
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the relevance of inhomogeneity in saturated fatty acid compositions for the crystallization kinetics of fat blends†
Julia
Seilert
 *a,
Tim
Hoffmann
a,
Michael
Rappolt
b and
Eckhard
Flöter
a
*a,
Tim
Hoffmann
a,
Michael
Rappolt
b and
Eckhard
Flöter
a
aDepartment of Food Process Engineering, Technische Universität Berlin, Straße des 17. Juni 135, Berlin 10623, Germany. E-mail: julia.seilert@tu-berlin.de
bSchool of Food Science and Nutrition, Food Colloids and Bioprocessing Group, University of Leeds, LS2 9JT, UK
First published on 13th May 2025
Abstract
In this contribution, the crystallization kinetics of model fat blends containing trisaturated and mono-unsaturated TAGs of different saturated fatty acid composition are investigated via differential scanning calorimetry (DSC) and time-resolved small and wide angle X-ray scattering (SAXS/WAXS). The effect of inhomogeneity in saturated fatty acid composition was studied by varying between palmitic and stearic sources for trisaturated (POSt – palm stearin and FHRO – fully hydrogenated rapeseed oil) and using palm mid fraction (PMF) and shea stearin (ShSt) sources for mimicking the mono-unsaturated TAGs (PMF – palm mid fraction and ShSt – shea stearin). The kinetics were followed via SAXS/WAXS upon cooling at different rates (1 and 10 °C min−1) and during the isothermal hold at 0 °C (273 K) for 60 min. The blends revealed a multiple step crystallization with co-existing polymorphic forms and a slow restructuring process. Generally, mono-unsaturated triglycerides tended to impede the polymorphic transition of the trisaturated fraction. The trisaturated triglyceride fraction formed an α phase at the beginning of the crystallization. Quickly after, co-existing β′ and β phases were recorded. In each case, the subsequent crystallization events were dictated by the mono-unsaturated fraction with ShSt affecting the crystallization more drastically than PMF. Differences were diminished when a low cooling rate was applied. Further, the low cooling rate allowed the formation of a β phase in FHRO-containing samples before an additional β′ phase was recorded. The differences in explicit fatty acid composition, TAG diversity and minor components are discussed as primary potential sources of differences in crystallization behavior. Calorimetric experiments capturing the crystallization and melting behavior of the industrial grade binary blends were compared to that of blends based on pure TAGs diluted in rapeseed oil, namely the combinations PPP–POP, SSS–SOS, PPP–SOS, and SSS–POP.
1. Introduction
The demand of finding suitable alternatives for palm oil (PO) in fat continuous products (e.g., margarine and shortenings) satisfying product and process qualities remains an ongoing challenge.1 PO is a rather cost-effective natural fat and the only substantial vegetable source of natural high melting triglycerides (e.g., tripalmitin, PPP). Substituting other tropical fats like shea stearin or cocoa butter for PO or reengineering PO fractions using hydrogenated oils and other oil modification techniques pose new challenges as both pathways generate different triglyceride (TAG) compositions. This affects the melting and crystallization characteristics2 and influences the granular crystal formation.3 Those granular crystals typical for palm oil have been shown to be composed of trisaturated triglycerides-rich mixed crystal in their most stable polymorphic form β and mono-unsaturated TAGs as surrounding layer.4 Even though PO has been studied intensively, the interplay of different TAGs – trisaturated and mono-unsaturated – during crystallization still remains insufficiently understood. To deepen this understanding is not only beneficial for the application of PO or its fractions but can also be instrumental for the design of alternative structuring fat compositions. Important parameters to be considered in this context are the differences between palmitic and stearic acid and the interaction between fully saturated and mono-unsaturated TAGs.Reviewing the crystallization literature reveals that simply translating observations from broadly studied PO5–9 including the formation of molecular compounds10,11 into stearic-acid counterparts is not straightforward. Attempts to compare blends of similar composition of trisaturated (high-melting) and mono-unsaturated (low-melting) fractions but different fatty acids (palmitic versus stearic acid) revealed significant differences. These occur in nucleation kinetics, heterogeneous crystallization and fractionation in the stearic blends versus mixed crystal formation in the palmitic blends.12 Even though little differences in the solid fat content profiles were found, the isothermal crystallization data indicate that PPP was able to facilitate the crystallization of POP, whereas no such effect could be found for the stearic-based blends. Also, PPP crystallizes faster in the presence of POP than SSS in the presence of SOS. Although no XRD studies were performed in the aforementioned reference, the difference in interaction might be explained by the lattice structure the pure components assume. Monoacid saturated TAGs (like PPP and SSS) assume the 2L stacking irrespective of polymorphic form. The molecular stacking of mono-unsaturated (symmetric) TAGs as POP and SOS depends on the chain lengths and the polymorphic form.13 POP assumes 3L in the most stable polymorphic form β, whereas a 2L stacking was reported for both β′ and the least stable form α.14 For SOS, on the other hand, a 2L stacking was only observed for the α1-form.15 Possibly, this relates to the seeding function of PPP reported by Vereecken et al. The term “seeding agent” might be displaced in this context assuming a seed is of similar molecular arrangement, i.e., polymorphic form13 and lamellar stacking.16 Hence, the seeding effects described in their study indicate a tendency to form mixed crystals between PPP and POP. Only a limited number of studies investigated potential mixed crystal formation between SSS and SOS.17,18 It was reported that mixed crystal formation in SSS–SOS systems is limited, most prevalent in metastable forms. Generally, the time it takes to reach the equilibrium state,19,20i.e., demixed phases, is subject to a multitude of kinetic effects. For example, Lutton stated that a stabilized solid solution of SSS–SOS during melting initially releases low-melting SOS. During subsequent stabilization (i.e., annealing) the low-melting SOS was only re-integrated into the mixed crystal/solid solution to a limited extent. As a consequence, an exclusively SOS containing phase was formed.17
As indicated above, SSS does not facilitate the crystallization of SOS in the same way PPP does for POP. However, SSS still acts as crystallization surface, i.e., heterogeneous nucleation. Co et al.18 investigated the heterogeneous crystallization of SOS on SSS. It was reported that a small amount of SSS (1–2%) crystallizes initially. This crystalline SSS phase resulted in increased nucleation rates and reduced induction times for the crystallization of an SOS solid phase.
Overall, data on blends of mixed fatty acid systems are scarce. For the binary system PPP–SOS monotectic behavior was reported.19 Even though separate crystallization for both components can be assumed, the kinetics of concurrent crystallization are less clear. A study on the mixture of a saturated monoacid TAG (trilaurin) and the molecular compound forming mixture of SOS and SSO (MCSOS/SSO) suggested interactive crystallization21 of both components due to close melting point temperatures. Further, the polymorphic transition from one component was facilitated by the presence of the other. No data on mixtures of SSS and POP could be found.
Besides competitive, interactive, and concurrent polymorphic crystallization of pure triglycerides mixtures, impurities in form of diglycerides and free fatty acids also affect the crystallization kinetics. This was shown in studies on purified shea stearin22 and other polar components on industrial grade fats23 including diglycerides.24,25
In this contribution, an attempt to deepen the understanding of the crystallization of fats containing trisaturated (high-melting) and symmetric mono-unsaturated triglycerides (mono-unsaturated) is documented. Pseudo-binary blends were composed of palm stearin (POSt) and palm mid fraction (PMF) or fully hydrogenated canola oil (FHRO) and shea stearin (ShSt) all diluted in rapeseed oil. These blends contain either palmitic (P) or stearic (S) acid as dominant fatty acid (homogeneous blends). Additionally, mixed fatty acid (i.e., inhomogeneous) blends were composed of POSt–ShSt and FHRO–PMF mixtures, respectively. These contain palmitic and stearic acid in either the high melting or low melting fraction. The industrial grade fat blends were investigated by time-resolved SAXS/WAXS. The crystallization kinetics and polymorphic pathways are related to the inhomogeneity of the saturated fatty acid composition (stearic versus palmitic acid). The crystallization and melting behavior of the pseudo-binary blends (POSt–PMF, FHRO–ShSt, POSt–ShSt, and FHRO–PMF) was compared to that of blends based on pure TAGs diluted in rapeseed oil, namely the combinations PPP–POP, SSS–SOS, PPP–SOS, and SSS–POP, captured by DSC. The true binary blends contain the dominating TAGs in the industrial grade blends elucidating possible differences between industrial and academic studies. The differences in explicit fatty acid composition, TAG diversity and minor components are discussed as primary potential sources of differences in crystallization behavior.
2. Materials and methods
2.1 Materials
Fully hydrogenated rapeseed oil (FHRO) was kindly provided by ADM (Hamburg, Germany). Palm stearin with an iodine value of 12 (POSt), a palm mid fraction with an iodine value of 33 (PMF), and shea stearin (ShSt) were provided by AAK (Aarhus, Denmark). The diglyceride content is 3.9%, 0.8%, 2.9%, and 1.4%, respectively. Refined rapeseed oil (RP) was contributed by Gustav Heess GmbH (Leonberg, Germany). All materials were used without further modification. Pure triglycerides – tristearin (SSS), tripalmitin (PPP), 1,3-palmitin-2-olein glycerol (POP), and 1,3-stearin-2-olein glycerol (SOS), all >99% – were acquired from Larodan AB (Solna, Sweden).2.2 Fat blend formulation
In this study, the fat blends were formulated such that the amount of trisaturated triglycerides was 5% (w/w) and the amount of mono-unsaturated triglycerides was 15% (w/w). This makes a total content of structuring triglyceride groups of 20%. The general composition and temperature range was chosen so that it represents crystallization conditions and formulation of spreadable fats. The raw materials were mixed and diluted with rapeseed oil to reach the desired concentration. Sources for trisaturated triglycerides were FHRO (91.3% stearic acid and 8.7% non-stearic acid) and POSt (6.8% stearic acid and 93.2% palmitic acid in the trisaturated TAGs fraction). Sources for mono-unsaturated triglycerides were ShSt and PMF. Note that POSt also contains about 16.8% mono-unsaturated TAGs of which the saturated fatty acids are composed of 92.3% palmitic and 7.7% stearic acid. The triglyceride composition of FHRO was derived from the fatty acid profile of rapeseed oil according to Coleman26 and accounting for hydrogenation. Information of the triglyceride profiles of POSt, PMF, and ShSt were provided by the supplier. Table 1 gives the P/S distribution in the trisaturated TAG and the mono-unsaturated TAG fraction of all blends.| Blend | POSt–PMF | POSt–ShSt | FHRO–PMF | FHRO–ShSt |
|---|---|---|---|---|
| Trisaturated TAGs (5%) | ||||
| % P | 93.3 | 89.3 | 13.1 | 4.5 |
| % S | 6.7 | 10.7 | 86.9 | 95.5 |
| % other saturated fatty acids | 0 | 0 | 0.9 | 1.0 |
| Mono-unsaturated TAGs (15%) | ||||
| % P | 87.8 | 15.2 | 88.0 | 9.1 |
| % S | 11.1 | 82.2 | 11.4 | 88.2 |
| % other saturated fatty acids | 1.1 | 2.6 | 0.6 | 2.7 |
All blends were prepared by melting the materials completely and stirred before blending to ensure homogeneity. All blends were stored in the refrigerator and remelted before each experiment.
2.3 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, with 2θ being the scattering angle). The 1D scattering patterns were recorded with a Mythen microstrip X-ray detector (Dectris Ltd, Baden, Switzerland). Each fat blend sample was melted on a hot plate prior to injection into quartz disposable capillaries (Capillary Tube Supplies Ltd, Cornwall, UK) with an outside diameter of 1.5 mm and subsequently sealed with wax. SAXS/WAXS measurements were performed using the following temperature protocol: first, the samples were heated to 70 °C and held for 10 min to erase any crystal memory. Then, the samples were cooled via the Peltier element to 0 °C at a rate of 10 or 1 °C min−1. The samples were held isothermally for 60 min. Afterwards, the samples were melted at a scan rate of 2 °C min−1. The X-ray pattern were collected during the cooling phase, the isothermal holding time, and melting. Since no continuous data collection during cooling and heating was possible, i.e., a step-wise temperature-time profile mimicking the continuous cooling was applied, see our previous manuscript for details.27,28 During the isothermal phase exposure times of 30 s were applied every minute, guaranteeing an optimum counting rate.
sinθ/λ, with 2θ being the scattering angle). The 1D scattering patterns were recorded with a Mythen microstrip X-ray detector (Dectris Ltd, Baden, Switzerland). Each fat blend sample was melted on a hot plate prior to injection into quartz disposable capillaries (Capillary Tube Supplies Ltd, Cornwall, UK) with an outside diameter of 1.5 mm and subsequently sealed with wax. SAXS/WAXS measurements were performed using the following temperature protocol: first, the samples were heated to 70 °C and held for 10 min to erase any crystal memory. Then, the samples were cooled via the Peltier element to 0 °C at a rate of 10 or 1 °C min−1. The samples were held isothermally for 60 min. Afterwards, the samples were melted at a scan rate of 2 °C min−1. The X-ray pattern were collected during the cooling phase, the isothermal holding time, and melting. Since no continuous data collection during cooling and heating was possible, i.e., a step-wise temperature-time profile mimicking the continuous cooling was applied, see our previous manuscript for details.27,28 During the isothermal phase exposure times of 30 s were applied every minute, guaranteeing an optimum counting rate.
SAXSTreat software (Anton Paar GmbH, Graz, Austria) and SAXSQuant software (Anton Paar GmbH, Graz, Austria) were used for setting the primary beam position to zero and normalizing for X-ray flux fluctuation and sample transmission, respectively. The normalized pattern of an empty capillary was subtracted from each normalized sample pattern. Additionally, the scattering contribution from molten triglycerides was subtracted from the total pattern:
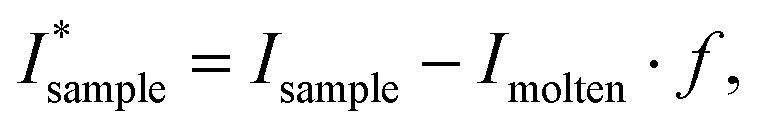 | (1) |
 | (2) |
 denotes the signal intensity corrected for the liquid contribution and Imolten denotes the signal intensity obtained for the molten sample. Here, a signal obtained without any solid material present, yet, close to the crystallization temperature of 0 °C was chosen (here: POSt–ShSt at 5 °C after cooling with a rate of 10 °C). Note, that the actual molten phase fraction is estimated in the region from q = 9.4 to 10.2 nm−1 (eqn (2)), where all X-ray scattering pattern only display diffuse scattering. Further, due to the line collimation focus of the instrument, the resulting patterns are smeared, mostly affecting the small-angle region, approx. from q = 0 to q = 2.5 nm−1 (first order reflection). This affected peak fitting greatly. Further, some peaks showed shoulders and asymmetrical shapes indicating two phases present at the same time. Therefore, d-spacings from the first order reflection were determined from the second derivative method29 as some peaks showed a shoulder indicating two phases present. Note, this second derivative method results in ‘2nd derivative peaks’ that have roughly half the width of the original peaks, i.e., spanning from inflection to inflection point of the recorded diffraction peak. The first order reflections were used to determine the d-spacings, which were additionally corrected for peak smearing.27,28 Further, peak analysis was performed in the WAXS region. The degree of crystallinity was determined by integrating the WAXD intensities,
denotes the signal intensity corrected for the liquid contribution and Imolten denotes the signal intensity obtained for the molten sample. Here, a signal obtained without any solid material present, yet, close to the crystallization temperature of 0 °C was chosen (here: POSt–ShSt at 5 °C after cooling with a rate of 10 °C). Note, that the actual molten phase fraction is estimated in the region from q = 9.4 to 10.2 nm−1 (eqn (2)), where all X-ray scattering pattern only display diffuse scattering. Further, due to the line collimation focus of the instrument, the resulting patterns are smeared, mostly affecting the small-angle region, approx. from q = 0 to q = 2.5 nm−1 (first order reflection). This affected peak fitting greatly. Further, some peaks showed shoulders and asymmetrical shapes indicating two phases present at the same time. Therefore, d-spacings from the first order reflection were determined from the second derivative method29 as some peaks showed a shoulder indicating two phases present. Note, this second derivative method results in ‘2nd derivative peaks’ that have roughly half the width of the original peaks, i.e., spanning from inflection to inflection point of the recorded diffraction peak. The first order reflections were used to determine the d-spacings, which were additionally corrected for peak smearing.27,28 Further, peak analysis was performed in the WAXS region. The degree of crystallinity was determined by integrating the WAXD intensities,  (eqn (1)), from q = 12 to q = 18 nm−1. A B-spline interpolation and its first derivative were applied achieving turnover curves.30 Here, peaks indicate changes in the polymorphic state. Both, liquid phase subtraction and peak analysis was performed in Matlab2020B and OriginLab2022b, respectively.
(eqn (1)), from q = 12 to q = 18 nm−1. A B-spline interpolation and its first derivative were applied achieving turnover curves.30 Here, peaks indicate changes in the polymorphic state. Both, liquid phase subtraction and peak analysis was performed in Matlab2020B and OriginLab2022b, respectively.
3. Results
3.1 Small- and wide-angle X-ray scattering
Time-resolved SAXS/WAXS studies were performed in which the evolution of crystalline phases during cooling and the isothermal holding time was followed. The experiments were performed on all pseudo-pure samples of industrial grade, i.e., POSt–PMF, POSt–ShSt, FHRO–PMF, and FHRO–ShSt. The polymorphic forms were identified in the WAXS region (12 nm−1 < q < 17 nm−1). The long spacings were derived from the first order reflection in the SAXS region. Fig. 1 gives the SAXD and WAXD pattern for FHRO–ShSt crystallized at 1 and 10 °C min−1 as example. The pattern of the other samples can be found in the ESI.† Details on long and short spacings of some TAGs used as reference for this study can be found in the ESI.† In general, the long spacing are correlated with the carbon number, e.g., 40.6 and 45 Å for tripalmitin (PPP) and tristearin (SSS), respectively.31 Also, the long spacing decreases with increasing stability of the polymorph from α to β′ to β, if the lamellar stacking does not change, e.g., 50.6, 46.8, and 45 Å for SSS,31 in which all phases display a 2L-stacking. In the presence of unsaturated fatty acids, TAGs tend to assume either the 2L lamellar stacking in less stable polymorphic forms as POP or can quickly transition into the 3L structure as reported for SOS, see Table S2 (ESI†). | ||
| Fig. 1 SAXD and WAXD patterns after subtraction of the liquid contribution (obtained at 5 °C for POSt–ShSt cooled at 10 °C min−1) of POSt–ShSt during cooling at 1 °C min−1 (left) and 10 °C min−1 (right) to 0 °C and subsequent isothermal holding time of 60 min. 1st and 3rd order reflections in the SAXS region and reflections in the WAXS region are indicated as identified. CR = cooling rate. | ||
 | ||
| Fig. 2 Long spacings and polymorphs as identified per time frame acquired during cooling at 10 °C min−1 (filled symbols) to 0 °C and during the subsequent isothermal holding time of 60 min. The start of the isothermal period was set to be zero min. Polymorphs as identified from WAXS are color-coded: α (orange), β′ (blue), and β polymorph (green). Overlapping regions indicate co-existing crystalline phases. | ||
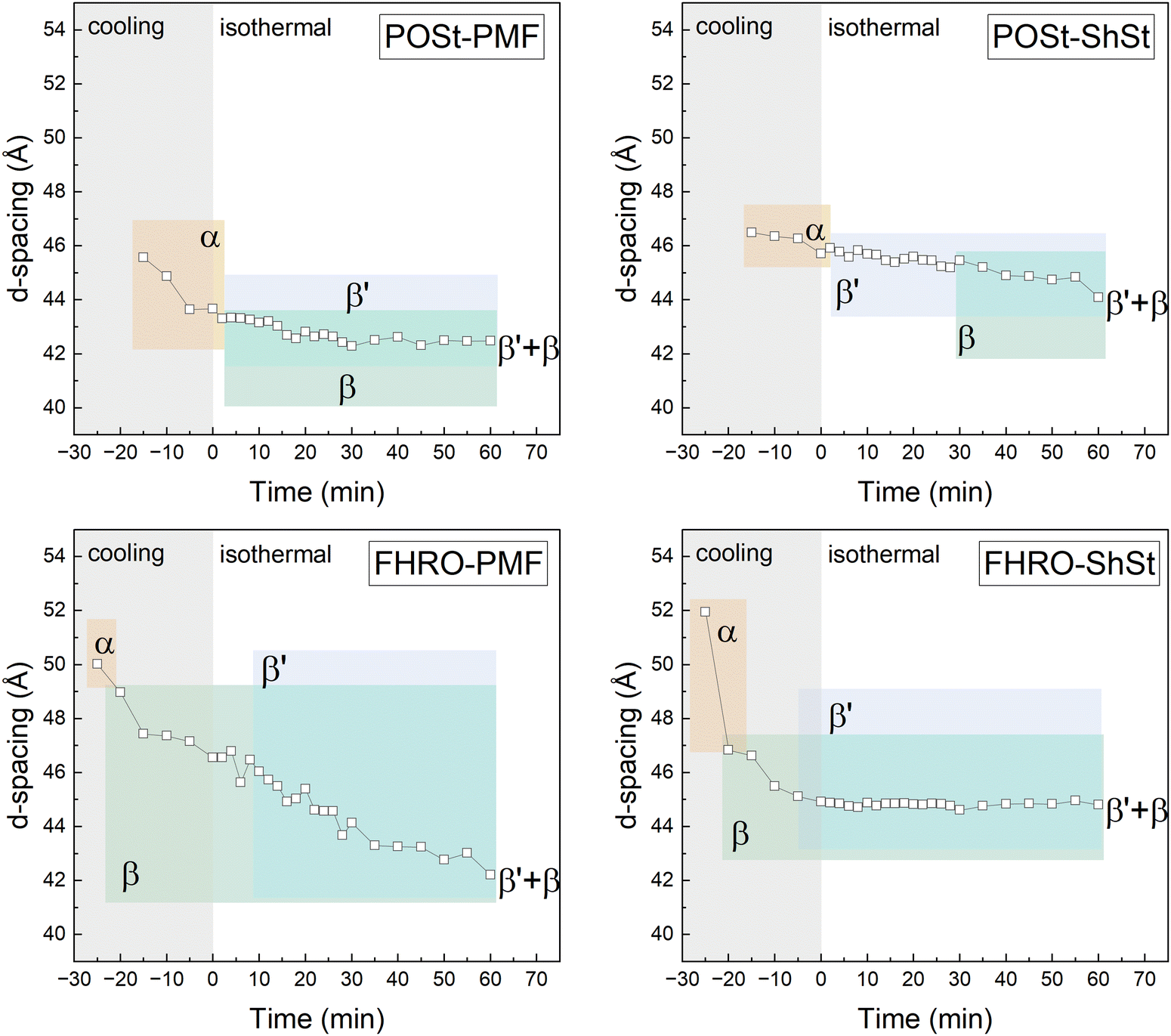 | ||
| Fig. 3 Long spacings and polymorphs as identified per time frame acquired during cooling at 1 °C min−1 (empty symbols) to 0 °C and during the subsequent isothermal holding time of 60 min. The start of the isothermal period was set to be zero min. Polymorphs as identified from WAXS are color-coded: α (orange), β′ (blue), and β polymorph (green). Overlapping regions indicate co-existing crystalline phases. | ||
| Blend | Cooling rate (°C min−1) | Time/temperature | Polymorphs | Long spacing (Å) | Short spacings (Å) |
|---|---|---|---|---|---|
| POSt–PMF | 10 | 0 min/0 °C | α | 47.0 | 4.17 |
| 60 min/0 °C | β′ + β | 42.1 | 4.62, 4.34, 3.88 | ||
| 1 | −15 min/15 °C | α | 45.6 | 4.14 | |
| 60 min/0 °C | β′ + β | 42.5 | 4.62, 4.34, 3.90 | ||
| POSt–ShSt | 10 | 0 min/0 °C | α | 47.6 | 4.17 |
| 60 min/0 °C | β′ + β | 45.2 | 4.56, 4.32, 4.15, 3.87 | ||
| 1 | −15 min/15 °C | α | 46.5 | 4.19 | |
| 60 min/0 °C | β′ + β | 44.1 | 4.58, 4.35, 4.17, 3.87, 3.81 | ||
| FHRO–PMF | 10 | −1 min/10 °C | α | 51.3 | 4.17 |
| 60 min/0 °C | β′ | 50.6, 42.1 | 4.41, 4.25, 3.90 | ||
| 1 | −25 min/25 °C | α | 50.0 | 4.19 | |
| 60 min/0 °C | β′ + β | 42.2 | 4.55, 4.53, 4.16, 3.92 | ||
| FHRO–ShSt | 10 | −1 min/10 °C | α | 51.8 | 4.16 |
| 60 min/0 °C | β′ | 50.1 | 4.32, 4.15, 3.89 | ||
| 1 | −25 min/25 °C | α | 52.0 | 4.16 | |
| 60 min/0 °C | β′ + β | 44.8 | 4.58, 4.32, 4.14, 3.88 | ||
In each experiment (cooling rate of 10 °C min−1 and 1 °C min−1), an α-phase was formed at the onset of crystallization. When cooled at 10 °C min−1, the POSt-containing systems (i.e., system that is dominated by the crystallization PPP) show a short-lived α phase, which is followed by a β′ phase occurring early in the isothermal holding phase. Both systems further exhibit a β phase starting from 40 min (POSt–PMF) and 20 min (POSt–ShSt). The FHRO-containing samples also a short-lived α and a β′ phase of prolonged stability. Contrary to the POSt-containing blends, the FHRO-containing blends show no traces of a β phase at the end of the isothermal holding time. A subsequent heating scan applying a scan rate of 2 °C min−1 revealed that a β phase fully formed early during heating (data shown in the ESI,† Fig. S7). Further, the first order reflection of FHRO–PMF showed a shoulder, to which two d-spacings were assigned, indicating two co-existing β′ phases.
When cooled at 1 °C min−1, again at first an α phase is formed which is followed by a β′ phase in POSt-containing systems. However, polymorphic pathways differ slightly. POSt–PMF exhibits co-existing β′ and β phases shortly after the α phase has vanished. In the POSt–ShSt system, a β phase was only observed from 30 min onwards. In contrast, the FHRO-containing samples all show a β phase very early on, at the beginning of the isothermal phase and in the cooling phase. The transition into β is followed by the formation of an β′ phase early during the isothermal hold that remains co-existing with the earlier formed β-phase throughout the experiment. This hints towards the fraction of the trisaturated TAGs (here FHRO) undergoing their preferred polymorphic phase transition unperturbed by the presence of mono-unsaturated TAGs.
Overall, when a high cooling rate is applied POSt-based samples show a β phase earlier than their FHRO-based counterparts. This observation is reversed when a low cooling rate is applied. The FHRO-based blends, however, show only little sensitivity to the change of the fatty acid in the mono-unsaturated TAG fraction for any cooling rate. The POSt-based samples are more sensitive to the added mono-unsaturated TAG fraction. More precisely, it influences the occurrence of the β-phase. This occurs earlier in mixture with ShSt than PMF when cooled at 10 °C min−1, yet, this observation is reversed once a low cooling rate of 1 °C min−1 is applied.
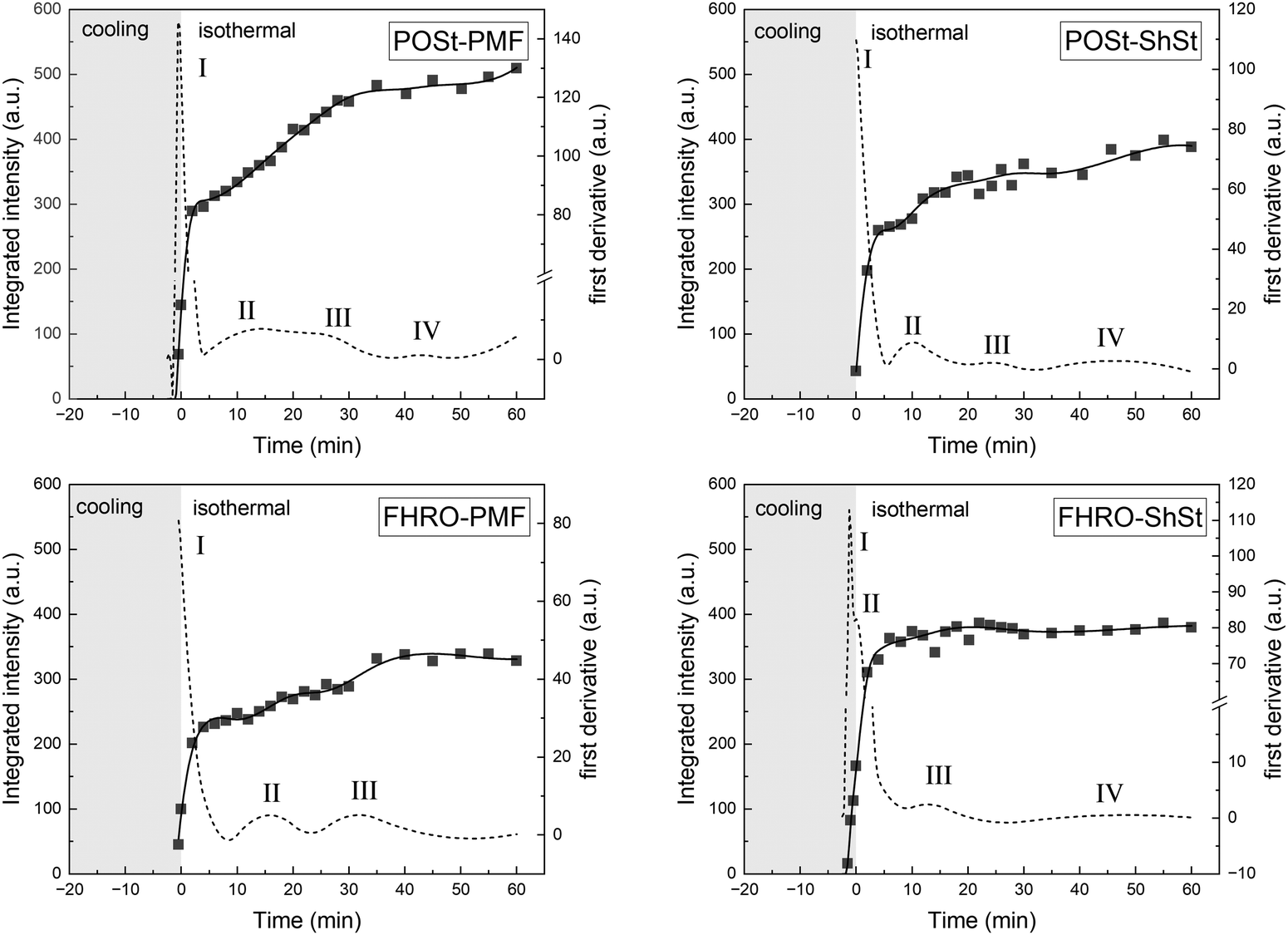 | ||
| Fig. 4 Integrated WAXD intensity per time frame obtained during cooling at 10 °C min−1 to 0 °C and during the subsequent isothermal holding time of 60 min. The start of the isothermal period was set to be zero min. The original data are given as symbols, a spline fit is given as solid line, and the first derivative of the integrated intensity is given as dashed line. | ||
 | ||
| Fig. 5 Integrated WAXD intensity per time frame obtained during slow cooling at 1 °C min−1 to 0 °C and during the subsequent isothermal holding time of 60 min. The start of the isothermal period was set to be zero min. The original data are given as symbols, a spline fit is given as solid line, and the first derivative of the integrated intensity is given as dashed line. | ||
All turnover curves show four transitions except for FHRO–PMF cooled at 10 °C min−1. The first transition is in line with a swift crystallization of an α phase from the liquid. The following transitions, II, III, and IV either mark the occurrence of a more stable polymorphic form (β′ or β) from the liquid or a transition from an existing phase and in some cases mark a change in the d-spacing (001), but no additional polymorphic form was identified. A summary is given in Table 3.
| Sample | 10 °C min−1 | 1 °C min−1 | |
|---|---|---|---|
| n.d. = not detected. | |||
| POSt–PMF | I | Liquid → α | Liquid → α |
| II | → β′ | → β′/β | |
| III | → β′; decrease in d-spacing | → β′/β | |
| IV | → β | Decrease in d-spacing | |
| POSt–ShSt | I | Liquid → α | Liquid → α |
| II | → β′ | Decrease in d-spacing | |
| III | → β | → β′ | |
| IV | Increase in d-spacing with β′ + β co-existing | → β | |
| FHRO–PMF | I | Liquid → α | α → β |
| II | → β′ | → β′ | |
| III | → β′; second d-spacing emerges | → β′ | |
| IV | n.d. | Increase in d-spacing with β′ + β co-existing | |
| FHRO–ShSt | I | Liquid → α | Liquid → α |
| II | → β′ | → β | |
| III | → β′; slight drop in d-spacing | → β′ | |
| IV | Increase in d-spacing | Increase in d-spacing with β′ + β co-existing | |
For both, FHRO–PMF and FHRO–ShSt, a third transition (Table 3) can be linked to a second d-spacing emerging. In the FHRO–PMF blend, this was observed as shoulder in the 001 reflection. On the contrary, the FHRO–ShSt did not show a shoulder but a broader peak region presumably due to two peaks merged that are very close in their peak center. This was documented by a slight drop in d-spacing in Fig. 2.
Albeit the signal intensity was very low given the amount of crystalline material at experiment conditions, no evidence of a γ-phase or a sub-α phase or any phase in 3L stacking was found, which are reported for mono-unsaturated TAGs such as SOS15 and POP.14 It is necessary to discuss the short spacings and their assignment to the polymorphic phases. Short spacings in the range of 4.1–4.2 Å are typical for an α phase but were also reported for some β′ phase, e.g., for POP (in β′2-2L) and SOS (β′-3L) (see Table S2 in the ESI†). Unambiguous assignments of polymorphic states in complex fat systems on the basis of pure component data is challenging. The low amount of crystalline material in the present study resulted in a low signal intensity in the q-range of the 3rd order reflection which allows to distinguish two phases of different composition (here: α and β′ potentially co-existing). In these dilute systems, an α-phase that remains stable for 60 min of isothermal crystallization at 0 °C in addition to the time during cooling seems rather unlikely. Further, in some cases, a reflection in this region was observed after the first α phase transitioned into a β-phase (see Fig. 1, CR 1 °C min−1). In these cases, additional low-melting material is unlikely to form an α-phase given the low supercooling and pre-crystallized material acting as heterogeneous surface. Therefore, the aforementioned short spacings were assigned to a β′ phase at the end of isothermal holding phase. This could be further validated by the turnover curves and the observed transitions (i.e., referring to the peaks of the first derivative curves given in Fig. 4 and 5).
Overall, the integrated intensities in the WAXD pattern reveal that all samples restructure and undergo multiple transitions during the isothermal hold. When a cooling rate of only 1 °C min−1 is applied, the samples can already crystallize during the cooling process depending on the respective dissolution temperature. This is well documented in Fig. 5. As the data suggest, all samples start crystallizing more than 20 min before the start of the isothermal period. Here, the PMF-containing samples take longer to crystallize. In contrast, the samples containing ShSt reach the plateau of maximum crystallinity already early in the isothermal period, yet, the turnover curves reveal further transitions to take place.
3.2 Isothermal crystallization via DSC
Calorimetric experiments were performed on the industrial grade blends (POSt–PMF, FHRO–ShSt, POSt–ShSt, and FHRO–PMF) and their binary counterparts (PPP–POP, SSS–SOS, PPP–SOS, and SSS–POP). Each blend contains 5% trisaturated TAGs (binary: PPP or SSS) and 15% mono-unsaturated TAGs (binary: POP or SOS) in rapeseed oil. The isothermal DSC signals obtained during cooling at 10 °C min−1 and 1 °C min−1 and isothermal holding time are given in Fig. 6 and 7, respectively. The DSC signal of rapeseed oil was subtracted for better visualization of the switch from cooling to isothermal phase. The raw data are given in the ESI† (Fig. S1 and S2). Table 4 summarizes the occurrence of the crystallization events.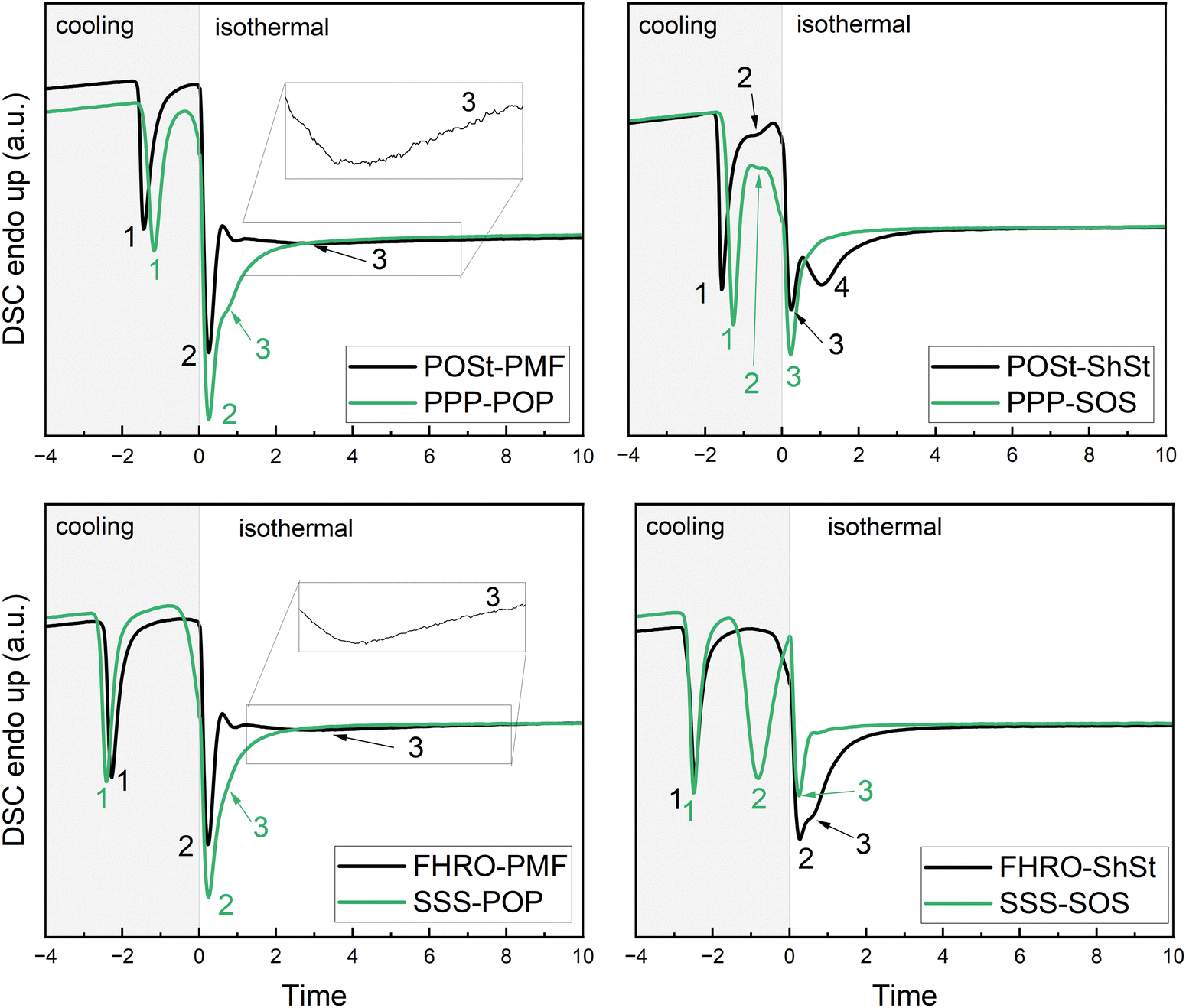 | ||
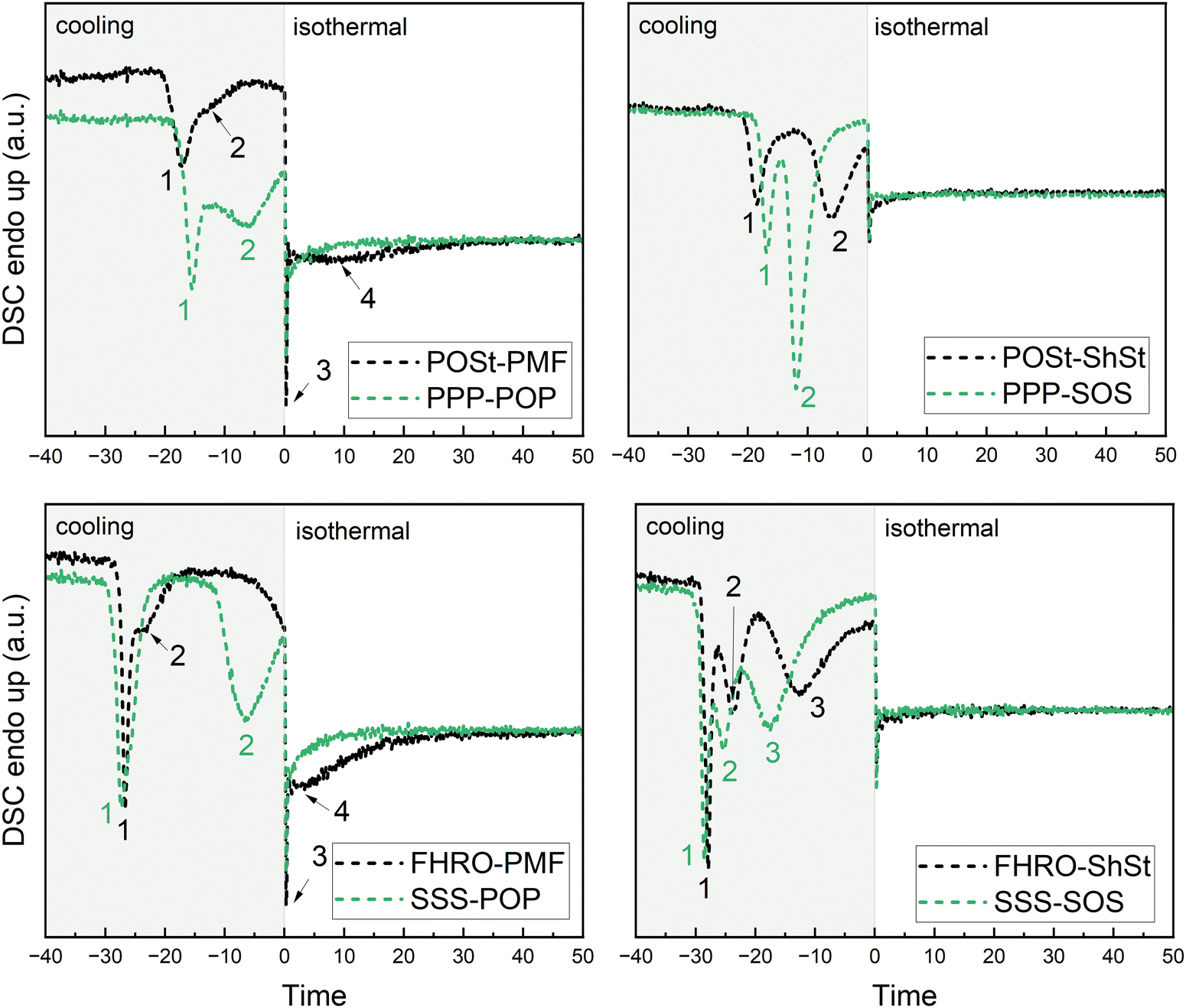
| Fig. 6 DSC signal obtained during cooling at 10 °C min−1 and isothermal holding time of 60 min. Time zero is set to the starting point of isothermal phase. Pseudo-binary (black), and true-binary blends (green) are presented. For clarity around the switch of cooling to isothermal phase, the signal obtained for rapeseed oil not showing any exothermic nor endothermic event was subtracted. The numeration of exothermic events relates to Table 4. | ||
 | ||
| Fig. 7 DSC signal obtained during cooling at 1 °C min−1 and isothermal holding time of 60 min. Time zero is set to the starting point of isothermal phase. Pseudo-binary (black), and true-binary (green) blends are presented. For clarity around the switch of cooling to isothermal phase, the signal obtained for rapeseed oil not showing any exothermic nor endothermic event was subtracted. The numeration of exothermic events relates to Table 4. | ||
| Cooling rate | Event # | 10 °C min−1 | 1 °C min−1 | Cooling rate | Event # | 10 °C min−1 | 1 °C min−1 |
|---|---|---|---|---|---|---|---|
| Sample | Time (min) | Time (min) | Sample | Time (min) | Time (min) | ||
| n.d. = not determined | |||||||
| POSt–PMF | 1 | −1.4 | −17.3 | PPP–POP | 1 | −1.2 | −15.5 |
| 2 | 0.3 | −11.8 | 2 | 0.2 | −6.5 | ||
| 3 | 2.9 | 0.3 | 3 | 0.8 | n.d. | ||
| 4 | n.d. | 9.8 | |||||
| POSt–ShSt | 1 | −1.6 | −18.6 | PPP–SOS | 1 | −1.3 | −16.8 |
| 2 | −0.6 | −6.1 | 2 | −0.6 | −11.9 | ||
| 3 | 0.2 | n.d. | 3 | 0.2 | n.d. | ||
| 4 | 1.1 | n.d. | SSS–POP | ||||
| FHRO–PMF | 1 | −2.3 | −26.7 | 1 | −2.4 | −27.4 | |
| 2 | 0.2 | −23.6 | 2 | 0.3 | −6.5 | ||
| 3 | 2.9 | 0.3 | 3 | 0.8 | n.d. | ||
| 4 | n.d. | 4.5 | |||||
| FHRO–ShSt | 1 | −2.5 | −28.1 | SSS–SOS | 1 | −2.5 | −28.5 |
| 2 | 0.3 | −24.2 | 2 | −0.9 | −25.3 | ||
| 3 | 0.6 | −13.0 | 3 | 0.2 | −17.6 | ||
Generally, in all blends the crystallization is dominated by the trisaturated fraction, i.e., FHRO and POSt, and only mildly affected by the mono-unsaturated fraction, i.e., PMF and ShSt. When cooled at 10 °C min−1, the time of the first crystallization event differs slightly, when either ShSt or PMF are added to POSt, i.e., appearing at −1.6 and −1.4 min, respectively. For FHRO these events occur earlier at −2.5 and −2.3 min, respectively. Differences can be observed for the second crystallization event. This event is only slightly affected, when PMF is added to either POSt or FHRO, but more so, when ShSt is added. In this case, two crystallization events are visible after reaching the isothermal phase. The two merged exothermic events are closer in the blend with FHRO than in the blend with POSt.
Comparison of the true binary blends to the commercial counterparts reveals similar crystallization pathways. The third (and fourth) crystallization events are most different in all blend comparisons: the first event occurs at around the same time as in the pseudo-binary blends, however, the third one occurs much faster in the PPP–POP than in the POSt–PMF blends, i.e., at 0.8 versus 2.9 min. A similar effect of sample purity can be observed when comparing SOS and ShSt. Interestingly, mixing POP with either PPP or SSS does not dramatically change the crystallization events. However, when mixing SOS with either PPP or SSS, a substantial difference is observed. This is in line with the observations made for the industrial grade blends. The second event is barely visible in the PPP–SOS mixture. It must be noted that the nature of the second event remains unclear from DSC data alone. It is possible to interpret this as the polymorphic transition of the source for trisaturated TAGs or the crystallization of mono-unsaturated TAGs in a lesser stable polymorph followed by a rapid transition into a more stable crystalline phase (third event). This, however, is only valid when we assume completely separate crystallization, i.e., fractionation.
When crystallizing the blends at a cooling rate of 1 °C min−1 almost all crystallization events occur during the cooling phase with exception of the PMF/POP dominated blends. In general, the number of events and the interplay of the source of mono-unsaturated TAGs with the source of trisaturated TAGs are similar to the ones described for 10 °C min−1. When applying a low cooling rate, the effect of mono-unsaturated TAGs on the initial crystallization (first event) of trisaturated TAGs becomes more apparent. When adding ShSt to either POSt or FHRO, the first event is recorded about a minute earlier than when adding PMF. Also, the POSt-blends crystallize faster than the true binary counterparts containing solely PPP. This can be attributed to the purity of POSt which contains also stearic acid (6.7%, see Table 1). Noteworthy is also a small shoulder (marked as second event) for POSt–PMF and FHRO–PMF directly following the first crystallization events which is not observed in the true binary blends.
Similarly to cooling at 10 °C min−1, the PMF/POP-blends exhibit a smeared out third exothermic event during the isothermal phase. In this subset of blends, FHRO–PMF shows this event earlier than POSt–PMF, at 4.5 versus 9.8 min (see Table 4). The third crystallization event observed in FHRO–ShSt when cooled at 1 °C min−1 is not found in POSt–ShSt. It seems reasonable to assume the presence of a third event also for POSt–ShSt following the interpretation for 10 °C min−1. This also holds for the true binary blends. As for all blends and cooling rates, the crystallization is facilitated in the true binary blends of highest purity.
3.3 Melting properties
Fig. 8 displays the melting curves obtained after crystallization at a cooling rate of 10 °C min−1 for the pseudo-binary blends and the accompanying binary blends. | ||
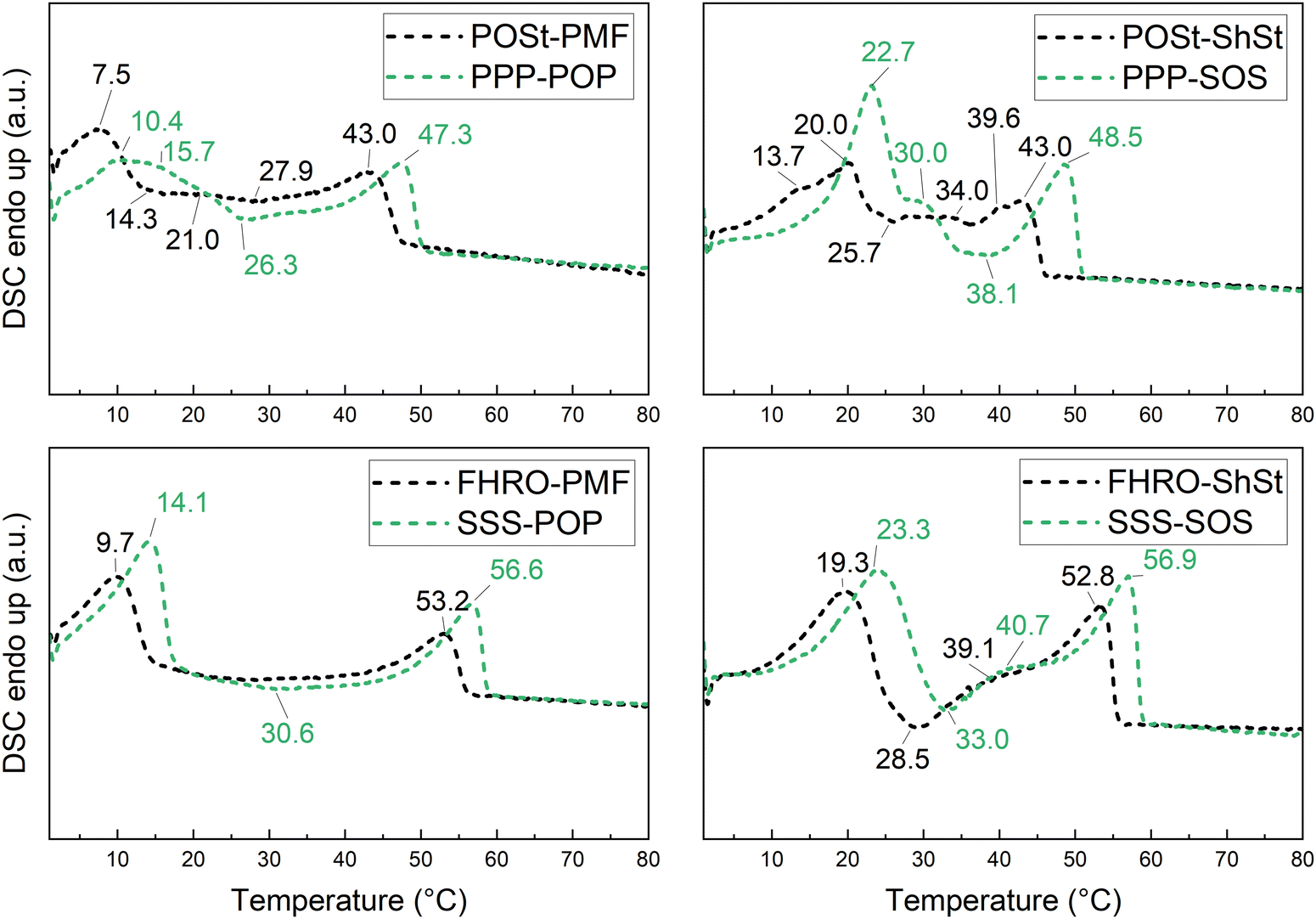
| Fig. 8 DSC melting curves for the pseudo-binary (black) and true binary blends (green). Crystallized to 0 °C at 10 °C min−1, isothermal period of 60 min. Heating rate of 2 °C min−1. Peak temperatures in °C are indicated. | ||
After cooling at a rate of 10 °C min−1 all blends exhibit a high-melting peak and a low-melting region. The high-melting peak can be assigned to the dominant fraction of trisaturated TAGs recrystallizing into the highest stable polymorphic form. The low-melting region is characterized by recrystallization during melting as for FHRO–PMF and FHRO–ShSt or relates to multiple melting events as for POSt–PMF and POSt–ShSt. The binary blends PPP–POP, SSS–POP, and SSS–SOS show similar melting behavior as the pseudo-binary blends. The comparison of pseudo-binary to binary blends illustrates that higher dissolution temperatures are consistently found for binary TAG-based blends. The most pronounced difference is observed for POSt–ShSt versus PPP–SOS. Here, the high-melting peak of POSt–ShSt shows as two merged peaks (at 40.9 and 43 °C) in contrast to the single high-melting peak found for PPP–SOS.
Fig. 9 gives the melting curves obtained after cooling at a rate of 1 °C min−1.
 | ||
| Fig. 9 DSC melting curves for the pseudo-binary blends (black) and true binary blends (green). Crystallized to 0 °C at 1 °C min−1, isothermal period of 60 min. Heating rate of 2 °C min−1. Peak temperatures in °C as indicated. | ||
As observed for the melting curves obtained after cooling at a higher rate, all blends show a high-melting peak and a low-melting region characterized by multiple melting events and recrystallization, e.g., observed for FHRO–ShSt. It must be noted that all melting curves were obtained after an isothermal holding period of 60 min following the cooling step. This means that during cooled at 1 °C min−1 from 80 °C to 0 °C crystallization already occurs during the cooling process, see Fig. 5. This becomes apparent when comparing the low-melting regions which can be assigned to the mono-unsaturated TAG fraction. In Fig. 8, cooling at 10 °C min−1, at least two melting events in addition of a recrystallization event can be clearly seen for FHRO–PMF (at 11.2 and 23 °C) and FHRO–ShSt (at 17.1 and 26.3 °C), whereas in Fig. 9 (cooling rate of 1 °C min−1) the recrystallization during melting, if present at all, cannot clearly be assigned. However, in the blends containing ShSt, one could argue that the events at 25.7 °C in POSt–ShSt and at 28.5 °C in FHRO–ShSt are related to recrystallization. Also, at the low cooling rate the difference between POSt–ShSt and PPP–SOS (industrial versus pure) remains most pronounced. Even the double high-melting peak is still present, though less pronounced and at 39.6 °C and 40.9 °C.
Interestingly, the melting curves are very similar for PPP–SOS and SSS–SOS, so SOS-dominating binary systems irrespective of the cooling rate applied. The low-melting peak temperatures in particular are very similar. This indicates that the crystallization of SOS is only mildly affected by the presence of trisaturated TAGs either PPP or SSS. The POP-based binary systems, PPP–POP and SSS–POP show differences in the low-melting region, e.g., peak shape, when comparing the different cooling rates. The greatest difference is observed in the binary system SSS–POP which shows a recrystallization during melting when crystallized at 10 °C min−1. The pseudo-binary blends are more sensitive to differences in cooling rate than their binary blend counterparts especially in the low-melting region. Overall it has to be noted that the relatively complicated melting patterns found, particularly in PMF/POP containing systems, are observed in both industrial grade materials and the pure TAG systems.
4. Discussion
4.1 Effect of cooling rate on the crystalline phases
X-ray diffraction experiments were performed on all pseudo-pure samples of industrial grade, i.e., POSt–PMF, POSt–ShSt, FHRO–PMF, FHRO–ShSt. When cooled at a high cooling rate of 10 °C min−1, all samples showed a short-lived α phase. Differences in its stability was only observed at a low cooling rate. This could not be detected with the DSC measurements, where differences between the blends POSt–ShSt and FHRO–ShSt after the first crystallization event were only minute. When a slow cooling rate of 1 °C min−1 was applied, a separate crystallization of trisaturated TAGs was observed. This is illustrated by the pronounced crystallization peaks observed in Fig. 7 during cooling and the initial d-spacings shown in Fig. 3. The polymorphic pathways clearly show that in the blends FHRO–PMF and FHRO–ShSt a β phase is detected before an additional β′ phase is observed. This indicates a polymorphic phase transition involving trisaturated TAGs and TAGs originating from either PMF or ShSt. In the latter, thus a strictly S-containing system, this happens faster than in the inhomogeneous FHRO–PMF system. The additional crystallization of a β′ phase can be related to the slow decrease in d-spacing. Here, the turnover curves reveal more than one transition following the appearance of a β phase (Fig. 5). Both phases, β′ and β, are dominated by the mono-unsaturated TAGs. This behavior is also observed in the POSt–PMF system – a strictly P-based system. When cooled slowly, first an α phase crystallizes. A β follows that co-exists with the β′ phase until the isothermal period is completed. The POSt–ShSt system behaved differently. Here, the growth of the β phase was observed later compared to the other blends at slow cooling. Yet, the turnover curves suggest an additional transition between the occurrence of β′ and β. Nevertheless, at the end of the crystallization process, two co-existing phases, β′ and β, were found irrespective of the applied cooling rate for most blends. For FHRO–PMF and FHRO–ShSt crystallized at a high cooling rate of 10 °C min−1 a β phase could only form during the melting. Here, the high cooling rate resulted in supersaturation of multiple crystalline phases of different composition that inhibited the transition into a β phase. The data gathered confirm that slow cooling rates favor phase separation because high-melting material is subjected to supersaturation for elongated periods.4.2 Effect of the TAG type on the polymorphic transition and mixed crystal formation
Overall, the initial occurrence of an α phase was not affected by the TAG type of the mono-unsaturated TAG fraction (PMF or ShSt) neither for POSt nor for FHRO (Fig. 3). This is in line with the minor differences observed in the isothermal DSC signals (Fig. 6). The observation suggests that initially a separate crystallization of the trisaturated TAGs is monitored. Further on, multiple crystallization events (at least three) could be identified. These are related to the step-wise decrease in d-spacing over time and the transitions identified in the turnover curves (Fig. 4).Trisaturated and mono-unsaturated TAGs differ profoundly in their polymorphic crystallization behavior. Monoacid trisaturated TAGs undergo a rapid polymorphic transition into β when crystallizing on their own. This also goes for systems dominated by PPP and SSS, such as POSt32,33 and FHRO.27 POP and SOS exhibit a richer polymorphic crystallization behavior. For both, multiple polymorphic forms were reported. Further, the lamellar stacking changes with the polymorphic form. While POP assumes the 2L stacking even in metastable states, for SOS also a α-3L15 form, and for both, a γ-3L form were reported.34 No 3L stacking was observed for the pseudo binary blends under the experimental conditions of this study. This eludes to an interactive crystallization in early stage crystallization that entraps the mono-unsaturated TAGs in a 2L structure and at least delays transformation into 3L. Interactive polymorphic transitions including promoting and inhibitory effects are known. For example, the metastable phase β′ of PPP can be triggered when adding mixed-acid saturated TAGs, e.g., PPP mixed with PSP35 and POP as mono-unsaturated TAG36 linked to mixed crystal formation, i.e., co-crystallization. Other trisaturated TAGs remain in a metastable phase β′ longer due to chain length mismatches such as PPS, PSS.37 POSt and FHRO used in this study contain small amounts of other saturated fatty acids (see Table 1). In mixtures, structural differences of the composing TAG molecules can alter the polymorphic pathway. Another study of our group showed that the impurities of FHRO did not inhibit the formation of a β phase, also no β′ phase was detected.27 Mixed crystals in binary mixtures have been observed for PPP–POP with a POP content exceeding 50%36 and temperature-dependent for SSS–SOS17 and SSS–SSO.38 However, we do note the limited information on the lamellar stacking (2L or 3L) of mixed or separate phases in existing studies on the phase behavior of the dominant TAGs in this study. In some instances, e.g., FHRO–PMF crystallized at 10 °C min−1, we observed two co-existing β′ phases with different d-spacings (see Fig. 2).
Components of relatively similar melting points assuming the same lamellar stacking have been reported to influence each other's polymorphic pathway, e.g., in a more recent study, the presence of the molecular compound of SOS/OSO (MCSOS/OSO) facilitated the polymorphic transition of trilaurin (LaLaLa) from β′ into β2. This was described as interactive polymorphic crystallization between LaLaLa and the MCSOS/OSO.21
Further, the tendency of pure components to assume a certain lamellar stacking in a polymorphic state influences mixed crystal formation. For example, SSS–SOS exhibit even in metastable forms only limited miscibility.17,18 This can be attributed to the mismatch in lamellar stacking, i.e., SSS assuming the 2L stacking SOS preferring the 3L stacking.15 Similarly, despite proximity of the melting points, for PPP–SOS monotectic behavior was reported.19,38 Little is known about SSS and POP, both favor 2L staking with the exception of POP in the β-form. The pure component behavior of the TAGs in this study suggests limited miscibility over the concentration range. Hence, sequential crystallization and polymorphic transition next to partial miscibility can be assumed. Following this, the trisaturated TAG fraction poses as heterogeneous nucleation site, i.e., crystallization surface, for the mono-unsaturated TAG fraction. Since the crystallization surface is of different composition than the outer layer this is not to be confused with the seeding effect. The presence of trisaturated TAGs clearly inhibited the mono-unsaturated TAG fraction to assume their preferred polymorphic states in their preferred stacking as no 3L reflections were found. It also becomes apparent that mono-unsaturated TAGs affect the polymorphic pathways of trisaturated TAGs differently, e.g. ShSt added to POSt furthered the occurrence of a β phase contrary to PMF added to POSt when cooled at 10 °C min−1. This was reversed when cooled at 1 °C min−1. Further, limited supersaturation and competing crystallization seems to prevent mono-unsaturated TAGs to fully segregate into 3L assuming phases. Here one could argue that when crystals of miscible TAGs crystallize separately on top of each other due to process-induced fractionation, a TAG exchange (diffusion) process takes place. This could hence generate a mixed crystal.
4.3 Effect of the inhomogeneity in saturated fatty acid composition
The data gathered confirm that the impurities present in industrial fats slow down crystallization. This was earlier shown by others.22 Especially diglycerides have profound effects on fat crystallization serving as nucleation site for heterogenous crystallization.24,25 The material used in this study came with little diglyceride content and considering the dilution factors, it is reasonable to disregard the effects of diglycerides here. The inhomogeneity in saturated fatty acid composition that is inherent to the industrial material slows down crystallization. Furthermore, it was found that the disturbance caused by different saturated fatty acids in the trisaturated and mono-unsaturated TAG fraction is stronger, if longer fatty acids are present in the mono-unsaturated TAG fraction. This was most evident for the inhomogeneous blends, POSt–ShSt and FHRO–PMF, and their respective binary replicates. Concluding, the calorimetric study revealed differences related to the fatty acid composition of the trisaturated and mono-unsaturated TAGs. The comparison to mixtures of pure TAGs diluted in rapeseed oil showed only minor differences.
The greatest difference between industrial and academic blends was observed for POSt–ShSt versus PPP–SOS. POSt–ShSt shows as two merged high-melting peaks (at 40.9 and 43.0 °C) in contrast to the single high-melting peak observed for the PPP–SOS blend. This is most likely accounted for by the deviation form exclusively palmitic acid as constituent of the trisaturated TAG fraction of POSt (Table 1) and the presence of impurities such as diglycerides. These differences diminish with lower cooling rates (1 °C min−1). After slow cooling, the blends POSt–ShSt and FHRO–ShSt show comparable melting behavior. This indicates that the crystallization of ShSt is more dependent on the supersaturation and ultimate time given for crystallization rather than the material it crystallizes on.
The pseudo-binary blends are more sensitive to differences in cooling rate than their binary blend counterparts especially in the low-melting region. For ShSt this can be related to the presence of SLS (1,3-distearoyl-2-linoleoyl-sn-glycerol) known to co-crystallize with SOS.39 Additionally, the stronger influence of cooling rate on the crystallization and melting of the industrial grade blends is assumed to relate the inherent presence of minor components.
Again, despite recrystallization events that were observed during melting (especially in the low-melting region), SAXS data showed no occurrence of a 3L structure during melting (data not shown). This indicates that the mono-unsaturated TAG fraction remained in the 2L stacking throughout the experiment.
5. Conclusion
In this study, the crystallization pathways of fat blends of defined TAG composition were evaluated in regards to the saturated fatty acid composition and its effect on recrystallization, polymorphic transition and fractionation, i.e., separate crystallization. Blends of homogeneous distribution of saturated fatty acids, POSt–PMF and FHRO–ShSt, and inhomogeneous distribution, POSt–ShSt and FHRO–PMF were studied using DSC and time-resolved SAXS/WAXS. The raw materials were from industrial origin. Additionally, a calorimetric study was performed on these pseudo-binary blends and their binary counterparts, i.e., mixtures of pure TAGs diluted in rapeseed oil, namely PPP–POP, SSS–SOS, PPP–SOS, and SSS–POP.All industrial grade blends showed lower melting points than their binary counterparts due to inhomogeneities in their saturated fatty acid profiles. For all mixtures, regardless of sample purity, the crystallization appears to be initially dominated by the trisaturated TAG fraction, that crystallizes into an α phase. The effect of the mono-unsaturated TAG fraction on the initial crystallization was negligible. However, the later crystallization and melting events illustrate a clear effect of the mono-unsaturated TAG fraction. The influences were more pronounced in the ShSt-containing blends, FHRO–ShSt and POSt–ShSt. This was also confirmed by studying the blends containing pure TAGs.
SAXS/WAXS analyses revealed that the polymorphic pathways were mostly affected by the source of trisaturated TAGs, e.g., POSt versus FHRO, than the source of mono-unsaturated TAGs. Generally, the α phase was only observed for a short time, which was confirmed by the DSC data. The occurrence of a β phase differed greatly for the POSt- and FHRO-containing samples. Surprisingly, the FHRO-containing samples stayed in a metastable β′ form for a longer period. During slow cooling, FHRO transitioned separately from a short-lived α form directly into a β phase. This transition proceeded faster in the presence of ShSt than of PMF. Here, the mono-unsaturated TAGs slowed down the phase transition of the trisaturated TAGs in FHRO blends. At the end of the isothermal crystallization a β′ and β phase coexisted regardless of the cooling rate and blend composition. Throughout, no 3L structures were detected despite being typical for mono-unsaturated TAGs, especially in more stable polymorphic forms. Hence, in mixtures with trisaturated TAGs, mono-unsaturated TAGs are forced into the 2L stacking and appear to remain therein for a considerable period of time. This was further captured in the evolution of d-spacing and turnover curves, which revealed slow restructuring processes.
This study documents the phase transitions observed in blends of trisaturated and mono-unsaturated triglycerides of homogeneous and inhomogeneous saturated fatty acid composition. Overall, the crystallization processes involve changes of the polymorphic form, slow lamellar restructuring processes, and time-dependent changes in composition.
Data availability
Data for this article, including raw data of short- and wide angle X-ray scattering experiments and differential scanning calorimetry experiments, are available at Zenodo at https://doi.org/10.5281/zenodo.14680033.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our sincere appreciation to Jeppe Hjorth and team, from AAK (Aarhus, Denmark), for their expert assistance in performing the diglyceride analysis on our raw materials.References
- N. Hinrichsen, Commercially Available Alternatives to Palm Oil, Lipid Technol., 2016, 28(3–4), 65–67, DOI:10.1002/lite.201600018.
- C. Himawan, V. M. Starov and A. G. F. Stapley, Thermodynamic and Kinetic Aspects of Fat Crystallization, Adv. Colloid Interface Sci., 2006, 122(1–3), 3–33, DOI:10.1016/j.cis.2006.06.016.
- N. Viet, R. Tom, T. Vinh, D. Koen and B. Filip Van, Granular Crystals in Palm Oil Based Shortening/Margarine: A Review, Cryst. Growth Des., 2020, 20(2), 1363–1372 CrossRef.
- L. Tanaka, K. Tanaka, S. Yamato, S. Ueno and K. Sato, Microbeam X-Ray Diffraction Study of Granular Crystals Formed in Water-in-Oil Emulsion, Food Biophys., 2009, 4(4), 331–339, DOI:10.1007/s11483-009-9131-x.
- W. S. Kevin, Crystallization of Palm Oil and Its Fractions. In Crystallization processes in fats and lipid systems, ed. N. Garti, Dekker, New York, NY, 2001, pp. 357–380 Search PubMed.
- X. Zhang, L. Li, H. Xie, Z. Liang, J. Su, G. Liu and B. Li, Comparative Analysis of Thermal Behavior, Isothermal Crystallization Kinetics and Polymorphism of Palm Oil Fractions, Molecules, 2013, 18(1), 1036–1052, DOI:10.3390/molecules18011036.
- C. Sainlaud, O. Taché, F. Testard, J. Saiter, M. C. Bohin and G. Coquerel, Impact of Cooling Profile on Refined Palm Oil Crystallization: Microscopic and Small and Wide-Angle X-Ray Scattering Investigations, Eur. J. Lipid Sci. Technol., 2022, 124(2), 2100045, DOI:10.1002/ejlt.202100045.
- O. Zaliha, H. Elina, K. Sivaruby, A. R. Norizzah and A. G. Marangoni, Dynamics of Polymorphic Transformations in Palm Oil, Palm Stearin and Palm Kernel Oil Characterized by Coupled Powder XRD-DSC, J. Oleo Sci., 2018, 67(6), 737–744, DOI:10.5650/jos.ess17168.
- C. W. Chen, O. M. Lai, H. M. Ghazali and C. L. Chong, Isothermal Crystallization Kinetics of Refined Palm Oil, J. Am. Oil Chem. Soc., 2002, 79(4), 403–410, DOI:10.1007/s11746-002-0496-4.
- L. Bayés-García, T. Calvet, M. À. Cuevas-Diarte, S. Ueno and K. Sato, Phase Behavior of Binary Mixture Systems of Saturated-Unsaturated Mixed-Acid Triacylglycerols: Effects of Glycerol Structures and Chain-Chain Interactions, J. Phys. Chem. B, 2015, 119(12), 4417–4427, DOI:10.1021/acs.jpcb.5b00673.
- L. Bayés-García, J. Macridachis, T. Calvet and K. Sato, In Situ Analyses of Crystallization Behavior of 1,2,3-Tripalmitoyl Glycerol under Static and Dynamic Thermal Conditions, J. Therm. Anal. Calorim., 2024, 149, 5215–5227, DOI:10.1007/s10973-024-13196-3.
- J. Vereecken, I. Foubert, K. W. Smith and K. Dewettinck, Effect of SatSatSat and SatOSat on Crystallization of Model Fat Blends, Eur. J. Lipid Sci. Technol., 2009, 111(3), 243–258, DOI:10.1002/ejlt.200800150.
- K. Sato, Crystallization Behaviour of Fats and Lipids – a Review, Chem. Eng. Sci., 2001, 56, 2255–2265 CrossRef CAS.
- L. Bayés-García, T. Calvet, M. Àngel Cuevas-Diarte, S. Ueno and K. Sato, In Situ Observation of Transformation Pathways of Polymorphic Forms of 1,3-Dipalmitoyl-2-Oleoyl Glycerol (POP) Examined with Synchrotron Radiation X-Ray Diffraction and DSC, CrystEngComm, 2013, 15(2), 302–314, 10.1039/C2CE26522B.
- O. O. Mykhaylyk and I. W. Hamley, The Packing of Triacylglycerols from SAXS Measurements: Application to the Structure of 1,3-Distearoyl-2-Oleoyl- Sn-Glycerol Crystal Phases, J. Phys. Chem. B, 2004, 108(23), 8069–8083, DOI:10.1021/jp0379704.
- K. Smith, Cocoa Butter and Cocoa Butter Equivalents, in Structured and Modified Lipids, ed. F. Gunstone, Bedfordshire, 2001, pp. 401–422 Search PubMed.
- E. S. Lutton, Phase Behavior of Triglyceride Mixtures Involving Primarily Tristearin, 2-Oleyldistearin, and Triolein, J. Am. Oil Chem. Soc., 1955, 32(2), 49–53 CrossRef CAS.
- E. D. Co, S. M. Ghazani, D. A. Pink and A. G. Marangoni, Heterogeneous Nucleation of 1,3-Distearoyl-2-Oleoylglycerol on Tristearin Surfaces, ACS Omega, 2019, 4(4), 6273–6282, DOI:10.1021/acsomega.9b00147.
- L. H. Wesdorp, J. van Meeteren, S. Jon, R. Giessen, P. Overbosch, P. Grootscholten, M. Struik, E. Royers, A. Don and T. Loos, Liquid-Multiple Solid Phase Equilibria in Fats: Theory and Experiments, in Structure and Properties of Fat Crystal Networks, ed. A. G. Marangoni and L. H. Wesdorp, CRC Press, Boca Raton, 2013, pp. 241–418 Search PubMed.
- J. Macridachis-González, L. Bayés-García and A. T. Calvet, An Insight into the Solid-State Miscibility of Triacylglycerol Crystals, Molecules, 2020, 25(19), 4562, DOI:10.3390/molecules25194562.
- S. Yoshikawa, S. Watanabe, Y. Yamamoto, F. Kaneko and K. Sato, Interactive Polymorphic Crystallization Behavior in Eutectic Triacylglycerol Mixtures Containing Molecular Compound Crystals, Cryst. Growth Des., 2022, 22(3), 1753–1763, DOI:10.1021/acs.cgd.1c01340.
- J. Ray, K. W. Smith, K. Bhaggan, Z. K. Nagy and A. G. F. Stapley, Crystallization and Polymorphic Behavior of Shea Stearin and the Effect of Removal of Polar Components, Eur. J. Lipid Sci. Technol., 2013, 115(10), 1094–1106, DOI:10.1002/ejlt.201200434.
- K. W. Smith, K. Bhaggan, G. Talbot and K. F. van Malssen, Crystallization of Fats: Influence of Minor Components and Additives, J. Am. Oil Chem. Soc., 2011, 88(8), 1085–1101, DOI:10.1007/s11746-011-1819-7.
- W.-L. Siew and W.-L. Ng, Influence of Diglycerides on Crystallisation of Palm Oil, J. Sci. Food Agric., 1999, 79(5), 722–726, DOI:10.1002/(SICI)1097-0010(199904)79:5<722::AID-JSFA242>3.0.CO;2-W.
- I. F. De Oliveira, R. Grimaldi and L. Ap. G. Gonçalves, Effect of Diacylglycerols on Crystallization of Palm Oil (Elaeis Guineensis), Eur. J. Lipid Sci. Technol., 2014, 116(7), 904–909, DOI:10.1002/ejlt.201300231.
- M. H. Coleman, The Unsolved Problems of Triglyceride Analysis, J. Am. Oil Chem. Soc., 1965, 42 Search PubMed.
- J. Seilert, M. Rappolt, G. Dol and E. Flöter, Interplay of Polymorphic Transition and Mixed Crystal Formation in Model Fat Systems, Cryst. Growth Des., 2024, 24(3), 1146–1158, DOI:10.1021/acs.cgd.3c01164.
- J. Seilert, M. Rappolt, G. Dol and E. Flöter, Influence of Monounsaturated Triglycerides on the Crystallization Pathway of Fully Saturated Triglycerides, Cryst. Growth Des., 2024, acs.cgd.4c01138, DOI:10.1021/acs.cgd.4c01138.
- E. Simone, M. Rappolt, H. Ewens, T. Rutherford, S. Marty Terrade, F. Giuffrida and C. Marmet, A Synchrotron X-Ray Scattering Study of the Crystallization Behavior of Mixtures of Confectionary Triacylglycerides: Effect of Chemical Composition and Shear on Polymorphism and Kinetics, Food Res. Int., 2024, 177, 113864, DOI:10.1016/j.foodres.2023.113864.
- M. Rappolt and G. Rapp, Simultaneous Small- and Wide-Angle X-Ray Diffraction during the Main Transition of Dimyristoylphosphatidylethanolamine, Ber. Bunsenges. Phys. Chem., 1996, 100(7), 1153–1162, DOI:10.1002/bbpc.19961000710.
- E. S. Lutton, Triple Chain-Length Structures of Saturated Triglycerides, J. Am. Oil Chem. Soc., 1948, 70, 248–254 CAS.
- W. MacNaughtan, I. A. Farhat, C. Himawan, V. M. Starov and A. G. F. Stapley, A Differential Scanning Calorimetry Study of the Crystallization Kinetics of Tristearin-Tripalmitin Mixtures, J. Am. Oil Chem. Soc., 2006, 83(1), 1–9, DOI:10.1007/s11746-006-1167-1.
- M. Kellens, W. Meeussen, R. Gehrke and H. Reynaers, Synchrotron Radiation Investigations of the Polymorphic Transitions of Saturated Monoacid Triglycerides. Part I: Tripalmitin and Tristearin, Chem. Phys. Lipids, 1991, 58, 131–144 CrossRef CAS.
- K. Sato, T. Arishima, Z. H. Wang, K. Ojima, N. Sagi and H. Mori, Polymorphism of POP and SOS. I. Occurrence and Polymorphic Transformation, J. Am. Oil Chem. Soc., 1989, 66(5), 664–674, DOI:10.1007/BF02669949.
- K. Bhaggan, K. W. Smith, C. Blecker and S. Danthine, Binary Mixtures of Tripalmitoylglycerol (PPP) and 1,3-Dipalmitoyl-2-Stearoyl-Sn-Glycerol (PSP): Polymorphism and Kinetic Phase Behavior, Eur. J. Lipid Sci. Technol., 2018, 120(3), 1700306, DOI:10.1002/ejlt.201700306.
- A. Minato, S. Ueno, J. Yano, Z. H. Wang, H. Seto, Y. Amemiya and K. Sato, Synchrotron Radiation X-Ray Diffraction Study on Phase Behavior of PPP-POP Binary Mixtures, J. Am. Oil Chem. Soc., 1996, 73(11), 1567–1572, DOI:10.1007/BF02523526.
- K. Bhaggan, K. W. Smith, C. Blecker and S. Danthine, Polymorphism and Kinetic Behavior of Binary Mixtures of Trisaturated Triacylglycerols Containing Palmitic and Stearic Acid Under Non-Isothermal Conditions, Eur. J. Lipid Sci. Technol., 2018, 120(9), 1800072, DOI:10.1002/ejlt.201800072.
- K. Wijarnprecha, R. West and D. Rousseau, Temperature-Dependent Mixing Behavior of Tristearin and 1,2-Distearoyl-3-Oleoyl-Rac-Glycerol (SSO), Cryst. Growth Des., 2023, 23(7), 4807–4814, DOI:10.1021/acs.cgd.2c01514.
- M. Takeuchi, S. Ueno, E. Floeter and K. Sato, Binary Phase Behavior of 1,3-Distearoyl-2-Oleoyl-Sn-Glycerol (SOS) and 1,3-Distearoyl-2-Linoleoyl-Sn-Glycerol (SLS), J. Am. Oil Chem. Soc., 2002, 79(7), 627–632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Non-treated time-resolved DSC data of the industrial grade and binary blends. SAXD/WAXD pattern after liquid subtraction. WAXS pattern during heating for FHRO-PMF and FHRO-ShSt. Summary of literature data on long and short spacings of pure triglycerides. See DOI: https://doi.org/10.1039/d5sm00418g |
| This journal is © The Royal Society of Chemistry 2025 |