
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignocellulose saccharification: historical insights and recent industrial advancements towards 2nd generation sugars†
Jorge
Bueno Moron
ab,
Gerard P. M.
van Klink
ab and
Gert-Jan M.
Gruter
 *ab
*ab
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1090 GD Amsterdam, The Netherlands. E-mail: g.j.m.gruter@uva.nl
bAvantium Support BV, Zekeringstraat 29, 1014 BV Amsterdam, The Netherlands
First published on 27th January 2025
Abstract
This study explores the initial industrial development of saccharification technologies, with a primary focus on hydrochloric acid (HCl) saccharification of biomass, particularly wood chips. It traces the historical progress from early 20th-century research to modern advancements, emphasizing the challenges, failures and successes in scaling up these processes. The work details the structural composition of wood, i.e. cellulose, hemicellulose, and lignin, and explains the mechanisms of their hydrolysis. Additionally, it reviews various methods for hydrolyzing wood chips into saccharides, including besides HCl-based methods also sulfuric acid hydrolysis, as well as other methods such as enzymatic hydrolysis and more recent technologies. This review highlights the industrial attempts to bring these technologies to scale, providing insights into the technological advancements and hurdles faced. As developers of Avantium's DAWN Technology, we introduce our optimized hydrochloric acid saccharification process, which enhances efficiency and addresses historical challenges. This comprehensive overview not only documents the historical and technical aspects of biomass saccharification but also underscores the importance of continued innovation in this field.
 Jorge Bueno Moron | Jorge Bueno Moron is currently a PhD fellow in the Industrial Sustainable Chemistry at the University of Amsterdam. He is also a team leader at Avantium, responsible for the development of their biorefinery program. He has a background on sugar chemistry, with experience in saccharification and scale-up technologies. |
 Gerard P. M. van Klink | Gerard van Klink has worked in the field of homogeneous catalysis and organic synthesis at various universities. Since 2014, he works at Avantium as scientist investigating new research lines. His current research interests include the valorization of waste materials. |
 Gert-Jan M. Gruter | Gert-Jan M. Gruter has a background in Polyolefin catalysis (DSM; now Sabic) and ws Professor of Polymer Catalysis at Eindhoven University of Technology. At Avantium he was appointed as CTO in 2004 to develop FDCA/PEF, MEG and biorefining technologies, Gert-Jan currently is also professor Industrial Sustainable Chemistry at the University of Amsterdam, working on the plastic transition. |
Sustainability spotlightThe plastic production volume has seen huge growth, from 15 million ton in 1964 to 400 million ton in 2021 and is expected to pass the 1 Gigaton per year mark (1000 million ton) before 2050. The total global CO2 footprint of this 400 million ton of plastics is approximately 1000 million ton (climate action; SDG 13). Only in a situation that by 2030 most of the volume growth will come from renewable plastics, the transition can be significant by 2050. This will ofcourse require sugar production on an extremely large scale. If on the other hand we will not make a transition to renewable polymers in 2050, the total plastics CO2 footprint for plastics will be approaching 3 Gt (3000 million tons) by 2050, which will be incompatible with the Paris climate agreement targets. For a transition of fossil feedstock to biomass for our plastics, access to high quality, low cost non-food sugars (Zero hunger; SDG 2) will be extremely important. Today, second generation (2G) sugars are not yet commercially available for large scale polymer production. Our review summarizes 100 years of lignocellulosic sugar biorefinery development and discusses recent developments towards lower cost options. It emphasizes the importance of the following UN sustainable development goals: zero hunger (SDG2), industry, innovation, and infrastructure (SDG 9), responsible (production and consumption; SDG 12) and climate action (SDG 13). |
1. Introduction
The journey of biomass saccharification from conceptualization to industrial application spans over two centuries, with significant milestones that have shaped its current state in the chemical industry. Saccharification, defined as the hydrolysis of carbohydrate-containing polymeric materials into monomeric carbohydrates or units with low degrees of polymerization, has its roots in early 20th-century efforts to industrialize the conversion of lignocellulosic materials into fermentable sugars.1The initial foray into biomass saccharification involved acid hydrolysis, leveraging the straightforward approach and availability of acids. Early researchers recognized the potential of converting wood and other lignocellulosic materials into valuable sugars through hydrolysis.2 Nature itself provided a blueprint for this process, as the decomposition of trees often occurs under mild acidic conditions through a process known as autohydrolysis. Organic acids within the hemicellulose and lignin structures of wood catalyze its gradual hydrolysis, a mechanism that was mimicked in early industrial processes.1,3
In the chemical industry, saccharification emerged as a critical process for extracting inherent sugars from biomass structures, essential for transforming materials like wood chips into fermentable sugars.4 This conversion is pivotal for producing biofuels and valuable biochemicals. Historically, the industrialization of biomass saccharification began in the early 20th century.5 Researchers focused on acid hydrolysis due to its simplicity and the availability of acids, but the process faced significant challenges. Equipment corrosion, low sugar yields, and the formation of inhibitors were major obstacles that hindered the large-scale adoption of acid hydrolysis. These inhibitors, generated during the acid hydrolysis process, pose significant challenges for subsequent enzymatic conversions by interfering with enzyme efficiency, thereby reducing the overall yield of fermentable sugars.6–9 Despite these issues, early developments in acid hydrolysis laid the groundwork for future advancements in the field. The mid-20th century saw significant progress with the advent of enzymatic hydrolysis.10 The discovery of efficient cellulases and advancements in recombinant DNA technology allowed for the production of tailored enzyme cocktails, enhancing hydrolysis efficiency. However, the cost of some enzymes and their chemical stability remain a considerable barrier for certain processes. To address this, recent developments have integrated pretreatment processes such as steam explosion, alkaline pretreatment, and ammonia fiber explosion. These methods disrupt the lignin–carbohydrate complex and increase the porosity of biomass and the accessible surface area of the polysaccharides, thereby improving saccharification efficiency.11
The utilization of biomass as a renewable energy source is crucial for addressing global energy needs and environmental challenges. Biomass, including agricultural residues, forestry waste, and dedicated energy crops, offers a sustainable alternative to fossil fuels, reducing greenhouse gas emissions and reliance on non-renewable resources while promoting rural economic development.12 Among biomass resources, wood is particularly significant due to its abundance, relative stability and established supply chains. Forests cover approximately 30% of the Earth's land area, providing a continuous supply of raw material for various applications.13 A sustainable management could ensure a reliable wood feedstock for biomass conversion processes, and wood processing by-products and residues, such as sawdust, chipped branches and bark, to be utilized effectively, minimizing waste and enhancing overall efficiency.
Wood, as a primary source of lignocellulosic biomass, is composed mainly of cellulose, hemicellulose, and lignin. Cellulose forms the structural backbone of plant cell walls with long chains of glucose units. Hemicellulose, a heterogeneous group of polysaccharides, surrounds cellulose microfibrils, adding flexibility and strength. Lignin, a complex aromatic polymer, fills the spaces between these components, providing rigidity and resistance to microbial attack.14–16 The hydrolysis of lignocellulosic biomass to produce 2nd generation glucose involves several stages. Initially, the less crystalline and thus less recalcitrant hemicellulose is hydrolyzed, releasing mono- and oligosaccharides and acetic acid. As the process progresses, cellulose becomes more accessible, and its hydrolysis produces glucose oligosaccharides. Over time, these oligosaccharides are further broken down into monomeric sugars. Lignin, being the most resistant component, undergoes significant changes only under severe acidic or basic conditions, producing phenolic compounds.17
As the developers of Avantium's DAWN technology, we have made significant advancements in biomass saccharification, specifically building upon the historical use of concentrated hydrochloric acid for the hydrolysis of wood chips.18–21 As the authors of this review and the driving force behind the development of Avantium's biorefinery technology, we aim to reflect a commitment to advancing the chemical industry by providing historical insights and showcasing the progress made by the scientific community in optimizing biomass conversion processes to obtain 2nd generation glucose.
The following sections delve into the structure and composition of wood, the various hydrolysis methods, and the historical efforts that have driven the industrialization of saccharification technologies. These insights highlight the critical role of saccharification in unlocking the full potential of lignocellulosic biomass and its implications for the chemical industry.
2. The structure and composition of wood
2.1 Wood formation in trees
Wood formation supports the growth and structural development of trees, contributing to the expansion of the crown, bole, and root system. The crown, consisting of leaves, twigs, and branches, aids photosynthesis and nutrient production. The bole (trunk) provides mechanical stability and serves as the conduit for water and nutrients between roots and crown. The roots, often as extensive as the crown, anchor the tree and absorb water and minerals, sometimes enhancing nutrition through symbiosis with mycorrhizae, which improve their phosphorus uptake.22–24Internally, the trunk features sapwood, which actively transports water and nutrients, and heartwood, which becomes decay-resistant through the deposition of substances during its transformation (Fig. 1). The vascular tissue, consisting of xylem (water transport) and phloem (sugar transport), is critical to tree function. Growth occurs in the cambium layer, which generates new cells for repair and development.26
 | ||
| Fig. 1 Transversal cut of the trunk, reproduced from ref. 25 with permission from the USDA, © 2025. | ||
2.2 Wood classification
Wood is classified into softwoods and hardwoods, distinguished by their cellular structure (Fig. 2):27 | ||
| Fig. 2 Transverse and tangential-longitudinal faces of softwood (left) and hardwood (right), reproduced from ref. 27 with permission from Springer, © 2006. | ||
• Softwoods are primarily composed of tracheids, which serve both structural support and water conduction functions. They also contain ray parenchyma cells, which are involved in the storage and lateral transport of nutrients. Softwoods lack vessels, resulting in a more uniform and simpler structure compared to hardwoods. This composition makes softwoods suitable for structural applications like construction and paper production. They generally have a higher lignin content, contributing to their strength and durability.
• Hardwoods have a more complex structure, containing a variety of cell types, including vessels (pores) for water conduction, fibers for structural support, and both axial and ray parenchyma cells for storage and transport. The presence of vessels allows for efficient water conduction, while the fibers provide mechanical strength. This diverse cellular structure makes hardwoods less uniform but adds versatility. Hardwoods are particularly valued for furniture and fine paper production due to their aesthetic and functional properties.
The tissue composition and structure of wood vary significantly across species and environmental conditions, influencing its suitability for specific industrial applications. This variability makes softwoods preferable for strength and uniformity, while hardwoods are favored for applications that require aesthetic uses like fine woodwork.27
2.3 Chemical composition of wood
Wood comprises approximately 50% carbon, 44% oxygen, and 6% hydrogen, with trace amounts of nitrogen and inorganics.13,15,28 The three primary biopolymers in wood (Fig. 3) are: | ||
| Fig. 3 Wood chemical composition. | ||
• Cellulose (40–50%): a highly crystalline polymer forming the backbone of cell walls. Its degree of polymerization (DP) and crystallinity index (CI) vary across species, influencing its mechanical properties and resistance to hydrolysis.29–31
• Hemicellulose (20–30%): a more amorphous polymer that bridges cellulose and lignin, enhancing flexibility and strength. Its composition varies by species, with mannose-rich hemicellulose in softwoods and xylose-rich variants in hardwoods.32–34
• Lignin (18–35%): an aromatic polymer that binds cellulose and hemicellulose, providing rigidity and microbial resistance. Softwoods generally have higher lignin content than hardwoods.35–37
Extractives (4–13%) and ash (less than 1%) are minor components. Extractives, often phenolic or aliphatic compounds, protect trees from environmental stress, while ash contains minerals such as calcium and potassium. These components vary based on species and environmental conditions, with tropical woods typically containing more ash.38–40 A more detailed account of the chemical composition of wood, including a compilation of literature on the structure of hemicellulose, cellulose, and lignin, can be found in the ESI.†
3. The mechanism of acid hydrolysis
The acid hydrolysis of wood has been investigated for various tree species, including aspen, birch, paper birch, willow, pine, spruce, eucalyptus, loblolly pine, poplar, yellow poplar, oak, and red oak. Among these, aspen wood has shown the highest conversion to sugars.41 Rasmussen et al.17 reviewed the hydrolysis of lignocellulosic materials and the mechanism of sugar dehydration under acidic conditions. During the hydrolysis of lignocellulosic biomass, the primary products from the breakdown of its sugar-bearing polymers are xylose, mannose, arabinose, galactose, and glucose.The hydrolysis of wood typically proceeds through three distinct stages. Initially, hemicellulose, which encases the cellulose, is more accessible and less crystalline, making it more susceptible to hydrolysis. During this first stage, hemicellulose dissolves, releasing its oligosaccharides into the acidic solution, along with acetic acid produced from the hydrolysis of acetyl ester groups in the hemicellulose structure. At this point, the cellulose remains largely intact, but the acid begins to disrupt its crystallinity and close packing, leading to a more amorphous cellulose structure.
In the second stage, as most of the hemicellulose is removed, the hydrolysis of cellulose commences, releasing glucose oligosaccharides into the solution. Prolonged acid exposure in the third stage results in the hydrolysis of these oligosaccharides into monomeric sugars. If these monosaccharide hexoses are not promptly removed, they can further dehydrate to form 5-hydroxymethylfurfural (5-HMF). Additionally, furfural is produced from the dehydration of pentose sugars, such as xylose and arabinose, present in solution after the hydrolysis of the hemicellulose wood structure.42
Moreover, these furan compounds can undergo ring-opening and polycondensation reactions in the presence of acids, resulting in the formation of humins from both furfural and HMF, or producing levulinic acid and formic acid from HMF (Fig. 4). After hydrolysis, the remaining structure of the wood is the lignin. It is the most recalcitrant structure from the three biopolymers present in wood, but under severe acidic or basic conditions its structure can be altered, producing phenolic compounds or acetic acid.
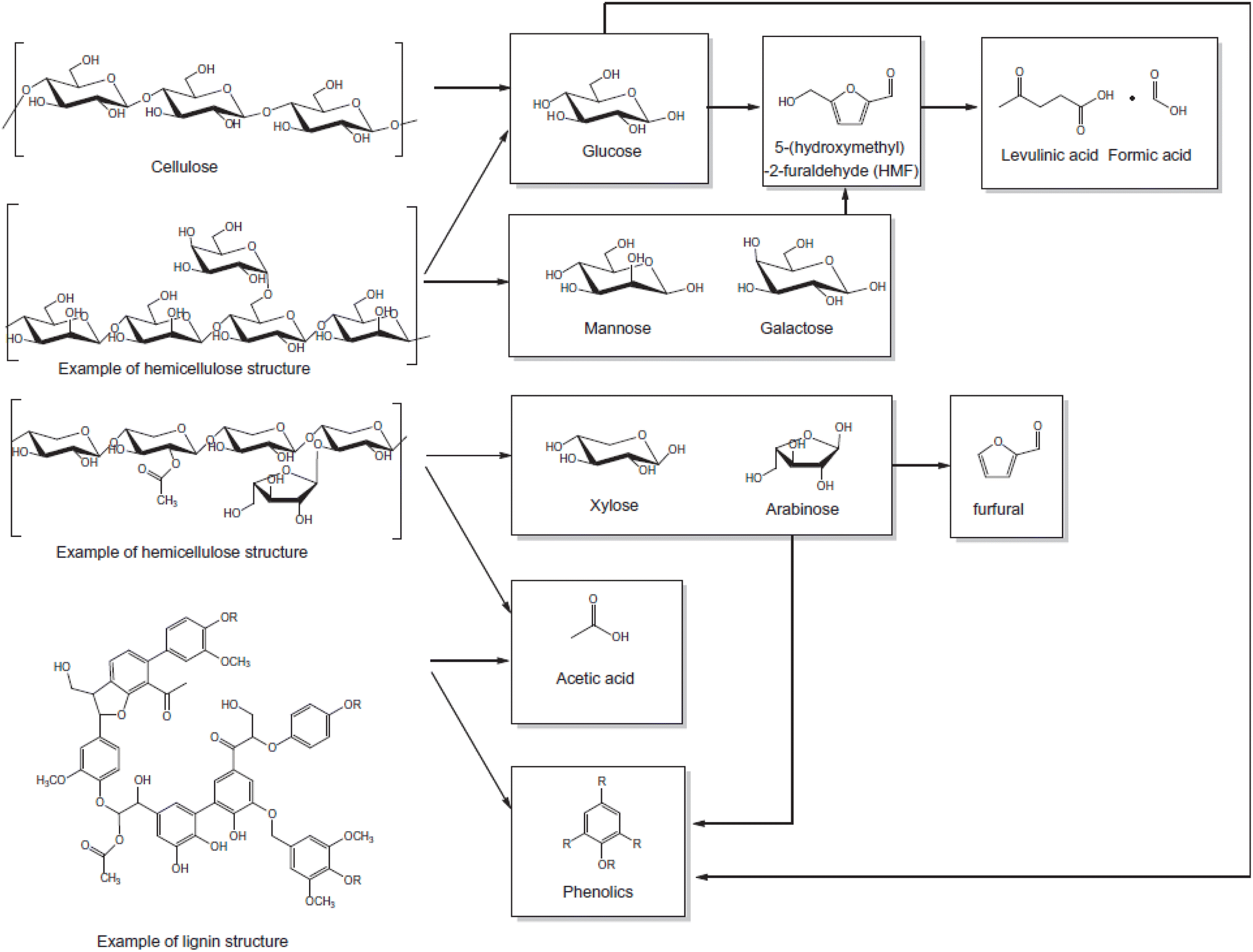 | ||
| Fig. 4 Classes of compounds obtained from the hydrolysis of wood under acidic conditions, reproduced from ref. 17 with permission from Elsevier, © 2014. | ||
In the chemical industry, well-established methods exist to dissolve lignin and produce wood pulp, primarily using sulfite and Kraft pulping technologies. The acid sulfite process, characterized by slow heating, relatively long reaction times, and moderate temperatures (around 130 °C), involves treating wood chips with sulfite and bisulfite ions to convert lignin into soluble lignosulfonates, which can then be separated from the cellulose fibers. However, this method is not suitable for all types of wood.43 In contrast, Kraft pulping is a more prevalent form of chemical pulping, accounting for 80% of the current chemical pulping in the industry.44 Kraft pulping involves the digesting of wood chips at 150–180 °C under pressures ranging from 690 kPa (100 psi) to 1380 kPa (200 psi) in ‘white liquor’, a water solution of sodium sulfide and sodium hydroxide. The lignin binding the cellulose fibers together in the wood is chemically dissolved by this liquor, obtaining the corresponding wood pulp.45
During the hydrolysis of wood the following factors limit the conversion to sugars; the physical properties of the wood itself, which vary across different tree families, the different reactivity of the carbohydrates in the holocellulose, and the structure and properties of the less reactive lignin material46 (Fig. 5). In terms of reactivity, the biopolymers composing the holocellulose have different degrees of crystallinity, with cellulose being the most crystalline and therefore the most chemically resistant to hydrolysis. Hemicellulose, which firmly attaches to cellulose microfibrils, acts as a physical barrier, impeding the initial hydrolysis of cellulose and delaying the release of glucose.46 Within cellulose, the accessibility of acid to different polymer regions is crucial for determining hydrolysis efficiency. Areas more covered by hemicellulose and lignin require higher temperatures, acid concentrations, and longer reaction times for complete hydrolysis. Hemicellulose, being less crystalline and more accessible, is more prone to produce sugar decomposition products. Consequently, the cellulose-to-hemicellulose ratio is a key factor in optimizing hydrolysis conditions (temperature, pressure, acid concentration, and time) to effectively hydrolyze both materials while minimizing sugar decomposition.
 | ||
| Fig. 5 Main factors influencing the hydrolysis of lignocellulosic biomass, reproduced from ref. 46 with permission from Elsevier, © 2021. | ||
Homogeneous hydrolysis is typically faster and requires milder conditions than heterogeneous hydrolysis, making the solubility of cellulose and hemicellulose in the reactive medium important for efficient hydrolysis. The solubility of each sugar polymer in an acidic medium can vary significantly and is influenced by temperature, pressure, acid concentration, and reaction time. Cellulose, present as allomorph β in lignocellulosic materials, has the lowest solubility in mineral acids compared to the other biopolymers in wood.46
Yoon et al.47 studied the effect of lignin on the hydrolysis of cellulose. For this, the hydrolysis of pure holocellulose was compared with the hydrolysis of lignocellulosic biomass. For the same given conditions, the pure holocellulose hydrolyzed faster but also produced more sugar decomposition compounds (formic and levulinic acid). This is explained by the structure of lignin, which interacts with and wraps the cellulose and hemicellulose, decreasing the accessibility of the acid and requiring longer reaction times to effectively hydrolyze the material. Baucher et al.48 also found that cell walls of plants with low(er) S/G (syringyl/guaiacyl) ratios in the lignin structure were easier to hydrolyze. Other researchers found that selectively pre-treating the lignocellulosic materials to remove some of the non-condensed lignin facilitated the overall hydrolysis of the (hemi)cellulosic material. The reason behind is that non-condensed lignin provide higher coverage over cellulose due to its linear shape.46
The physical properties of biomass such as particle size, cell wall thickness, pore size and pore volume have an impact on the hydrolysis of the material. Large chip thicknesses slow the acid hydrolysis of aspen wood, and xylose yields from the hydrolysis of the hemicellulose decrease overall.49 Particle size gave contrasting results for its effect on the hydrolysis of lignocellulosic material. It has a minor effect when a high liquid-to-wood ratio is used, but when this ratio is lowered and less acid is employed, a fraction of xylan could not be recovered after hydrolysis and remained within the particle matter.50 This latter effect becomes crucial when considering the economic feasibility for an industrial application.
Rinaldi et al.51 reviewed the mechanisms involved in cellulose hydrolysis within lignocellulosic materials. The first report on the hydrolysis of 1,4-β-glucans was by Freudenberg and Blomqvist in 1935.52 They demonstrated that the hydrolysis rate of 1,4-β-glucans in H2SO4 at 18 and 30 °C significantly decreased as the degree of polymerization (DP) increased. The activation energy for cellulose hydrolysis was found to be 125 kJ mol−1, slightly higher than that for 1,4-β-glucans with lower DP (114–121 kJ mol−1).51 Saeman first reported the kinetics of cellulose hydrolysis in dilute acid at 170–190 °C in 1945.53 It was determined that cellulose hydrolysis follows a first-order reaction with respect to H3O+ concentration, with an apparent activation energy of 179 kJ mol−1. The overall rate equation for the homogeneous acid hydrolysis of glycosides is presented in Fig. 6.
 | ||
| Fig. 6 Rate of the homogeneous acid hydrolysis of glycosides, reproduced from ref. 53 with permission from the American Chemical Society, © 1945. | ||
Where S, SH+, P, and KSH represent the substrate, the protonated substrate, the reaction product, and the equilibrium constant of the protonation step, respectively. The overall reaction rate of the homogeneous hydrolysis of glycosides clearly shows that the rate of hydrolysis depends directly on the acidity of the catalyst employed. Regarding the protonation of the glycosidic oxygen, it is expected that a strong acid should be more successful in the protonation of the glycosidic oxygen than a weaker acid because the glycosidic oxygen is weakly basic.51
The hydrolysis of cellulose is assumed to follow a similar mechanism as the acid-catalyzed hydrolysis of glycosides which was studied in detail by Nevell and Comstock in 1976 and 1979, respectively.54,55 As shown in Fig. 7, the first step of this mechanism involves the protonation of either the glycosidic oxygen (pathway I) or the pyranic oxygen (pathway II). In principle, taking into account the nature of H3O+ species and the conformational restriction of the cellulosic chains along the glycosidic bond, a simultaneous protonation of both oxygen atoms would be possible as well (inset in Fig. 7).

At this step, either a cyclic (pathway I) or acyclic (pathway II) route occurs.56 Zhou et al.46 explained that the formation of the cyclic intermediate in pathway I requires a conformational change to a half-chair configuration, where C1, C2, C5, and O5 lie in the same plane. This step is energetically demanding due to the rotational constraints imposed by inter- and intramolecular hydrogen bonds within the supramolecular structure of solid cellulose. However, in homogeneous hydrolysis, where cellulose is dissolved in solvents that disrupt hydrogen bond networks, these constraints are relaxed, resulting in much higher hydrolysis rates compared to heterogeneous conditions. Thus, pathway I is considered the dominant pathway for homogeneous acid-catalyzed hydrolysis.54 The role and extent of the alternative pathway II in cellulose hydrolysis remain unclear.56
During cellulose hydrolysis, the first barrier is accessing and hydrolyzing the most crystalline domains. After this initial phase, the rate of hydrolysis levels off, reaching a steady state that slows the reaction.41 Given the generally low solubility of cellulose in various solvents, its hydrolysis has been extensively studied under both heterogeneous and homogeneous conditions.57 Under heterogeneous conditions (Fig. 8, left), D-anhydroglucan layers on the cellulose surface are gradually released and hydrolyzed in the solution. In contrast, under homogeneous conditions (Fig. 8, right), cellulose dissolution and hydrolysis likely occur simultaneously. With longer reaction times in homogeneous conditions, the degree of polymerization (DP) of the oligomers in solution decreases, eventually forming monomeric glucose.58
 | ||
| Fig. 8 Representation of the hydrolysis of cellulose under heterogeneous and homogeneous conditions, reproduced from ref. 51 with permission from Wiley, © 2009. | ||
A breakthrough in lignocellulosic saccharification was the discovery by Rogers et al. that ionic liquids (IL) like [C4mim]Cl are able to fully dissolve untreated wood.59 In contrast to wood delignification methods that rely on the insolubility of cellulose in certain solvents, the IL has the ability to dissolve both lignin and polysaccharides simultaneously. Ionic liquids present new approaches to the hydrolysis of lignocellulosic materials, but their high costs require quantitative recovery and recycling of the reactants, which makes it difficult when these technologies are upscaled in size and applied in a biorefinery-based industry.60
In their review, Mäki-Arvela et al. discussed the hydrolysis of hemicellulose and the factors affecting the hydrolysis of its various heteropolymers.41 Hemicelluloses, present in all plant cell walls, consist of different monosaccharide units, which contribute to their structural complexity and varied reactivity during hydrolysis. The rate of acid hydrolysis for these heteropolymers is influenced by the anomeric configuration of the sugars (α- or β-anomer) and their ring form (furanose or pyranose). β-Anomers generally react faster than α-anomers, and furanose forms hydrolyze more readily than pyranose forms due to higher angle strain. Specifically, arabinose, predominantly in the furanose form, hydrolyzes more rapidly compared to xylose, which is mainly in the pyranose form. This structural dependence highlights the variability in hemicellulose hydrolysis rates and underscores the necessity of understanding these factors to optimize hydrolysis conditions for efficient biomass conversion.
Another key factor in hemicellulose hydrolysis efficiency is its backbone pattern. Hemicellulose contains various substituents, such as acetyl groups, 4-O-methylglucuronic acid, and arabinosyl residues, that significantly influence recalcitrance. Acetylation, for instance, increases hydrophobic interactions, creating steric hindrance and limiting enzymatic access.61 These acetyl groups are also hydrolyzed during processing, releasing acetic acid, which can inhibit enzymatic activity and sometimes reduce overall hydrolysis efficiency.62 Similarly, glucuronidation, such as the incorporation of 4-O-methylglucuronic acid residues, adds negatively charged groups that form strong interactions with lignin, further contributing to resistance. Arabinofuranosyl substitutions, commonly found in arabinoxylans, act as cross-linking points with lignin or other polysaccharides, also compounding recalcitrance. Recent research63 highlights that removing glucuronic acid from xylan significantly increases fermentable sugar release in Arabidopsis plants without the need for chemical pretreatment. Additionally, identifying a gymnosperm enzyme responsible for xylan glucuronosylation presents a promising target for genetically improving forestry trees, enhancing biomass efficiency for sustainable industrial applications.
Overall, the hydrolysis mechanism for hemicellulose follows a similar pathway to that of cellulose, involving either protonation of the glycosidic bond or the pyranic oxygen.41 The hydrolysis rate of hemicellulose is influenced by the anhydro-sugar structure, such as whether the sugar is an α- or β-anomer, and whether it is in a furanose or pyranose form. β-Anomers hydrolyze faster than α-anomers. Additionally, acid hydrolysis proceeds more quickly for furanose forms compared to pyranose forms; for example, arabinose (primarily in the furanose form) hydrolyzes faster than xylose (primarily in the pyranose form). This is due to the higher structural angle strain in the five-membered furanose ring, whereas the six-membered pyranose ring is more stable and less strained. However, both furanose and pyranose rings are almost strain-free. The increased hydrolysis rate of furanose may also be attributed to differences in ring conformation and the inter- and intramolecular stabilizations that are more pronounced in six-membered pyranose rings due to their extensive hydrogen bonding networks.41 Organic acids present beneficial properties during hemicellulose hydrolysis compared to mineral acids, as they are milder and can selectively hydrolyze hemicellulose sugars. An example of this technology includes the selective hydrolysis of arabinose units in arabinoxylans and arabinogalactan.64
The activation energy for the hydrolysis of hemicelluloses varies, with galactoglucomannan having a higher activation energy than arabinogalactan due to the nature of their ether linkages and polymer structures.41 Studies on model compounds indicate that glucosides and mannosides are harder to hydrolyze than galactosides. Within the same hemicellulose polymer, such as arabinogalactan, the activation energy for hydrolyzing arabinose side-chains is lower than for galactose units in the backbone, making lower acid concentrations more effective for the selective scission of arabinose. This variation is attributed to differences in anomeric configurations and stabilization mechanisms within the polymer structures.41 In studying the hydrolysis of xylose hemicellulose, terms like fast- and slow-reacting xylan are often used. Fast-reacting xylans, typically part of side chains in xyloglucans rather than composing the backbone as in glucuronoxylans, exhibit about 8–23% lower activation energy and are more prone to hydrolysis.3
4. The evolution of the biomass saccharification industry
4.1 Early developments
The initial developments in understanding and isolating sugars date back to 1747 when Andreas Marggraf isolated glucose from beets using alcohol, marking a significant departure from the traditional extraction of sugars solely from sugarcane. This discovery was pivotal for biomass saccharification in Europe, setting a foundational technique for extracting sugars from non-traditional sources. Notably, the term “sugars” historically referred primarily to sucrose, the common table sugar derived from sugarcane and, later, sugar beets.5 These historical nuances are essential for understanding the evolution of sugar processing and the broadening of the term ‘sugar’ to include various saccharides like sucrose, glucose, and fructose, each with unique industrial and culinary roles.Subsequently, in 1789, Franz Karl Achard, a student of Marggraf, developed an industrial process to extract and isolate sugars from beets, leading to the construction of various plants. With support from King Friedrich Wilhelm III, Achard opened the first sugar beet refinery in Kunern, Silesia, in 1801 (Fig. 9).66 By 1802, this refinery processed 400 tons of beets per year, achieving sugar yields of 4%. Despite being destroyed during the Napoleonic Wars in 1807, the plant was rebuilt on a smaller scale in 1810 to support the Prussian government during sugar embargoes.67 Despite this setback, the government of Prussia supported the reconstruction of Achard's plant in 1810 to provide sugars for the country amidst supply embargoes.
 | ||
| Fig. 9 First Beet-Sugar Factory in the World—Built at Kunern, Silesia, 1801.65 | ||
Sugar-import merchants faced increasing difficulties competing with the lower price of sugars after Achard's discovery. These first industrial efforts made sugars accessible to everyone, and Achard's work was eventually declared a necessity by the government. The growth of sugar beets surged across the country, but the defeat of Napoleon at Waterloo and the consequent ending of the sugar embargo caused a decline in sugar prices. This downturn made the new beet sugar industry unable to compete, leading to the bankruptcy of most of Achard's refineries by 1815.65,68
In 1811, the Russian chemist Gottlieb Kirchhoff hydrolyzed starch for the first time, by the action of sulfuric acid under heating,1 and the isolated sugar was for the first time referred as glucose.69 The chemist Jöns Jacob Berzelius recognized this reaction as one of the first catalytic reactions in (organic) chemistry.70 In 1819, the French chemist Henri Braconnot accomplished the hydrolysis of cellulose using sulfuric acid,71 but the employment of large amounts and the inherent challenges for the recovery of the acid could not make the process economically attractive. In 1815, Anselme Payen, a French chemist in charge of a borax-refining plant,72 broke the Dutch monopoly on borax (most of which was mined in the Dutch East Indies) by developing a process to produce it from soda and boric acid.73 In 1820, he joined the refining beet-sugar industry, introducing two years later the use of activated charcoal to remove colored impurities from industrial sugar solutions. In 1833, he discovered and isolated diastase, an enzyme present in plants to hydrolyze starch into maltose. Hereafter, Payen studied the analytical composition of wood that culminated in the discovery of the carbohydrate polymer cellulose.74 He became professor of industrial and agricultural chemistry in 1835 at the Central School of Arts and Manufactures, Paris. Among his other contributions, Payen developed new processes to refine sugar, starch and alcohol from potatoes.72
Fructose, another important sugar, was identified later by the French chemist Augustin-Pierre Dubrunfaut in 1847.75 The distinction between different types of sugars, including sucrose, glucose, and fructose, became clearer as these compounds were identified and their properties understood over time. Dubrunfaut's work helped delineate fructose as a distinct sugar, commonly known as fruit sugar due to its common occurrence in fruits and other natural sources.
In 1855, G. F. Melsens built upon Braconnot's work by employing diluted sulfuric acid to hydrolyze cellulose, applying high temperatures around 200 °C under significant pressure, though exact pressure details are not specified in the sources.4,76 While these conditions facilitated cellulose breakdown, the resulting sugar solutions were too dilute for economical industrial applications, prompting a need for processes yielding more concentrated sugar solutions. This necessity arose even though sugar, notably sucrose, was widely available by then, thus requiring economically viable concentrations for industrial profitability.
In 1878, the German chemist A. Mitscherlich developed the idea to use sugars for fermentation processes.77 Mitscherlich observed that sugars in the waste liquor during the production of sulfite pulp could be fermented into ethyl alcohol, which was more valuable than sugars. In 1879, A. Béchamp set the foundation for the upcoming saccharification revolution when he discovered that fuming aqueous solutions of super-concentrated hydrochloric acid (HCl) above 37 wt% could hydrolyze cellulose, obtaining sugar solutions in (much) higher concentrations than before.78
In 1880, E. S. Dauziville used for the first time HCl gas for the saccharification of wet sawdust.79 The HCl was recovered using a vacuum pump and the slurry formed by the solid residue and sugars in solution was treated with water and heated to complete the hydrolysis of the oligosaccharides in solution. This last step is later commonly referred to in literature as post-hydrolysis or second hydrolysis.80
In, 1883, E. Fleschsig accomplished the hydrolysis of cotton using concentrated sulfuric acid.81 For the last stage, the sugar–acid solution was diluted and heated to accomplish the second hydrolysis of the sugar oligomers in solution. The hydrolysis of the sugars was almost quantitative but the big volumes of acid required and the low concentration of the sugar acidic solutions made the process unattractive.82 However, Fleschsig's research set the basis for the isolation of lignin by developing a process to quantitatively hydrolyze all the sugars from the biomass, leaving the lignin almost unaffected.82
In 1892, Lindsey and Tollens extended the method of Fleschsig with extensive research on the role of sulfonate groups in sulfite pulping by determining the sulfur and sulfonic acid content of lignin.83,84 After hydrolyzing the biomass, the amount of sulfonate groups in the lignin was shown to impact the quality and properties of the lignin. Since the hydrolysis of biomass started to become industrialized, the amount of lignin from saccharification plants was increasing, so it became important to have a simple and reliable method for the determination of sulfonate content of lignin in pulp to find new applications.
Between 1898 and 1900 new patent applications for the saccharification of biomass with sulfuric acid were filed, soon followed by the construction of new biomass hydrolysis plants.84 The biggest challenge was in the recovery of the large volumes of sulfuric acid required to fully hydrolyze the biomass, which resulted in very dilute sugar solutions. Also, the energy required to concentrate these solutions and separate the sugars from the acid was a serious scale-up issue. R. F. Ruttan patented a new process to ferment these sugars into ethanol, which was easier to recover.85 The chemist E. Simonsen worked also on these fermentation processes using sulfuric acid for the saccharification of sawdust. During his work, he used dilute acid (0.5–1% sulfuric acid) under high pressure (12–20 bar), allowing him to produce 7.6 liters of ethanol per 100 kg of dry sawdust fed into the system.86 A small experimental plant was erected in Oslo but the large quantities of water to be separated from the sugars, along with the acid, failed to make the process economically attractive, so the plant soon ceased operation.84 Nevertheless, these efforts were later considered as a land mark in the upscaling of saccharification technologies.84
In 1898, A. Mitscherlich also developed a process to obtain paper glue from waste liquor during sulfite pulping, which, along with his earlier fermentation processes, was implemented on an industrial scale in Hof, Germany.77
At the end of the 19th century, more researchers started to direct their efforts to use wood waste as feedstock for saccharification purposes. At this time, most of the construction materials were based on wood and in the manufacturing of lumber more than 50% of the tree was usually wasted as sawdust, shavings, slabs and edgings. The sawmill waste was estimated to be about one ton for each thousand board feet of lumber produced (around 1.5 ton).82
The plant was built to process 2 tons of dry sawdust per day in order to raise funds from American capitalists. The results were satisfactory and a larger plant was erected at Hattiesburg, Mississippi. The plant cost was 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dollars (equivalent to about $7.5 million today, indicating a large-scale operation) but it never successfully operated. It was one of the largest plants ever built for this purpose and served as a striking example of a technology that produced reliable results at laboratory scale but faced serious challenges when magnified in size.89 The feedstock chosen was wood waste, and the hydrolysis was achieved inside digesters made of iron with lead liners to prevent corrosion. The temperature inside the reactors was about 145 °C and the pressure around 7 bar. The reaction time ranged from 4 to 6 hours and the sulfurous acid and steam were later blown off into absorbing tanks to partially recover the acid. The main challenges were the big volumes of acid used, its recovery, and the prolonged action of high temperature and pressure on the sugars, resulting in undesired sugar decomposition reactions. It was reported that the lead lining also could not sustain through several runs, which forced the factory to frequently stop for longer time periods.84
000 dollars (equivalent to about $7.5 million today, indicating a large-scale operation) but it never successfully operated. It was one of the largest plants ever built for this purpose and served as a striking example of a technology that produced reliable results at laboratory scale but faced serious challenges when magnified in size.89 The feedstock chosen was wood waste, and the hydrolysis was achieved inside digesters made of iron with lead liners to prevent corrosion. The temperature inside the reactors was about 145 °C and the pressure around 7 bar. The reaction time ranged from 4 to 6 hours and the sulfurous acid and steam were later blown off into absorbing tanks to partially recover the acid. The main challenges were the big volumes of acid used, its recovery, and the prolonged action of high temperature and pressure on the sugars, resulting in undesired sugar decomposition reactions. It was reported that the lead lining also could not sustain through several runs, which forced the factory to frequently stop for longer time periods.84
In 1909 Malcolm F. Ewen and G. H. Tomlinson, two chemical engineers associated with Classen at Highland Park, were granted a patent for an industrial process to hydrolyze large quantities of lignocellulose materials using aqueous hydrochloric and/or sulfuric acid either separately or in combination.90 The goal was to produce large quantities of fermentable sugars and quickly convert them into alcohols. Malcolm and Tomlinson developed a method in which the biomass in contact with the hydrolyzing agent was quickly heated and cooled to reduce undesired sugar decomposition reactions.90 The same year, J. K. George patented, under the Classen rights, a process to remove organic acids formed during the hydrolysis of biomass inside the digesters (mainly formic and acetic acid) to avoid undesired reactions during the fermentation processes.91 After several years of research, Malcolm and Tomlinson were convinced that Classen's method needed modifications in order to attract investors. Their feedstock of choice was sawdust, and their focus was on shortening the hydrolysis time from 6 h to 45 min by using direct steam and then quickly removing the sugars from the reactor to avoid their decomposition.
The patents for this modified Classen method were purchased by the Wood Waste Products Company and a commercial scale factory at Chicago Heights was built to prove the technology.84 The plant was built in workshops on the grounds of a large machinery manufacturer and a small quantity of sulfur dioxide gas (1% in weight of the biomass) was used to hydrolyze the biomass at 7 bars of pressure by applying steam.84 After 10 to 15 min, the steam was cut off and the digesters revolved slowly for 40 to 45 min, keeping temperature and pressure constant inside the reactors. The Wood Waste Products Company (now called Standard Alcohol Corporation) erected in 1910 a new plant at Georgetown, South Carolina, to keep testing the modified Classen method from Malcolm and Tomlinson. In the meantime, the Classen Lignum Company that bought the rights in 1903 from Classen continued its efforts to make their original Classen process profitable by building another factory at Port Hadlock, Washington.84
The new plant at Georgetown, from the Standard Alcohol Corporation, used waste materials from sawmills to produce ethanol. The plant produced 6.4 to 9.5 liters of alcohol per 100 kg of wood waste using the modified Classen method, and the annual output of the factory was about 2000000 liters of ethanol. The factory also applied the conditions recommended by Simonsen earlier in the 1890s.86 This successful plant was later acquired by E. I. du Pont de Nemours and Company which operated it for a few years only. After disposing the plant to DuPont, the Standard Alcohol Corporation built another factory in Louisiana. However, during World War I, the amount of material available for saccharification was significantly reduced, and this, together with a significant decrease in price for alcohol and sugars, resulted in both plants ceasing operation.84 These plants, and the research and development undertaken, were recognized as one of the best developed single-stage batch wood waste hydrolysis on commercial scale. The process had the advantage of great simplicity, and, with some improvements, it is still the method of choice for certain situations.
Wallace P. Cohoe also published work on the recovery of organic volatile aromatic compounds present in wood which are responsible for its characteristic wood fragrance. These compounds are easily hydrolyzed even at mild conditions and, therefore, were never isolated before. Cohoe patented a process to split the hydrolysis of sawdust with several pre-treatments using steam to remove these volatiles first. Then, the acetic acid formed was recovered during the pre-hydrolysis. Then both the hemicellulose and the cellulose were fully hydrolyzed using steam and vaporized 35% HCl (0.5–2 wt% HCl relative to the wood).92 The idea of applying consecutive steps to hydrolyze different components of the biomass in separate stages was later replicated in industry to increase the purity of the product streams.
4.2 The saccharification industry during the first world war
Bergius looked for interest to commercialize this process on industrial scale. The German chemist and businessman Hans Goldschmidt, the son of Theodor who founded the Chemische Fabrik Th. Goldschmidt in Berlin (Fig. 10), offered Bergius his resources to invest in his technology. Th. Goldschmidt AG, founded in 1911, acquired a chemical plant in Mannheim-Rheinau to have its own supply of inorganic chemicals such as hydrochloric acid and sulfuric acid, reducing the dependency on other larger corporations. The Mannheim-Rheinau plant had the capacity to produce mineral acids in large quantities, becoming crucial for the upcoming work of Bergius. At the beginning of World War I, in 1914, Bergius accepted the offer from Theodor Goldschmidt to move his laboratories to Mannheim-Rheinau and assume the position of firm director at this plant. In the beginning, the goal was to commercialize his work related to the liquefaction of coal to supply the country with alternatives sources of fuel in order to reduce the dependency of German imports. However, soon his work turned completely to the saccharification of wood waste.
 | ||
| Fig. 10 Workers at the Chemische Fabrik Theodor Goldschmidt in Berlin.96 | ||
The commercialization of synthetic fuel required ten years of research and the expenditure of 3 million dollars before its practical success was assured.97 After World War I, the demand for gasoline decreased, and this technology was attracting fewer investors. Despite that, several factories were built throughout Germany, reaching an annual production peak of 300000 tons of motor fuel through this coal-hydrogenation process. Later this technology also arrived in the US, and several factories were built when the rights were purchased by Standard Oil of New Jersey.98
Before this early work in the Goldschmidt firm, Bergius already expressed his concerns about the volumes of wood waste produced by the pulp and wood distillation industries, defining these industries as highly inefficient in terms of mass balance and wood utilization. At industrial scale, only 50% of the wood was transformed into pulp and the distillation process of wood produced less than 40% of useful materials. Additionally, the manufacturing of timber was similarly inefficient and big quantities of waste wood were produced in sawmilling and similar operations.99,100 This, together with the beginning of the World War I, producing a scarcity of food in Germany, made Bergius turn his attention to use the highly available wood waste from the timber and construction industry to alleviate this issue. For that, Bergius adapted the saccharification of wood for the production of a concentrated and digestible sugar material, or mixture of sugars, fit for human consumption in the form of pure glucose or mixed carbohydrates to be used as cattle feed.101 The foundation of Bergius's work was the work of Richard Willstätter, who was awarded the Nobel Prize in Chemistry in 1915 for his research on chlorophyll, and László Zechmeister in 1913 (Fig. 11),102 in which they successfully hydrolyzed cellulose using fuming HCl at low temperatures.
 | ||
| Fig. 11 (Top left) Prof. Richard Willstätter. (Top right) Prof. László Zechmeister. (Bottom) Willstätter and László Zechmeister laboratories in 1913.102 | ||
This technology simplified the hydrolysis by almost quantitatively hydrolyzing the sugars within wood at room temperature and atmospheric pressure by the action of highly concentrated HCl solutions. Wohl and Krull also reported in 1921 a 60.9% yield of D-glucose from pine shavings when using fuming HCl at 20 °C for 5 hours.82,103 Zeno Ostenberg was granted a patent in 1917 for the hydrolysis of cellulose at low temperatures with hydrochloric acid in concentrations up to 39%, combined with either sulfuric or phosphoric acid. Ostenberg remarked the importance of the hydrochloric acid solution concentration to effectively hydrolyze all of the cellulose.104 Also in 1917, Ostenberg was granted a patent obtaining similar results by dissolving sodium or calcium chloride in sulfuric or phosphoric acid.105
The plant at Mannheim-Rheinau supplied the required volumes of supersaturated hydrochloric acid to commercialize the process, and Bergius convinced the Swedish chemist Erik Hägglund to unite forces and create Deutsche Bergin A.-G. fur Kohle-und Erdölchemie. Hägglund was an experienced organic chemist, production engineer, and wood and forest product specialist, primarily focused on lignin and lignosulfonic acids. He joined forces with Bergius to focus his work on the saccharification of wood waste using fuming hydrochloric acid at low temperatures. During this time, Hägglund also studied the effect of concentrated sulfuric acid for the hydrolysis of sawdust. In 1915, he reported that using a 1:1 mass ratio of biomass to sulfuric acid, a 55% sugar yield could be achieved.106
In 1914, Francis E. Gallagher obtained a patent for recycling the acidic sugar solution after processing sawdust, referred to as hydrolysate, to hydrolyze fresh biomass.107 Gallagher discovered that the acidic sugar solution after the hydrolysis of sawdust still had the required acid concentration to continue hydrolyzing new biomass, and therefore, this acidic sugar stream could be reused several times before separating the sugars from the acid. In this way, the sugar concentration in the acidic solution increased after every recycle, improving the overall economic aspect of the process. This concept was further developed by Hägglund, who found that reusing the same acidic sugar solution for the saccharification of new batches of wood led the hydrolysate to reach an equilibrium where the hydrolysis stopped, potentially due to a loss of HCl from undesired reactions of the acid with sugars and lignin.108
The eventual outcome of this work was the Bergius–Rheinau or Bergius–Willstätter–Zechmeister process, now referred to in literature as the consecutive hydrolysis of biomass in reactors connected in series by the action of fuming hydrochloric acid at low temperatures in one single hydrolysis step, with the principles and details patented in 1917.109,110 During this time, Henry Ford, founder of the Ford Motor Company, also showed interest in developing this technology in the US, but never proceeded, possibly due to economic viability and technical challenges in scaling the technology.111
The Darboven process offered a simplified alternative, suspending wood chips in carbon tetrachloride to act as a heat-absorbing medium during HCl gas hydrolysis. This design minimized sugar decomposition by efficiently removing heat and eliminated the need for complex moving components. The hydrolyzed material was dried to remove residual HCl and water after the carbon tetrachloride was recovered.114
The Herneng process further evolved the technology, employing wet wood chips in poly(vinyl chloride)-lined reactors with 40 inclined trays.115 Pre-hydrolysis with a 30 wt% HCl solution separated hemicellulose sugars, which were filtered and partially recycled to increase sugar concentration to 20–25 wt%. Subsequent cellulose hydrolysis used 41–42 wt% HCl, with HCl gas introduced to enhance acid concentration during the reaction. This process concluded with the separation of sugars from lignin, followed by post-hydrolysis to maximize fermentable sugar yields.
Additionally, E. C. Sherrard made significant contributions to saccharification research, investigating the use of concentrated HCl and sulfuric acid for cellulose hydrolysis. His studies on the effect of chloride salts, such as zinc and barium chlorides, demonstrated that these compounds improved cellulose swelling and enhanced overall hydrolysis efficiency when combined with mineral acids. Although some of Sherrard's findings were later refined, his work influenced 20th-century developments in saccharification technology.116,117
These technologies and studies highlighted innovative approaches to saccharification, though many faced practical challenges such as material durability, scalability, and process optimization.
 | ||
| Fig. 12 (Left) Erik Hägglund. (Right) Friedrich Bergius in his laboratory.122,123 | ||
Bergius then devoted himself to the saccharification of biomass and contributed with publications and several patents to optimize the process.99,124,125 Bergius, as managing director of the firm and very active in the media, brought articles to the spotlight and explained how the world could feed on converted wood100 and that food from waste wood is a problem of German chemists.126 His work in the corporation was focused on collecting funds to explore and scale-up the hydrolysis technology. Bergius convinced Dutch capitalists and Scotch distillers showing how his sugar production could meet the demands of (for instance) the chocolate candy industry, assuring they could provide 450 pounds of edible glucose sugar per ton of dry wood fed into the system, which was an idealistic output compared to the sugar industry production at that time.
Dutch capitalists and Scotch distillers eventually invested 1 million dollars in the Bergin corporation and Bergius started running industrial tests in Switzerland. The first pilot plant was already under construction in 1925, which aligns with the industrial developments mentioned in 1927, followed by the first, large scale plant in 1932 in Mannheim-Rheinau.127 In 1935, a small plant based on the Bergius–Rheinau process started operations in England with a capacity of 7000 hectoliters of alcohol per year.128 These accomplishment depleted the funds, but Bergius pursued the investors to further support the project with an additional million dollar, concluding that only perseverance, faith, and a great deal of optimism, not to mention time, are necessary to develop a successful industry of this kind. The foreign funds not only helped Bergius to restore his shattered finances, but came in handy to the Nazi war program that took over his technology in 1936.98,129
The Bergius–Rheinau process utilized forestry residues as feedstock, which were shredded, dried (moisture reduced from 40% to 5–10%), and mixed with sawdust. Hydrolysis was conducted using a countercurrent system in reactors connected in series, using a 40 wt% HCl solution at 15–20 °C. This approach dissolved two-thirds of the wood, achieving near-complete saccharification, while one-third remained as lignin, which could be used as fuel or converted into charcoal.
Sugar recovery involved washing lignin with water and concentrating the solution using evaporators. The system, designed to minimize sugar degradation and equipment corrosion, employed vacuum distillation and spray drying to separate HCl and recover sugar syrups with 55–65% sugars and residual acid. Post-hydrolysis of oligomeric sugars yielded fermentable monosaccharides suitable for alcohol production. The sugar distribution in the hydrolysate was approximately 60% glucose, 17–21% mannose, and smaller proportions of galactose, xylose, and fructose.
For every 100 kg of wood processed, the outputs included 33 kg of lignin, 2–2.5 kg of acetic acid, and 50 liters of alcohol from fermentable sugars. Non-fermentable sugars, such as xylose and galactose, were concentrated, crystallized, and used as fodder. A complete and detailed description of the Bergius–Rheinau process, including reactor configurations, hydrolysis parameters, and separation techniques, is available in the ESI.†
Several modifications were applied to the Bergius–Rheinau process. Tanchyna suggested to use oil as heating medium during the post-hydrolysis and sugar recovery.8 The sugar oligomers in the acidic hydrolysate solution were found to be cellotriose, cellotetraose and cellohexaose, indicating that no five-membered oligomers were present.130 When using pine wood, the sugar composition after post-hydrolyzing the hydrolysate was found to be 56% D-glucose, 18% D-mannose, 2.5% D-galactose, 1.5% D-fructose, 7% D-xylose, 2% lignin and 11% of undetermined substances.131 Bergius also worked on the recovery and recycling of the hydrochloric acid,9 and he finally suggested that their sugar product should be used only for cattle feed.
Some of these findings were applied in a second larger plant built in 1939 in Regensburg, Germany, at a cost of about 8 million dollars. The plant, referred as Süddeutsche Holzverzuckerungswerke A. G., had the capacity of about 130 tons of wood per day and 60% sugar yields were claimed.82 At the end of World War II, the British intelligence department reported that the real total capacity was lower and never fully reached.7 The feedstock was mainly spruce logs and production of finished products during one war-year were 3000 tons of sugar for food and animal fodder, which is about 25% of the claimed productivity, and 3000 tons of lignin. In later literature, the plant was reported to have a capacity of 36000 tons of sugar per year.127 The plant was very similar to the factory at Rheinau-Mannheim, only larger and more modern. The British intelligence reported the concentration of the hydrochloric acid used for the hydrolysis of biomass was 51 wt%. The acid was usually produced in the same saccharification plant by burning chlorine in an atmosphere of coal gas in a quartz combustion chamber. When the chlorine could not be produced, commercial hydrochloric acid was purchased and re-concentrated. Also, calcium chloride was added to the dilute acid solution in order to increase the chloride concentration.132
During 20 years of Bergius leading this technology, the capacities at the Rheinau and Regensburg plants achieved peaks of only 400 and 1600 tons of raw sugar production per year, respectively.133 However, the factories were operational as long as they were funded by the government, and at the end of the war with the finance of the German government shattered, both plants ceased operation. Industrial interests were shifting for investors after the oil industry appeared on the scene while the scarcity of food was declining, and with it, the reasons to invest in the Bergius technology. Feed-yeast production could barely compete with the increasing oil-seed growth throughout the world and the petrochemical industry could also provide alcohol from ethylene obtained by petroleum cracking. Moreover, the Bergius–Rheinau process at industrial scale lacked sufficient investigations of various operation steps. The recovery of the HCl hydrolyzing agent was only 82%, and challenges inherent to build a factory resistant to fuming hydrochloric acid drained the funds. Most of the plant had to be covered or lined with quartz and platinum, which made the installation very expensive. In 1945, Bergius sought permission from the American authorities to resume production of sugar from waste wood after the World War II, but the request was not granted.134
In 1948, the plant in Rheinau was rebuilt to run a modified Bergius process, later known as the Udic-Rheinau process. This process was developed by Theodor Riehm in 1948, and patented in the 1950s.135 It was designed to increase the purity of the dextrose product stream during the hydrolysis of the cellulose by introducing a preceding step, referred as pre-hydrolysis, to first remove the hemicellulosic fraction of the wood. This represented one of the first detailed descriptions of a pre-hydrolysis method.
The key innovation was the introduction of a pre-hydrolysis step using low-concentration HCl and elevated temperatures (e.g., 130 °C with 1 wt% HCl or 20 °C with 32 wt% HCl in later iterations111) to remove hemicellulose before cellulose hydrolysis. This adjustment allowed the hemicellulose sugars to be separately utilized for fermentation (e.g., producing alcohol or furfural) while preparing the cellulose for subsequent hydrolysis.
In the main hydrolysis, cellulose was treated with 41 wt% HCl at 20 °C in reactors connected in series, achieving high sugar recovery. Innovations in lignin washing significantly reduced water usage and enabled direct recycling of the acid. Acid recovery was enhanced through a three-stage distillation system, reducing acid losses to 5–6% (compared to 18–20% in the earlier process) and minimizing energy consumption.133
Additional modifications included:
• Simplified reactor designs for pre- and main-hydrolysis, enabling the use of finely divided feedstock like sawdust without prior drying.
• Optimized acid recovery using vacuum distillation and thin-layer evaporators, lowering steam consumption by nearly 70%.
• Efficient heat recovery and water recycling, further improving the process's energy efficiency.
The final glucose syrup was filtered, decolorized, and crystallized, achieving purities of 99% glucose. From 100 kg of wood, 22 kg of crystalline glucose was obtained after a single crystallization step.133 A complete and detailed description of the Udic-Rheinau process, including mass balances, equipment configurations, and operational specifics, is available in the ESI.†
In 1960, this plant achieved semi-work status with a yearly production of 1200 tons of sugars, with a recovery of 94–95% of the acid by vacuum distillation and its further reconcentration in two columns operating at different pressures. Two unique features of this process were the inclusion of a fermentation process for the product sugar solutions and plans to use the lignin as a material to use for the manufacturing of plastic-like material.111,132,136
Scholler explained that for longer runs, the temperature inside the reactors had to be reduced and the flow rate increased in order to minimize the degradation of sugars inside the vessels, although this could potentially lead to lower sugar concentration.137 Scholler also patented various improvements on his percolator technique. These improvements focused on the recovery of sugars from inside the lignocellulosic residue after hydrolysis, by washing this residue with more diluted sulfuric acid at 10–20 °C below the temperature of the hydrolysis reactors. The actual temperature of reaction in the percolators was typically around 120–150 °C, and the pressure ranged from 5 to 10 bar. The contact of this colder and more diluted acid with the warmer and more concentrated sulfuric acid inside the biomass produced a sudden decrease in pressure, which caused steam to develop inside the pores of the biomass. This steam expelled the hydrolysate along with the sugars in it, from inside the solid residue to the outside, increasing the recovery of the sugars and improving the overall technoeconomic aspect of the process.138 Additionally, this steam could also help disintegrate the solid material into smaller pieces. However, several difficulties were reported to also be inherent in the Scholler process as it was practiced in Germany; the intense labor required, the extensive use of water and the rather diluted final sugar solution were factors that slowed commercialization of the process.140
In 1931, the first Scholler commercial plant was built at Tornesch, Germany, with a production capacity of 13000 tons per year. In Dessau and Holzminden, Germany, two commercial plants were operating with a yearly production of 42000 and 24000 tons, respectively. In Ems, Switzerland, a plant with a yearly production of 35000 tons was built.140 In 1936, a plant was built in Manchuria, and later in 1938 one in Korea and three more in Italy would follow. All these plants used wood as feedstock with an estimation that, in 1941, at least twenty such plants were already in operation in Nazi Germany and its ally countries. In addition to alcohol, these plants produced glycerol and feed yeast from these sugar hydrolysates.132,141 After World War II, the oil industry made most of this technology and plants less attractive to investors, and faced many difficulties to survive. As a result, most of the Scholler plants in Germany ceased operation.140
000 gallons of ethyl alcohol per day from 300 tons of wood flour and sawdust fed, corresponding to roughly a 22% conversion of all sugars to ethanol.142 During World War II, German submarines were sinking transport ships from Cuba containing molasses and ethanol. Therefore, the US Government decided to invest on these technologies to use wood for the production of ethanol in their synthetic rubber program.140 The Defense plant corporation, an organization of the US government, awarded a contract to the Vulcan Copper and Supply Company to design and construct a facility to produce ethanol. This plant was based on modifications of the Scholler process, previously researched at the Forest Products Laboratory (FPL) of the US Department of Agriculture in Madison, Wisconsin.140 Based on early work by Ritter and Sherard, FPL built a pilot facility that operated this improved Scholler technology, referred in literature as the Madison–Scholler process.143 In this modification, the residence time inside the reactors was shortened, minimizing the formation of decomposition products from sugars, with the prospect of increasing the production of ethanol up to 45%.143 Original schemes of the process can be seen in Fig. 13. Although the construction stopped at the end of World War II, the Defense plant corporation decided to eventually complete the facilities and test this saccharification process. The plant was completed in 1946, and test runs were initiated with the Vulcan Copper and Supply Company providing technical management.
 | ||
| Fig. 13 Drawing of the Madison–Scholler process from the 1940s, reproduced from ref. 143 with permission from the American Chemical Society, © 1946. The wood chips were loaded into the Hydrolyzer where diluted sulfuric acid at high pressures and temperatures would transform the biomass into its oligomeric sugars. The sulfuric acid would then be de-pressurized and recovered in the recycling tank to separate from the sugar solutions and the lignin. Later, the sugars were neutralized and fermented in three stages to finally produce ethanol. | ||
Despite problems with tar formation and calcium sulfate deposits, from neutralization of the sulfuric acid used to hydrolyze the cellulose, the designed production rate was achieved. It was claimed that the sugars produced from a ton of dry bark-free Douglas fir wood waste were sufficient for the production of 244 l of 95% pure ethanol.51 Dow Chemical Co. opened, in 1944 a pilot plant in Marquette, Michigan, and by 1944 already 155 plants were based on this modified Scholler process for the hydrolysis of wood.144 Most of the work was done on Douglas fir and southern pine, which refers to pine species from the southern United States. Sugar yields of 50% based on the dry wood material were reported by using 0.5 wt% sulfuric acid. However, the process eventually proved too costly to compete with petroleum-derived synthetic ethanol by the end of the World War II.
There were efforts to utilize the facilities for the production of waxy products, but these were not designed for this type of operation and the plants had to be shut down and be dismantled.140 Eventually, with the prevalence of cheap-petroleum ethanol and other chemical products, there was no economic incentive to pursue biomass saccharification technologies in the US.140
4.3 The Soviet Union and the saccharification industry
In 1935, the first industrial scale plant for the production of ethanol was built in Leningrad. The Soviet Union developed a 1-stage percolation process using wood chips mixed with sawdust in a 1:1 mass ratio, and using concentrated sulfuric acid at different temperatures. The process stages and parameters of this percolation process can be found in Table 1.
| Stage | Duration, min | H2SO4, % | Pressure, MPa | T, °C | Hydrolysate, m3 |
|---|---|---|---|---|---|
| Sawdust + chips (1:1) loading 145 kg m−3 |
40 | 0.8–1 | 0.0 | 25–90 | Circulation |
| Steaming | 30 | — | 0.9 | 175 | — |
| Percolation | 80 | 0.7–0.85 | 1.2 | 187 | 40–43 |
| Washing | 10 | — | 1.25 | 190 | 9–10 |
| Squeezing out | 30 | — | 1.3 | 195 | 7–7.5 |
| Lignin discharge | 10 | — | 0.6–0.7 | 160 | — |
| Total | 200 | 3.3–3.8% RS in hydrolysate | 57–60 |
The 1-stage USSR modification of the Scholler process produced 45–46% sugar yield from “oven dry wood”, which corresponded with 60–70% of the original polysaccharide content in sawdust. After the percolation hydrolysis, the residual lignin still contained 15–25% residual cellulose. The annual production of lignin reached 1.5 M tons by the end of 1980s, but only 30% of this lignin was used in further applications. Whereas the rest formed giant landfills around the hydrolysis plants (Fig. 14), with the inherent risk of self-ignition. Part of the sulfuric acid could not be recovered and the sugar hydrolysate contained about 2% of glucose, 3% of other hexoses, 1% of pentoses, 0.4% mixed oligosaccharides and up to 0.6% of sugar decomposition products, including furfural, HMF and levulinic acid.145 Pseudo–lignin compounds were also obtained in the hydrolysate, most likely from the condensation of sugars, furfural and phenolic compounds present in the lignin, turning the sugar solutions dark. Among minor contaminants, methanol, formaldehyde and soluble phenolics were also reported.145
 | ||
| Fig. 14 Lignin Pile in the modern day Russia. | ||
In a later modification of this percolation process, a separate, milder preceding step to remove the less recalcitrant hemicellulosic fraction of the wood was introduced. These studies were based on the relative decomposition rates of the sugars produced during the hydrolysis of biomass, which followed as the order: D-glucose < D-galactose < D-mannose < L-arabinose < D-xylose.145 This pre-hydrolysis was accomplished using 0.5 wt% sulfuric acid at 130–145 °C to obtain a 3.5–4.2% sugar hydrolysate with a C5:C6 (pentose:hexose) sugar ratio of 7:3. This preliminary hydrolysis quantitatively removed most of the pentose fraction in the wood and was later followed by an additional, major percolation step. In this step, 0.8–1.0 wt% sulfuric acid was used at 185–190 °C to recover 75% of all the hexoses present in the biomass. This main percolation stage produced a sugar solution of 2.5–3.2 wt% with a C5:C6 sugar ratio of 1:4. The amount of ethanol, and other compounds formed during this process are detailed in Table 2.
| Ethanol | Fodder yeast | Furfural | Hydrolysis lignin | CO2 | Humins | Distillation waste |
|---|---|---|---|---|---|---|
| 160–175 | 30–40 | 5–7 | 350–400 | 70–90 | 150–225 | 100–150 |
Chalov and Korotkov published their results on pine wood saccharification using different hydrochloric acid concentrations at various temperatures (Table 3).146 Wood chips with a moisture content of ∼0.5 wt% were submerged in 30–36% HCl solutions inside pre-heated vessels at 20–40 °C. Once the hydrolysis was complete, the HCl concentration was diluted to 2% and heated at 100 °C for 3 h to carry out what is nowadays referred as the second hydrolysis or post-hydrolysis step, not to be confused with a pre- and main-hydrolysis step. It was noted that a 32 wt% aqueous HCl solution was obtained when washing the lignin after the hydrolysis and this solution could directly be reused for the hemicellulose hydrolysis of a new batch of biomass, improving the economical aspect of the process.146
| Temperature (°C) | HCl concentration (%) | Time for hemicellulose hydrolysis (h) |
|---|---|---|
| 35 | 30 | 8 |
| 32 | 4 | |
| 34 | 2 | |
| 36 | 1 | |
| 40 | 30 | 4 |
| 32 | 2 | |
| 34 | 1 – partial degradation of sugars | |
| 36 | 1 – partial degradation of sugars |
Chalov reported that at 20 °C, the HCl concentration required to be 36 wt% or higher to quantitatively hydrolyze the hemicellulose within 10 h. When the reactors were heated to 30 °C, 36 wt% HCl could hydrolyze the hemicellulose within 2 h and some disruption of the cellulose crystallinity was already observed.146 At 30 °C, HCl concentrations lower than 36 wt% could already hydrolyze the hemicellulose using longer reaction times, with reports showing this could be achieved within 16 hours.146
Odincovs and Kudinovs also studied the decomposition of sugars in the hydrolysate solution at different temperatures.147 They found that heating the sugar solutions at temperatures higher than 60 °C led to the decomposition of monomeric glucose in concentrated HCl at a rate 1.5–2 times faster than the sugar oligomers present in the hydrolysate. Applying temperatures below 60 °C to the hydrolysate resulted in less than 10 wt% sugar loss.147 Odincovs and Kudinovs concluded that the velocity of saccharification in wood using concentrated HCl solutions relied mainly on two factors: (a) diffusion of the saccharide solution through the biomass, and (b) hydrolysis velocity. Elevating temperatures from 20 to 40 °C resulted in a 6-fold increase in the polysaccharide hydrolysis velocity.
In 1961, Lebedev and Bannikova reviewed the effects of temperature on wood hydrolysis on an industrial scale.148 They replicated experiments earlier described by Chalov and Korotkov,146 in which the hydrolysis reactors were heated to study the hydrolysis of biomass, this time using a 40 wt% aqueous HCl solution only. Table 4 shows that sugar yields rapidly increased with temperature until reaching a plateau of around 48% sugar yield after 40 min at 60 °C, after which the sugar yield did not increase further148 as sugar decomposition starts to become important. At temperatures lower than 50 °C, decomposition is not prominent with the 60 min reaction time reported and mainly the hemicellulose is hydrolyzed (∼32% at 20 °C) plus the least recalcitrant fraction of the cellulose (up to 43% sugar yield after 1 h at 50 °C). The biomass was reported to contain 47.4% recalcitrant polysaccharides, very close to the reported 50.1% cellulose.
| Time (min) | Sugar yield (wt% of the dried wood starting material) | ||||
|---|---|---|---|---|---|
| 20 °C | 30 °C | 40 °C | 50 °C | 60 °C | |
| 10 | 30.21 | 31.30 | 32.55 | 37.18 | 43.71 |
| 20 | 32.29 | 32.51 | 35.55 | 40.08 | 47.44 |
| 30 | 32.20 | 33.81 | 38.39 | 40.14 | 47.20 |
| 40 | 31.49 | 34.71 | 38.74 | 42.09 | 48.12 |
| 50 | 31.66 | 36.68 | 37.08 | 42.26 | 47.76 |
| 60 | 30.97 | 39.41 | 38.80 | 43.22 | 45.77 |
Lebedev and Bannikova also studied the hydrolysis velocity and diffusion of the oligomeric sugars through the wood particle.148 They found that after hydrolysis of the wood carbohydrates into sugar oligomers, the diffusion velocity of the oligomeric sugars from inside to outside of the wood chip was substantially slower than the hydrolysis velocity.148 This was noted when obtaining higher sugar yields when high stirring speeds were applied to the reactors. Lebedev and Bannikova reported that in order to facilitate the diffusion of the dissolved oligosaccharides to the outside of the wood particle, several parameters were of importance, namely; (a) reduction of the particle size by using finely divided material, such as sawdust, (b) using a continuous countercurrent of fresh acid together with a modest increase in HCl concentration and (c) reducing the viscosity of the hydrolysate by elevating the temperature.148
These conclusions were later supported by Chalov and Goryachikh after their discovery that pinewood hemicellulose hydrolysis efficiency relied on the particle size of the applied lignocellulosic material.149 For bigger particles, the diffusion was slower and required longer times for complete hydrolysis. HCl concentrations below 40 wt% could only hydrolyze cellulose under dynamic conditions, which meant that the sugars had to be continuously removed from the medium fast enough in a countercurrent manner. Petkevich and Ochneva reported that compared to wood chips, the utilization of sawdust (reduced the hydrolysis time) and the velocity with which the sugars were washed out of the residue, which allowed to reduce the number of hydrolysis reactors in the plant.150 In 1964, N. V. Chalov and L. B. Paasikivi reviewed this work and concluded that the diffusion of hydrolysis products inside the particles was the rate-limiting step and it was several times slower than the velocity of the polysaccharide hydrolysis.151
In 1966, Chalov and Leshchuk found that using a 6-fold (in weight) excess of acid during wood hydrolysis and decanting the final hydrolysate from the reactor resulted in 43% of the hydrolyzed sugars not being recovered as they were still retained inside the biomass. This retention is why centrifugation was used in the Bergius process to extract these sugars.152 Glazkova and Kunina found that an increase in temperature led to a better mass transfer coefficient by a factor of 1.35, whereas doubling the amount of water for the lignin washing, increased it by a factor of 1.2.153 Extracts of maximum concentrations were obtained with counter-current washings with a smaller liquid-to-solid ratio and at a temperature of 80 °C.153
Chalov also studied the interactions between HCl, sugars, and lignin.154 The effective HCl concentration inside the wood particle was found to be higher than in the immediate surrounding outside the wood particle. This phenomenon was attributed to the interactions of HCl with the dissolved polysaccharides and lignin dissolved in the hydrolysate, which created a concentration gradient. After hydrolyzing biomass with 41 wt% HCl, the amount of HCl bound to the lignin was estimated to be between 17.5 and 23.5% of the starting lignin weight.154 Chalov and Leshchuk reported that the hydrolysis of wood polysaccharides using 38 to 50% HCl reached an equilibrium in the polysaccharides-HCl system at which the hydrolysis reaction stopped.155 After interruption of the hydrolysis reaction, it was found that 1 g of sugar oligosaccharides in solution could bind up to 0.486 g of HCl. Based on these data, the chemical composition of the HCl-oligosaccharide adduct was estimated to be C6H12O6·2.4HCl. Chalov re-investigated these results and later published that the formation of a HCl adduct with the reaction product (i.e. glucose) was more accurately estimated to be 2C6H12O6·3HCl.156 The HCl–sugar and HCl–lignin interactions would significantly slow down or even stop the hydrolysis process when the HCl concentration reached values below 37 wt% at 20 °C, which was in alignment with earlier research described by Hägglund in the 1920s.108 Chalov replicated these experiments to evaluate the HCl interaction with lignin, and found that the hydrolysis of pure cellulose in concentrated aqueous HCl required less HCl compared to the hydrolysis of lignocellulosic material, due to the absence of lignin in the microcrystalline cellulose.156 In 1974, Glazkova and Kunina observed that raising the temperature increased the hydrolysis rate, so the equilibrium understood as the point where the hydrolysis of cellulose stopped due to the HCl–sugar–lignin interactions, was reached faster.153 For example, at an acid:wood ratio of 8 (w/w), approaching the equilibrium at 80–90 °C was observed in 2–3 h, while at 20 °C it only occurred after 9 h.
In these years, several pilot plants testing modifications on the Bergius–Rheinau process were built all over the Soviet Union. Improvements in the downstream process were also reported, focusing on the recovery of HCl together with new drying techniques for the lignin residue. Korotkov and Glazkova published a continuous 2-step evaporation to improve the recovery of the hydrochloric acid.157 The viscosity of the hydrolysate increased during the removal of the acid and as a result, the heat transfer coefficient of the acid–sugar mixture substantially decreased. The heat exchanger materials of the evaporator had to be compensated for this and quickly remove the excess of heat from the evaporation to minimize degradation of the sugars. The expenses to manufacture a single evaporator that could meet these requirements to remove HCl at all concentrations and increasing viscosities were rather high.157 Another challenge using one-step evaporators was related to the heating elements, which would become completely covered with resinous sugar degradation compounds during the second half of the evaporation. This in turn led to an increase in sugar loss besides several stop times in order to clean these pieces of equipment.157 Korotkov and Glazkova concluded that the first evaporation should be carried until a concentration of solids was achieved around 30 to 50%, followed by a second evaporator specifically designed to efficiently remove the acid from a more dense, concentrated hydrolysate with lower HCl concentrations.157 Under these conditions, minimal capital and operational costs could be achieved.
Marone and Zaytseva studied the kinetics of lignin air-drying.158 After hydrolyzing wood chips with gaseous HCl, the lignin was dried in electric ovens under convection. The wet lignocellulosic residues were pierced with a needle and a thermocouple was placed into the hole so that its tip was located in the center of the particle. During the experiments, temperature and air flow velocities varied and the concentration of water, HCl and sugars were measured in the center of the particles using standard analytical methods. Marone and Zaytseva found that oven drying was an effective method to dry the lignin, but a small amount of HCl (2–5%) remained inside the residual lignin up till the end of the experiments.157 The drying efficiency increased at higher temperatures until reaching a plateau of 70 °C, after which drying slowed down until reaching 100 °C, where further drying was not investigated.157 This drying efficiency was limited by the slow diffusion of the acid inside the wood particles, which did not improve with higher air flow velocities. The heat transfer coefficient of the wet lignin also did not improve by modifying the air flow velocities, and the formation of sugar degradation compounds was not observed when drying the lignin at temperatures below 60 °C.
Mikhailovich also studied the drying of lignin but from an industrial perspective by using drum dryers.159 The effects of the drum rotation speed, the initial temperature of the drying agent (air, 550–700 °C), and the speed of the conveyor belt feeding lignin to the dryer (2.1–6.3 m s−1) were studied. In order to quantify the impact of these parameters, the moisture content (HCl and water), specific fuel consumption, temperature of the drying agent and average particle size of the dried pulverized lignin were measured. It was found that the speed of the conveyor belt feeding the dryer was the most impactful parameter. Increasing it reduced the contact time of drying agent (air) with the lignin and as a result, the moisture content increased while the fuel consumption and the temperature of the drying agent decreased. Mikhailovich concluded that conveyer belt drying with hot air in a rotating drum was suitable to dry the lignin to moisture contents of 5–10 wt%.159
The main-hydrolysis was a continuous process in reactors connected in series. One batch reactor was assigned to the hydrolysis of the hemicellulose (pre-hydrolysis), two were commissioned for the continuous hydrolysis of the cellulose (main-hydrolysis) and two for the water-washing of the residual lignin. The last reactor was used for the concomitant operations, namely wood chips uploading or lignin unloading.
For the hydrolysis of hemicellulose in the modified Bergius process, an intermediate hydrolysate stream was sometimes used instead of fresh 41 wt% HCl. This hydrolysate, referred to in literature160 as “intermediate hydrolysate”, was collected during the first stage of the main hydrolysis, just after the hydrolysate product was taken off. Although this intermediate had a lower sugar concentration, it still contained 41 wt% HCl, making it effective for the hydrolysis process. The intermediate hydrolysate provided a means to remove residual sugars after the main hydrolysis step and recycle hydrochloric acid while maintaining the necessary acid concentration for effective hydrolysis.149
When commissioning the pilot setup during its first year of operation, several challenges were encountered in the hydrolysis section. The sugar concentration in the hydrolysates was only 9–11%, and the total sugar yield amounted to just 37–41% of the wood's original content.150
When sawdust was used as feedstock, Petkevich noted that most of the material floated inside the reactors, leaving the bottom primarily filled with HCl solution. During the stationary pre-hydrolysis, the sugar concentration was significantly lower compared to the pre-hydrolysis of wood chips. This lower concentration was likely due to the 1.75 times shorter incubation time of sawdust with the intermediate hydrolysate compared to wood chips. This difference was also attributed to the substantially lower packing density of sawdust in the reactor, which was 105 g l−1 compared to 156–180 g l−1 for wood chips. Consequently, the weight-to-weight ratio of intermediate hydrolysate to sawdust nearly doubled compared to wood chips.
In 1961, a modification to the hydrolysis process was introduced to improve efficiency by separating hemicellulose and cellulose hydrolysis. This process included a pre-hydrolysis step in which wood chips were dried with 2 wt% HCl at 100 °C for 3 hours. This step partially hydrolyzed hemicellulose and disrupted cellulose's crystalline structure, enhancing accessibility for subsequent hydrolysis. The pre-hydrolysate contained 2–4% glucose derived from cellulose.
The main hydrolysis used 41 wt% HCl in a series of reactors, yielding a hydrolysate with 18.1% sugars and 31.7% HCl. After 24 hours, no polysaccharides remained in the hydrolyzed material, achieving a 96% theoretical sugar yield and leaving lignin free of polysaccharides. HCl losses were minimized to 0.8% of the feedstock weight. A parallel development at the Kansk pilot plant introduced a three-stage process for hemicellulose and cellulose hydrolysis and lignin washing. Hemicellulose hydrolysis was performed with 37 wt% HCl, while cellulose hydrolysis utilized 41 wt% HCl, achieving 94% sugar conversion and hydrolyzing 65% of the starting wood mass.
Key challenges included incomplete separation of hemicellulose and cellulose hydrolysates, leading to increased HCl consumption by 15–20%. Further optimization was proposed by increasing the volume of 37 wt% HCl by 1.5 times, which would raise total HCl consumption by 25–30%. A detailed description of these modifications, including reactor configurations and sugar compositions, is provided in the ESI.†
Until 1985, 18 ethanol, 16 fodder yeast, and 15 furfural/xylitol hydrolysis plants using percolation and Bergius–Rheinau processes were operating in the forest regions of Belorussia, the Ural, Eastern and Far East Siberia, as well as Central Russia, Ukraine, and Uzbekistan, utilizing various agricultural wastes as feedstock.145 Over the years, these plants suffered from irregular feedstock supply, and after government or local authority support ceased, many plants became unprofitable and had to be sold to other industries at prices ranging from 1 to 7 million dollars.145
4.4 Saccharification in Japan: The Hokkaido and the Noguchi-Chisso process
In the 1960s, the Japanese government also developed new saccharification technologies. The Hokkaido two-stage hydrolysis used sulfuric acid for the saccharification of wood in industrial tests which would culminate in a full commercial plant with a capacity to process 100 tons per day of wood in 1963.161,162 The dried powdered wood was sprayed with a concentrated acid solution at high pressures and short times to minimize sugar decomposition reactions. The acid recovery was around 80% and this was accomplished by diffusion dialysis using an ion exchange resin. The overall sugar yields were reported as 280 kg of crystalline glucose from 1 ton of dry wood. This process was modified after research by T. Kobayashi.163,164 Instead of directly spraying the high concentrated acid, the wood was first immersed in diluted sulfuric acid and heated to 40–50 °C to hydrolyze some of the material while re-concentrating the acid.Additionally, Japan explored the use of hydrogen chloride gas instead of aqueous HCl solutions for biomass saccharification.162 This method, called the Noguchi-Chisso process, was operational in a pilot plant from 1958 to 1962. This technology was a further development of the Prodor, Darboven, and Herneng processes (see Section 4.2), which also utilized HCl. In the Noguchi-Chisso process, wood was first dried, and anhydrous HCl gas was introduced into the system. Steam at 100–130 °C was then applied to accelerate the hydrolysis of hemicellulosic sugars. In the subsequent step, a counter-current stream of water extracted these sugars. The solid residue was then partially dried and cooled to 10 °C before cellulose hydrolysis. Anhydrous HCl was applied, adsorbing into the remaining moisture of the lignocellulose to achieve concentrations of 42 wt% HCl within the wood particles. The reactors were rapidly heated to 40–45 °C, completing cellulose hydrolysis in less than 30 minutes, depending on particle size. The sugar solution product was flash-heated to recover most of the HCl gas quickly, followed by post-hydrolysis to obtain monomeric sugars by diluting the remaining acid to 3% and heating to 70–80 °C for approximately 1–2 hours. The Noguchi-Chisso process claimed to achieve a 90% conversion of the wood's available carbohydrate content into sugars. Its main advantages lies in the simplicity of making a 42 wt% HCl solution in situ inside the biomass, reduced corrosion issues, and higher sugar concentration compared to previous similar processes.51,132,165,166
4.5 Lessons from historical processes: scaling challenges and innovations
The historical evolution of saccharification technologies reveals critical insights into the challenges of scaling processes from laboratory experiments to pilot and industrial scales. Many early methods, such as the Bergius–Rheinau process, demonstrated impressive efficiencies under controlled conditions but encountered significant scalability issues due to limitations in the materials of construction, energy inefficiencies, and difficulties in acid recovery. For example, the reliance on expensive materials like tantalum for reactors in early designs besides the high acid losses and energy consumption, limited their commercial viability.A key takeaway from these early processes is the importance of adaptable designs that account for material durability, energy efficiency, and cost-effectiveness. Modern technologies can learn from this by integrating sustainable materials like poly(vinyl chloride) and employing advanced heat recovery systems, as seen in later modifications. These adjustments significantly reduced operational costs while improving acid recovery and sugar yield.
Another important insight is the significance of process integration, particularly in acid recovery and sugar purification. While processes that incorporated multiple stages—such as pre-hydrolysis for hemicellulose removal—have demonstrated improved yields for cellulose hydrolysis, they also present challenges in separating the liquid streams of hemicellulose and cellulose hydrolysates. Since the transition between these stages is not clearly defined, the mixing of hydrolysates complicates sugar separation and downstream processing. Future research should aim to refine these integration strategies, focusing on minimizing waste, reducing energy input, and enhancing the efficiency of sugar recovery while addressing the separation challenges inherent to multi-stage processes.
Looking ahead, advancements in reactor materials, automation, and computational modeling hold promise for overcoming scalability barriers. By simulating process dynamics at various scales, researchers can predict challenges before pilot trials, accelerating the pathway to commercialization.
5. The saccharification industry after the oil crisis
On October 6, 1973, during Yom Kippur, the holiest day in Judaism and Samaritanism, the fourth Arab-Israeli conflict began.167 The Arab coalition, led by Egypt and Syria, launched a surprise attack against Israel. Throughout the war, the United States and the Soviet Union supported opposing sides, bringing the two superpowers to the brink of direct confrontation.168 The Yom Kippur War lasted from October 6 to October 24, but its ramifications significantly impacted the global economy and political landscape for years to come.In 1974, following Israel's victory in the Yom Kippur War, the Arab countries were dissatisfied with the terms imposed on them. In retaliation for U.S. support of Israel, the Arab members of OPEC, led by Saudi Arabia, decided to cut their oil production by 5% each month. This embargo resulted in a massive spike in the price of refined petroleum products. Between 1972 and 1975, consumers experienced an average increase of 57% in gasoline prices.169 To address the resulting petrol-supply crisis and explore alternatives to oil-based industries, the U.S. government established the Department of Energy (DOE) in 1977 and launched new comprehensive R&D programs.170 The oil embargo's domino effect compelled chemical industries worldwide to seek alternative sources for producing materials from above-the-ground carbon.
5.1 Revisiting the Scholler and Bergius–Rheinau process
During the 1980s, the US and New Zealand reopened several biorefineries based on the Scholler's percolation processes and other HCl-based technologies, but eventually failed to deliver a scalable technology.124 The Solar Energy Research Institute in the US proposed a batch reactor system that retained the simplicity of the percolation process but used a counter-current flow of liquids to minimize the degradation of sugars. Further testing of this system however, did not produce substantial sugar yield improvements. Dow Chemical, in the US, also opened a pilot plant for the saccharification of biomass using HCl, but eventually closed it due to the inability to efficiently recover the hydrochloric acid.124In 1987, the US company Arkenol Inc. obtained a patent for their saccharification of biomass using concentrated sulfuric acid with plans to build a commercial plant (Fig. 15).173,174 Arkenol and Masada Resource Group (Birmingham, AL) developed an ion-exchange-based chromatographic separation of the acid from sugar solutions and new processes for the valorization of solid waste streams. They were granted a patent for their OxyNol® process and donated one pilot plant to the university of Auburn, Alabama. Under the agreement with Masada Resource Group, researchers in Auburn's Department of Chemical Engineering developed a series of patent families that converted waste streams from pulp and paper mills into commodity chemicals. This study opened new market opportunities for the Masada Resource Group, which grew in 2009 from one domestic waste-to-energy project in New York to 19 strategic partnerships, covering market and project development activities in 47 international markets.175 Masada Resource Group also developed technologies to convert plastic and rubber portions of waste streams into diesel fuels.176
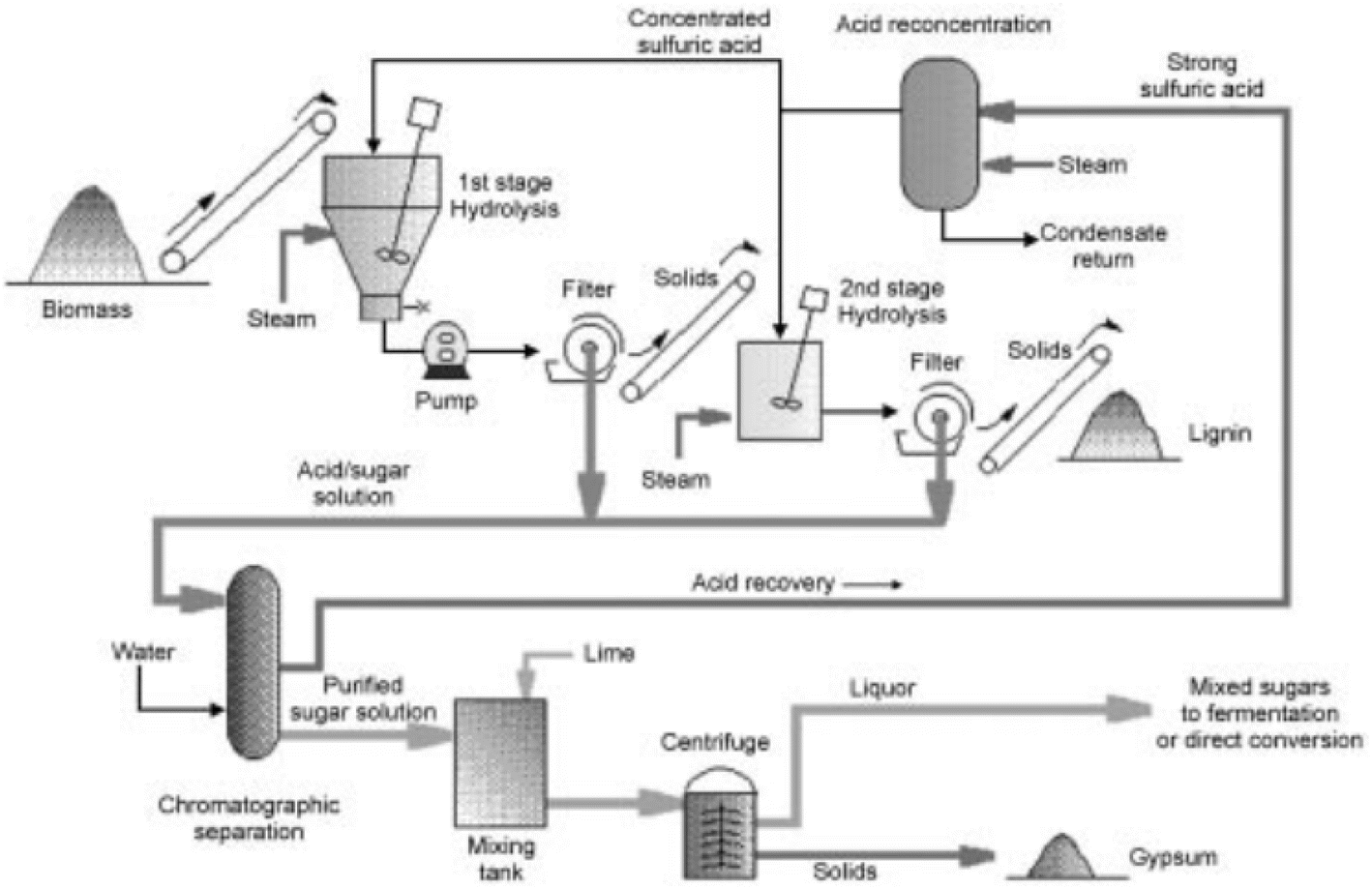 | ||
| Fig. 15 Simplified flow diagram of the Arkenol process for the saccharification of biomass with sulfuric acid, figure adapted from ref. 171 and 172. | ||
5.2 Arkenol process, Biosulfurol and HCl CleanTech
BlueFire Renewables continued operation of the Arkenol saccharification process in the US, but currently the company has shifted its operations to Japan.177 BlueFire's technology is being used at the Izumi Biorefinery, which is owned and operated by JGC Corporation. This facility is located in Izumi, a small industrial town in southern Japan, several hours southwest of Tokyo. The plant, which started operations in September 2002, is situated next to a 35-year-old NEDO ethanol purification facility. The Izumi Biorefinery is under contract to NEDO through 2007 to produce sugar for ethanol production. JGC Corporation planned to use this plant as a platform to scale the technology to various capacities, marketing it to its client companies in Japan and Southeast Asia. BlueFire ferments sugars obtained from the saccharification of cellulosic waste materials, including agricultural residues, paper mill sludge, municipal solid waste, wood, and green wastes such as leaves, grass clippings, and fruit waste. Using concentrated sulfuric acid, the cellulosic material is first hydrolyzed, followed by the fermentation of these sugars into alcohols.177At BlueFire's pilot plant facilities, the biomass feedstock is first cleaned and ground to reduce particle size before hydrolysis. The material is dried, and the cellulose and hemicellulose are dissolved together using sulfuric acid, resulting in one solution. The next step involves post-hydrolysis of the recovered materials to produce oligomeric hexose and pentose sugar solutions. Both the separation and hydrolysis steps use sulfuric acid, with the insoluble lignin being recovered by filtration and pressing. The acid is then recovered from the hydrolysate by chromatographic separation. The separated sulfuric acid is re-circulated and re-concentrated to the required level for the separation and hydrolysis of new biomass. The small quantity of acid left in the sugar solution is neutralized with lime to produce hydrated gypsum (CaSO4·2H2O), which is readily separated from the sugar solution and used as an agricultural soil conditioner177
In 2006, the Dutch company Biosulfurol Energy developed a biorefinery based on hydrolyzing lignocellulosic feedstocks with 70 wt% sulfuric acid.178 Water is added to the mixture to enhance the hydrolysis by the in situ formation of heat inside the reactor. After the hydrolysis, the oligomeric sugars are fermented yielding ethanol, while the sulfuric acid is partly recovered using anion-selective membranes and partly via biological sulfate reduction in an anaerobic waste water treatment plant. The sulfide produced is recovered as H2S gas and burned to produce SO2 and SO3 to again produce sulfuric acid. In this unique approach, the sulfuric acid is recycled to a large extent. The Biosulfurol process is able to handle a wide variety of lignocellulosic biomass, both grass and willow wood were tested as model substrates.178
In 2007, HCl CleanTech was founded in Israel to address the downstream limitations of earlier saccharification technologies developed by Bergius and Hägglund using 42 wt% HCl. To improve the costly, complex, and inefficient acid recovery in the Bergius–Rheinau process, HCl CleanTech patented a cold extraction process.179,180 This process involves bringing the aqueous HCl solution into contact with an immiscible organic solvent containing an oil-soluble amine and an oil-soluble organic acid with a pKa above 4.4, such as caproic, caprylic, or lauric acid. Both water-insoluble components are dissolved in dodecane. In the presence of NaCl, this amine and acid selectively form an adduct with HCl, followed by an acid–base couple extraction (ABC). During ABC extractions, the interactions between the lipophilic amines and acids in the organic phase result in the formation of new complexes, some of which involve proton transfer.181 The weak carboxylic acid coordinates with the amine to activate it, enhancing complexation with HCl (Fig. 16). This combination of the aqueous HCl solution and the immiscible organic solvent with both the amine and the organic acid forms an HCl-carrying extractant. The HCl is then recovered as HCl gas after passing the organic mixture with HCl over xylene vapors at 170 °C (Fig. 17).
 | ||
| Fig. 16 (Left) Activation of the amine by the carboxylic acid. (Right) Complexation of HCl with the alkyl amine during the recovery of the acid, figure from ref. 181. | ||
 | ||
| Fig. 17 Stripping of the acid after its extraction from the aqueous solution, figure from ref. 179. | ||
In an illustrative example, extractants were prepared as described above using two different acids and amines to observe their impact on the final acid recovery. The acids chosen were a strong, linear alkylbenzene sulfonic acid (LAS) and the weak, carboxylic acid capric acid (CAP). Both combined with two different amines with a branched alkyl structure: the amine JM-T – a primary amine in which the alkyl chain (R) is attached to the nitrogen by a tertiary carbon R–C(CH3)2NH2, and TEHA – a tertiary amine in which three identical branched alkyl chains are attached to the nitrogen – tris(2-ethylhexylamine) (CH2(CH2)3CH(C2H5)CH2)3N. A sample of each extractant was in contact with aqueous HCl at concentration levels of 5 and 10%, and the loaded extractants thus obtained were subjected to 165 °C for 20 min to recover the HCl gas. The results are shown in Table 5.180
:linear alkylbenzene sulfonic acid. CAP:capric acid. JM-T: is a commercial mixture of highly branched primary aliphatic amines, predominantly within the C16 to C22 carbon range. TEHA:tris(2-ethylhexylamine), a tertiary amine with three identical branched alkyl chains, ((CH2(CH2)3CH(C2H5)CH2)3N)
| Extractants acid:base 1:1 molar |
LAS:JMT |
GAP:JMT |
LAS:TEHA |
CAP:TEHA |
|---|---|---|---|---|
| Molar HCl loading by 5% HCl | 0.11 | 0.91 | 0.08 | 0.96 |
| Residual HCl after stripping | 0.02 | 0.31 | 0.01 | 0.21 |
| % HCl stripped | 81.8 | 65.9 | 87.5 | 78.1 |
| Extractant per mol of recovered HCl in liters | 11 | 2 | 14 | 1 |
| Molar HCl loading by 10% hydrochloric acid | 0.23 | 1.01 | 0.11 | 1.0 |
| Residual HCl after stripping | 0.04 | 0.26 | 0.005 | 0.22 |
| % HCl stripped | 82.6 | 74.3 | 95 | 78 |
| Extractant per mol of recovered HCl | 5 | 1 | 2 | 1 |
HCl CleanTech used a wide variety of wet cellulosic feedstock without drying or pre-treatment prior to hydrolysis, claiming minimal water and energy consumption and high sugar yields. Initially planning to develop their fermentation processes, HCl CleanTech ultimately focused on licensing their saccharification technology. In 2009, the company received its first venture capital funding from Burrill & Co. and Khosla Ventures.182 In 2012, HCl CleanTech rebranded to Virdia, securing $30 million in private financing and a $75 million loan package from the Mississippi Development Authority to build plants producing biofuels from cellulosic sugars.183 Virdia developed the CASE process, publishing several patents,184,185 aiming to modernize the Bergius–Rheinau process for industrial-scale application.186 Virdia opened a pilot plant in Danville, Virginia, in 2012 (Fig. 18), and expanded their technology to produce renewable fuels and fuel intermediates, including diesel, jet fuel, ethanol, and butanol. They also produced renewable chemicals and materials like biosurfactants, lubricants, plastics, and synthetic rubber, along with nutritional additives such as baker's yeast and amino acids for the animal feed industry.186 Additional details about the Virdia process can be found in the ESI.†
 | ||
| Fig. 18 Top side of the hydrolysis reactors at the Virdia pilot plant in Danville, the yellow conical funnels feed the wood material into the digesters. | ||
5.3 Lessons from post-oil crisis developments: toward sustainable saccharification technologies
The oil crisis of the 1970s significantly shifted the trajectory of saccharification technologies, compelling industries and governments to seek alternative methods for converting biomass into valuable products. The adaptations and innovations in this period reveal key lessons for current and future research in the field.One prominent insight is the role of economic and material efficiency in determining the viability of saccharification technologies. For example, processes like those developed by Arkenol, Biosulfurol, and HCl CleanTech introduced improvements in acid recovery and recycling, demonstrating the necessity of reducing resource consumption to make saccharification commercially feasible. Technologies like the Arkenol process, which leveraged chromatographic acid recovery and valorized by-products such as gypsum, highlights the importance of circularity in process design. Similarly, HCl CleanTech's advancements in acid extraction and CASE process optimization show how modern adaptations of historical methods can address prior inefficiencies.
Another lesson is the critical importance of feedstock flexibility. Post-oil crisis developments increasingly focused on utilizing a broad range of lignocellulosic materials, including municipal solid waste, agricultural residues, and forestry by-products. Processes like the Biosulfurol biorefinery and Virdia's CASE technology demonstrated the potential of adapting saccharification methods to accommodate heterogeneous feedstocks. This adaptability ensures resilience in regions with varying biomass availability and opens opportunities for waste valorization.
The innovations of this era also emphasizes the value of integrated systems. The co-location of biorefineries, as seen with the Izumi Biorefinery in Japan, allowed for the simultaneous production of bioethanol, renewable fuels, and chemicals. Such integrated approaches enhance economic viability by diversifying output streams and optimizing resource utilization.
Lastly, these developments reaffirm the need for scalable solutions. Many technologies from this period, such as BlueFire Renewables and Virdia, successfully scaled pilot processes to commercial plants by addressing historical challenges, including acid recovery and sugar separation. However, they also revealed persistent challenges, such as high capital costs and the need for continuous process optimization.
Looking ahead, these lessons remark the importance of designing systems that prioritize sustainability, scalability, and adaptability. By building on the advances of the post-oil crisis era, the saccharification industry could continue to evolve, creating more efficient, cost-effective, and environmentally friendly processes for biomass conversion.
6. Current technologies
In 2014, Stora Enso, headquartered in Finland, acquired Virdia and its saccharification technology, known as the CASE process, in a $62 million deal.187 Stora Enso remains a leading global provider of renewable solutions in packaging, biomaterials, wood construction, and paper.187 Some of their most notable patents are based on paper production and downstream processes for biomass hydrolysate materials.188–190However, as of January 2021, Stora Enso decided to permanently close its US-based Virdia operations. This included a research center in Danville, Virginia, and a demonstration plant in Raceland, Louisiana.191 This strategic move aligns with Stora Enso's focus on driving growth and value creation from forest-based materials, shifting their innovation efforts further down the value chain instead of being a raw material supplier. The closure process is stepwise and impacts approximately 65 employees and 18 contractors. The intellectual property gained from Virdia's research will be used by Stora Enso's different business units in future applications.192
The Norwegian company Weyland produced industrial sugars and lignin at a pilot scale. The Statoil-backed Weyland ethanol pilot plant in Bergen, Norway, began production in 2010,193 producing 200000 liter of ethanol from second-generation (2G) biomass, paving the way for large commercial scale. The company also had plans to license their technology worldwide.194 Their saccharification process is based on mineral acid hydrolysis and Weyland patented improvements in the acid recovery compared to Bergius and Scholler's processes. Based on their patents from 2010,195 their saccharification process is based on crystallizing the sugars in the hydrolysate product solution using anti-solvents or a combination of anti-solvents such as methyl ethyl ketone, acetone, methanol, ethanol, n-propanol and iso-propanol. The overall saccharification process comprises the following steps:
(1) Hydrolyzing the lignocellulosic material using sulfuric or phosphoric aqueous acid solutions.
(2) Separating the acid from the hydrolysate with an extraction solvent to yield (a) a first aqueous acidic solution containing the extraction solvent and (b) a residue containing oligosaccharides. This step involves crashing the sugars out rather than merely extracting the acid.
(3) Subjecting the residue to an oligosaccharide cleavage (depolymerization/hydrolysis) reaction to yield an aqueous solution of fermentable sugars.
The preferred acid is concentrated sulfuric acid or a mixture of sulfuric and phosphoric acid in about a 2:1 volume ratio. The cellulosic feedstock includes grass, straw, wood (e.g., sawdust or wood shavings), paper, or corn husks. The acid hydrolysis is performed continuously with water cooling to maintain the mixture at 50–55 °C. The acid is added in a 2:1 to 4:1 weight ratio (acid) and the hydrolysis lasts about 2 hours. The cellulose is broken down to produce oligosaccharides, which can be precipitated by the extraction solvent to yield a lignin/sugar slurry. The extraction step is done at 0.5 bar and 50 °C. The slurry residue is post-hydrolyzed by diluting the acid concentration to 1 wt% at about 140 °C and 5–6 bars for about 2 hours. The resulting hydrolysate is neutralized to a pH suitable for fermentation. Weyland's process recovers 98.5% of the acids and solvents used, producing approximately 1 liter of ethanol for every 3.6 kg of raw biomass, depending on the feedstock.194
The Swedish chemical company Sekab published a patent application in 2008 for their Sekab-e technology, focusing on saccharification of lignocellulosic materials, particularly wood chips, using diluted sulfuric acid at high temperatures in two stages.196 In the first hydrolysis step, 1 wt% H2SO4 at 180 °C and 10 bars of pressure dissolves the hemicellulose and partially disrupts the cellulose structure. In the second step, the acid is diluted to 0.1 wt% and heated to 200 °C under 20 bar of pressure. The entire hydrolysis process takes only 25 minutes. The wet lignin residue is then processed through a membrane filter press to remove the retained liquid and separate the hydrolysate from the lignin.
Recent information from Sekab's website details their updated process, which includes four main steps: pre-treatment, enzymatic hydrolysis, fermentation, and reprocessing. The raw material undergoes acid and steam pre-treatment at 200 °C, releasing sugars from hemicellulose. The slurry is then neutralized, and enzymes are added to break down the cellulose into glucose. This glucose can be used in various processes. For ethanol production, yeast is added to ferment the sugars. The entire slurry is distilled to purify ethanol, leaving solid lignin, which is filtered and dried for use as biofuel. Remaining solutes in the water are converted into biogas, providing energy for the plant or being sold.197
In addition to their work with lignocellulosic materials, Sekab has also patented a method for the pre-treatment of non-wood lignocellulosic material containing less than 5% (w/w) starch or sugar, using organic acid-producing bacteria and aimed at improving the efficiency of ethanol production from these materials. The method is particularly suitable for materials such as agricultural residues, which typically have lower lignin content compared to wood. The patent details that no, or substantially no, inorganic acid or base, including SO2, is added in the method, which enhances its environmental benefits and cost-effectiveness.198
Green Sugars GmbH, later known as Green Sugar AG, was a prominent entity in the field of saccharification technologies, founded on 2007 by Matthias Schmidt, with its headquarters in Meiβen, Germany.199 This company specialized in developing innovative methods for converting lignocellulosic biomass into valuable biochemicals using a series of patented technologies.
Green Sugar made significant contributions to the field of saccharification technologies, particularly through their innovations in using super concentrated hydrochloric acid (HCl) for biomass conversion. Their focus on using high concentrations of HCl (37% and above) distinguishes their methods from other saccharification techniques, which typically use diluted acids or enzymatic processes. This comprehensive approach to saccharification allowed them to tap into approximately 70% of plant biomass, converting it into a variety of products including sugars like glucose, xylose, and arabinose, as well as biochemicals such as ethanol and furfural.200
One of their notable patents describes a process that utilizes hydrochloric acid in concentrations exceeding 37% for the saccharification of lignocellulosic biomass.201 This method is advantageous because it allows for the hydrolysis of cellulose at relatively low temperatures, reducing energy consumption while maximizing the yield of sugars. This process not only enhances the efficiency of saccharification but also simplifies the subsequent steps of sugar recovery and purification.201 The company also developed modular hydrolysis reactor systems, which are adaptable and scalable to meet different processing needs.202 This modularity is essential for handling various types of biomass, offering flexibility across different production scales. Complementing these systems are optimized biomass pretreatment methods described in their patents.203,204 These methods improve the effectiveness of the hydrolysis process by enhancing the accessibility of cellulose and hemicellulose, which are critical for efficient sugar recovery.
Efficient acid recovery techniques aimed to mitigate some of the economic and environmental concerns associated with using high-concentration acids.205 These methods were designed to recycle and reuse acids within the process, a crucial step towards minimizing waste and operational costs. However, it's important to note that while these techniques intended to address these challenges, they may not have completely resolved all issues related to the use of high-concentration acids. Advanced methods for controlling hydrolysis conditions206 allowed for the precise adjustment of critical parameters such as temperature and acid concentration, optimizing the conditions for saccharification to enhance the yield and quality of the resulting sugars.
Unfortunately, despite their technological prowess and innovative contributions to green chemistry, Green Sugar AG ceased operations due to financial difficulties. The company filed for insolvency on February 18, 2020, leading to the dissolution of the company.200,207 Collectively, Green Sugar AG's technologies had the potential to revolutionize industries that rely on bio-based products, such as biofuels, bioplastics, and biochemicals. Despite the company's eventual dissolution, the legacy of their technological innovations continues to offer valuable insights and tools for sustainable biomass utilization, presenting ongoing opportunities for other companies to adopt and refine these pioneering methods for broader commercial use.
6.1 The DAWN technology, a modified Bergius saccharification process
The Dutch chemical company Avantium, established in 2000, specializes in bio-based plastics. Their key product is 2,5-furandicarboxylic acid (FDCA), which is essential for their plant-based polymer poly(ethylene furanoate) (PEF).208–210 For Avantium it was clear in 2010 that with serious fossil feedstock replacement by biomass the world cannot rely on 1st generation (1G) sugars for plastic production (500 million tons virgin plastics per year). Gruter and McKay started by evaluating different biorefinery options for producing pure glucose (required for FDCA production). Many technologies (most of them discussed in this review) were evaluated and ranked with respect to business case viability for pure glucose production.The resulting ranking is obviously clearly different than when ranking biorefinery options for ethanol production, in which case the intermediate sugars do not need to be separated and isolated. Gruter and McKay identified low temperature processes (using concentrated HCl or H2SO4 or the liquid salt ZnCl2. xH2O·xHCl) as best options as with these technologies it was possible to combine high polysaccharide conversion with high glucose yield (by limiting glucose decomposition) and the option to apply staged hydrolysis to separate hemicellulose and cellulose sugars (glucose from the cellulose fraction). As HCl can be separated and recycled overhead, Avantium selected the Bergius-type technology as having the highest potential. After a failed attempt to merge the Avantium plans with German-based start-up Green Sugar in 2013, Avantium started the development of its own Bergius-type concentrated HCl saccharification process.211,212 In 2018, at the opening of its Pilot plant (20 tons of dry wood chips annually, Fig. 19),213 Avantium created the DAWN Technology™, with a focus to convert lignocellulosic biomass into second-generation sugars.18–21,214 This technology employs a two-stage HCl hydrolysis process to produce a pure glucose stream, starting with pre-hydrolysis using a 37 wt% HCl solution to break down hemicellulose, followed by main-hydrolysis of the cellulose with a 42 wt% HCl solution.
 | ||
| Fig. 19 The Avantium DAWN™ Pilot plant in Delfzijl, The Netherlands with 8 × 230 l “Fixed bed” reactors for operation in Simulated Moving Bed (SMB) mode (for SMB mode operation, see Fig. S27†). | ||
Compared to past iterations of the Bergius process, the modifications by Avantium involved some new concepts:
• Modern materials of construction: moving from tantalum or rubber to PVC-lined reactors, a material that can withstand the action of concentrated hydrochloric solutions for longer times (also reported by Green Sugar).
• Immiscible displacement fluid: intended to separate the pre- and main-hydrolysates by displacing the hydrolysate from the wood particles when shifting from 37%HCl to 42% HCl (from pre- to main hydrolysis) or from 43% HCl to water (for washing the lignin).
• Optimization of hydrolysis rate: this involves improving sugar recovery and product concentration by addressing the physical changes in wood during each hydrolysis stage. In the first stage, wood's porosity is influenced by lignin, hemicellulose, and cellulose. In the second stage, after hemicellulose is removed, the wood's porosity increases and the acid penetrates more easily. To keep residence times consistent across both stages, a higher flow rate is needed in the first stage, or a lower flow rate in the second. This balance can be managed using a displacement fluid to adjust the HCl flow between the stages.
• Optimization of the feedstock: feedstock varies in particle size, with smaller chips improving acid access but costing more. Larger chips or in practise larger chip size distributions are more available but can hinder hydrolysis due to mass transfer issues in the larger chips. Using a higher acid-to-biomass ratio can address this while still achieving high sugar yields. Balancing market availability, biorefinery economics (especially HCl-sugar separation and HCl recycling costs), and optimal particle size for efficient hydrolysis is an ongoing focus for the development.
• Optimization of the biorefinery flowsheet: addressing the balance of either using more reactors connected in series or investing in chip size reduction equipment, increasing CAPEX, or; producing hydrolysate with lower sugar concentrations, which increases the OPEX of the plant as higher energy input is required to evaporate the additional acid (e.g., via steam consumption).
The DAWN saccharification process begins with de-barking and drying wood chips to below 10 wt% moisture to prevent dilution of the concentrated acids. In the first hydrolysis stage, hemicellulose is recovered as an acidic solution of C5/C6 oligomeric sugars in 37 wt% HCl. The second stage uses 42 wt% HCl to hydrolyse cellulose, which is then collected as an acidic solution of monomeric and oligomeric glucose. Lignin is washed and pressed to recover retained acids and sugars, then dried to remove moisture and residual HCl before pelletization. A wiped-film evaporator separates sugars from the acid, using immiscible oil at high temperatures to collect sugars that crash out of solution in the oil when boiling off HCl/water. Both hydrolysates undergo post-hydrolysis to convert oligomers into monomeric sugars. A schematic of the process is shown in Fig. 20 and additional details about the DAWN process can be found in the ESI.†
 | ||
| Fig. 20 DAWN Technology™ process block flow diagram: starting from the top-left, the process includes de-barking and drying wood chips, followed by two-stage hydrolysis using different HCl concentrations. The sugar solutions from each hydrolysis stage are collected separately and then post-hydrolyzed to produce monomeric sugars in solution. Lignin is pressed, pelletized, and the sugars are purified before being transported to customers. | ||
After the completion of the hydrolysis, the reactor containing the residual lignin with displacement fluid is emptied and transported to a Hastelloy screw press, where the solids are mechanically pressed to reduce the liquid content. The recovered hydrolysate and the displacement fluid from inside the lignin are separated in a decanter, after which the hydrolysate is sent to a storage tank.
Additionally, separating sugars from the acidic solution proved challenging. The high temperatures required for evaporation led to sugar decomposition, similar to issues encountered in previous versions of the Bergius process. The wiped-film evaporator used in the lab could not be scaled up for industrial applications, complicating the separation process further. Another issue was HCl recovery, as the process experienced significant HCl losses, making recovery inefficient and troublesome. This affected the overall economic viability of the process, necessitating a more streamlined approach.
Due to these challenges, Avantium transitioned to a one-stage hydrolysis process using hardwood, which produces a mixed sugar stream containing both xylose and glucose. This single hydrolysis stage simplifies the process and addresses some of the issues faced in the two-stage method. In this modified approach, the concentration of HCl was increased to above 43% to ensure efficient hydrolysis. The slight increases in HCl concentration above 42% does not have significant negative economic implications but ensures that the concentration remains sufficiently high to hydrolyse the cellulose across all reactors. Maintaining a high HCl concentration is essential, as HCl can interact with lignin and react with sugars and other compounds, lowering its concentration (vide supra). However, it's worth noting that the hydrolysis of biomass consumes water (one equivalent per soluble sugar moiety), which increases the HCl concentration.
6.2 Pilot tests on hydrogen fluoride saccharification technology
The development of hydrogen fluoride (HF) saccharification technology emerged from the need to overcome the inherent challenges of efficiently hydrolyzing lignocellulosic biomass. Traditional saccharification methods struggled with low sugar yields and scalability, particularly due to the recalcitrance of biomass and process design. HF offered distinct advantages, including its ability to disrupt both cellulose and hemicellulose structures effectively, promising improved efficiency in biomass conversion.One key advantage of HF is its ability to achieve high sugar conversion rates under mild conditions compared to other high-concentration acid processes.132 For example, a patent filed in 1937 by Australian chemists Hoch and Bohunek utilized 95% gaseous HF under reduced pressure, achieving a 90% sugar yield, with 80% of the sugars recovered as product. A German pilot plant, based on this process, operated successfully for six months, maintaining hydrolysis temperatures of 35–40 °C and pressures of 40 mbar for 30 minutes. The recovery and recycling of HF were also straightforward, with the acid being recovered by heating at 62 °C, reducing operational costs and environmental impact. Furthermore, studies highlighted that the low boiling point of HF simplified its recovery process compared to other mineral acids, making it more practical in continuous operations.
In 1979, Hawley and Selke studied the use of HF for biomass saccharification, reporting sugar yields of 70–85% from aspen wood chips, with total fluoride retention in the sugar and lignin of approximately 4 mg g−1 of wood.132,215 This process differed from earlier methods by using different biomass types and potentially different forms of HF (gaseous vs. aqueous). The use of HF also demonstrated reduced corrosion of construction materials compared to other high-concentration acid processes, contributing to its appeal for industrial applications.
Despite these advantages, the technology also faced critical challenges that hindered its widespread adoption. The extreme toxicity of HF posed major safety risks, requiring stringent handling protocols and specialized materials of construction, which increased capital and operational costs. Exposure to HF can cause severe health issues, including blindness, respiratory failure, and even death. Furthermore, while HF recovery was efficient, residual HF in the sugar solution and lignin required additional purification steps, complicating downstream processing. During the 1980s, the chemical company Hoechst in Germany utilized anhydrous HF for cellulose hydrolysis, achieving around 45% yields of fermentable sugars with more than 99% of the HF effectively recovered. However, the presence of residual fluoride in the sugar solution limited its immediate application, and the high operational risks associated with HF led to the eventual discontinuation of this technology. Hoechst eventually merged with the French pharmaceutical company Sanofi, after which reports on biomass saccharification ceased.140
In conclusion, while HF saccharification offered significant benefits in terms of sugar conversion efficiency and acid recovery, the safety hazards, operational complexity, and costs ultimately outweighed these advantages. These challenges emphasize the importance of balancing technical performance with safety and economic feasibility in the development of future saccharification technologies.
6.3 The industrialization of the enzymatic hydrolysis
The initially destructive potential of the fungus Trichoderma reesei, first isolated from rotting US Army equipment on the Solomon Islands during World War II, turned to be a revolutionary discovery in the saccharification of biomass.216 This method that relies on the secretion of cellulases to decompose the cellulose, is now currently a well-established technology that drastically reduced the cost production of cellulosic ethanol during the 1980s.10 The Department of Energy (DOE) in the US promoted enzymatic hydrolysis as a key element for development. Despite that the conversion of lignocellulosic materials through enzymatic hydrolysis offered the potential for higher yields, higher selectivity, lower energy costs and milder operating conditions than chemical processes, such technology, however, was judged to be too high risk for industry to pursue at that time.217In the 1980s, St. Lawrence Technologies (headquarters in Canada) collaborated with the Tennessee Valley Authority (TVA) in the USA, and the Swedish Ethanol Development Foundation to develop a two-stage hydrolysis referred as the CASH process (Canada, America, Sweden Hydrolysis). This program focused on the conversion of wood feedstock into ethanol on industrial scale. The CASH process combined diluted sulfuric acid at moderate temperatures followed by the action of hydrochloric acid, which was an idea originally suggested by the Soviet Union during their early saccharification research during the 1920s. In 2015, Jung et al. studied the CASH process and its evolution.111 Since 1995, the CASH process has been used to convert softwoods into ethanol. However, the high production costs compared to other alcohol production industries, including methanol, and the increased attention into new alternative fuels reduced the funds and investments.
In the 1990s, many researchers investigated the effects of acid pretreatment to enhance the saccharification of biomass.111 One of these ideas was a continuous and integrated process using dilute acids prior to the enzymatic hydrolysis to enhance the latter. As part of such an effort, simultaneous saccharification and fermentation (SSF) was evaluated and compared with separate hydrolysis and fermentation. Several plants were established for pretreatment and fermentation together; however, it was not easy to successfully demonstrate the increased efficiency of the process.111
The U.S. Department of Energy (DOE) funded the scale-up and commercialization of these processes. A prototype pilot plant, which integrated feedstock handling, pretreatment, solid–liquid separation, fermentation and distillation, was installed at the National Renewable Energy Laboratory (NREL), US, for the DOE. NREL favored and techno-economically analyzed combinations of dilute acid pretreatments with enzymatic hydrolysis. Lignocellulosic biomass was submerged in 1 wt% sulfuric acid at 140–160 °C for several minutes. The effect of dilute acid pretreatment was studied with a focus on the enzymatic digestibility of pretreated lignocellulose using a variety of woody crops and agricultural residues in a single- or two-stage process. Also, the operating parameters for minimizing hemicellulose loss and maximizing hemicellulose recovery were also investigated.111 Entities like DOE and NREL incentivized enzymatic biorefineries, and the use of enzymes gained momentum, becoming one of the most replicated saccharification processes to hydrolyze biomass under mild conditions.
Sweetwater in the US also adopted and patented several processes related to the field of enzymatic hydrolysis of lignocellulosic sugars. A patent was granted in 2013 (ref. 218) for their process comprising reduction of the particle size, pretreatment of the material with a weak acid to pre-hydrolyze the hemicellulose under heating followed by enzymatic hydrolysis to recover the pentose and hexose sugars.218 Other commercial approaches based on enzymatic hydrolysis are summarized in the ESI (Table S4),† based on a market analysis of Anuj K. Chandel et al. in 2021.11
Celtic Renewables is another example of enzymatic biorefinery technology that transforms low-value byproducts, residues, and waste into high-value green chemicals such as bioacetone, biobutanol, and bioethanol.219 Their process starts with the preparation and hydrolysis of biomass feedstock to break down complex carbohydrates into fermentable sugars.
These sugars undergo Acetone–Butanol–Ethanol (ABE) fermentation, where Clostridium bacteria convert them into acetone, butanol, and ethanol under optimized conditions. After fermentation, the resulting mixture is subjected to advanced separation and purification processes, including distillation and solvent extraction, to isolate and purify the desired chemicals. Celtic Renewables' flagship facility in Grangemouth, Scotland, processes local byproducts and waste to produce these high-value green chemicals, demonstrating the commercial viability of their technology. This facility has an initial production capacity of 1000 tons per annum, with plans for additional full-scale biorefineries capable of producing up to 8000 tons annually.220
6.4 More recent technologies
Shell Oil Company published a patent application in 2012 for the utilization of α-hydroxysulfonic acids to hydrolyze lignocellulosic biomass.221 The recovery of the α-hydroxysulfonic acids appeared to be easier compared to mineral acids. Also, these acids seemed to be stronger than HCl, since an aqueous solution of the adduct had been reported to react with NaCl and liberating the weaker acid, HCl.192 The α-hydroxysulfonic acids are prepared by the combination of an organic carbonyl compound (such as acetone), SO2 and water in a reaction illustrated in Fig. 21. | ||
| Fig. 21 General synthesis of α-hydroxysulfonic acids. | ||
The reaction in Fig. 21 illustrates an equilibrium process, which allows for the facile regeneration of the acid. When the reaction mixture is heated, the equilibrium shifts towards the starting carbonyl compound, sulfur dioxide, and water. If the volatile components (e.g., sulfur dioxide) are removed from the reaction mixture through vaporization, the equilibrium completely shifts to the left, and the solution becomes neutral. By increasing the temperature and lowering the pressure, sulfur dioxide can be driven off, causing the reaction to completely reverse. If the carbonyl compound is volatile (e.g., acetaldehyde), it is also easily removed in the vapor phase221
Shell Oil Company found that these acids produce very few of the undesired byproducts, such as furfurals, which are commonly produced by other mineral acids during biomass saccharification. Additionally, since the acids are effectively removed from the reaction mixture, neutralization with base and the formation of salts are substantially avoided. The ability to reverse and recycle these acids allows the use of higher concentrations that would otherwise be economically or environmentally impractical. This equilibrium is influenced by the nature of the carbonyl compound employed, with steric and electronic effects having a strong influence on the thermal stability of the acid. More steric bulk around the carbonyl group favors a lower thermal stability of the acid form.221 Thus, one can tune the strength of the acid and the temperature of facile decomposition by selecting the appropriate carbonyl compound.
Following the process shown in Fig. 22, lignocellulosic biomass is fed into the hydrolysis reactor together with a recycled stream of α-hydroxysulfonic acid. After hydrolysis, the acid is recovered and regenerated after purification, separating it from other small fractions of volatile compounds in the gas stream. The sugar solution product is then filtered out from the lignin residue and fermented into alcohols.221
 | ||
| Fig. 22 Block flow diagram of the Shell Oil Company patented process, figure from ref. 221. | ||
Archer-Daniels-Midland Company (ADM) is currently one of the largest nutrition companies focused on both human and animal food production from crops. They obtained a patent in 2011 for the production of sugars using a combination of organic and mineral acids to stage and separate the hydrolysis of hemicellulose and cellulose.222 This invention concerns a method combining weak organic acids, such as acetic or formic acid, to selectively remove the hemicellulose first before hydrolyzing the cellulose. After the initial hydrolysis (or pre-hydrolysis) with a steam formed by the weak organic acid at high temperatures, the material is dried and pelletized. The weak acid-processed biomass is washed to separate the solubilized and depolymerized hemicellulose. Subsequently, the cellulosic fraction is submerged in a second, strong mineral acid solution, preferably sulfuric acid. This two-stage hydrolysis process allows for a more efficient separation and conversion of hemicellulose and cellulose into fermentable sugars, improving overall yields and reducing the formation of undesirable byproducts.
Vertoro, founded in May 2017, developed a technology to process biomass on an industrial scale to separate lignin, cellulose, and hemicellulose into different streams, producing biofuels, paper, pulp, and biobased materials. Vertoro is a spin-off of the Chemelot Institute for Science & Technology, a Dutch public-private research and technical validation institute established by Eindhoven University of Technology, Maastricht University, Royal DSM, and the Brightlands Chemelot Campus. Vertoro obtained a patent for their Goldilock® process, described in Fig. 23.223
 | ||
| Fig. 23 Flow diagram of the Vertoro patented Goldilock process for the fractionation of biomass, figure from ref. 223. | ||
This technology is based on the fractionation of lignocellulosic feedstock (wood chips or sawdust, birch hardwood, or Douglas softwood) by mixing the feedstock with methanol and sulfuric acid. The acid supports the cleaving of the cellulosic component from the lignin, which helps the lignin dissolve into the methanol. By applying a compressed stream of nitrogen, the methanol remains in the liquid phase and effectively dissolves the lignin. Nitrogen or hydrogen is used at 10 to 30 bar and temperatures ranging from 140–200 °C for 30–120 minutes.
After the dissolution of the lignin, the mixture is subjected to vacuum filtration to separate the crude liquid lignin oil from the cellulose pulp. This liquid lignin oil contains low molecular weight oligomeric lignin fragments and some polysaccharides. As plant cells in woody biomass contain lignin–carbohydrate interlinkages such as phenyl glycoside, benzyl ether, and γ-ester bonds, an efficient cleavage is required for lignin extraction and valorization. However, the release of sugars cannot be prevented, and they can convert to furfurals, which may cause undesired repolymerization reactions due to lignin-furfural condensation reactions.
When sulfuric acid is used in combination with methanol, most of the hemicellulosic sugars are converted into methylated sugars such as methyl-pentapyranoside, methyl-D-gluconpyranoside, methyl-D-xylopyranoside, methyl 3-O-acetylpentopyranoside, and dimethyl-4-O-methyl-hexanopyranoside.223 Methylated sugars are separated from the lignin by liquid–liquid extraction using ethyl acetate or water. A high degree of delignification is always accompanied by a large extent of C5 (pentose) sugars release.
Bloom Biorenewables, a Swiss biorefinery founded in 2019, developed a biomass valorization process on an industrial scale, focusing on the flavors and fragrances (F&F) market by delivering vanillin and eugenol through the extraction of lignin from lignocellulosic feedstock.224 Currently, their pilot plant operates with a production capacity of 50 tons per year of starting feedstock. The process fractionates lignocellulosic biomass into its three major components—lignin, cellulose, and hemicellulose— using formaldehyde. This fractionation is in cooperation with Luterbacher from the University of Lausanne.225 Formaldehyde is used to protect the lignin during extraction by converting free diols on the lignin side chains into acetals. This reaction prevents the elimination of the benzylic alcohol on the side chain during the acidic extraction of lignin. When applied to beech wood, this process results in extracted lignin, providing monomers with a 47% (w/w) yield after hydrogenolysis.226
Renmatix is a U.S.-based company that specializes in the hydrolysis of lignocellulosic biomass using supercritical water.227–229 Supercritical water, which is water heated and pressurized above its critical point (374 °C and 22.1 MPa), has unique properties that facilitate various chemical reactions, including rapid biomass hydrolysis. In this state, water's density, viscosity, and dielectric constant decrease significantly, making it an effective medium for hydrolysis. Supercritical hydrolysis is a process where water in its supercritical state is used to achieve rapid and efficient biomass saccharification.230 During this process, the phenolic components of biomass, such as lignin, are converted into a water-insoluble liquid mixture of low molecular weight phenols, while the polysaccharides are hydrolyzed into simple sugars with near-quantitative yields. This technology's continuous operation is crucial for industrial scalability, given the extremely short reaction times required. Renmatix remains active and continues to advance its technology platform. The company's proprietary Plantrose® process is at the core of their operations, enabling the cost-effective conversion of biomass into useful sugars and other bio-based products.231
The concept of CMF production from carbohydrate and HCl was first reported by Haworth in 1944, achieving a 21.3% molar yield of CMF by mixing carbon tetrachloride with a saturated concentrated HCl solution of fructose at 40 °C.233 This approach has been further investigated and refined by Mascal and later by our research team.232,234–241
Mascal's approach, often utilizing concentrated hydrochloric acid in biphasic setups, has enabled the selective extraction and purification of CMF, mitigating the degradation pathways common with other furfurals like HMF (5-hydroxymethylfurfural). This technique not only enhances the stability and yield of CMF but also exemplifies the practical application of biorefinery technologies in transforming agricultural and wood-based wastes into valuable chemicals.235 This work underscores the integration of chemical innovation with environmental sustainability, promoting the broader adoption of biomass-derived chemicals in various industries. This synergy between biorefinery technologies and the production of CMF illustrates a promising path towards reducing reliance on fossil-based resources, aligning with global sustainability goals.
Micromidas was founded in 2008 in West Sacramento, California, and was one of the first companies to develop an innovative chemical process to produce a variety of commodity chemicals from cellulosic biomass and ethylene.242 Origin Materials has developed a two-step process for producing 5-(chloromethyl)furfural (CMF) from starch or lignocellulosic biomass.232,243,244 Initially, the biomass is exposed to HCl gas in a fluidized bed reactor, facilitating rapid hydrolysis. The process can optionally include the addition of Lewis acidic metal chlorides. Fluidization within the reactor ensures thorough and consistent mixing of the biomass and the gaseous acid. The reactor operates at elevated temperatures, specifically 220 °C and a pressure of 15 atm, since lower temperatures result in lignin capturing some of the HCl gas.243 The reactor maintains a residence time of approximately 2 minutes, after which the gas and solid mixture exits the reactor due to the pressure differential between the reactor interior (15 atm) and the exterior (4 atm). This mixture then undergoes solid–gas separation using a cyclone. The gaseous HCl is passed through a desiccant to eliminate any present water before being recycled, offering a cost advantage over conventional acid–water separation methods like azeotrope shifting, which are more energy-intensive and costly232 (Fig. 24).
 | ||
| Fig. 24 The biphasic liquid–gas reactor for the continuous production of CMF developed by Origin Materials.243,244 | ||
The remaining solid mixture, which contains hydrolyzed sugars, CMF, lignin, and metal salts, is combined with dichloromethane (DCM) or toluene to form a solid/liquid mixture. Chloride salts in the mixture drive the reaction equilibrium through nucleophilic substitution of 5-(hydroxymethyl)furfural (HMF) to produce CMF, which dissolves in the DCM or toluene, separating it from the remaining solids that contain lignin and other by-products. In trials with biomass as feedstock, CMF was detected but not quantified. However, when fructose was used as the feedstock with CaCl2 (12 mole equivalents) and AlCl3 (0.2 mole equivalents) suspended in DCM, a 35% CMF yield was achieved. Hydrothermal carbon (HTC) is the main product from the process, of important commercial interest to Origin Materials for application as carbon black in tires. Other soluble side products like HMF, furfural, levulinic acid (LA), and formic acid (FA) are obtained in the organic phase. The DCM or toluene solvent is recovered by distillation and continuously recycled to the solvent feed.
In 2013, they secured financing and opened their pilot plant. By 2017, Micromidas had secured more than $80 million and changed their name to Origin Materials.245 By 2021, Origin Materials had secured $990 million and went public on the NASDAQ.246 Origin's journey from a college project to a publicly traded company on NASDAQ highlights their significant growth and the confidence investors have in their technology and business model. In February 2023, Origin Materials and Avantium N. V., a leading technology company in renewable chemistry, announced a partnership to accelerate the mass production of FDCA and PEF for use in advanced chemicals and plastics.247 The partnership aims to bring the technology platforms of both companies together in order to produce FDCA from sustainable wood residues on an industrial scale. FDCA (furandicarboxylic acid) is the key building block for the biopolymer PEF (polyethylene furanoate) – a 100% plant-based, fully recyclable plastic material, with superior functionality and a significantly reduced carbon footprint compared to conventional plastics. PEF can be used in a wide range of applications such as bottles, packaging, films, fibers and textiles, which represent major end-markets.
As of 2024, the company's first plant, Origin 1, located in Sarnia, Ontario, has been commissioned and started-up. This plant uses cornstarch as the initial feedstock, with plans to introduce wood handling later in 2024. Origin 2, the company's second manufacturing plant, is pursuing an asset-light strategy to optimize scale-up synergies and reduce project execution risks.248
 | ||
| Fig. 25 Lignocellulosic conversion to furanics in an integrated 2-step process developed by Avantium.239 | ||
The Yukon process leverages the presence of glucose and xylose in the acidic solution, derived from the hydrolysis of hardwood, where xylose is the predominant saccharide in hemicellulose and glucose is the main component of cellulose. Upon heating, glucose dehydrates to form 5-hydroxymethylfurfural (HMF), which, in the presence of HCl, undergoes halogenation to produce CMF. The lower polarity of CMF allows it to be extracted from the acidic solution using immiscible organic solvents, simplifying downstream product separation from the acid. Xylose, present in the mixed sugar stream is converted to furfural through acid-catalyzed dehydration. Similar to CMF, furfural is continuously extracted into an immiscible organic solvent, reducing its contact time with the acid and minimizing decomposition.
Furfural and CMF are obtained in high purity, and the organic solvent can be evaporated and recirculated back into the reactor. The use of biphasic systems for converting saccharides into CMF in high yields is a well-established method234–236,249,250 but in our case we use the DAWN™ hydrolysis as a front-end for separating the sugars (in the form of monomers and oligomers) from the lignin and using the hydrolysate directly, thereby eliminating the acid/sugar separation and the post hydrolysis.
Although a staged hydrolysis is still possible, both CMF and furfural can also be produced simultaneously from a single stage hydrolysis, although furfural typically requires lower temperatures and shorter reaction times compared to CMF production from glucose (Fig. 25).
As shown in Fig. 26, high yields of furfural and CMF are achieved and it is observed that furfural is produced in higher yields at shorter reaction times compared to CMF formation from glucose. Specifically, a 96.8% molar yield of furfural is reached after 1.5 hours, whereas achieving a similar molar yield for CMF (93.6%) takes about 4 hours. Notably, furfural begins to decompose after 1.5 hours, resulting in black solid deposits in the reactor. The conversion of xylose to furfural is more efficient and faster because it involves only one dehydration step, while glucose requires a longer reaction time due to the additional steps needed for its conversion to CMF.
 | ||
| Fig. 26 The molar yields of furfural and CMF were obtained at 80 °C over a period of 4 hours. The reaction took place in a biphasic batch system containing fluorobenzene (organic phase) and DAWN hydrolysate (aqueous phase). The results are represented in terms of CMF molar yield (green) and furfural molar yield (blue), reproduced from ref. 239 with permission from the American Chemical Society, © 2023. | ||
The residual lignin after hydrolysis and removal of the acid has been tested for various applications. Although industrial drying (to remove HCl/water) reduces the solid material to a residual moisture content of 0.5–5%, some aqueous HCl and intracellular moisture remains. Additionally, some chloride remains on the surface as precipitated salt, and small amounts of organic chloride compounds (derivatives from lignin, sugar (e.g. CMF or CMF ending up in humins), or extractives) are present in the lignin. The potential for lignin to replace bitumen in asphalt is promising,251 with the world's first test road made with lignin produced by DAWN Technology™ in the Netherlands in 2021.224
Overall, the shift to a one-stage hydrolysis process, along with the integrated production of CMF and furfural, significantly improved the economic viability and scalability of lignocellulosic biomass conversion, enabling more efficient and sustainable production of bio-based chemicals. On the other hand, furfural and CMF are a strategic fit for Avantium's FDCA business but is not a solution for supplying the world with 2 G sugars as feedstock for a bio-based economy (e.g. monomers for plastics such as glycols (ethylene-, propylene- and butylene glycol), lactic acid, succinic acid, sorbitol/isosorbide, glycolic acid. etc.). For that reason, Avantium will continue to focus on developing an economically viable 2nd generation sugar production from biomass.
These waste materials are typically combinations of cellulose and plastics, such as PET in textiles. By applying hydrolysis and CMF reaction conditions that ensure non-cellulosic components like PET or polyolefin plastics remain unaffected, their recovery and recycling are facilitated. While this process is in its early development stages, initial results have shown the recovery of approximately 60–95% of the starting cellulosic material present in waste textiles as CMF. The highest CMF yields were observed for cigarette butts and cleaning wipes, reaching up to 93.2% and 85.9%, respectively.241
Although the CMF yield from waste textiles was not as high as previously achieved from pure hydrolysate, the hydrolysis yield for this material remained around 80%, indicating high efficiency in breaking down the cellulosic content.240 These findings highlight the potential for refining this process to maximize CMF production from various cellulosic waste streams, paving the way for more sustainable waste management and chemical production solutions.
This concept has been recently explored and optimized in work by Leenders et al., who selectively hydrolyzed waste textile cotton using 43% hydrochloric acid, achieving a significant glucose yield from the cellulose content of the textile (Fig. 27).252 This targeted hydrolysis not only maximizes sugar recovery but also facilitates the separation of polyester, which remained largely unaltered and intact. The intact polyester was then subjected to glycolysis, converting it into bis(2-hydroxyethyl) terephthalate (BHET) with high efficiency and purity. This dual approach not only exemplifies high material recovery but also underlines the process's scalability and applicability to industrial recycling practices.
 | ||
| Fig. 27 SEM images to display the structure of the polycotton waste textile prior to acid hydrolysis. The image on the right highlights a significant decrease in cotton fibers, demonstrating the effective selective hydrolysis of the cotton component.252 | ||
6.5 Perspectives on advancing current saccharification technologies
The evolution of saccharification technologies discussed in this chapter reflects significant progress in harnessing biomass for the production of biofuels, biochemicals, and renewable materials. However, as industries continue to adapt these technologies for modern applications, several critical lessons and forward-looking strategies emerge that can guide future research and development.7. Conclusions
This review examined the evolution of saccharification methods from their initial development to modern industrial applications, illustrating how historical milestones have driven contemporary innovations. Early 20th-century acid hydrolysis, despite challenges such as equipment corrosion and sugar degradation, laid a critical foundation for the development of advanced techniques like enzymatic hydrolysis and pretreatment strategies. These advancements have successfully addressed technical barriers, highlighting the importance of adaptable processes to enhance efficiency and overcome practical challenges.Recent advancements, including steam explosion, alkaline pretreatment, and ammonia fiber explosion, have significantly improved saccharification by breaking down the lignin–carbohydrate complex. These innovations emphasize the value of integrated approaches for optimizing results and reflect steady progress in achieving scalability and cost-efficiency in enzymatic hydrolysis.
The role of biomass as a renewable resource has been firmly established, with wood-based biomass offering a particularly promising feedstock due to its abundance and supply chain infrastructure. The effective utilization of wood residues and by-products not only minimizes waste but also aligns with the global shift toward a circular bioeconomy. This highlights the broader significance of these technologies in achieving sustainable energy and material production. The development of Avantium's DAWN technology and its Yukon evolution for waste valorization showcases the integration of historical insights into industrial applications, effectively tackling challenges like acid recovery and sugar concentration. This progress highlights how innovative strategies can resolve economic and technical barriers, driving the industrial adoption of these processes.
Overall, these advancements over time shows the effort and problem-solving required to tackle challenges in the chemical industry. These advancements have paved the way for more efficient and sustainable biomass conversion methods. Moving forward, interdisciplinary collaboration and sustained research investments will be essential to unlocking the full potential of lignocellulosic biomass, contributing to a future defined by renewable energy and sustainable material production.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Partially funded by IMPRESS project, under the European Union's Horizon 2020 program and grant agreement no. 869993.References
- Scherer, Bulletin des Neuesten und Wissenswürdigsten aus der Naturwissenschaft, so wie den Künsten, Manufakturen, technischen Gewerben, der Landwirthschaft und der bürgerlichen Haushaltung, ed. Amelang, Carl Friedrich Amelang, Berlin, 1811, pp. 262–263 Search PubMed.
- F. K. Achard, Procédé d'extraction du sucre de bette, Ann. Chem., 1799, 32, 163–168 Search PubMed.
- F. Carrasco and C. Roy, Kinetic study of dilute-acid prehydrolysis of xylan-containing biomass, Wood Sci. Technol., 1992, 26, 189–207 CAS.
- G. Melsens, Process for converting the products obtained by distillation of resins into saleable oils, Dinglers Polytech, 1856 Search PubMed.
- A. Marggraf, Experiences chimiques faites dans le dessein de tirer un veritable sucre de diverses plantes, qui croissent dans nos contrées, Histoire de l’académie royale des sciences et belles-lettres de Berlin, 1747, pp. 79–90 Search PubMed.
- R. N. Gurram and T. J. Menkhaus, Continuous enzymatic hydrolysis of lignocellulosic biomass with simultaneous detoxification and enzyme recovery, Appl. Biochem. Biotechnol., 2014, 173(6), 1319–1335 CrossRef CAS PubMed.
- (BIOS), British Intelligence Objectives Subcommittee, Bergius Wood Sugar Plants at Mannheim-Rheinau and Regensburg. British Intelligence Objectives Sub-Committee, 1945 Search PubMed.
- J. V. Tanchyna and H. M. Fanto, “Viscosity as a qualitative probe”, Chem. Sheets Sci. Ind. Organ Czech Chem. Soc. Sci. Ind., 1930, vol. 1, 24, pp. 105–106 Search PubMed.
- F. Bergius, Chem. Trade J., 1933, 93, 356 CAS.
- B. Yang, Z. Dai, S.-Y. Ding and C. E. Wyman, Enzymatic hydrolysis of cellulosic biomass, Biofuels, 2011, 2(4), 421–449 CrossRef CAS.
- A. K. Chandel, M. B. S. Forte, I. S. Gonçalves, T. S. Milessi, P. V. Arruda, W. Carvalho and S. I. Mussatto, Brazilian biorefineries from second generation biomass: critical insights from industry and future perspectives, Biofuels, Bioprod. Biorefin., 2021, 15, 1190–1208 CrossRef CAS.
- M. Morales, J. Quintero, R. Conejeros and G. Aroca, Life cycle assessment of lignocellulosic bioethanol: Environmentalimpacts and energy balance, Renewable Sustainable Energy Rev., 2015, 42, 1349–1361 CrossRef CAS.
- R. Rowell, R. Pettersen and M. Tshabalala, Cell Wall Chemistry, in Handbook of Wood Chemistry and Wood Composites, ed. R. M. Rowell, CRC Press, 2012, pp. 33–72 Search PubMed.
- K. Świątek, S. Gaag, A. Klier, A. Kruse, J. Sauer and D. Steinbach, Acid Hydrolysis of Lignocellulosic Biomass: Sugars and Furfurals Formation, Catalysts, 2020, 10, 437 CrossRef.
- T. Stevanovic, Chemical Composition and Properties of Wood, in Lignocellulosic Fibers and Wood Handbook, John Wiley & Sons, Ltd, 2016, pp. 49–106 Search PubMed.
- P. Singh, H. Duarte, L. Alves, F. Antunes, N. L. Moigne, J. Dormanns, B. Duchemin, M. P. Staiger and B. Medronho, From Cellulose Dissolution and Regeneration to Added Value Applications — Synergism Between Molecular Understanding and Material Development, in Cellulose – Fundamental Aspects and Current Trends, InTech, 2015 Search PubMed.
- H. Rasmussen, H. R. Sørensen and A. S. Meyer, Formation of degradation compounds from lignocellulosic biomass in the biorefinery: sugar reaction mechanisms, Carbohydr. Res., 2014, 385, 45–57 CrossRef CAS PubMed.
- S. H. Russell, J. B. Morón, M. Kersbulck, B. McKay and G.-J. M. Gruter, Controlled process for the conversion of particulate matter comprising hemicellulose, cellulose and lignin, WO2021018559, 2021.
- M. Kersbulck, B. McKay and G.-J. M. Gruter, Process for the Conversion of a Solid Material Containing Hemicellulose, Cellulose and Lignin, WO2019149833A1, 2019.
- M. Kersbulck, B. McKay and G.-J. M. Gruter, Process for Acidic Hydrolysis of a Particulate Solid Material Containing Cellulose, Lignin, and Hemicellulose, Wherein the Latter Has a High Content of Xylose, WO2020239730A1, 2020.
- A. L. Jongerius, S. H. Russel, K. J. Damen and B. McKay, Processed lignin and process to prepare such, WO2020259991A1, 2020.
- C. Plomion, A. Stokes, C. P. France and L. De Rhe, Wood Formation in Trees, Plant Physiol., 2015, 127(4), 1513–1523 CrossRef.
- K. Mohammadi, S. Khalesro, Y. Sohrabi and G. Heidari, A Review: Beneficial Effects of the Mycorrhizal Fungi for Plant Growth, J. Appl. Environ. Biol. Sci., 2001, 1(9), 310–319 Search PubMed.
- Forest Products Laboratory, Forest Service, Differences between Heartwood and Sapwood, U.S. Department of Agriculture, 1966 Search PubMed.
- Forest Service U.S. Department of Agriculture, Anatomy of a Tree, online, available, https://www.fs.usda.gov/learn/trees/anatomy-of-tree, accessed 05 August 2024 Search PubMed.
- D. M. Barry, Chem-Is-Tree, J. Chem. Educ., 1997, 74, 1175 CrossRef CAS.
- B. Butterfield, The structure of wood: form and function, in Primary Wood Processing, Springer, 2006, pp. 1–22 Search PubMed.
- K. Luostarinen and K. Hakkarainen, Chemical composition of wood and its connection with wood anatomy in Betula pubescens, Scand. J. For. Res., 2019, 34, 577–584 CrossRef.
- E. Montet, Investigation of the consequences of the use of ozone in the bleaching of cellulosic fibres, Doctoral thesis, Univ. Grenoble Alpes, CNRS, Grenoble INP (Institute of Engineering Univ. Grenoble Alpes), LGP2, 2021 Search PubMed.
- R. Gemci and C. Çebiçci, Examining the effect of mercerization process applied under different conditions via the degree of whiteness and color efficiency, J. Appl. Polym. Sci., 2011, 121, 202–209 CrossRef CAS.
- J. Gawron, M. Szczęsna, T. Zielenkiewicz and T. Gołofit, Cellulose Crystallinity Index Examination in Oak Wood Originated from Antique Woodwork, 2012 Search PubMed.
- J. C. F. Walker, Primary Wood Processing, Principles and Practice, Springer, 2006 Search PubMed.
- J. Berglund, Wood Hemicelluloses – Fundamental Insights on Biological and Technical Properties, Doctoral thesis, Wallenberg Wood Science Centre (WWSC), Department of Fiber and Polymer Technology, School of Chemical Engineering, Royal Institute of Technology KTH, SE-100 44 Stockholm, Sweden, 2018.
- N. N. Deshavath, V. D. Veeranki and V. V. Goud, Chapter 1 – Lignocellulosic feedstocks for the production of bioethanol: availability, structure, and composition, in Sustainable Bioenergy, ed. M. Rai and A. P. Ingle, Elsevier, 2019, pp. 1–19 Search PubMed.
- Z. Börcsök and Z. Pásztory, The role of lignin in wood working processes using elevated temperatures: an abbreviated literature survey, Eur. J. Wood Wood Prod., 2020, 79, 511–526 CrossRef.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, The Catalytic Valorization of Lignin for the Production of Renewable Chemicals, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- M. Y. Capanema and E. A. Chang, Characterization of Lignocellulosic Materials, Blackwell Publishing, 2008 Search PubMed.
- G. T. Kirker, A. B. Blodgett, R. A. Arango, P. K. Lebow and C. A. Clausen, International Biodeterioration & Biodegradation: The role of extractives in naturally durable wood species, Int. Biodeterior. Biodegrad., 2013, 82, 53–58 CrossRef CAS.
- M. Morel-Rouhier, Wood as a hostile habitat for ligninolytic fungi, in Wood Degradation and Ligninolytic Fungi, Elsevier, 2021, pp. 115–149 Search PubMed.
- D. Smołka-Danielowska and M. Jabłońska, Chemical and mineral composition of ashes from wood biomass combustion in domestic wood-fired furnaces, Int. J. Environ. Sci. Technol., 2021, 19, 5359–5372 CrossRef.
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Y. Murzin, Synthesis of Sugars by Hydrolysis of Hemicelluloses – A Review, Chem. Rev., 2011, 111, 5638–5666 CrossRef PubMed.
- A. T. Hoang, S. Nizetic, H. C. Ong, C. T. Chong, A. E. Atabani and V. V. Pham, Acid-based lignocellulosic biomass biorefinery for bioenergy production: Advantages, application constraints, and perspectives, J. Environ. Manage., 2021, 296, 113194 CrossRef CAS PubMed.
- R. G. Aravamuthan, Chemical Pulping, in Encyclopedia of Forest Sciences, Elsevier, 2004, pp. 904–910 Search PubMed.
- N. P. Cheremisinoff and P. E. Rosenfeld, Sources of air emissions from pulp and paper mills, in Handbook of Pollution Prevention and Cleaner Production, Elsevier, 2010, pp. 179–259 Search PubMed.
- E. Ahmad and K. K. Pant, Lignin Conversion: A Key to the Concept of Lignocellulosic Biomass-Based Integrated Biorefinery, in Waste Biorefinery, Elsevier, 2018, pp. 409–444 Search PubMed.
- Z. Zhou, D. Liu and X. Zhao, Conversion of lignocellulose to biofuels and chemicals via sugar platform: An updated review on chemistry and mechanisms of acid hydrolysis of lignocellulose, Renewable Sustainable Energy Rev., 2021, 146, 111169 CrossRef CAS.
- S. Y. Yoon, S.-H. Han and S.-J. Shin, The effect of hemicelluloses and lignin on acid hydrolysis of cellulose, Energy, 2014, 77(1), 19–24 CrossRef CAS.
- M. Baucher, M. A. Bernard-Vailhe, B. Chabbert, J.-M. Besle, C. Opsomer, M. V. Montagu and J. Botterman, Down-regulation of cinnamyl alcohol dehydrogenase in transgenic alfalfa (Medicago sativa L.) and the effect on lignin composition and digestibility, Plant Mol. Biol., 1999, 39(3), 437–447 CrossRef CAS PubMed.
- L. Tillman, A. E. Araseed, Y. Y. Lee and R. Torget, Effect of transient variation of temperature on acid hydrolysis of aspen hemicellulose, Appl. Biochem. Biotechnol., 1989, 20, 107–117 CrossRef.
- E. L. Springen, Prehydrolysis of hardwoods with dilute sulfuric acid, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24(4), 614–623 CrossRef.
- R. Rinaldi and F. Schüth, Acid Hydrolysis of Cellulose as the Entry Point into Biorefinery Schemes, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- K. Freudenberg and G. Blomqvist, Die Hydrolyse der Cellulose und ihrer Oligosaccharide, Ber. Dtsch. Chem. Ges., 1935, 68(11), 2070–2082 CrossRef.
- J. F. Saeman, Kinetics of Wood Saccharification – Hydrolysis of Cellulose and Decomposition of Sugars in Dilute Acid at High Temperature, Ind. Eng. Chem., 1945, 37(1), 43–52 CrossRef CAS.
- T. P. Nevell and W. R. Upton, The hydrolysis of cotton cellulose by hydrochloric acid in benzene, Carbohydr. Res., 1976, 49, 163–174 CrossRef CAS.
- M. J. Comstock, Hydrolysis of Cellulose: Mechanisms of Enzymatic and Acid Catalysis, Adv. Chem., 1979, 127–143 Search PubMed.
- L.-T. Fan, M. M. Gharpuray and Y.-H. Lee, Acid Hydrolysis of Cellulose, in Cellulose Hydrolysis, Springer, Berlin Heidelberg, 1987, pp. 121–148 Search PubMed.
- S. Naz, N. Ahmad, J. Akhtar, N. M. Ahmad, A. Ali and M. Zia, Management of citrus waste by switching in the production of nanocellulose, IET Nanobiotechnol., 2016, 10, 395–399 CrossRef PubMed.
- A. T. Hoang, S. Nizetic, H. C. Ong, C. T. Chong, A. E. Atabani and V. V. Pham, Acid-based lignocellulosic biomass biorefinery for bioenergy production: Advantages, application constraints, and perspectives, J. Environ. Manage., 2021, 296, 113194 CrossRef CAS PubMed.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Can ionic liquids dissolve wood? Processing and analysis of lignocellulosic materials with 1-n-butyl-3-methylimidazolium chloride, Green Chem., 2007, 9, 63–69 RSC.
- S. Zhu, Use of ionic liquids for the efficient utilization of lignocellulosic materials, J. Chem. Technol. Biotechnol., 2008, 83(6), 777–779 CrossRef CAS.
- E. Heinone, Structural basis for the recalcitrance and molecular packing of hemicelluloses, Doctoral thesis, KTH Royal Institute of Technology, Stockholm, 2024.
- S. Pramod, M. Latha Gandla, M. Derba-Maceluch, L. J. Jönsson, E. J. Mellerowicz and S. Winestrand, Saccharification Potential of Transgenic Greenhouse- and Field-Grown Aspen Engineered for Reduced Xylan Acetylation, Front. Plant Sci., 2021, 12, 704960 CrossRef PubMed.
- J. J. Lyczakowski, K. B. Wicher, O. M. Terrett, N. Faria-Blanc, X. Yu, D. Brown, K. B. R. M. Krogh, P. Dupree and M. Busse-Wicher, Removal of glucuronic acid from xylan is a strategy to improve the conversion of plant biomass to sugars for bioenergy, Biotechnol. Biofuels, 2017, 10, 224 CrossRef PubMed.
- S. Hizukuri, Nutritional and Physiological Functions and Uses of L-Arabinose, J. Appl. Glycosci., 1999, 46, 159–165 CrossRef CAS.
- G. M. Rolph, Something about Sugar: its History, Growth, Manufacture and Distribution, John J. Newbegin, 1917 Search PubMed.
- G. F. von Schoenberg, Handbuch der politischen Oekonomie: Volkswirtschaftslehre, H. Laupp, Tübingen, 1896 Search PubMed.
- H. Laupp, Handbuch der politischen Oekonomie, H. Laupp, Tübingen, 1896, vol. 2 Search PubMed.
- G. Woddfall, The National Cyclopædia of Useful Knowledge, Charles Knight, 1847, vol. 1 Search PubMed.
- G. Kirchhoff, Bulletin des Neuesten und Wissenswürdigsten aus der Naturwissenschaft, so wie den Künsten, Manufakturen, technischen Gewerben, der Landwirthschaft und der bürgerlichen Haushaltung, ed. Amelang, Carl Friedrich Amelang, Berlin, 1811, p. 27 Search PubMed.
- J. J. Berzelius, Årsberättelsen Om Framsteg I Fysik Och Kemi, Royal Swedish Academy of Sciences, 1835 Search PubMed.
- H. Braconnot, Ann. Chim. Phys., 1819, 12, 172 Search PubMed.
- Britannica, “Anselme Payen”, 2022, online, available, https://www.britannica.com/biography/Anselme-Payen, accessed 21 August 2024.
- A. Payen and J. F. Persoz, Mémoire sur la diastase, les principaux produits de ses réactions et leurs applications aux arts industriels, Ann. Chim. Phys., 1833, 53, 73–92 Search PubMed.
- A. Payen, Mémoire sur la composition du tissu propre des plantes et du ligneux, C. R. Hebd. Seances Acad. Sci., 1838,(7), 1052–1056 Search PubMed.
- J. White, H. F. C. S. Sucrose, and Fructose: History, Manufacture, Composition, Applications, and Production, in Fructose, High Fructose Corn Syrup, Sucrose and Health, Humana Press, 2014, pp. 13–33 Search PubMed.
- J. Kim, Y. Lee and R. Torget, Cellulose hydrolysis under extremely low sulfuric acid and high-temperature conditions, Appl. Biochem. Biotechnol., 2001, 91, 331–340 CrossRef PubMed.
- W. R. Pötsch, Lexikon Bedeutender Chemiker, Leipzig: Bibliogr. Inst, 1988 Search PubMed.
- A. Béchamp, Ann. Chim. Phys., 1856, 48, 327 Search PubMed.
- E. S. Dauziville, Verfahren der Umwandlung von Holzmasse in Glucose und Alkohol und zugehörige Apparate, DE11836C, 1880.
- H. Chen, Chapter 3 – Lignocellulose biorefinery feedstock engineering, in Lignocellulose Biorefinery Engineering, ed. H. Chen, Woodhead Publishing, 2015, pp. 37–86 Search PubMed.
- E. Fleschsig and Z. physik, Chem, 1883, 7, 523 Search PubMed.
- E. E. Harris, Wood Saccharification, in Advances in Carbohydrate Chemistry, 1949 Search PubMed.
- R. P. Beatson, Determination of Sulfonate Groups and Total Sulfur, Springer, Berlin Heidelberg, 1992, pp. 473–484 Search PubMed.
- J. B. Lindsey and B. Tollens, Ueber Holz-Sulfitflüssigkeit und Lignin, Justus Liebigs Ann. Chem., 1892, 267(2–3), 341–366 CrossRef.
- R. F. Ruttan, Ethyl alcohol from sawdust and other wood waste, J. Soc. Chem. Ind., London, 1909, 28, 1290–1294 CrossRef.
- E. Simonsen, Spiritus aus Cellulose und Holz, Zeit. f Angw. Chem., 1898, 195–196(219–228), 1007–1012 CrossRef.
- S. Karp, New Technologies for Bioethanol Production: Patents and Innovation, in Liquid Biofuels: Bioethanol, 2022, pp. 489–515 Search PubMed.
- A. Classen, Process of converting cellulose into sugar, US Pat., US654518, 1900 Search PubMed.
- F. W. Kressmann, The Manufacture of Ethyl Alcohol from Wood Waste, U.S Department of Agriculture, 1922 Search PubMed.
- E. Malcolm F. and G. Herbet Tomlinson, Process of producing from ligno-cellulose fermentable sugar, GB190924589A, 1909.
- J. K. George, Process of forming a food product, US Pat., US981634, 1909 Search PubMed.
- W. P. Cohoe, Method of Making glucose-like product from cellulosic and ligneous materials, US Pat., US985726, 1911 Search PubMed.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, 2010, pp. 337–340. Search PubMed.
- F. Bergius, Die Anwendung hoher drucke bei chemischen Vorgängen und eine nachbildung des Entstehungsprozesses der Steinkohle, ed. W. Knapp, 1913 Search PubMed.
- F. Bergius, Process for producing liquid or soluble organic combination from hard coal and the like, DE856825, 1918.
- F. Bergius, New Uses for Coal and Wood, 1929, vol. 140, pp. 322–324 Search PubMed.
- R. E. Oesper, Friedrich Bergius (1884–1949), J. Chem. Educ., 1949, 26, 508 CrossRef CAS.
- M. Detroit, Encyclopedia of World Biography, 2009, p. 30 Search PubMed.
- F. Bergius, Conversion of Wood To Carbohydrates, Ind. Eng. Chem., 1937, 29, 247–253 Search PubMed.
- F. Bergius, World Will Feed on Converted Wood, 1928, vol. 14, p. 323 Search PubMed.
- F. Bergius, The Production of Food-stuffs, Alcohol and Glucose from Wood by means of the Bergius-Rheinau Process, Curr. Sci., 1937, 5, 632–637 Search PubMed.
- R. Willstätter and L. Zechmeister, Zur Kenntnis der Hydrolyse von Cellulose I, 1913, vol. 46, pp. 2401–2412 Search PubMed.
- A. Wohl and H. Krull, Cellulosechemie, 1921, 2, 1 CAS.
- Z. Ostenberg, Process of dissolving cellulose, US Pat., US1218954, 1917 Search PubMed.
- Z. Ostenberg, Process of preparing solutions of cellulose, US Pat., US1242030, 1917 Search PubMed.
- E. Hägglund and J. Prakt, Chem, 1915, 91, 358 Search PubMed.
- F. E. Gallagher, Process of producing fermentable sugars, US Pat., US1091327, 1914 Search PubMed.
- E. Hägglund, Chapter IV. The Decomposition of Wood by Acids, in The Chemistry of Wood, Academic Press INC., 1951, pp. 390–413 Search PubMed.
- A. E. Comyns, Encyclopedic Dictionary of Named Processes in Chemical Technology, 4th edn, CRC Press, 2014 Search PubMed.
- T. Goldschmidt and E. Hägglund, Verfahren zur Herstellung von Sulfitsprit, AT83876B, 1916.
- Y. H. Jung and K. H. Kim, Acidic Pretreatment (Chapter 3) in Pretreatment of Biomass; Processes and Technologies, A. Pandey Ed. Elsevier, 2015, pp. 27–50 Search PubMed.
- W. R. Ormandy, Sugar from wood, J. Soc. Chem. Ind., 1926, 45, T267–T288 CrossRef.
- E. Boullanger, Distillerie agricole et industrielle: eaux-de-vie de fruits – rhums, California, J.-B. Baillière, 1925 Search PubMed.
- H. Terrisse, Process for converting cellulose and cellulose-yielding matter into dextrine and glucose, US Pat., US1511786, 1924 Search PubMed.
- J. Perl, Process for the saccharification of cellulose-bearing material, US Pat., US1677406, 1928 Search PubMed.
- E. C. Sherrard and A. W. Froehlke, The Action of Concentrated Hydrochloric Acid on Different Celluloses, J. Am. Chem. Soc., 1923, 45, 1729–1734 Search PubMed.
- E. C. Sherrard and W. H. Gaugr, Effect of Acid and Salts upon the Hydrolysis of Wood, Ind. Eng. Chem., 1923, 15, 1164–1165 CrossRef CAS.
- E. Hägglund, Sven. Kem. Tidskr., 1923, 35, 2 Search PubMed.
- E. Hägglund, Die Hydrolyse der Zellulose und des Holzes, Sammlung chemischer und chemisch-technischer Vorträge, 1915 Search PubMed.
- E. Hägglund, F. Koch and N. Loefman, Verfahren zur Herstellung von konzentrierten Kohlehydratloesungen durch Aufschliessen von zellulosehaltigem Material mit Salzsaeure, DE382463C, 1920.
- E. Hägglund, Papierfabrikant, 1927, vol. 25, p. 52 Search PubMed.
- Wikimedia Commons, Erik Hägglund, 1919, online, available, https://commons.wikimedia.org/wiki/File:Erik_Hägglund,_1919.jpg, accessed 13 August 2024.
- Alamy, Friedrich Bergius in his laboratory, 1928, online, available, https://www.alamy.com/stock-photo-friedrich-bergius-in-his-laboratory-1928-37016703.html, accessed 13 August 2024.
- Z. Fang, Pretreatment Techniques for Biofuels and Biorefineries, in Chapter 7.8: the Hydrochloric Acid Process, Springer, 2013, pp. 138–150 Search PubMed.
- F. Bergius, Method of treating products of hydrolysis of cellulose, US Pat., US1547893, 1925 Search PubMed.
- F. Bergius, Food from Waste Wood Is Problem of German Chemist, Sci. News Lett., 1936, 30, 180–191 CrossRef.
- B. Kamm, Biorefineries-their scenarios and challenges, Pure Appl. Chem., 2014, 86, 821–831 CAS.
- United States Office of International Trade, World Chemical Developments, U.S. G.P.O., Washington, 1935 Search PubMed.
- F. Bergius, New Uses for Coal and Wood, Sci. Am., 1929, 140, 322–324 CrossRef.
- L. Zechmeister and G. Toch, Zur Kenntnis der Hydrolyse von Chitin mit Salzsäure (I. Mitteil.), Ber. Dtsch. Chem. Ges., 1931, 64(8), 2028–2032 CrossRef.
- E. Hägglund, Űber den Zellulose-gehalt und die Hydrolysierbarkeit der ver-schiedenen Hemicellulosen des Fichtenholzes, Tek. Tidskr., Upplaga C, 1933, 63, 65 Search PubMed.
- M. C. Hawley, S. M. Selke and D. T. A. Lamport, Comparison of hydrogen fluoride saccharification of lignocellulosic materials with other saccharification technologies, Energy Agric., 1983, 2, 219–244 CrossRef CAS.
- K. Schoenemann, The New Rheinau Saccharification Process, Food and Agriculture Organization of the United Nations, 1953 Search PubMed.
- E. Glesinger, The Coming Age of Wood, Simon and Schuster, Inc, 1949 Search PubMed.
- T. Riehm, Process for the saccharification of softwood sawdust, US Pat., US2945777A, 1957 Search PubMed.
- H. Wenzl, The Acid Hydrolysis of Wood, in The Chemical Technology of Wood, New York, Academic Press, 1970 Search PubMed.
- H. Scholler, Process and device for the saccharification of cellulose and the like, US Pat., US1890304A, 1932 Search PubMed.
- H. Scholler, Apparatus for the saccharification of cellulose, US Pat., US2086963A, 1937 Search PubMed.
- P. Kolachov and L. W. Nicholson, Carbohydrate sources for ethyl alcohol production, Econ. Bot., 1951, 5, 60–81 CrossRef CAS.
- R. Katzen and D. J. Schell, Lignocellulosic Feedstock Biorefinery: History and Plant Development for Biomass Hydrolysis, in Biorefineries-Industrial Processes and Products, Wiley-VCH Verlag GmbH, 2006, pp. 129–138 Search PubMed.
- S. C. Prescott and C. G. Dunn, Industrial Microbiology, McGraw-Hill, 1959 Search PubMed.
- R. Katzen and D. F. Othmer, Wood Hydrolysis. A Continuous Process, Ind. Eng. Chem., 1942, 34, 314–322 CrossRef CAS.
- E. E. Harris and E. Beglinger, The Madison Wood-Sugar Process, Ind. Eng. Chem., 1946, 9, 890–895 CrossRef.
- W. L. Faith, Development of the Scholler Process in the United States, Ind. Eng. Chem., 1945, 37, 9–11 CrossRef CAS.
- M. L. Rabinovich, Wood Hydrolysis Industry in the Soviet Union and Russia: A Mini-Review, Russian Academy of Sciences, 2010, vol. 44, pp. 173–186 Search PubMed.
- N. V. Chalov and N. V. Korotkov, Hydrolysis of hemicellulose components of pinewood with 30-36% hydrochloric acid at 30-40 °C, Gidroliz. Lesokhim. Prom-st., 1968, 21, 4–6 Search PubMed.
- P. N. Odincovs, K. V. Kudinovs and A. G. Ignatjuks, The Influence of Temperature on the Hydrolysis of Wood and Cellulose with Concentrated Hydrochloric Acid, 1951, vol. 2, pp. 68–82 Search PubMed.
- N. V. Lebedev, A. A. Bannikova and L. B. Paasikivi, Hydrolysis of wood with concentrated hydrochloric acid solutions at different temperatures, Sb. Tr., Gos. Nauchno-Issled. Inst. Gidroliz. Sul'fitno-Spirt. Prom-sti., 1961, 9, 20–35 CAS.
- N. V. Chalov, E. F. Goryachikh and A. E. Lashchuk, Hydrolysis of wood with concentrated hydrochloric acid, Gidroliz. Lesokhim. Prom-st., 1959, 12, 3–5 CAS.
- A. A. Petkevich, I. N. Ochneva, N. V. Korotkov, E. D. Revzina, N. V. Chalov, A. E. Leshchuk and E. F. Goryachikh, Testing of a new hydrolysis of wood with concentrated hydrochloric acid in a pilot battery of diffusors, Sb. Tr., Gos. Nauchno-Issled. Inst. Gidroliz. Sul'fitno-Spirt. Prom-sti., 1960, 8, 47–65 CAS.
- N. V. Chalov and L. B. Paasikivi, Hydrolysis of lignocellulose with 38-41% hydrochloric acid at 20 °C, State Research Institute of Hydrolysis and Sulfite-Alcohol Industry, 1964, 7 Search PubMed.
- A. E. Leshchuk and N. V. S. Chalov, Penetration of concentrated hydrochloric acid into the pores of wood particles and the formation of hydrolyzates within the particles, Gosudarstvennyi Nauchno-issledovatel'skii Institut Gidroliznoi i Sul'fitnospirtovoi Promyshlennosti, 1966, p. 15 Search PubMed.
- E. L. Glazkova and A. I. Kunina, Effect of Temperature on the Extraction of Prehydrolysis Products from Lignocellulose Chips, 1974, 27, pp. 12–13 Search PubMed.
- N. V. Chalov and L. B. Passikivi, Hydrolysis of polysaccharides of pinewood with 38-41% hydrochloric acid at 20 °C, Gosudarstvennyi Nauchno-issledovatel'skii Institut Gidroliznoi i Sul'fitnospirtovoi Promyshlennosti, 1962, p. 35 Search PubMed.
- A. E. Leshchuk and N. V. Chalov, Continuous hydrolysis of plant tissue with 45-48% hydrochloric acid. Equilibrium in the system polysaccharides-hydrolysis products-hydrochloric acid, Gosudarstvennyi Nauchno-issledovatel'skii Institut Gidroliznoi i Sul'fitnospirtovoi Promyshlennosti, 1965, vol. 18, pp. 10–13 Search PubMed.
- N. V. Chalov, A. E. Leshchuk and I. V. Zemel, Equilibrium state in the system cellulose-hydrogen chloride-water-hydrolysis products, Sb. Tr., Vses. Nauchno-Issled. Inst. Gidroliza Rastit. Mater., 1968, 17 Search PubMed.
- N. V. Korotkov, E. L. Glazkova, N. V. Chalov and L. Y. Konovalova, Selection of an optimal scheme of hydrochloric acid recovery from wood hydrolysates, Sbornik trudov VNIIGS, 1964, vol. 13, pp. 203–218 Search PubMed.
- I. Y. Marone, G. I. Zaytseva, L. U. Stepchenkova, Z. A. Kozlova, N. Smirnova and O. M. Stukalina, Kinetics of Airdrying of Hydrochloric Acid Hydrolysate Mass, 1974, 27, pp. 21–22 Search PubMed.
- V. S. Mikhailovich, Improvement of Environmental Characteristics for the Processes of Plant Raw Materials Conversion, 1997 Search PubMed.
- I. Rafiqul, A. Sakinah and A. Zularisam, Hydrolysis of Lignocellulosic Biomass for Recovering Hemicellulose: State of the Art, in Waste Biomass Management – A Holistic Approach, 2017, pp. 73–106 Search PubMed.
- E. G. Locke and E. Garnum, Working party on wood hydrolysis, For. Prod. J., 1961, 11, 380 Search PubMed.
- F. Higgins and G. E. Ho, Hydrolysis of cellulose using HCl: A comparison between liquid phase and gaseous phase processes, Agric. Wastes, 1982, 4(2), 97–116 CrossRef CAS.
- T. Kobayashi, Advances in Technique of Wood Saccharification, J. Agric. Chem. Soc. Jpn., 1956, 30(3), 30–40 Search PubMed.
- Food and Agriculture Organization (FAO), An International Review of Forestry and Forest Products, Unasylva, 1956, 10(1) Search PubMed.
- R. H. Farmer, Chemistry in the Utilization of Wood: Pergamon Series of Monographs on Furniture and Timber, Elsevier, 2013 Search PubMed.
- J. Karchesy and P. Koch, Energy Production from Hardwoods Growing on Southern Pine Sites, U.S. Department of Agriculture (Forest Service), 1979 Search PubMed.
- R. Bolia, Overreliance on Technology in Warfare: the Yom Kippur War as a Case Study, Parameters, 2004, vol. 34, p. 12 Search PubMed.
- W. B. Quandt, Peace Process: American Diplomacy and the Arab-Israeli Conflict since 1967, Brookings Institution Press, 2010 Search PubMed.
- G. Covi, Puzzling Out The First Oil Shock. History, Politics and the Macroeconomy in a Forty-Year Retrospective, History of Economic Thought and Policy, 2015, 57–91 Search PubMed.
- V. Lauber, Political Economy of Renewable Energy, in International Encyclopedia of the Social Behavioral Sciences, Elsevier, 2015, pp. 367–373 Search PubMed.
- A. Shahbazi and B. Zhang, Chapter 5 – Dilute and concentrated acid hydrolysis of lignocellulosic biomass, in Bioalcohol Production, K. Waldron, ed. Woodhead Publishing, 2010, pp. 143–158 Search PubMed.
- P. Harmsen, S. Lips and R. Bakker, Pretreatment of Lignocellulose for Biotechnological Production of Lactic Acid, Food & Biobased Research, Wageningen UR, 2013, Rapport 1384 Search PubMed.
- N. A. Glaeser, Saccharification process using concentrated sulfuric acid, US Pat., US4663108, 1987 Search PubMed.
- N. A. Glaeser, H. W. Blanch and C. R. Wilke, Ion-exchange-based chromatographic separation of acid from sugar solutions, US Pat., US5338431, 1992 Search PubMed.
- Advance Local Media LLC, “Birmingham-based Masada Resource Group looks at global projects”, 2009, online, available, https://www.al.com/businessnews/2009/10/birmingham-based_masada_resour.html, accessed 22 July 2024.
- D. V. Watkins, “Masada Signs Contract with Canadian Firm for Clean Fuels Project in South Africa”, 2023, online, available, https://www.donaldwatkins.com/post/masada-signs-contract-with-canadian-firm-for-clean-fuels-project-in-south-africa, accessed 22 July 2024.
- BlueFire Renewables, Production Plant, online, available, https://bfreinc.com/, accessed 22 July 2024.
- J. W. van Groenestijn, J. H. O. Hazewinkel and R. R. Bakker, Pre-treatment of lingo-cellulose with biological acid recycling (the Biosulfurol process), Int. Sugar J., 2006, 689–692 Search PubMed.
- A. Baniel and R. Jansen, Method for the extraction and recovery of HCl from aqueous solutions, US Pat., US 7169320B2, 2007 Search PubMed.
- A. Baniel and A. Eyal, A process for the recovery of HCl from a dilute solution thereof and extractant composition for use therein, WO2009125400, 2009.
- N. Syzova, A. M. Eyal, A. Vitner and B. Hazan, Interactions between the Components of ABC Extractants and Extraction of Monocarboxylic Acids by These Extractants, Solvent Extraction and Ion Exchange, 2004, vol. 22, pp. 31–49 Search PubMed.
- M. Voith, HCL Cleantech Wins Venture Funding, 2009, online, available, https://cen.acs.org/articles/87/web/2009/06/HCL-Cleantech-Wins-Venture-Funding.html.
- C. Zimmerman, HCL CleanTech Changes Name, Announces Financing, 2012, online, available, https://energy.agwired.com/2012/03/06/hcl-cleantech-changes-name-announces-financing/.
- A. Eyal and R. P. Jansen, Lignocellulose conversion processes and products, US Pat., US9512495, 2016 Search PubMed.
- A. Eyal and R. P. Jansen, Methods and systems for processing a sucrose crop and sugar mixtures, US Pat., US10760138B2, 2020 Search PubMed.
- R. Jansen, P. Travisano, L. Madsen, N. Matis, J. Alan, N. Lapidot, A. Eyal, T. Allen, Z.-V. Belman, B. Hallac and M. Zviely, Methods for treating lignocellulosic materials, US Pat., US11053558, 2021 Search PubMed.
- J. Lane, Stora Enso acquires Virdia in (up to) 62M deal, 2014, online, available, https://www.biofuelsdigest.com/bdigest/2014/06/23/stora-enso-acquires-virdia-in-up-to-62m-deal/.
- I. Heiskanen, L. Axrup and R. Laitinen, Process for the production of a paper or board product and a paper or board product according to the process, WO2011056135A1, 2011.
- I. Heiskanen, L. Axrup, M.-A. Norborg and I. Knoos, Process for producing a dispersion comprising ananoparticles and a dispersion produced according to the process, US Pat., US9365978B2, 2013 Search PubMed.
- J. Kavakka and M. Granstrom, Method for treating lignocellulosic materials, WO2016125067A1, 2016.
- S. Enso, Stora Enso will close its Virdia operations in the United States, 29 January 2021, online, available, https://www.storaenso.com/en/newsroom/regulatory-and-investor-releases/2021/1/stora-enso-will-close-its-virdia-operations-in-the-united-states, accessed 24 July 2024.
- W. J. Wilson and B. S. Das, Production of alkali metal sulfites or bisulfites, US Pat., US3549319A, 1970 Search PubMed.
- Biofuels International, Weyland opens Norwegian 2G plant, 2010, online, available, https://biofuels-news.com/news/weyland-opens-norwegian-2g-plant/, accessed 13 August 2024.
- K. R. Weydahl, Method of production of alcohol, WO2010038021A3, 2010.
- K. R. Weydahl, Process for the production of alcohol, WO2010128272A1, 2010.
- T. van der Meulen, G. Fransson, L. Sundlof and J. Lindsedt, Apparatus for extraction of saccharides from lignocellulose material by means of hydrolysis and use of a certain material in the apparatus, WO2008108709A1, 2008.
- Sekab, The Biorefinery Process technology, 2024, online, available, https://www.sekab.com/en/this-is-how-it-works/biorefinery-demo-plant/our-process/, accessed 22 July 2024.
- L. Bjornsson, S.-E. Svensson, S. Monavari, E. Kreuger and G. Zacchi, Pretreatment of non-wood lignocelluiosic material, US Pat., US 8835156B2, 2014 Search PubMed.
- N. Data, Green Sugar AG, Meißen, Germany (liq), online, available, https://www.northdata.com/Green+Sugar+AG,+Mei%C3%9Fen/Amtsgericht+Dresden+HRB+35748, accessed 06 August 2024.
- G. Sugar, Alles aus (Grün)zucker: Produktinnovationen aus Biomasse, 2008 Search PubMed.
- M. Schmidt and W. Reschetilowski, Extraktionsreaktor zur Hydrolyse von pflanzlichen Rohstoffen, EP1878480B1, 2008.
- F. Kose and M. Schmidt, Modular system for the customer-oriented design of hydrolysis reactors, WO2015136044A1, 2015.
- M. Schmidt, Verbesserter Extraktionsreaktor zur Hydrolyse von pflanzlichen Rohstoffen, DE102008036213A1, 2010.
- M. Schmidt, Verfahren zur Hydrolyse von pelletierfähigen Biomassen mittels Halogenwasserstoffsäuren, DE102012020166A1, 2012.
- F. Kose and M. Schmidt, Verfahren und Apparatur zur Entfernung von Halogenwasserstoff und Wasser aus Hydrolysaten, DE102010033925A1, 2011.
- F. Kose, Die folgenden Angaben sind den vom Anmelder eingereichten Unterlagen entnommen, DE102006032600A1, 2008.
- Insolvenz Portal, Instellung des Geschäftsbetriebs der Green Sugar AG, 20 May 2020, online, available, https://app.insolvenz-portal.de/Nachrichten/einstellung-des-geschaeftsbetriebs-der-green-sugar-ag/21788, accessed 06 August 2024.
- A. S. Vaguerido De sousa Dias, G.-J. M. Gruter and R.-J. van Putten, Process for the conversion of a carbohydrate-containing feedstock, US Pat., US2013324708A1, 2013 Search PubMed.
- C. Muñoz de Diego, M. A. Dam and G.-J. M. Gruter, Method for the preparation of 2,5-furandicarboxylic acid and for the preparation of the dialkyl ester of 2,5-furandicarboxylic acid, WO2011043661A1, 2011.
- L. Sipos, G.-J. M. Gruter, J. Kolstad and M. A. Dam, A process for preparing a polymer product having a 2,5-furandicarboxylate moiety within the polymer backbone to be used in bottle, film or fibre applications, WO2013062408A1, 2013.
- B. McKay and G.-J. M. Gruter, Process for the Production of Solid Saccharides from Lignocellulosic Biomass, WO2016099272, 2016.
- B. McKay and G.-J. M. Gruter, Process for the Preparation of a Saccharide-Containing Solution from a Torrefied Cellulosic Biomass, WO2016099273, 2016.
- Avantium, Avantium DAWN technology, 2018, online, available, https://avantium.com/technologies/dawn/, accessed 13 August 2024.
- B. McKay and G.-J. M. Gruter, Process for the Preparation of a Saccharide-containing Solution from a Torrefied Cellulosic Biomass, WO2016099273(A1), 2016.
- S. E. M. Selke, Kinetics of Hydrogen Fluoride Saccharification of Cellulose, PhD thesis, Michigan State University, 1984 Search PubMed.
- R. H. Bischof, J. Ramoni and B. Seiboth, Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei, Microb. Cell Factories, 2016, 15, 106 CrossRef PubMed.
- C. E. Wyman, Twenty Years of Trials, Tribulations, and Research Progress in Bioethanol Technology: Selected Key Events Along the Way, in Applied Biochemistry and Biotechnology, Humana Press, 2001, pp. 5–21 Search PubMed.
- S. Parekh and C. P. Felice, Methods and systems for saccharification of biomass, US Pat., US8563277B1, 2013 Search PubMed.
- J. S. White, K. A. Leiper, M. Tangney and S. Messenger, Process for the Manufacture of Butanol or Acetone, US Pat., US11046977B2, 2021 Search PubMed.
- Celtic Renewables, Biorefinery, 2024, online, available, https://www.celtic-renewables.com/biorefinery/, accessed 24 July 2024.
- R. L. Blackbourn and P. R. Weider, Treating biomass to produce materials useful for biofuels, WO2012061596A1, 2012.
- T. P. Binder, P. D. Bloom, P. H. Doane and C. Ma, Method of producing sugars using a combination of acids to selectively hydrolyze hemicellulosic and cellulosic materials, US Pat., US8057770B2, 2011 Search PubMed.
- M. D. Boot, P. Kouris, E. J. M. Hensen and X. Huang, A method for obtaining a lignin oil composition using a compressing gas and acid assisted process, EP3798286A1, 2021.
- The world's first test road with lignin produced in the Netherlands, https://www.uu.nl/en/news/the-worlds-first-test-road-with-lignin-produced-in-the-netherlands.
- L. Shuai, M. Talebi Amiri, Y. M. Questell-Santiago, F. Héroguël, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization, Science, 2016, 354(6310), 329–333 CrossRef CAS PubMed.
- M. T. Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Fractionation of lignocellulosic biomass to produce uncondensed aldehyde-stabilized lignin, Nat. Protoc., 2019, 14, 921–954 CrossRef PubMed.
- S. Kilambi, Solvo-thermal hydrolysis of xylose, US Pat., US 8119823, 2009 Search PubMed.
- S. Kilambi and K. L. Kadam, Solvo-thermal fractionation of biomass, US Pat., US 8282738, 2010 Search PubMed.
- J. Buffiere, P. Ahvenainen, M. Borrega, K. Svedström and H. Sixta, upercritical water hydrolysis: a green pathway for producing low-molecular-weight cellulose, Green Chem., 2016, 18, 6516–6525 RSC.
- D. V. Kazachkin, M. Colakyan and F. J. Moesler, Supercritical hydrolysis of biomass, WO2014012030A1, 2014.
- Renmatix, What We Do, Renmatix, 2024, online, available, https://renmatix.com/plantrose-process/, accessed 24 July 2024.
- J. Bueno Morón, F. Arbore, G. P. M. van Klink, M. Mascal and G.-J. M. Gruter, Industrial Routes from Sugars and Biomass to CMF and other 5-(Halomethyl)furfurals, ChemSusChem, 2024, 17, e202400495 CrossRef PubMed.
- W. N. Haworth and W. G. M. Jones, The conversion of sucrose into furan compounds. Part I. 5-Hydroxymethylfurfuraldehyde and some derivatives, J. Chem. Soc., 1944, 667–670 RSC.
- M. Mascal, High-yield conversion of cellulosic biomass into furanic biofuels and value-added products, US Pat., US20090234142A1, 2009 Search PubMed.
- M. Mascal and E. B. Nikitin, Dramatic Advancements in the Saccharide to 5-(Chloromethyl)furfural Conversion Reaction, ChemSusChem, 2009, 2(9), 859–861 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, Towards the Efficient, Total Glycan Utilization of Biomass, ChemSusChem, 2009, 2(5), 423–426 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, Direct, High-Yield Conversion of Cellulose into Biofuel, Angew. Chem., 2008, 120, 8042–8044 CrossRef.
- M. Mascal and E. B. Nikitin, Co-processing of Carbohydrates and Lipids in Oil Crops To Produce a Hybrid Biodiesel, Energy Fuels, 2010, 24, 2170–2171 CrossRef CAS.
- J. Bueno Moron, G. van Klink and G.-J. M. Gruter, Production and Downstream Integration of 5-(Chloromethyl)furfural from Lignocellulose, ACS Sustainable Chem. Eng., 2023, 11(49), 17492–17509 CrossRef CAS PubMed.
- J. Bueno Moron, G.-J. M. Gruter and G. P. M. van Klink, Method of extracting 5-Chloromethylfurfural with an organic solvent from cellulosic fibers and man-made non-cellulosic fibers hydrolysed together with hydrochloric acid, WO2023166122A1, 2023.
- J. Bueno Moron, G. P. M. van Klink and G.-J. M. Gruter, From rags to riches: Converting cellulose containing waste to 5-(chloromethyl)furfural (CMF), Waste Management Bulletin, 2024, 2(3), 58–68 CrossRef.
- E. Growth, Micromidas: Transforming a College Project into an Industry Leading Bio-Tech Company, 2013, online, available, https://earlygrowthfinancialservices.com/blog/micromidas-transforming-a-college-project-into-an-industry-leading-bio-tech-company/.
- M. Masuno, J. Bissell, R. L. Smith, B. Higgins, B. A. Wood and M. Foster, Utilizing a multiphase reactor for the conversion of biomass to produce substituted furans, WO2012170520A1, 2012.
- M. N. Masuno, J. Bisell, R. L. Smith, B. Higgins and A. B. Wood, Utilizing a multiphase reactor for the conversion of biomass to produce substituted furans, US Pat., US 2019/0270717 A1, 2019 Search PubMed.
- C. Anderson, West Sacramento startup secures 40 million investment, plus alliance with Nestlé, Danone, 2017, online, available, https://www.sacbee.com/news/business/biz-columns-blogs/cathie-anderson/article138360048.html.
- Origin Materials, Artius Announces Expected Closing of Business Combination with Origin Materials, 2021, online, available, https://investors.originmaterials.com/news-releases/news-release-details/artius-announces-expected-closing-business-combination-origin, accessed 24 July 2024.
- Avantium, Avantium and Origin Materials to Accelerate the Mass Production of FDCA and PEF for Advanced Chemicals and Plastics, 2023, online, available, https://avantium.com/avantium-and-origin-materials-to-accelerate-the-mass-production-of-fdca-and-pef-for-advanced-chemicals-and-plastics/.
- Origin Materials, Origin Materials, Inc. Reports Operating and Financial Results for First Quarter, 2024, online, available, https://investors.originmaterials.com/news-releases/news-release-details/origin-materials-inc-reports-operating-and-financial-results-2, accessed 24 July 2024.
- M. Mascal, 5-(Chloromethyl)furfural is the New HMF: Functionally Equivalent But More Practical in Terms of its Production From Biomass, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS PubMed.
- M. Mascal, 5-(Chloromethyl)furfural (CMF): A Platform for Transforming Cellulose into Commercial Products, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CrossRef CAS.
- C. Moretti, R. Hoefnagels, M. van Veen, B. Corona, S. Obydenkova, S. Russell, A. Jongerius, I. Vural-Gürsel and M. Junginger, Using lignin from local biorefineries for asphalts: LCA case study for the Netherlands, J. Clean. Prod., 2022, 343, 131063 CrossRef CAS.
- N. Leenders, R. M. Moerbeek, M. J. Puijk, R. J. Bronkhorst, J. Bueno Morón, G. P. M. van Klink and G.-J. M. Gruter, “Polycotton waste textile recycling by sequential hydrolysis and glycolysis”, Nat. Commun., 2025 DOI:10.1038/s41467-025-55935-6 , accepted..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00600c |
| This journal is © The Royal Society of Chemistry 2025 |