
Electrochemical activity for the ORR catalyzed by Pt/C PLS in comparison with Pt/C ref and bare GDL was measured under more realistic conditions using a half-cell setup. This kind of setup (Fig. S4†) has already been reported50 and can predict more accurately the catalyst performance of a fuel cell when compared to the standard setup in a three-electrode cell using a rotating disk electrode (RDE) and oxygen-saturated electrolyte. Polarization curves for the ORR were hence recorded using linear sweep voltammetry (LSV) in HClO4 2 M/air at 5 mV s−1, as presented in Fig. 6b. Both polarization curves present a profile with the oxygen reduction reaction starting at 0.9 V (vs. RHE), and the slight difference can be attributed to the higher capacitive current from the higher carbon content for Pt/C PLS. Fig. S8† shows a typical Tafel plot curves for the ORR catalyzed by the Pt catalyst; both electrodes present similar Tafel slopes which confirm similar kinetics and rate determining steps for the ORR, however Pt/C ref has a slight lower overpotential (30 mV) compared to the Pt/C PLS, which might be attributed to the higher Pt loading and lower capacitive current. However, overall, the electrochemical activity of Pt/C PLS for the ORR is comparable to that of the commercial Pt/C catalyst.
Polarization curves from a PEMFC H2/air with Pt/C PLS and Pt/C commercial are shown in Fig. 8. The fuel cell tests were performed at 80 °C with humidified H2 and air gases and flow rates of 300 and 600 mL min−1, respectively, back pressure of 2 bar and 100% relative humidity. During the fuel cell measurements, both the anode/cathode inlet and outlet were heated at 100 °C to avoid water condensation and the liquid water excess after the outlets were removed by using a water trap. The Pt/C ref MEA was prepared with a Pt loading of 0.5 mgPt−1 cm−2 on the anode and cathode sides. Similarly, the Pt/C PLS MEA was also prepared using a theoretical Pt precursor loading of 0.5 mgPt−1 cm−2 on both electrodes, however taking into account 50% of conversion from the Pt precursor salt into Pt metallic nanoparticles after PLS synthesis, the final Pt loading from Pt/C PLS MEA was 0.25 mgPt−1 cm−2. The comparison between Pt/C PLS and Pt/C ref was made considering both values, the nominal Pt loading deposited by printing and the effective Pt catalyst loading obtained after the synthesis measured by ICP-MS, which take place in the HOR and ORR reactions. To evaluate the practical catalyst performance, polarization curves were recorded by measuring the output cell voltage from different applied current densities from 0.08 to 1 A cm−2 as illustrated in Fig. 8. According to these polarization curves, Pt/C PLS presented fuel cell performance close to reference MEA Pt/C (0.5 mgPt−1 cm−2). At 0.9 A cm−2 the maximum power densities obtained for Pt/C PLS and Pt/C ref were 0.42 and 0.45 W cm−2, respectively; the slightly higher power density for Pt/C ref of 7% is likely related to the higher catalyst loading with 0.5mgPt−1 cm−2 compared to Pt/C PLS that presents half of that (0.25 mgPt−1 cm−2). The power density increases linearly until around 0.9–1 A cm−2, where a plateau is clearly observed, before decreasing at higher current densities (>1 A cm−2) due to the higher resistance associated with the mass transport limitation.
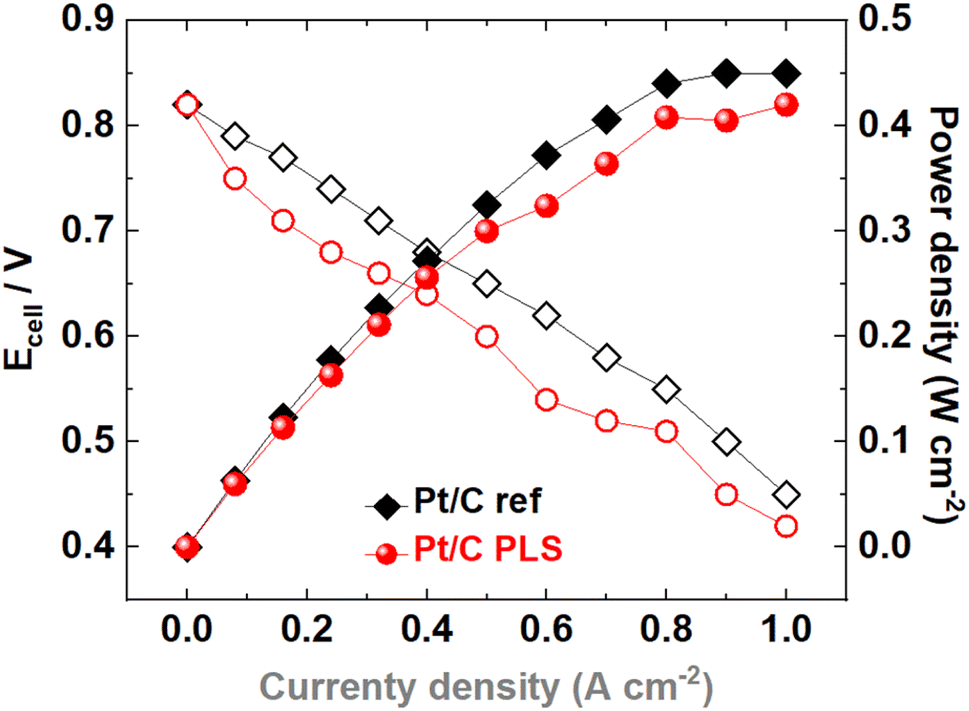 | ||
| Fig. 8 Polarization and power density curves obtained from the PEMFC H2/air at 80 °C, 100% relative humidity (RH), flow rates of 300/600 mL min H2/air, catalyzed by Pt/C PLS corrected by the Pt loading of 0.5 mgPt−1 cm−2 on the anode/cathode sides applied during the catalyst synthesis. Pt/C commercial with a metal loading of 0.5 mgPt−1 cm−2 was included for comparison. | ||
When both polarization curves are corrected by the effective Pt loading present in the gas diffusion electrodes, 0.5 mgPt−1 cm−2 for Pt/C ref and 0.25 mgPt−1 cm−2 for Pt/C PLS considering a conversion rate of 50% from the Pt salt to Pt nanoparticles obtained after PLS synthesis, the Pt/C PLS catalyst presents better cell performance or Pt utilization per gram of catalyst, in particular at higher current density (0.9–1 A cm−2) with two times higher current density per of Pt compared to the commercial Pt/C (Fig. 9). Therefore, while the PLS process requires further optimization to enhance the conversion efficiency from metal precursors to metal catalysts, it shows its suitability for producing GDEs. This strategy allows for the controlled and uniform deposition of catalyst layers, with well-dispersed nanoparticles over the carbon support of the gas diffusion layer. This results in a significant improvement in Pt utilization efficiency per unit of generated power density. Thus, print-light-synthesis has several advantages such as the fast and low-cost process and scalability since inkjet and flash lamps can be used to print and flash on areas from mm2 to m2 catalysts and layers/gradients, etc., allowing a large-scale production of GDEs with applications in several energy conversion devices such as fuel cells, batteries, electrolyzers, etc.
 | ||
| Fig. 9 Polarization curves (V vs. A gPt−1) obtained from the PEMFC H2/air at 80 °C, 100% relative humidity (RH), flow rates of 300/600 mL min H2/air, catalyzed by Pt/C PLS corrected by the effective Pt loading of 0.25 mgPt−1 cm−2 for both electrodes obtained after the catalyst synthesis. Pt/C commercial with a metal loading of 0.5 mgPt−1 cm−2 was included for comparison. | ||
Conclusions
In this study, we reported the manufacturing of electrocatalytically active gas diffusion electrodes containing small Pt nanoparticles by using two simple steps: (i) inkjet printing and (ii) high-intensity flash light irradiation from a xenon lamp. The precursor film was printed by inkjet over a commercial GDL from an ink with a metal loading of 0.5 mgPt−1 cm−2 and then sintered by flash light irradiation at 450 V-pulse and 45Hz for a total flash light irradiation of 100 ms. ICP-MS analysis evidenced a conversion ratio into metallic Pt nanoparticles of 50%, which corresponds to an effective metal loading of 0.25 mgPt−1 cm−2. XRD spectra showed characteristic Pt peaks of the face-centered cubic crystalline structure, which corroborate with typical Pt (111), (200), (220) and (311) planes. XPS analysis for Pt/C PLS also showed typical peaks attributed mainly to the metallic platinum Pt0 (82%) with the absence of chloride, which confirms the Pt salt conversion into metallic Pt nanoparticles with a particle size average of 5.0 ± 0.3 nm and uniform dispersion over the carbon support. Electrochemical characterization performed using half-cell and fuel cell setups for the oxygen reduction reaction and H2/air, respectively, showed typical performance with electrochemical activity for Pt/C PLS comparable to a commercial Pt/C catalyst. However, the Pt/C PLS catalyst presents better cell performance or Pt utilization per gram of catalyst with twice higher mass activity, at higher current density. Therefore, all findings presented in this work evidence PLS as a suitable approach for gas diffusion electrode manufacturing on a roll-to-roll basis with several advantages such as low precursor cost and large flexibility when manufacturing GDEs with different areas (mm2 to m2), surface design, catalyst type, metallic loadings, catalyst gradients/layers, etc. Further experiments are in progress to improve even more the flash light irradiation parameters to get higher Pt catalyst conversion during the synthesis, which might improve the power density per g of catalyst and then minimize the cost technology.Data availability
All the data have been included in the ESI.†Author contributions
All authors designed the experiments collectively, discussed the results, and contributed to the manuscript. W. O. S. performed the electrochemical, PLS and XRD experiments and their respective data analyses. A. M. performed inkjet printing and SEM experiments and related data analysis. L. B. performed the TEM analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from Innosuisse – Swiss Innovation Agency grant 100.229 IP-ENG – Smarter electrodes for improved fuel cells. We also thank CIME EPFL (Interdisciplinary Centre for Electron Microscopy – École Polytechnique Fédérale de Lausanne) and Dr Emad Oveisi for HAADF-STEM analysis.Notes and references
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- S. Campanari, G. Manzolini and F. Garcia de la Iglesia, J. Power Sources, 2009, 186, 464–477 CrossRef CAS.
- Z. Wang, Z. Liu, L. Fan, Q. Du and K. Jiao, Energy Rev., 2023, 2, 1–12 Search PubMed.
- M. M. Tellez-Cruz, J. Escorihuela, O. Solorza-Feria and V. Compañ, Polymers, 2021, 13, 1–54 CrossRef PubMed.
- L. Osmieri, J. Park, D. A. Cullen, P. Zelenay, D. J. Myers and K. C. Neyerlin, Curr. Opin. Electrochem., 2021, 25, 1–12 Search PubMed.
- M. N. Islam, A. B. Mansoor Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- Y. Peng, J. Y. Choi, T. Fürstenhaupt, K. Bai, Y. Zhang and D. Banham, J. Mater. Chem. A, 2021, 9, 13471–13476 RSC.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- D. Banham, J. Zou, S. Mukerjee, Z. Liu, D. Yang, Y. Zhang, Y. Peng and A. Dong, J. Power Sources, 2021, 490, 1–18 CrossRef.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- M. M. Rahman, K. Inaba, G. Batnyagt, M. Saikawa, Y. Kato, R. Awata, B. Delgertsetsega, Y. Kaneta, K. Higashi, T. Uruga, Y. Iwasawa, K. Ui and T. Takeguchi, RSC Adv., 2021, 11, 20601–20611 RSC.
- X. Zhang, H. Li, J. Yang, Y. Lei, C. Wang, J. Wang, Y. Tang and Z. Mao, RSC Adv., 2021, 11, 13316–13328 RSC.
- A. Singh and K. Miyabayashi, RSC Adv., 2019, 10, 362–366 RSC.
- L. Du, Y. Shao, J. Sun, G. Yin, J. Liu and Y. Wang, Nano Energy, 2016, 29, 314–322 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- K. Wuttikid, S. Shimpalee, J. W. Weidner and K. Punyawudho, Fuel Cells, 2017, 17, 643–651 CrossRef CAS.
- D. Van Dao, G. Adilbish, T. D. Le, I. H. Lee and Y. T. Yu, RSC Adv., 2019, 9, 15635–15641 RSC.
- Y. Li, Z. Wu, C. Wang, X. Yu, W. Gao, B. Wang, C. Wu, Y. Yao, J. Yang and Z. Zou, Adv. Funct. Mater., 2024, 34, 1–10 Search PubMed.
- M. Chen, C. Zhao, F. Sun, J. Fan, H. Li and H. Wang, eTransportation, 2020, 5, 1–26 CrossRef.
- H. Liu, L. Ney, N. Zamel and X. Li, Appl. Sci., 2022, 12, 1–41 Search PubMed.
- J. Zhao, H. Liu and X. Li, Electrochem. Energy Rev., 2023, 6, 1–61 Search PubMed.
- Z. Turtayeva, F. Xu, J. Dillet, K. Mozet, R. Peignier, A. Celzard and G. Maranzana, Int. J. Hydrogen Energy, 2022, 47, 16165–16178 CrossRef CAS.
- H. Rezk, T. Wilberforce, E. T. Sayed, A. N. M. Alahmadi and A. G. Olabi, Energy Rep., 2022, 8, 6181–6190 CrossRef.
- I. A. Soomro, F. H. Memon, W. Mughal, M. A. Khan, W. Ali, Y. Liu, K. H. Choi and K. H. Thebo, Membranes, 2023, 13, 1–14 Search PubMed.
- N. Zhao, Y. Chu, Z. Xie, K. Eggen, F. Girard and Z. Shi, Fuel Cells, 2020, 20, 176–184 CrossRef CAS.
- B. Karthikeyan, P. Ramasamy, M. Pandi Maharajan, N. Padmamalini, J. Sivakumar, S. Choudhury and G. F. Savari, Sustainability, 2024, 16, 1–21 CrossRef.
- J. A. Salva, A. Iranzo, F. Rosa, E. Tapia, E. Lopez and F. Isorna, Int. J. Hydrogen Energy, 2016, 41, 19713–19723 CrossRef CAS.
- G. Tsotridis, A. Pilenga, G. De Marco and T. Malkow, EU Harmonised Test Protocols for PEMFC MEA Testing in Single Cell Configuration for Automotive Applications, JRC Sci. Policy Rep., 2015, pp. 1–60 Search PubMed.
- H. E. Kim, J. Kwon and H. Lee, Chem. Sci., 2022, 13, 6782–6795 RSC.
- L. Huang, M. Wei, R. Qi, C. L. Dong, D. Dang, C. C. Yang, C. Xia, C. Chen, S. Zaman, F. M. Li, B. You and B. Y. Xia, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- Z. Liu, Y. Yin, D. Yang, C. Zhang, P. Ming, B. Li and S. Yang, RSC Adv., 2020, 10, 6287–6296 RSC.
- Y.-S. Li, D. Menga, H. A. Gasteiger and B. Suthar, J. Electrochem. Soc., 2023, 170, 1–19 Search PubMed.
- G. Yang, C. H. Lee, X. Qiao, S. K. Babu, U. Martinez and J. S. Spendelow, Electrochem. Energy Rev., 2024, 7, 1–46 CrossRef.
- N. Seselj, S. M. Alfaro, E. Bompolaki, L. N. Cleemann, T. Torres and K. Azizi, Adv. Mat., 2023, 35, 1–28 CrossRef PubMed.
- Z. Zhang, M. Guo, Z. Yu, S. Yao, J. Wang, D. Qiu and L. Peng, Energy, 2022, 239, 1–10 Search PubMed.
- S. Zhang, S. Liu, H. Xu, G. Liu and K. Wang, Energy, 2022, 239, 1–11 Search PubMed.
- X. Bai, L. Luo, B. Huang, Q. Jian and Z. Cheng, Energy, 2022, 246, 1–11 CrossRef.
- A. Ganesan and M. Narayanasamy, Mater. Renew Sustain. Energy, 2019, 8, 1–14 CrossRef.
- S. Liu, S. Li, R. Wang, Y. Rao, Q. Zhong, K. Hong and M. Pan, J. Electrochem. Soc., 2019, 166, 1308–1313 CrossRef.
- M. A. Derendyaev, D. V. Koryakin, E. M. Filalova, A. B. Yalmaev, M. Z. Galin, E. V. Gerasimova, A. E. Antipov, A. V. Levchenko and Y. A. Dobrovolsky, Nanotechnol. Russ., 2020, 15, 797–806 CrossRef CAS.
- C. Lei, F. Yang, N. Macauley, M. Spinetta, G. Purdy, J. Jankovic, D. A. Cullen, K. L. More, Y. S. Kim and H. Xu, J. Electrochem. Soc., 2021, 168, 1–11 Search PubMed.
- A. Willert, F. Z. Tabary, T. Zubkova, P. E. Santangelo, M. Romagnoli and R. R. Baumann, Int. J. Hydrogen Energy, 2022, 47, 20973–20986 CrossRef CAS.
- C. A. G. Bezerra, L. J. Deiner and G. Tremiliosi-Filho, J. Electrochem. Soc., 2020, 167, 1–8 CrossRef.
- H. Maleki and V. Bertola, Catal. Sci. Technol., 2020, 10, 3140–3159 RSC.
- S. Shukla, D. Stanier, M. S. Saha, J. Stumper and M. Secanell, J. Electrochem. Soc., 2016, 163, 677–687 CrossRef.
- A. Lesch, Adv. Mater. Technol., 2018, 3, 1–10 Search PubMed.
- V. Costa Bassetto, M. Mensi, E. Oveisi, H. H. Girault and A. Lesch, ACS Appl. Energy Mater., 2019, 2, 6322–6331 CrossRef CAS.
- W. O. Silva, V. Costa Bassetto, D. Baster, M. Mensi, E. Oveisi and H. H. Girault, ACS Appl. Electron. Mater., 2020, 2, 927–935 CrossRef CAS.
- N. Schmitt, M. Schmidt, G. Hübner and B. J. M. Etzold, J. Power Sources, 2022, 539, 1–12 CrossRef.
- B. Derby, Annu. Rev. Mater. Res., 2010, 40, 395–414 CrossRef CAS.
- B. D. James, J. M. Huya-Kouadio, C. Houchins and D. A. Desantis, Mass Production Cost Estimation of Direct H2 PEM Fuel Cell Systems for Transportation Applications: 2018 Update, Strategic Analysis Inc., 2018 Search PubMed.
- H. Liu, J. Li, X. Xu, F. Wang, J. Liu, Z. Li and J. Ji, Electrochim. Acta, 2013, 93, 25–31 CrossRef CAS.
- D. H. Kim, J. H. Cha, S. Chong, S. H. Cho, H. Shin, J. Ahn, D. Jeon, J. Kim, S. Y. Choi and I. D. Kim, ACS Nano, 2023, 17, 23347–23358 CrossRef CAS PubMed.
- M. Jeyaraj, S. Gurunathan, M. Qasim, M. H. Kang and J. H. Kim, Nanomaterials, 2019, 9, 1–41 CrossRef PubMed.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- M. Favaro, C. Valero-Vidal, J. Eichhorn, F. M. Toma, P. N. Ross, J. Yano, Z. Liu and E. J. Crumlin, J. Mater. Chem. A, 2017, 5, 11634–11643 RSC.
- N. C. Röttcher, Y. P. Ku, M. Minichova, K. Ehelebe and S. Cherevko, J. Phys Energy, 2023, 5, 1–13 Search PubMed.
- A. A. Alekseenko, V. E. Guterman, N. Y. Tabachkova, O. I. Safronenko and J. Solid, State Electr., 2017, 21, 2899–2907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04837g |
| This journal is © The Royal Society of Chemistry 2025 |