
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 transport behavior in poly(fluoroalkyl acrylate) and poly(fluoroalkyl methacrylate): a comparative study of fluoropolymer structure–property relationships†
Sinan
Feng
a,
Yucheng
Zhang
a,
Nobuyuki
Otozawa
b,
Shinichi
Murata
a and
Atsushi
Takahara
 *a
*a
aResearch Center for Negative Emissions Technologies, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: takahara.atsushi.150@m.kyushu-u.ac.jp
bNew Business Development Department, Development Office, Electronic Materials General Division, Electronics Company, AGC Inc., 1-5-1, Marunouchi, Chiyoda-ku, Tokyo 100-8405, Japan
First published on 24th June 2025
Abstract
This study examines the CO2 transport behavior of poly(fluoroalkyl acrylate) (PFA) and poly(fluoroalkyl methacrylate) (PFMA) thin films. Using time-resolved attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) and a quartz crystal microbalance (QCM), we quantified CO2 diffusivity and solubility, linking these properties to the polymer side-chain architecture. The results demonstrate that PFMA exhibits lower CO2 permeability than PFA at comparable side-chain lengths, owing to restricted chain mobility caused by the α-methyl backbone of PFMA. Additionally, longer fluorinated side chains increase CO2 diffusivity while simultaneously reducing solubility owing to weaker polar interactions with CO2. Overall, the CO2 permeability of PFA surpasses that of the PFMA series because of its higher diffusivity. These findings highlight the intricate balance between diffusivity and solubility governed by the molecular structure.
Introduction
Excessive CO2 emissions into the atmosphere are a primary driver of global climate change, necessitating urgent strategies to mitigate greenhouse gas emissions. Among these strategies, direct air capture (DAC) has emerged as a promising technology for capturing CO2 directly from the atmosphere.1 DAC offers significant advantages, including the ability to capture diffuse CO2 emissions and retrofit capture units in decentralized locations.2 Membrane-based separation technologies have gained attention as a cost-effective and energy-efficient alternative for CO2 capture.3 These technologies rely on selective materials that facilitate CO2 transport while minimizing the passage of other gases. Polymers represent a significant class of materials for such applications owing to their tunable properties and ease of fabrication. However, the performance of membrane-based systems depends critically on the interplay between gas permeability and selectivity, which is governed by the chemical structure and physical properties of the polymer.Fluorinated polymers represent a significant class of high-performance materials, valued for their unique combination of thermal stability, chemical inertness, low surface energy, and tunable optical properties. Polyfluoroalkyl acrylates represent a unique class of functional polymers that combine the advantageous properties of fluorinated side chains with the structural versatility of acrylate backbones. The strong electron-withdrawing nature and low polarizability of the C–F bond impart exceptional thermal stability, chemical resistance, and notably low surface energy to these materials, making them ideal candidates for non-wetting coatings, anti-fouling surfaces, and advanced membranes.4,5 Recent advances in controlled radical polymerization techniques, such as ATRP and RAFT, have enabled precise manipulation of molecular weight, architecture, and composition, allowing for the tailored design of polyfluoroalkyl acrylates with tunable surface and bulk properties.6,7 Moreover, the self-assembly behavior driven by the incompatibility between fluoroalkyl side chains and hydrocarbon segments leads to microphase-separated structures that further enhance their performance in demanding environments.8,9 Fluorinated polymers have emerged as a cornerstone material class for CO2-selective applications, particularly in supercritical CO2 (scCO2) applications, thus revolutionizing various industrial processes in recent decades.10–12 They are well known for their affinity for scCO2 and exhibit higher solubility than non-fluorinated polymers.10,13 Their low surface energy and chemical resistance make them ideal for use in aggressive scCO2 environments, such as in separation, coating, and extraction processes. In particular, poly(fluoroalkyl acrylate) (PFA) and poly(fluoroalkyl methacrylate) (PFMA) have attracted attention because of their tunable side-chain architectures and robust performance in aggressive scCO2 environments.14 The low cohesive energy density of fluorinated polymers, resulting from weak van der Waals interactions between polymer chains, facilitates easier CO2 penetration and diffusion. Additionally, Lewis acid–base and quadrupole-induced dipole interactions between CO2 and fluorinated segments enhance CO2 solubility. However, increasing regulatory restrictions on per- and polyfluoroalkyl substances (PFAS)—driven by the extreme persistence and toxicity of bioaccumulative long-chain variants (e.g., C8 compounds like PFOA/PFOS)—demand material innovation aligned with “essential use” principles.15,16 Our molecular design prioritizes side-chain stability and mineralization pathways to mitigate environmental impact while retaining critical CO2 affinity and chemical resistance—advancing fluoropolymers that balance functionality with regulatory and sustainability mandates.
Our previous studies investigated the structural and transport behaviors of PFA and PFMA in CO2-rich environments, particularly the influence of the fluoroalkyl side-chain length on aggregation and crystallite formation.4,17 In scCO2 applications, PFA and PFMA demonstrated exceptional stability and performance. Their ability to swell in the presence of CO2 while maintaining their structural integrity, is critical for their use under such conditions. We also analyzed the structure of a PFA-C8 polymer brush subjected to scCO2 using grazing incidence wide-angle X-ray diffraction (GI-WAXD) and neutron reflectivity (NR) measurement.18 Fluoroalkyl acrylates with CF2 chains longer than 8 carbons exhibit crystallinity owing to the ordered packing of sidechains.4 Furthermore, scCO2 plasticizes semicrystalline fluoropolymer brushes by disrupting crystallites at pressures >4.1 MPa, enabling structural reorganization akin to high-temperature annealing. Understanding these phenomena is essential for optimizing PFA and PFMA in scCO2-related processes. However, broadening the use of PFA and PFMA beyond specialized scCO2 applications requires exploring their behaviors at atmospheric pressure. The structural insights gained from high-pressure studies—such as the effect of the fluoroalkyl side-chain length on crystallinity and swelling—can help us understand how these polymers perform under milder conditions. This knowledge is critical for applications such as CO2 capture, where efficient gas separation occurs at atmospheric pressure, and for designing packaging materials and environmental sensors with tailored gas-permeation properties. Furthermore, studying permeability under atmospheric conditions provides valuable insights into the fundamental interactions between CO2 and fluorinated polymers, thus guiding the development of novel materials with enhanced CO2-philic properties for both low- and high-pressure applications.
Building on our prior work, this study systematically evaluates the CO2 transport behavior in PFA and PFMA, focusing on the interplay among the polymer structure, CO2 solubility, diffusivity, and permeability. We examined the structural characterization of PFA and PFMA with varying fluoroalkyl side-chain lengths and CO2 transport properties, including permeability, solubility, and diffusivity in PFA and PFMA under atmospheric conditions. This study provides a comprehensive understanding of the factors influencing CO2 transport in fluorinated polymers, with implications for their application in membrane-based separation systems. By correlating the structural properties with the gas transport performance, we sought to establish design principles for next-generation materials for CO2 capture and separation.
Experimental section
Materials
2,2,2-Trifluoroethyl methacrylate (TFEMA, FMA-C1), 2,2,3,3,3-pentafluoropropyl methacrylate (PFPMA, FMA-C2), 2-(perfluorobutyl)ethyl methacrylate (PFBEMA, FMA-C4), 2,2,2-trifluoroethyl acrylate (TFEA, FA-C1), and 2,2,3,3,3-pentafluoropropyl acrylate (PFPA, FA-C2) were provided by AGC Inc. (Japan) and used as received. Monomers were labeled according to the length of carbon–fluorine (Rf) sidechain for clarity. 2,2′-Azobis(2-methylbutyronitrile) (AMBN) was obtained from TCI (Tokyo, Japan) and recrystallized from methanol before use. AK-225 (a mixture of 3,3-dichloro-1,1,1,2,2-pentafluoropropane and 1,3-dichloro-1,1,2,2,3-pentafluoropropane) was provided by AGC Inc. and used without further purification. HPLC-grade methanol was obtained from Fujifilm Wako Chemical Corporation (Osaka, Japan). A two-component PDMS kit (Sylgard184) was purchased from Dow Corning (USA). Poly(methyl methacrylate) was obtained from TCI (Tokyo, Japan).Polymer synthesis
The synthesis procedure was identical for all the monomers as shown in Scheme 1. Herein, we describe the synthesis of poly(2,2,2-trifluoroethyl methacrylate) (PFMA-C1) as a representative example. In a typical synthesis, FMA-C1 (10 g, 58.8 mmol) was dissolved in AK-225 (20 g) in a 50-mL round-bottom flask equipped with a magnetic stir bar. AMBN (0.02 g, 0.104 mmol) was then added to the solution. The flask was sealed with a rubber septum, and the mixture was purged with nitrogen for 20 min to remove dissolved oxygen. The flask was then immersed in an oil bath preheated to 60 °C, and the polymerization was allowed to proceed for 24 h with constant stirring. After the reaction, the polymer solution was cooled to room temperature and precipitated in 300 mL of cold methanol. The precipitate was collected via filtration, washed thoroughly with methanol, and vacuum-dried at room temperature for 24 h to yield PFMA-C1 as a white powder (yield of 85%). Identical conditions were applied to synthesize PFMA-C2, PFMA-C4, PFA-C2, and PFA-C4 using their respective monomers with yields varying between 78 and 90%. | ||
| Scheme 1 Chemical structure of poly(fluoroalkyl acrylates) (PFA-Cy) and poly(fluoroalkyl methacrylates) (PFMA-Cy), where x and y − 1 are the numbers of methylene and fluoromethylene repeating units, respectively. | ||
Characterization of PFA and PFMA thin films
The synthesized polymers were characterized by gel permeation chromatography (GPC) using a Tosoh HLC-8320 GPC system with a evaporative light scattering detector (ELSD). The separation was performed using an Agilent PLgel 5 μm Mixed-C column (300 × 7.5 mm) at 40 °C. The mobile phase was a mixture of HFE-7300 (1,1,1,2,2,3,4,5,5,5-decafluoro-3-methoxy-4-(trifluoromethyl)pentane) and HFIP (1,1,1,3,3,3-hexafluoroisopropanol) (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at a flow rate of 1.0 mL min−1. The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (Đ) were determined using PMMA calibration standards. The chemical structure of the synthesized PFMA and PFA was confirmed using 1H-NMR spectroscopy. Samples were dissolved in a 50/50 vol% mixture of deuterated trifluoroacetic acid and CDCl3 (5 mg mL−1). 1H NMR spectra were recorded on a Bruker AVANCE III 600 MHz spectrometer (room temperature). The samples were referenced to tetramethylsilane (TMS) internal standard. Thermal properties of the polymers were analyzed using differential scanning calorimetry (DSC) on a Thermo Plus EVO2 DSC vesta equipped with an automated liquid–nitrogen cooling system (Rigaku Corp., Japan) under an N2 atmosphere, with a heating/cooling rate of 20 °C min−1 in the range of −80 °C to 150 °C.
:1, v/v) at a flow rate of 1.0 mL min−1. The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (Đ) were determined using PMMA calibration standards. The chemical structure of the synthesized PFMA and PFA was confirmed using 1H-NMR spectroscopy. Samples were dissolved in a 50/50 vol% mixture of deuterated trifluoroacetic acid and CDCl3 (5 mg mL−1). 1H NMR spectra were recorded on a Bruker AVANCE III 600 MHz spectrometer (room temperature). The samples were referenced to tetramethylsilane (TMS) internal standard. Thermal properties of the polymers were analyzed using differential scanning calorimetry (DSC) on a Thermo Plus EVO2 DSC vesta equipped with an automated liquid–nitrogen cooling system (Rigaku Corp., Japan) under an N2 atmosphere, with a heating/cooling rate of 20 °C min−1 in the range of −80 °C to 150 °C.
The wide-angle X-ray scattering (WAXS) measurements were conducted at the SPring-8 synchrotron radiation facility using beamline BL38B1 with an incident wavelength of 1 Å. A PILATUS3X 2 M detector (Dectris Ltd) with 1475 × 1679 pixels of 172 × 172 μm2 was placed 260 mm away from the sample. The samples were mounted between two Kapton films (20 μm each) and exposed to an X-ray beam for 10 s. CeO2 was used as the standard for calibration of the detector geometry and image integration using the azimuthal integration tool pyFAI.19 The scattered intensities were azimuthally averaged and recorded as a function of the scattering vector q, where q = 4πλ−1sinθ, and θ represents half the scattering angle. The q value was in the 2–36 nm−1 range.
The polymers were analyzed using a Bruker Vertex70 FTIR spectrometer equipped with horizontally attenuated total reflectance accessories (PIKE). Thin films of the polymers were prepared by casting their solutions onto attenuated total reflection (ATR) crystals (45° Ge, length of 8 cm, and thickness of 4 mm). Spectra were recorded in the range of 4000–600 cm−1 with a resolution of 2 cm−1. CO2 (or N2) was introduced into the flow cell at a rate of 200 sccm (standard cubic centimeters per minute) using a mass-flow controller (KOFLOC, 3660). For time-resolved data acquisition, 1000 spectra were collected in an interval of 78 ms with a resolution of 2 cm−1, accumulating 16 scans for each spectrum.
To determine the CO2 diffusivity in the polymer using ATR–FTIR spectroscopy, the concentration was correlated with the experimental absorbance using the Beer–Lambert law. Barbari et al. integrated the equation for the evanescent field strength using the Beer–Lambert law to establish an ATR Fickian diffusion model based on certain assumptions.20,21 A comprehensive derivation of the equation was conducted in previous studies.20 The simplified equation for diffusion is as follows:
 | (1) |
The CO2 sorption capacity was examined at room temperature using a quartz crystal microbalance (QCM922A, Seiko EG&G). CO2 (or N2) was introduced into the flow cell at a flow rate of 50 sccm using a mass-flow controller (KOFLOC, BR-2C). The QCM crystals (QA-A9M-AU(M)(SEP), Seiko EG&G) were cleaned through UV/ozone treatment (ASM401OZ, ASUMI GIKEN) for 10 min. Thin films of the polymers were spin-coated (MS-B100 Spin-Coater, Mikasa) onto QCM crystals from their solutions (1 wt% in AK-225). The frequency changes were monitored in response to exposure to certain atmospheres (sorption and desorption). The Sauerbrey equation (eqn (2)) was used to relate the frequency change (ΔF) to the added mass (Δm) on the crystal, either the deposited polymer (mp) or the gas sorption (mg).22 The CO2 sorption capacity is defined as mg/mp (mg g−1).
 | (2) |
The permeability (P) of CO2 was determined at 25 °C at a feed pressure of 150 cmHg and atmospheric permeate pressure using the constant-pressure/variable-volume method.23 The samples were coated onto microporous polyacrylonitrile (PAN) support membranes (UF 010104, SolSep BV) and sealed inside a stainless steel filter holder (Millipore XX4404700). The permeation flow was measured using a mass flow meter (Agilent ADM Flow Meter, G6691A). P was calculated using the following equation:
 | (3) |
Results and discussion
Characterization of PFA and PFMA thin films
Poly(fluoroalkyl acrylates) (PFA-Cy) and poly(fluoroalkyl methacrylates) (PFMA-Cy) with varying Rf lengths were synthesized via radical polymerization. Rf was chosen to be short to yield an amorphous polymer. Polymers with C2 chains were soluble in THF, whereas longer C4 chains were dissolved in hydrochlorofluorocarbon solvents (AK-225). As shown in Fig. 1, the 1H NMR spectra of PFA and PFMA showed similar proton signals corresponding to methylene groups in the backbone (Ha) as well as the sidechain (Hc), which appeared at 2.4 ppm and 4.5 ppm, respectively. PFMA also showed proton signals corresponding to the methyl groups (Hb) at 0.8–1.2 ppm. All the integral results were consistent with the stoichiometric ratio of the molecular structure. The GPC results (Table 1) indicated that all the samples exhibited Mn in the range of 20–100 kDa and Đ < 2.0. This high degree of polymerization suggests that structural integrity is necessary for membrane applications. | ||
| Fig. 1 1H NMR spectra of PFA and PFMA. | ||
| M n (kDa) | M w (kDa) | PDI | T g (K) | |
|---|---|---|---|---|
| PFA-C2 | 75.3 | 147.0 | 1.95 | 258 |
| PFA-C4 | 94.4 | 166.7 | 1.76 | 249 |
| PFMA-C2 | 21.0 | 28.3 | 1.35 | 332 |
| PFMA-C4 | 45.2 | 58.9 | 1.30 | 294 |
The glass transition temperatures (Tg) of the polymer samples were obtained from the DSC traces, as shown in Fig. 2. Our findings indicate that both side-chain length and the presence of an α-methyl group on the backbone played crucial roles in determining Tg, with notable differences between fluorinated polymers and their non-fluorinated counterparts. The side-chain length emerged as a primary factor influencing Tg in these systems. In both the PFA and PFMA series, shorter side chains resulted in higher Tg values, which can be attributed to the stronger intermolecular interactions between the polymer backbones.24–26 This trend was also observed in non-fluorinated analogs.27 Notably, longer side chains lowered Tg by increasing the free volume and acting as internal plasticizers, reducing the energy required for segmental motion. Additionally, the presence of a methyl group on the backbone, which distinguishes methacrylates from acrylates, consistently led to higher Tg values.
 | ||
| Fig. 2 Glass transition temperature (Tg) of PFA and PFMA as well as nonfluorine counterparts PA and PMA. | ||
A comparison of the fluorinated (meth)acrylates with their non-fluorinated analogs revealed consistently higher Tg values in the fluorinated versions. This can be attributed to a combination of factors. The high electronegativity of fluorine atoms creates strong C–F dipoles, leading to enhanced intermolecular dipole–dipole interactions between chains. Furthermore, the larger atomic radius of fluorine compared to hydrogen results in increased steric hindrance and segmental stiffness, which restricts chain mobility. Together, these effects elevate the energy barrier for segmental motion, resulting in a higher Tg compared to their non-fluorinated analogs. This effect was more pronounced for PFA with shorter side chains, suggesting a complex interplay between fluorination effects and side chain dynamics.
These structure–property relationships have significant implications for the CO2 transport behavior. Polymers with Tg values near or below the intended operating temperature are likely to exhibit enhanced CO2 permeability owing to the increased chain mobility in the rubbery state. Tuning the Tg of fluorinated (meth)acrylates via careful control of the side chain length, backbone substitution, and degree of fluorination could optimize the CO2 transport properties.
All samples exhibited broad, diffuse peaks in their WAXS profiles (Fig. 3), with no sharp crystalline peaks, confirming their amorphous nature. This finding is consistent with previous studies and is advantageous for applications requiring gas permeability, as amorphous polymers typically possess a higher free volume than semi-crystalline polymers, facilitating molecular diffusion.4
 | ||
| Fig. 3 WAXS 1d profiles of PFA and PFMA. | ||
For PFMAs, three characteristic peaks were observed at approximately 1.2 Å−1 (qI), 2.2 Å−1 (qII), and 2.8 Å−1 (qIII). The detailed d-spacing (d) values were calculated from qpeak using the Bragg equation (d = 2π/q) and summarized in Table 2. The qI peak with dI ≈ 5 Å arose from the packing of fluoroalkyl sidechains.28 The qII peak corresponded to an intramolecular methyl–methyl correlation, while the third broad maximum qIII peak corresponded to the average repeat-unit correlation, averaged from the C–C distances along the backbone chains, which has the shortest correlation distance.29 This profile resembled that of the nonfluorine counterpart PMMA, but with noticeable variations in peak positions. The qI peak of PMMA at 0.94 Å−1 arose from the intermolecular backbone–backbone, backbone–ester, and backbone–methyl correlations.29,30 The qII and qIII peaks were almost identical to those of PFMA, which corresponded to the average repeat-unit correlation in the (meth)acrylate backbone.
| q I (Å−1) | d 1 (Å) | q II (Å−1) | d 2 (Å) | q III (Å−1) | d 3 (Å) | |
|---|---|---|---|---|---|---|
| d-Spacing (d) is calculated from Bragg equation. | ||||||
| PFA-C2 | 1.29 | 4.88 | — | — | 2.77 | 2.27 |
| PFA-C4 | 1.20 | 5.26 | — | — | 2.80 | 2.24 |
| PFMA-C2 | 1.21 | 5.19 | 2.23 | 2.81 | 2.91 | 2.16 |
| PFMA-C4 | 1.17 | 5.38 | 2.24 | 2.81 | 2.81 | 2.24 |
In contrast, PFAs exhibited only two prominent broad peaks at qI = 1.2 Å−1 and qIII = 2.8 Å−1. Similar to PFMA, the qI peak of PFA arose from the packing of fluoroalkyl side chains. However, PFA showed a slightly reduced d1 spacing than PFMA. This is likely due to the presence of the α-methyl group in PFMA, which caused steric hindrance and disrupted close packing of the fluorinated side chains. The absence of a qII peak in PFA indicates the lack of an α-methyl group, eliminating methyl–methyl correlations observed in PFA.
A subtle increase in d1 was observed for both PFA and PFMA with increasing side-chain length (C4). This indicates that a longer side chain introduced greater flexibility and potentially higher free volume, which is consistent with the trends observed in Tg. The WAXS data provide insights into the molecular packing and structural organization of these fluorinated polymers, which are crucial for understanding their properties, including their interactions with CO2 and their potential performance in various applications.30
In addition, ATR-FTIR characterization of PFA and PFMA revealed key differences as well as similarities in their chemical structures (Fig. 4). The spectra indicated no presence of unreacted monomers, as evidenced by the absence of significant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching peaks around 1640 cm−1. This aligns with the GPC results, confirming the high degree of polymerization in both PFA and PFMA. Both polymers exhibited characteristic absorption bands corresponding to their fluorinated side chains and ester functional groups. All the (meth)acrylate samples exhibited a characteristic carbonyl stretching vibration band at 1730–1760 cm−1 corresponding to the ester functional groups in their backbones. The subtle variation in wavenumber resulted from the α-substituents and Rf groups, probably because of the differences in the chemical environment around the carbonyl group. Furthermore, the presence of spacer methylene groups and the α-methyl group that could alter the electron density and steric interactions may have contributed to the observed shifts. The strong absorption bands at νs (CF2) ∼1130 cm−1 and νas (CF2) ∼1200 cm−1 attributed to the C–F stretching vibrations of fluoroalkyl side chains, were prominent in both PFA and PFMA.31 A notable difference emerged when comparing the C4 and C2 variants: the C4 samples exhibited peaks at higher wavenumbers compared with the C2 samples (1135 cm−1vs. 1126 cm−1). This shift could be related to side chain aggregation because the increased length favors stable packing.32 Comparable peak profiles suggest that the fluorinated groups dominate the spectra in this region. However, a notable difference between the two polymers was observed in the absorption bands around 2950 cm−1, with PFMA exhibiting more pronounced bands. These bands were attributed to the methyl group associated with the methacrylate structure, which distinguishes PFMA from PFA.
C stretching peaks around 1640 cm−1. This aligns with the GPC results, confirming the high degree of polymerization in both PFA and PFMA. Both polymers exhibited characteristic absorption bands corresponding to their fluorinated side chains and ester functional groups. All the (meth)acrylate samples exhibited a characteristic carbonyl stretching vibration band at 1730–1760 cm−1 corresponding to the ester functional groups in their backbones. The subtle variation in wavenumber resulted from the α-substituents and Rf groups, probably because of the differences in the chemical environment around the carbonyl group. Furthermore, the presence of spacer methylene groups and the α-methyl group that could alter the electron density and steric interactions may have contributed to the observed shifts. The strong absorption bands at νs (CF2) ∼1130 cm−1 and νas (CF2) ∼1200 cm−1 attributed to the C–F stretching vibrations of fluoroalkyl side chains, were prominent in both PFA and PFMA.31 A notable difference emerged when comparing the C4 and C2 variants: the C4 samples exhibited peaks at higher wavenumbers compared with the C2 samples (1135 cm−1vs. 1126 cm−1). This shift could be related to side chain aggregation because the increased length favors stable packing.32 Comparable peak profiles suggest that the fluorinated groups dominate the spectra in this region. However, a notable difference between the two polymers was observed in the absorption bands around 2950 cm−1, with PFMA exhibiting more pronounced bands. These bands were attributed to the methyl group associated with the methacrylate structure, which distinguishes PFMA from PFA.
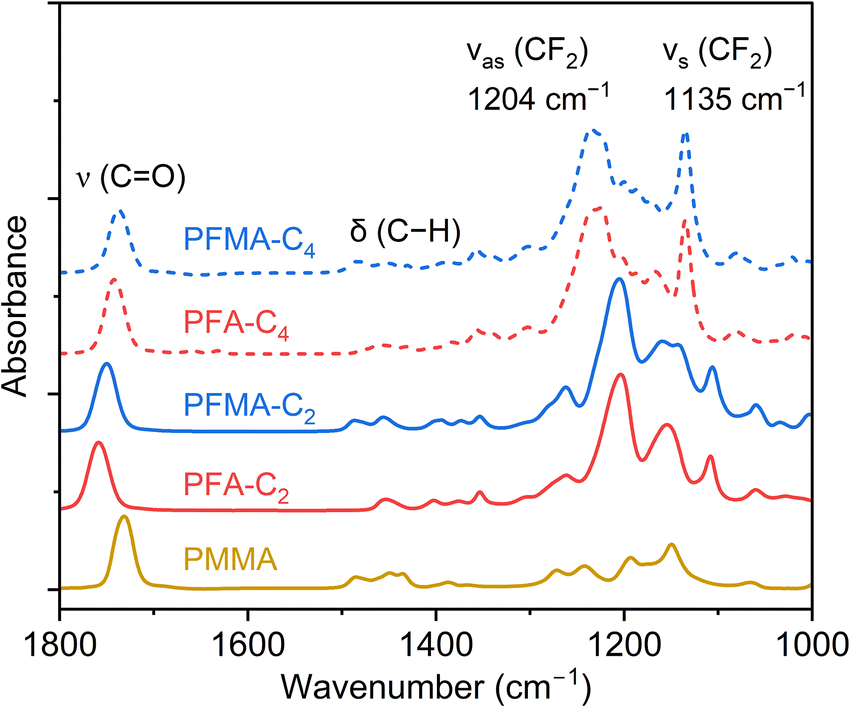 | ||
| Fig. 4 ATR-FTIR spectra of PFA and PFMA. | ||
The CO2 sorption by the polymer at equilibrium was demonstrated by ATR-FTIR spectra (Fig. 5). The CO2 that diffused into the polymer exhibited a distinct asymmetric stretching vibration (v3) band compared to gaseous CO2. This result aligns with previous literature on CO2 dissolved in various solvents and polymers. The v3 band for all fluoro(meth)acrylates appeared at an identical wavenumber of 2342 cm−1, which is different from that of PMMA at 2338 cm−1. This discrepancy could be attributed to the different interaction sites of CO2 with the fluorine and nonfluorine counterparts. For PMMA, CO2 mainly interacts with the carbonyl groups.33 In contrast, with the incorporation of Rf groups in PFMA, CO2 might have interacted with the side chain via weak Lewis base (Rf)–Lewis acid (CO2) interactions or via its quadrupole moment interacting with the induced dipoles of the Rf. The identical v3 band position at 2342 cm−1 across all fluoromethacrylates (regardless of chain length) suggests that the Rf groups dominated the CO2 interactions, creating a consistent sorption environment distinct from that of PMMA.
 | ||
| Fig. 5 ATR-FTIR spectra of CO2 in PFA and PFMA. | ||
In addition, time-resolved ATR-FTIR was used to analyze CO2 diffusion in both polymers. The concentration of CO2 was correlated to the experimental absorbance using the Beer–Lambert law. The absorbance (At) of the v3 band was tracked as a function of time during sorption and desorption. The experimental data were fitted using Fickian diffusion, a model that describes diffusion driven by concentration gradients. The diffusion coefficient (D) was derived from linear regression of ln(1 − At/A∞) vs. time (t) plot as shown in Fig. 6a. These plots reveal that CO2 diffused faster in PFA than in PFMA at comparable side chain lengths. The faster diffusion in PFA is likely due to the absence of the α-methyl group, which may reduce steric hindrance and enhance side chain mobility, thereby increasing free volume and facilitating CO2 transport. In addition, longer chain length induced higher D values for each polymer series.
 | ||
| Fig. 6 (a) Plot of ln(1 − At/A∞) for the CO2 asymmetric stretching band (ν3) as a function of time. (b) Relationship between diffusivity (D) and Tg for various polymers. | ||
The D values were plotted as a function of Tg for various polymer samples in Fig. 6b. This scatter plot, which is accompanied by a downward-sloping trend line, reveals an inverse relationship between D and Tg. Sylgard, with an exceptionally low Tg (170 K), exhibited a high D value (10−5 cm2 s−1), indicating significant chain mobility and free volume. In contrast, PMMA, which has a high Tg (380 K), exhibited a significantly lower D value (10−9 cm2 s−1), indicating its rigid structure with a minimal free volume. Compared with PFMA, PFA chains were more mobile and thus increased the free volume, facilitating faster gas diffusion, as reflected by the higher D values. As discussed earlier, the Tg values of the samples were significantly influenced by the fluoroalkyl (Rf) side-chain length. In both series, longer Rf chains induced higher D values. A side chain with faster dynamics increased the free volume, thus decreasing Tg and inducing faster CO2 diffusion.
The strong affinity of both polymers for CO2, inferred from the pronounced C–F stretching bands, suggests that these materials probably exhibit high CO2 solubility—a desirable property for gas separation applications.34,35 The CO2 sorption capacities of PFA and PFMA were evaluated using QCM. The Sauerbrey equation was used to calculate the mass of the deposited polymers and sorbed molecules in a certain atmosphere. The CO2 sorption data are summarized in Table 3 and Fig. 7. Compared with the n-alkyl methacrylates, both PFA and PFMA exhibited higher CO2 sorption capacities owing to stronger interactions between CO2 and Rf groups.
| T g (K) | D/10−8 (cm2 s−1) | S (cm3 cm−3) | P (Barrer) | |
|---|---|---|---|---|
| Measured at 297 K. | ||||
| PFA-C2 | 258 | 6.61 ± 0.28 | 2.82 ± 0.10 | 50 ± 3 |
| PFA-C4 | 249 | 16.5 ± 0.5 | 2.61 ± 0.25 | 121 ± 4 |
| PFMA-C2 | 332 | 1.45 ± 0.05 | 3.60 ± 0.48 | 14 ± 1 |
| PFMA-C4 | 294 | 2.25 ± 0.11 | 2.57 ± 0.37 | 20 ± 1 |
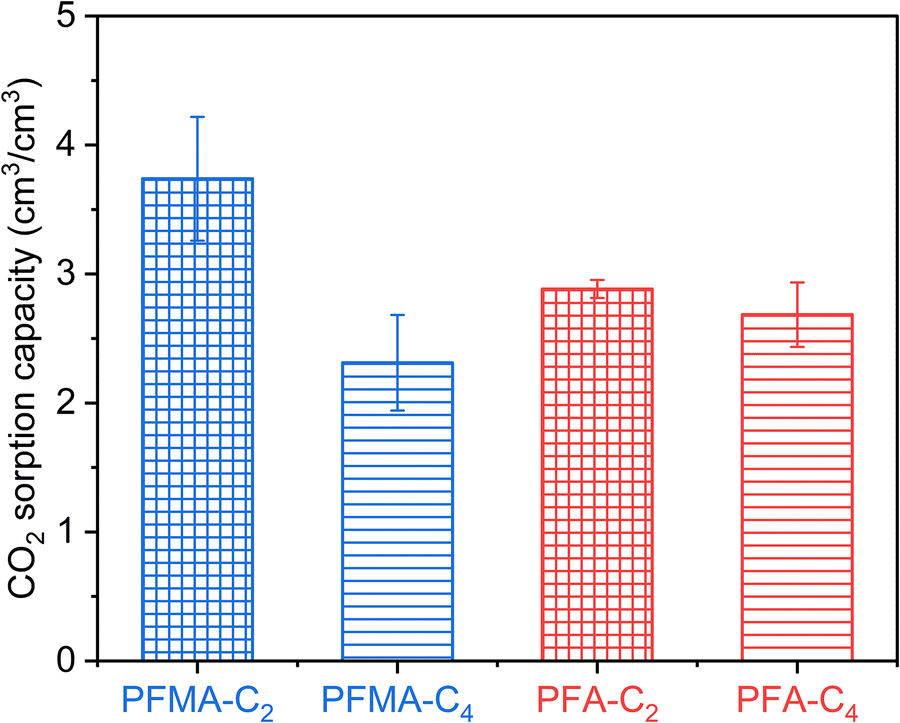 | ||
| Fig. 7 CO2 sorption of PFA and PFMA calculated from QCM results. | ||
In addition, PFMA exhibited higher CO2 sorption than PFA, which could be attributed to the additional unoccupied volume of the glassy polymers. The α-methyl group hindered the chain rotation, which limited the efficient packing of the Rf side chain. However, both PFMA and PFA exhibited a clear decrease in CO2 sorption as the Rf side-chain length increased from C2 to C4. This reduction was attributed to increased van der Waals interactions between the longer Rf chains, thereby weakening their interactions with CO2. Thus, Rf chains longer than C8 are expected to become semi-crystalline, exhibiting significantly lower gas sorption than the amorphous ones. This effect was also observed for the n-alkyl methacrylates with increasing side-chain length, as previously reported.36,37 The highest CO2 sorption capacity was observed in PFMA-C2 (5.8 mg g−1), where the α-methyl group and shorter C2Rf chains synergistically maximized free volume and CO2 interaction sites. The α-methyl group disrupted packing efficiency, while the shorter Rf chains minimized interchain interactions. In contrast, PFA-C4 exhibited the lowest sorption (3.5 mg g−1) owing to more efficient packing and stronger Rf–Rf interactions. It is crucial to note that this high CO2 affinity is a distinguishing feature of the fluorinated polymers. In their non-fluorinated counterparts, such as poly(n-alkyl methacrylates), gas sorption is significantly lower as it is primarily driven by weaker van der Waals forces. The enhanced solubility in PFA and PFMA is a direct consequence of the favorable CO2–Rf interactions, as PFMA surpasses PFA owing to the disruption of packing efficiency by the α-methyl group. Shorter Rf chains (C2) optimized sorption by reducing interchain interactions, whereas longer chains (C4) diminished sorption due to enhanced Rf–Rf interactions. The α-methyl group and shorter Rf length contributed together in achieving the inefficient packing of Rf to realize higher CO2 sorption.
The gas permeability through a polymer membrane is generally expressed as a product of its solubility and diffusivity (P = S × D). The interplay between solubility, diffusivity, and glass transition temperature (Tg) is complex, as structural features—such as the Rf length—simultaneously influence the free volume, chain flexibility, and intermolecular interactions. For PFA, P rose from 50 Barrer (PFA-C2) to 121 Barrer (PFA-C4), driven by a significant D increase (6.6 to 16.5 × 10−8 cm2 s−1) despite a slight S decrease (2.82 to 2.61 cm3 cm−3). This indicates that the increased diffusivity, resulting from lower Tg and higher free volume, outweighed any solubility reduction caused by Rf–Rf interactions. Compared with PFA, PFMA exhibited a significantly lower P value at comparable Rf lengths. This reveals that the decreased chain mobility outweighed the pronounced S increase. This nuanced interplay highlights the challenge of optimizing D and S to achieve high gas permeability, thereby guiding the design of advanced fluorinated materials for CO2 capture and gas separation membrane applications.
Conclusions
In this work, we have established critical structure–property relationships governing CO2 transport in PFA and PFMA polymers. Our findings demonstrate that while both polymer families show a strong affinity for CO2, their permeability is dictated by a delicate balance between solubility and diffusivity. The absence of an α-methyl group in the PFA backbone enhances chain mobility, leading to higher diffusivity and, consequently, superior CO2 permeability compared to the PFMA series. Meanwhile, extending the fluoroalkyl side-chain length acts as an internal plasticizer that lowers the glass transition temperature (Tg) and boosts diffusivity, albeit at the expense of solubility due to side-chain aggregation. This structure-guided approach provides a foundation for developing next-generation fluorinated membranes with tunable gas transport properties, enabling their application in direct air capture (DAC), post-combustion CO2 separation, and barrier materials for gas packaging and environmental sensing.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Acknowledgements
This study was based on the results obtained from a project (JPNP18016) commissioned by the New Energy and Industrial Technology Development Organization (NEDO). The synchrotron radiation experiments were performed at the BL38B1 beamline of SPring-8 (RIKEN). The authors thank Dr Hiroyasu Masunaga (Japan Synchrotron Radiation Research Institute) and Dr Sono Sasaki (Kyoto Institute of Technology) for their assistance in X-ray scattering measurement.References
- S. Fujikawa and R. Selyanchyn, MRS Bull., 2022, 47, 416–423 Search PubMed.
- S. Fujikawa, R. Selyanchyn and T. Kunitake, Polym. J., 2021, 53, 111–119 CrossRef CAS.
- M. Liu, M. D. Nothling, P. A. Webley, Q. Fu and G. G. Qiao, Acc. Chem. Res., 2019, 52, 1905–1914 CrossRef CAS PubMed.
- K. Honda, M. Morita, H. Otsuka and A. Takahara, Macromolecules, 2005, 38, 5699–5705 CrossRef CAS.
- Y. Liu, Y. Higaki, M. Mukai, N. Ohta, T. Kabe and A. Takahara, Polym. J., 2019, 51, 189–198 CrossRef CAS.
- W. Yao, Y. Li and X. Huang, Polymer, 2014, 55, 6197–6211 CrossRef CAS.
- S. Banerjee, B. V. Tawade, V. Ladmiral, L. X. Dupuy, M. P. MacDonald and B. Améduri, Polym. Chem., 2017, 8, 1978–1988 RSC.
- J.-M. Corpart, S. Girault and D. Juhué, Langmuir, 2001, 17, 7237–7244 CrossRef CAS.
- F. Montefusco, R. Bongiovanni, A. Priola and B. Ameduri, Macromolecules, 2004, 37, 9804–9813 CrossRef CAS.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–564 Search PubMed.
- M. Zhou, R. Ni, Y. Zhao, J. Huang and X. Deng, Soft Matter, 2021, 17, 5107–5115 Search PubMed.
- B. Améduri, Molecules, 2023, 28, 7564 Search PubMed.
- J. M. DeSimone, Z. Guan and C. S. Elsbernd, Science, 1992, 257, 945–947 Search PubMed.
- E. J. Beckman, Chem. Commun., 2004, 1885–1888 Search PubMed.
- B. Ameduri, Macromolecules, 2025, 58, 2781–2791 CrossRef CAS.
- I. T. Cousins, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier, L. Vierke, Z. Wang and J. C. DeWitt, Environ. Sci.: Processes Impacts, 2019, 21, 1803–1815 RSC.
- K. Honda, M. Morita, O. Sakata, S. Sasaki and A. Takahara, Macromolecules, 2010, 43, 454–460 CrossRef CAS.
- H. Yamaguchi, P. Gin, H. Arita, M. Kobayashi, S. Bennett, S. K. Satija, M. Asada, T. Koga and A. Takahara, RSC Adv., 2013, 3, 4778–4785 RSC.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed.
- G. T. Fieldson and T. A. Barbari, Polymer, 1993, 34, 1146–1153 CrossRef CAS.
- Y. A. Elabd, M. G. Baschetti and T. A. Barbari, J. Polym. Sci., Part B:Polym. Phys., 2003, 41, 2794–2807 CrossRef CAS.
- G. Sauerbrey, Z. Phys. Chem., 1959, 155, 206–222 CAS.
- H. Lin and B. Freeman, Springer Handbook of Materials Measurement Methods, Springer, Berlin, Heidelberg, 1st edn, 2006, pp. 371–387 Search PubMed.
- A. Arbe, A.-C. Genix, S. Arrese-Igor, J. Colmenero and D. Richter, Macromolecules, 2010, 43, 3107–3119 CrossRef CAS.
- F. Rindfleisch, T. P. DiNoia and M. A. McHugh, J. Phys. Chem., 1996, 100, 15581–15587 CrossRef CAS.
- S. Koizumi, K. Tadano, Y. Tanaka, T. Shimidzu, S. Kutsumizu and S. Yano, Macromolecules, 1992, 25, 6563–6567 CrossRef CAS.
- S. Rogers and L. Mandelkern, J. Phys. Chem., 1957, 61, 985–991 CrossRef CAS.
- V. V. Volkov, N. A. Platé, A. Takahara, T. Kajiyama, N. Amaya and Y. Murata, Polymer, 1992, 33, 1316–1320 CrossRef CAS.
- S. Eim, S. Jo, J. Kim, S. Park, D. Lee, T. P. Russell and D. Y. Ryu, ACS Macro Lett., 2024, 13, 1490–1494 CrossRef CAS PubMed.
- R. Lovell and A. H. Windle, Polymer, 1981, 22, 175–184 CrossRef CAS.
- M. Matsunaga, T. Suzuki, K. Yamamoto and T. Hasegawa, Macromolecules, 2008, 41, 5780–5784 CrossRef CAS.
- T. Hasegawa, in Quantitative Infrared Spectroscopy for Understanding of a Condensed Matter, ed. T. Hasegawa, Springer, Japan, Tokyo, 2017, pp. 165–193 Search PubMed.
- S. G. Kazarian, M. F. Vincent, F. V. Bright, C. L. Liotta and C. A. Eckert, J. Am. Chem. Soc., 1996, 118, 1729–1736 CrossRef CAS.
- M. J. Muldoon, S. N. V. K. Aki, J. L. Anderson, J. K. Dixon and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 9001–9009 CrossRef CAS PubMed.
- P. Raveendran and S. L. Wallen, J. Phys. Chem. B, 2003, 107, 1473–1477 CrossRef CAS.
- Z. Mogri and D. R. Paul, Polymer, 2001, 42, 7781–7789 CrossRef CAS.
- Z. Mogri and D. R. Paul, Polymer, 2001, 42, 7765–7780 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00413f |
| This journal is © The Royal Society of Chemistry 2025 |