
Surfactant-driven modifications in protein structure
Sugam
Kumar
 ab,
Debasish
Saha
c,
Debes
Ray
a and
Vinod K.
Aswal
*ab
ab,
Debasish
Saha
c,
Debes
Ray
a and
Vinod K.
Aswal
*ab
aSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: vkaswal@barc.gov.in
bHomi Bhabha National Institute, Mumbai 400 094, India
cJuelich Center for Neutron Science-4 (JCNS-4), Forschungszentrum Juelich GmbH, Juelich 52425, Germany
First published on 20th May 2025
Abstract
The interaction between proteins and surfactants has gained significant research interest due to its extensive applications across various fields, including the food industry, cosmetics, and medicine. Surfactants are known to unfold the proteins, where there are extensive models describing the basic mechanism of such unfolding and the resultant structure formed across micro-to-macro length scales. These models grounded on extensive experimental and simulation studies aim to predict the interaction dynamics based on several physicochemical parameters, such as surfactant properties (e.g., ionic character and tail length), protein characteristics (e.g., charge and isoelectric point), and solution conditions (e.g., pH, ionic strength, and temperature). Recently, there has been growing interest in the refolding of surfactant-induced unfolded proteins using combinations of ionic and nonionic surfactants and some mechanical procedures such as dilution, dialysis, etc. While the mechanisms of such refolding are still being explored, a general consensus suggests preferential binding of ionic surfactants with nonionic surfactants to form mixed micelles, rather than protein–surfactant complexes. It has also been demonstrated that the interaction of proteins with surfactants can be effectively utilized to guide the heat-induced gelation of proteins. This review article will summarize the fundamentals and recent updates on (i) protein interaction with surfactants; (ii) the phenomenon of protein unfolding and refolding, and (iii) the utilization of protein–surfactant interactions to direct heat-induced protein gelation.
 Sugam Kumar | Dr Sugam Kumar is a Scientific Officer in the Solid-State Physics Division at BARC and Assistant Professor at HBNI, Mumbai. He joined BARC in 2009 after completing his MSc from Aligarh Muslim University and earned his PhD from HBNI in 2015. He pursued postdoctoral research at Stockholm University, Sweden (2017–2019). His research focuses on the structure and interactions in multicomponent soft matter systems, with expertise in neutron scattering techniques. He has published extensively on protein gelation, surfactant-mediated self-assembly, and colloidal interactions in complex fluids. |
 Debasish Saha | Born in 1984, Debasish Saha holds a degree, Dr rer. nat., in Physical Chemistry from Westfälische Wilhelms-Universität Münster, Germany (2011). Before joining for his PhD, he completed his Master of Science in Physics from IIT Madras, India. He pursued his first post-doctoral study at CEA Saclay, France followed by the DST Inspire Faculty fellowship at the Bhabha Atomic Research Centre, India (2016–2022). He received the Marie Skłodowska-Curie fellowship under the GNeuS programme at Forschungszentrum Juelich, Germany (2022–2024). Currently, he is working at the same institute as a Junior Scientist in the EU RIANA project. His research focuses on different soft matter systems. |
 Debes Ray | Dr Debes Ray has been working as a Scientific Officer at Solid State Physics Division, BARC, Mumbai, since 2014. He did his MSc at Banaras Hindu University, Varanasi and PhD at HBNI, Mumbai (2012) before joining as a KSKRA Fellow at the BARC. He carried out his postdoctoral research at IBI-4, Forschungszentrum Jülich, Germany, during 2022–24. His research work focuses on neutron scattering techniques and their applications to multi-component soft matter, nanomaterials, and biological systems. As a beamline scientist at the SANS-I facility at the Dhruva reactor, he supports several user groups from across India. |
 Vinod K. Aswal | Dr Vinod K. Aswal is presently Head, Solid State Physics Division, Bhabha Atomic Research Centre, Mumbai. He is also Senior Professor, Homi Bhabha National Institute, Mumbai. He has been working as a scientist at the Bhabha Atomic Research Centre since 1993. He is MSc in Physics from IIT Bombay (1992), PhD from Bombay University (1999) and Post-doctorate from Paul Scherrer Institut, Switzerland (2001–2003). He is expert in the field of neutron scattering and applications to condensed matter research. He has also been involved in the development of neutron scattering facilities at research reactors, BARC and supporting the use of these national facilities among many university researchers. He has been listed in the world's top 2 percent of the most-cited scientists consecutively for 5 years (2020–2024) by Stanford University. |
1. Introduction
Proteins are among the most important biomolecules, playing an essential role in nearly all biological processes and supporting numerous functions in the human body as well as in our everyday lives. The functionality of a protein is intrinsically linked to its specific three-dimensional folded structure, which is sensitive to various factors, both natural and artificial. Changes in environmental conditions such as pH, temperature, and the presence of chemicals or additives can disrupt this delicate structure, leading to protein denaturation or unfolding. The amount of free energy required to disrupt the native structure of a protein is significantly low and hence the structure can be modified by minor perturbation in the surrounding components.1 One prominent area of research has focused on the interaction between proteins and surfactants. Surfactants, which are commonly used in both industrial and biological processes, often induce protein unfolding upon interaction, altering their structure and function.Protein–surfactant complexes have been studied for decades due to their significance in a wide range of fields, including pharmaceuticals, food science, and biotechnology.2,3 Despite the extensive research already conducted, studies on protein–surfactant interactions continue to grow, exploring new dimensions of this complex relationship.1–10 These investigations aim to deepen our understanding of how surfactants affect protein structures and functions and uncover new opportunities for leveraging these interactions in innovative applications.
The surfactant can perturb the native structure of the protein by modifying intra-protein hydrogen bonding and the interplay of electrostatic and hydrophobic forces (Fig. 1). Since the strength of these interactions depends on various physicochemical parameters, these factors can be strategically manipulated to influence different phases of protein solutions, including crystallization, coacervation, and gelation. The impact of surfactants on protein structure and phase behavior is governed not only by these solution conditions but also by the intrinsic properties of both the protein and the surfactant (Fig. 1). For instance, a protein's charge, shape, and molecular weight, along with the type, charge, and hydrophobicity of the surfactant, all play significant roles in determining the outcome of protein–surfactant interactions. As an example, ionic surfactants bind more strongly to proteins, often leading to pronounced conformational changes such as unfolding. In contrast, nonionic surfactants exhibit much lower binding affinity, typically preserving the protein's structure. Remarkably, recent studies have demonstrated that a combination of ionic and nonionic surfactants can reverse the unfolding caused by ionic surfactants alone.11,12 These findings present exciting possibilities for controlling protein stability and manipulating phase transitions in protein solutions, with potential applications across a range of industries. Given the complexity of the topic and the growing interest from multidisciplinary research areas, a comprehensive review of “surfactant-driven modifications in protein structure” is both timely and essential.
 | ||
| Fig. 1 Schematic representation of various physicochemical parameters influencing protein–surfactant interactions. Different classes of surfactants, including ionic, nonionic, zwitterionic, and double-chain surfactants, interact with proteins (both globular and fibrous) through a range of non-covalent interactions such as hydrophobic forces, electrostatic attractions, van der Waals interactions, and hydrogen bonding, under suitable solution conditions (e.g., pH, temperature, ionic strength, and the presence of co-solvents). | ||
In this review, we aim to cover the fundamental aspects of protein–surfactant interactions and their impact on protein structure, leading to complex assemblies. Emphasis will be placed on the ability of surfactants to induce protein unfolding and refolding. Additionally, we will provide a detailed discussion of the general dependence of protein–surfactant interactions on various physicochemical parameters. This review also explores surfactant-driven modifications in gelation. Lastly, we will highlight some of the emerging applications of protein–surfactant complexes in various fields.
2. Physics of protein–surfactant interactions
2.1. Surfactant binding to protein molecules
The binding curve of surfactants to proteins follows definite steps as described in Fig. 2.3,13,14 As the first step, surfactant monomers bind to the protein mostly at oppositely charged moieties, whereas their tails hide in the nearby hydrophobic segment of the protein. The surfactant with the anionic headgroup usually binds to the cationic site of the protein and vice versa. This region of the binding curve is known as the specific binding region, where the amount of surfactant binding to protein increases with increasing surfactant concentration. In this binding region, the surfactants do not cause significant conformational changes in the protein. The specific binding region is usually followed by a slow rising part or a plateau in the binding energy curve. The region is known as the non-cooperative region, where the further binding of surfactant molecules starts unfolding of proteins and formation of shared micellar clusters combining more than one protein molecule. A further rise in surfactant concentration initiates proper cooperative binding of the surfactant molecules, resulting in the formation of micelle-like clusters along the hydrophobic patches of the unfolded protein backbone. In this region, the binding curve shows typically linear behavior. Addition of surfactants beyond the cooperative binding region gives rise to the saturation region, where a plateau can be observed in the binding curve. In this region, excess surfactant molecules do not bind to proteins, rather form free micelles in the solution.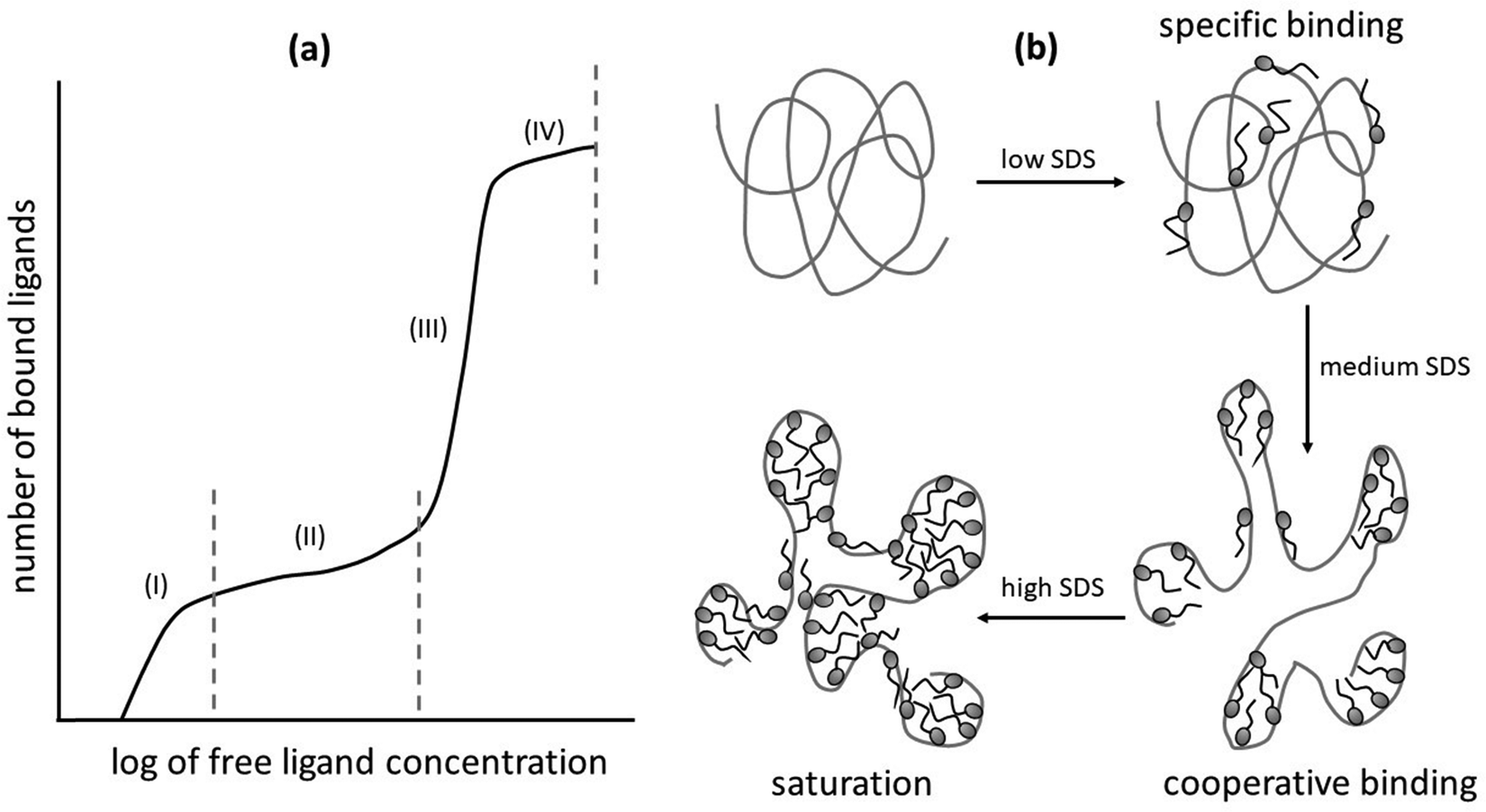 | ||
| Fig. 2 (a) Representative binding isotherm illustrating the interaction between a surfactant and protein, presented as the number of bound ligands (surfactant) per protein molecule plotted against the logarithm of free ligand concentration. The plot is divided into four characteristic regions: (i) specific binding region, where initial binding occurs at defined sites; (ii) non-cooperative binding region, with gradual, non-saturating interaction; (iii) cooperative binding region, marked by a sharp increase in binding; and (iv) saturation region, where all available sites are occupied. (b) A schematic representation of surfactant-induced protein unfolding, highlighting the role of increasing concentrations of sodium dodecyl sulfate (SDS) surfactant in promoting structural transitions in the protein. Reproduced from ref. 15 with permission from the Americal Chemical Society, copyright 1995. | ||
2.2. Interactions governing the surfactant binding to protein
Surfactants and proteins both share common properties of being amphiphilic and charged (except nonionic surfactants). Therefore, the interaction between the two components (surfactants and proteins) is mainly governed by hydrophobic interaction and electrostatic interaction.16,17 However, the extent up to which these two play a role is still under investigation. The ionic surfactants are known to strongly interact with the protein utilizing both the above stated forces, whereas nonionic surfactants usually do not interact significantly with the protein. The nonionic surfactants are belived to interact with hydrophobic sites available on the protein surface, and no further binding occurs after these sites are saturated.15,18,19 On the other hand, ionic surfactants are able to explore the hydrophobic patches of the protein molecules buried inside the core of molecular structure. This in turn causes the unfolding of the protein, giving rise to further binding of the surfactant molecules [as described in Section 4 and depicted in the schematic in Fig. 2(b)].17 The nonionic surfactants are therefore particularly of interest for membrane solubilization,20 while preserving the enzyme, receptor, or transporter function, whereas ionic surfactants are of importance in the applications requiring protein unfolding such as SDS-page. In addition, nonionic surfactants are also widely used in biotherapeutic formulations to protect proteins from interfacial damage by minimizing interfacial stress, thereby preserving the stability of the drug product.17 There are a number of studies, exploring and reporting the effect of these interactions on surfactant driven modifications in proteins.In a very interesting work conducted by Anna Stenstam et al., a rich phase behavior of the lysozyme protein as induced by the presence of the oppositely charged surfactant octyl sulfate (SOS) has been reported.21 The phase behavior covers solid, gel, and soluble complexes of the Ly(OS)8–SOS–water ternary system, as originated due to the interplay of hydrophobic and electrostatic interactions. In fact, pathways of association of SDS with ubiquitin (UBI), has been explored to understand the relative role of these two (hydrophobic and electrostatic) contributions.22–24 It has been realized that the binding strength of residues in UBI with SDS at sub-denaturing concentrations (<1.0 mM) is influenced by a combination of the residue's hydrophobicity and its local electrostatic environment. However, neither factor alone adequately explains the interaction of SDS with the folded UBI structure.
The effect of the chain length of surfactants has been extensively examined to illustrate the relevance of hydrophobic interaction in these systems.25–27 In addition to the hydrophobic and electrostatic interactions, hydrogen bonding has also been shown to play an important role in surfactant driven structural modification in proteins. It has been shown that the two surfactants differing in their role as a H-bond acceptor and/or donor interact differently with the protein.28 The surfactant, which is the H-acceptor, destabilizes the protein, whereas that which can act as both the H-bond acceptor and donor stabilizes the protein. Computer simulations have been used to study the interaction of the anionic, cationic, nonionic and zwitterionic surfactants with bovine serum albumin (BSA) protein.29 It has been shown that the hydrogen bonds formed by the nonionic surfactant are stronger than those formed by the ionic and zwitterionic surfactants.
The effects of cosolvent on the protein–surfactant interactions have been investigated to highlight the role of non-covalent interactions.30 Quantum chemistry calculations have shown that the main intermolecular interactions between BSA and the ionic surfactant (cetyl pyridinium chloride) in the presence of glycerol/DMSO are dominated by hydrogen bonding as well as van der Waals interactions, promoting micelle formation and allowing the supramolecular assemblies.31 Tuning of one or more than one of these interactions allows the characteristic changes in the protein–surfactant assemblies as well as the phase behaviour of proteins.32
3. Dependence of protein–surfactant interactions on physicochemical parameters
Protein–surfactant interactions are highly dependent on physicochemical parameters such as pH, ionic strength, temperature, concentrations and types of surfactants, presence of additives, etc. These factors influence the binding affinity, structural stability, and conformational changes in proteins upon interaction with surfactants. For instance, pH affects the ionization state of both proteins and surfactants, altering electrostatic interactions.3 Ionic strength causes the screening of the electrostatic interaction among proteins and surfactants.33 Temperature influences the hydrogen bonding as well as hydrophobic interactions, impacting the unfolding and refolding dynamics of proteins.34Fig. 3 demonstrates how concentration alters the folded nature of a simple α-helix protein (ACBP, acyl-coenzyme-A-binding protein) in the presence of SDS surfactant.35 We provide herein a brief account of general dependence of protein–surfactant interactions on these parameters, which is essential for applications of protein–surfactant complexes. | ||
| Fig. 3 Schematic illustration of the four distinct stages involved in the binding of sodium dodecyl sulfate (SDS) to bovine acyl-coenzyme-A-binding protein (ACBP), depicting the progressive denaturation process, stage A: ACBP binds 1 to 3 SDS molecules with no significant loss of its native structure. Stage B: approximately 37 SDS molecules associate with 2 ACBP molecules to form a protein-decorated micelle. Stage C: a total of around 40 SDS molecules bind to form a monomeric ACBP–SDS complex, resulting in a shell-like structure surrounding the protein. Stage D: a speculative model resembling a beads-on-a-string arrangement is proposed, representing extended binding at high SDS concentrations. The schematic is based on insights derived from a combination of experimental techniques, including fluorescence spectroscopy, circular dichroism (CD) spectroscopy, capillary electrophoresis, isothermal titration calorimetry (ITC), eluent gel permeation chromatography (GPC), and small-angle X-ray scattering (SAXS). Reproduced from ref. 35 with permission from Elsevier, copyright 2009. | ||
3.1. pH of the system
The dependence of protein–surfactant interactions on pH arises from its influence on the ionization states of both proteins and surfactants, which affects electrostatic interactions and protein stability. Proteins possess ionizable side chains whose charge varies with pH, leading to changes in the net charge and dipole moments of the protein. This variation affects the strength and nature of protein–surfactant binding. At the protein's isoelectric point (pI), where the net charge on protein is zero, hydrophobic interactions dominate.3 Conversely, at pH values significantly above or below the pI, proteins carry net positive or negative charges, dictating interactions with charged surfactants through electrostatic force.36 Additionally, extreme pH values destabilize protein structures, exposing hydrophobic cores and enhancing surfactant binding.34 The pH also influences surfactant ionization and micelle formation, which in turn modulate the availability of surfactant molecules for interaction with proteins.37,383.2. Ionic strength
The dependence of protein–surfactant interactions on ionic strength is again rooted in its effect on electrostatic forces, which are critical in determining the binding affinity and structural outcomes of these interactions. Ionic strength affects the screening of electrostatic charges on both proteins and surfactants. At low ionic strength, the electrostatic interactions between oppositely charged proteins and surfactant molecules are strong due to minimal charge shielding, promoting tighter binding.39 However, as ionic strength increases, the ionic environment reduces the effective charge of the interacting molecules, thereby screening their electrostatic fields and hence weakening the binding of oppositely charged proteins and surfactants.3 On the other hand, higher ionic strength can promote the binding of similarly charged proteins and surfactants. Moreover, high ionic strength also enhances binding in systems (interfacial adsorption) where hydrophobic interactions dominate, as ions may destabilize protein conformations and expose hydrophobic regions that interact more readily with surfactants.34 Additionally, the ionic strength affects micelle formation, altering the structure of micelles, surfactant aggregation behavior and availability for protein binding. From the protein point of view, as ionic strength increases, the repulsion between protein molecules decreases, which can lead to attractive interactions among them, finally resulting in protein aggregation. Ionic strength can enhance the interaction between an anionic surfactant and an anionic protein, when there is site-specific binding.40 These effects have significant implications in industrial and biotechnological applications, such as in protein purification and formulation stability.41–463.3. Temperature
The dependence of protein–surfactant interactions on temperature arises from its influence on molecular motions, hydrophobic forces, and the structural stability of proteins. Temperature affects the kinetic energy of molecules, altering the dynamics of binding and the strength of intermolecular interactions. At moderate temperatures, increased molecular motion enhances collisions between proteins and surfactants, potentially increasing binding rates.34 However, higher temperatures can destabilize the native conformation of proteins, exposing hydrophobic regions that enhance interaction with surfactants.3 This temperature-induced unfolding often leads to stronger binding but can also disrupt protein functionality. Additionally, temperature directly determines the critical micelle concentration (CMC) of surfactants, dictating their aggregation state and availability for molecular interactions.47 On the other hand, extreme temperatures may denature proteins completely, leading to irreversible aggregation and gelation. The protein gelation is shown to be affected by surfactants and is discussed in detail in Section 6.9,483.4. Type of protein
The type of protein significantly influences its interactions with surfactants due to variations in amino acid composition, structure, and surface properties. Proteins with a high proportion of hydrophobic amino acids tend to have stronger interactions with surfactants, as these residues facilitate binding through hydrophobic forces also.34 The globular proteins (e.g., bovine serum albumin, α-lactoglobulin, β-glucosidase etc.) with well-defined structures exhibit selective binding to surfactants at specific sites, whereas intrinsically disordered proteins (e.g., chaperones, α-synuclein, tau etc.), lacking a rigid structure, often interact more extensively across their surface.3 Surface charge and the distribution of polar and nonpolar regions determine the extent of electrostatic and hydrophobic interactions. For instance, proteins with a high net charge can engage more effectively with ionic surfactants of opposite charge.49 Moreover, post-translational modifications such as glycosylation or phosphorylation alter the protein's surface characteristics, impacting surfactant binding.393.5. Type of surfactant
The type of surfactant plays a critical role in determining the nature and strength of protein–surfactant interactions. Surfactants can be broadly classified into ionic (anionic and cationic), nonionic, and zwitterionic categories, and each type influences protein behavior differently due to the distinct physicochemical properties of the surfactants. Anionic and cationic surfactants tend to interact with oppositely charged positive and negative residues of proteins, leading to protein denaturation at higher surfactant concentration (Fig. 2).3,34,39 Zwitterionic surfactants, carrying both positive and negative charges on the same molecule, can interact with proteins through both hydrophobic and electrostatic forces, allowing for more complex and often less disruptive interactions.1 The surfactant's hydrophilic–lipophilic balance (HLB) also influences the interaction strength and mode of binding, with surfactants having high HLB values (more hydrophilic) typically causing less disruption to protein folding than those with lower HLB values (more hydrophobic).47 The type of surfactant, therefore, determines not only the extent of protein denaturation or stabilization but also influences the potential applications of proteins in detergents, drug delivery systems, and industrial processes.3.6. Chain length of surfactants
The length of the surfactant's hydrocarbon chain plays a significant role in modulating protein–surfactant interactions, influencing both the binding mechanism and the extent of protein denaturation. Surfactants with longer hydrocarbon chains tend to have a higher hydrophobicity, which enhances their ability to interact with hydrophobic regions of proteins. This results in stronger hydrophobic interactions, often leading to protein destabilization or conformational changes, especially when the surfactant concentration exceeds the protein's solubility limit in aqueous solutions.34 Longer-chain surfactants promote the formation of larger micelles that sequester protein molecules and alter their aggregation behavior.47 On the other hand, surfactants with shorter chain lengths are typically less hydrophobic and tend to interact further weakly with the hydrophobic regions of proteins, resulting in milder effects on protein structure.50 Additionally, the critical micelle concentration (CMC), which decreases with increasing surfactant chain length, dictates the availability of surfactant molecules for protein binding. As the surfactant chain length increases, the CMC decreases, meaning that a lower concentration of surfactant is required to form micelles that can interact with proteins, potentially increasing the rate and strength of interaction.1 The dependence of protein–surfactant interactions on the chain length is crucial for designing surfactant-based formulations in drug delivery, protein purification, and detergent applications, where controlling the extent of protein denaturation or stabilization is key.51,52It has been shown that the single chain surfactant interacts with proteins predominantly via electrostatic interaction, whereas the double chain surfactant interacts with proteins largely through hydrophobic interaction. Moreover, interaction of the double chain surfactant is stronger compared to the single chain surfactant due to higher hydrophobicity. Similarly, extensive theoretical and experimental investigations were presented on interaction of bovine α-lactalbumin (α-LA) with alkyl trimethylammonium bromides (CnTAB, n = 10, 12, 14, and 16).26 It has been shown that the enthalpy changes (ΔH) as well as area of the enthalpogram increased with increasing the chain length of CnTAB. It has been further inferred that C10TAB and C12TAB could partially unfold α-LA, whereas C16TAB gives rise to a molten globule state α-LA. The role of single and double chain surfactants has been compared in disrupting the protein structure.27
Gemini surfactants, also known as dimeric surfactants, contain two hydrophobic tails connected by a spacer group, which significantly alters their interactions with proteins compared to conventional monomeric surfactants. These surfactants exhibit enhanced surface activity and self-assembly properties due to their unique structure, which leads to stronger and more versatile protein–surfactant interactions. The presence of two hydrophobic tails allows Gemini surfactants to interact more effectively with the hydrophobic regions of proteins, often causing protein denaturation or aggregation at lower concentrations than their monomeric counterparts.53–55 Additionally, the spacer group between the hydrophobic tails influences the binding affinity and the extent of protein unfolding, as it modulates the flexibility and the surface charge of the surfactant.56,57 Gemini surfactants, therefore, offer greater control over protein stability and aggregation, making them valuable in biotechnological applications such as protein purification, drug delivery, and enzyme stabilization.58
3.7. Co-solvents and additives
The presence of co-solvents in protein–surfactant systems can significantly alter the nature and extent of interactions between proteins and surfactants, depending on the co-solvent's properties, concentration, and its interactions with both the surfactant and the protein. Co-solvents are often used to modify the solubility, stability, and functionality of proteins, as well as to tune surfactant micellization and aggregation behavior. Common co-solvents, such as alcohols, glycerol, or urea, can impact protein structure. For instance, glycerol, which is a stabilizing agent enhances protein solubility by preventing aggregation (increasing the viscosity of the solution) and preserving protein conformations, thereby modulating protein–surfactant interactions in a way that minimizes denaturation.59–61 On the other hand, urea, a common denaturant, is a chaotropic agent that destabilizes protein structure by disrupting hydrogen bonding and hydrophobic interactions and hence leads to enhanced protein denaturation in the presence of surfactants.62–64 Alcohols disrupt water–water interactions, leading to protein denaturation or aggregation effects that are amplified in the presence of surfactants. By reducing the hydration shell around proteins, alcohols facilitate surfactant binding to hydrophobic regions. The addition of alcohols such as ethanol or methanol either promotes protein unfolding or induces refolding by altering the surrounding water structure. These structural changes enhance or diminish surfactant–protein interactions in a concentration-dependent manner.65 The effect of co-solvents is also linked to the balance between hydrophilic and hydrophobic interactions. Co-solvents like polyethylene glycol (PEG) can promote micelle formation by surfactants, which in turn alters protein partitioning between the aqueous phase and the micelle. PEGs are often used in formulations to crowd the solution, promoting protein–surfactant interactions by reducing the effective volume available to the proteins, thus facilitating interaction.47,66–684. Unfolding and refolding of proteins
4.1. Unfolding of proteins
Depending on the solvent conditions and the physicochemical parameters that influence the protein–surfactant complex formation, proteins can go from their native state to unfolded/denatured state or refold back to their native state from the unfolded state.56–65 Knowledge of the folded (or unfolded) state of the protein is essential for efficient utilization in different applications such as in the pharmaceutical, food, and cosmetic industries.69–71 The application of protein–surfactant complexes was first discovered a long time back in the first century A.D. when the Egyptians showed the importance of interaction between proteins and amphiphilic molecules for personal washing. The first report on SDS interaction with protein molecules was published in 1936 by Baylis, in which he investigated the inactivation of diphtheria toxin by different surfactants including SDS.72 Since then, SDS received the utmost attention from researchers until today to study surfactant–protein interactions.The interactions between different proteins and amphiphilies/surfactants including SDS are governed by the aggregation state of the surfactants. The surfactant binding to the protein takes place following four steps with increasing surfactant concentration, as discussed in Fig. 2.18,19,73,74 In the cooperative binding region, during the process of micelle formation, the internally directed hydrophobic regions of the protein get exposed to the aqueous environment, which is entropically unfavorable as the hydrophobic region of the proteins is not stable in the aqueous environment.75 This leads to the unfolding of the protein via micelle-like cluster formation along the hydrophobic patches.
Experimental evidence shows that the unfolding of globular proteins typically occurs above the critical micelle concentration (CMC) of the surfactants. However, some recent studies show that the process of protein unfolding can start much lower than the CMC in aqueous dispersion (without protein) but above the critical aggregation concentration (CAC). The CAC of a specific protein–surfactant complex depends on many factors such as the protein concentration, binding affinity of the surfactant to the protein, pH of the system, etc.76,77 For example, the critical micelle concentration (CMC) of SDS is ∼7 mM in water15,78 but the CAC was observed to be as low as 2.2 mM in the presence of 1 wt% BSA protein at pH 5.4.77 A similar trend has been observed in α-synuclein–SDS complexes, where the CAC of SDS is much lower than the CMC of SDS in the aqueous solution.79 The argument is not only valid for anionic surfactants like SDS but also for cationic surfactants such as dodecyl trimethyl ammonium bromide (DTAB) and or longer alkyl chains. In a study of BSA with CnTAB (n = 10, 12, 14, and 16), it was observed that CnTAB surfactants denature the protein at concentrations, which are well below that of their CMC.12
The ionic surfactants denature proteins at very low concentrations compared to other denaturants such as urea or guanidinium chloride.72–75 However, the mixing of oppositely charged SDS and DTAB unfolds BSA differently compared to the individual surfactants. In this case, the unfolding of the protein is mainly dictated by the molar ratio between these two ionic surfactants.80 In another study, Lu et al. compared the interaction between different proteins with fluorinated and hydrogenated surfactants.81 The result shows that the fluorinated surfactants exhibited stronger interactions with proteins than hydrogenated ones.
It has been further noted that below the isoelectric point (IEP), proteins used to exhibit charge reversibility and thus interact differently with the surfactants. For example, SDS denatures the protein S6 more rapidly below pH 5.82 El Kadi et al. showed that the alpha helix content was 70% to 40% with decreasing pH from 4 to 2.83 Similarly, ionic liquid (IL) 1-butyl 3-methylimidazolium octyl sulfate ([BMIM][OSU]) was observed to interact very strongly with HSA protein at low pH even at submicellar concentrations.84
4.2. Unfolding to refolding
The interplay of hydrophobic and electrostatic interactions between the proteins, denaturants, and renaturation agents is the major factor that decides the final state of the protein. Understanding the state of the protein is crucial as the misfolding of proteins could lead to some diseases such as cancer, Alzheimer's, Parkinson's, etc.85,86Different strategies have been used to control the final state of proteins and refold them back to the folded/native state. However, the obtained yields are not very promising in many cases. The effective conditions for different proteins are different and difficult to rationalize, making finding the proper way of refolding very challenging. In this part of the review, we are aiming to provide insight into role of surfactants in refolding back the unfolded protein, apart from a brief description of some mechanical procedures such as dialysis, dilution, etc.
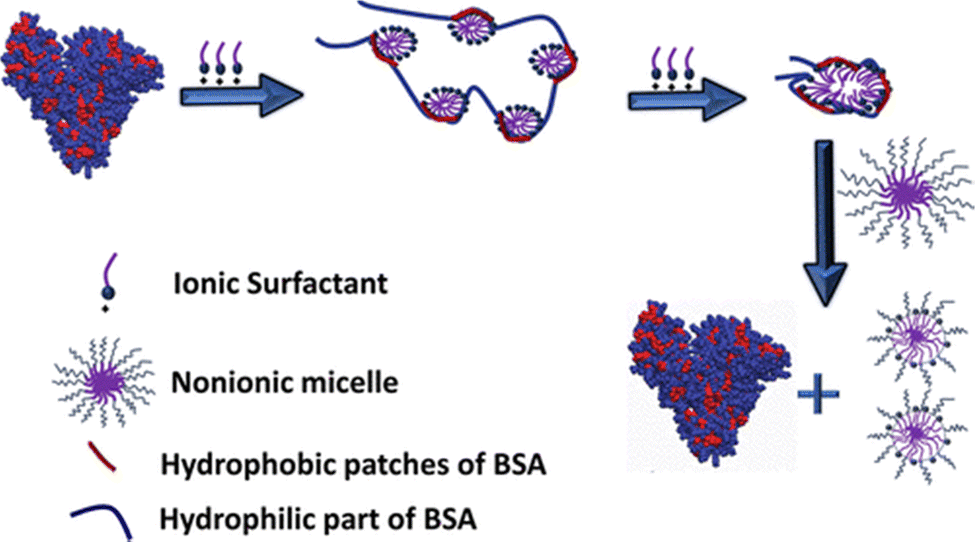 | ||
| Fig. 4 Schematic showing the unfolding of BSA protein in the presence of ionic (CnTAB; n = 10, 12, 14 and 16) surfactants and then refolding upon further addition of nonionic (C12E10) surfactants at pH 7. All tested cationic surfactants induce BSA unfolding, where the denatured state is depicted as a beads-on-a-string configuration, consistent with a random flight model. Upon subsequent addition of the nonionic surfactant, C12E10, mixed micelles are formed, which effectively sequester the ionic surfactant away from the protein. Reproduced from ref. 12 with permission from the Americal Chemical Society, copyright 2018. | ||
In contrast, once the hydrophobic interaction between ionic and nonionic surfactants dominates over the interaction between protein and ionic surfactants, the strip off of the ionic surfactants from the protein is inevitable, resulting in the formation of the refolded protein with ionic–nonionic surfactant mixed micelles. Similar concepts have been examined recently by Yadav et al. where they used mixed micelles of CTAB and different reagents, (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) hydrate (CHAPS) and two bile salts, namely, sodium cholate (NaC) and sodium deoxycholate (NaDC). The formation of mixed micelles results in the removal of CTAB molecules from the protein environment and allows the protein to retain its native-like structure.87 The mixed micelle-driven refolding of unfolded protein has also been achieved by using tri-block copolymers such as P123 or F127. The addition of P123 induces the recovery of ∼87% of its α-helical structure of human serum albumin (HSA) protein denatured by the SDS surfactant.88 The formation of mixed micelles by SDS-P123 allows the removal of bound SDS molecules from the hydrophobic patches of the unfolded HSA protein. Similar results were also found when P123 or F127 was added to the SDS-induced unfolded BSA protein solution. The recovery of the α-helix content was confirmed by different spectroscopic techniques. However, the major drawback of the surfactant-induced refolding is that such a strategy does not work for the protein denaturation caused by reagents like guanidine hydrochloride due to its non-interactive behavior with the polymer.87,89
The thermally denatured proteins can be refolded using temperature-responsive mixed-shell polymeric micelles. A mixed-shell polymeric micelle (MSPM), with a PLA core and a homogeneously mixed PEG and PNIAPM shell, was prepared by Liu et al. for this purpose. Above the lower critical solution temperature of PNIPAM, the MSPM evolves into a core–shell-corona micelle, as a functional state with hydrophobic PNIPAM domains on its surface. In such formation, the unfolded proteins are captured by the hydrophobic PNIPAM domains of the core–shell-corona micelle. During cooling, PNIPAM reverts into its hydrophilic state, thereby inducing the release of the bound unfolded proteins. The refolding process of the released proteins is spontaneously accomplished by the presence of PEG in the mixed shell.90
The denaturants can also be removed from the protein solution using the chromatography method.102,103 This method is very useful because of its simultaneous purification capabilities.
5. Models of protein–surfactant complexes
Ionic surfactants usually form protein–surfactant complexes, in which, the surfactant needs to have some charge on the surface but not necessarily on the protein.56–60,86 On the other hand, nonionic surfactants have very weak interactions (hydrophobic only) with some membrane proteins and a few globular proteins such as α-lactalbumin (αLA).110,111 The resultant complexes have structures that are revealed by employing several suitable experimental techniques and simulations.112 Scattering techniques particularly small-angle X-ray or neutron (SAXS or SANS) scattering are among the most used methods to extract structural features of protein–surfactant complexes. The modelling of scattering data obtained for protein–nonionic systems is relatively simple compared to the protein–ionic surfactant complexes. In most of the cases, the results can be taken as a linear superposition of the scattering contribution from individual components.11,12 In contrast, the modelling of the scattering results of protein–ionic surfactant complexes cannot be defined by such superposition of the individual contribution from each entity. For this reason, different structural models have been proposed to describe the structures of these complexes based on both experimental (e.g., scattering,12,35,79,113,114 binding isotherm,115,116 rheology,19 spectroscopy,3,15,116–118etc.) and computer simulation112 results, such as (a) a correlated “beads-on-a-string” model of unfolded protein, in which a cluster of micelle-like structures are stabilized by the extended protein structure.113,114 Although this model successfully explained many scattering results,11,119 it failed to satisfactorily describe some of the SAXS data of protein–SDS complexes.4 (b) “Protein-decorated micelles”, in which denatured protein covers the surface of the micelles. Intriguingly, this model is consistent with SAXS data.120 There can be changes in the architecture of the protein-decorated micelle with increasing protein size. In such structures, multiple small proteins can be accommodated on one micelle or large proteins can decorate several micelles. This model is similar to the so-called “random flight model”, which provides information about the size of the micelles organized along the protein chain, the number of micelles per cluster, and the separation between the centers of two nearest micelles.121 The “random-flight model” successfully explained some of the recent SANS results.12,80 (c) A “flexible capped helical cylindrical micelle” with the proteins wrapping around the micelle.122 Recently, a SANS study of BSA and OVA (ovalbumin) complexes with SDS led to the conclusion that the necklace and bead structure of protein surfactant aggregates accounted for the scattering behavior of the system. In fact, the scattering data allow the determination of the size of individual micelles bound to the protein under various conditions.123,124 However, for multimeric proteins, such as hemoglobin the situation might be different while interacting with surfactants. The tetrameric structure of hemoglobin consists of two identical α and two identical β subunits. The subunit dissociation of multimeric proteins could form different structures, which is beyond the scope of the above described models.125The structure formation of proteins with mixed ionic–nonionic micelles is different from the above-mentioned model and depends on the concentration of each surfactant. For example, the addition of a sufficiently high amount of nonionic C12E10 surfactant disrupts the formation of bead-on-string/random-flight structure formation of BSA–SDS or BSA–CnTAB systems by forming mixed surfactant micelles.11,12 This leads to the coexistence of the refolded protein with mixed nonionic and ionic surfactant complexes in the system, as discussed eariler. On the other hand, the combination of oppositely charged surfactants leads to the formation of different microstructures and interacts differently with globular proteins.126 In this case, unfolded proteins wrap the structures of mixed surfactants around their surface. Along with the connected protein-decorated micelle like structure, rod-like and bilayer vesicles of protein–surfactant complexes are formed at different molar fractions of mixed surfactants (Fig. 5).80 The structure of small proteins like lysozyme (Mw = 14 kDa) with ionic surfactants is rather different. At a low molar ratio of lysozyme to SDS, the system shows the formation of insoluble complexes. The increase in the surfactant concentrations leading to the formation of a hexagonal closed packed lattice can be seen from the Bragg peaks in the scattering data.108,109,127 Lysozyme shows a similar kind of interaction with cationic surfactants such as DTAB and CTAB above the isoelectric point (at pH = 13.0), leading to the fibril formation by these complexes.128
 | ||
| Fig. 5 Structures resulting from the interaction of BSA protein with mixed anionic SDS and cationic DTAB surfactants in varying molar ratios at pH 7 and in the presence of 0.2 M NaCl. Experiments were conducted with 1 wt% BSA and SDS–DTAB mixtures at a fixed total surfactant concentration of 50 mM. The DTAB molar fraction X = [DTAB]/([DTAB] + [SDS]) was systematically varied from 0 to 1. Individually, both SDS and DTAB interact with BSA forming connected protein-decorated micelle-like structures, wherein surfactant micelles are aligned along the unfolded protein chain. Upon mixing SDS and DTAB in varying molar fractions, the protein–surfactant interactions are modulated. At lower molar fractions of either surfactant, the mixed systems resemble the connected micellar structures seen with the pure components. However, near equimolar ratios, the BSA wrapps around mixed surfactant assemblies. Depending on the composition, diverse microstructures are observed, including rod-like and bilayer vesicle-like protein–surfactant complexes. Reproduced from ref. 80 with permission from the Royal Society of Chemistry, copyright 2021. | ||
The structural models (e.g., beads-on-a-string, protein-decorated micelles, etc.) being used to describe the protein–surfactant complexes have evolved through a rigorous experimental and theoretical framework and have significantly advanced our understanding of these systems. However, these models also exhibit notable limitations and present substantial opportunities for further development:
(i) These models often assume uniform binding and overlook the heterogeneous nature of protein surfaces, thereby neglecting domain-specific unfolding and localized interactions.
(ii) Most models offer static snapshots, whereas protein–surfactant systems are inherently dynamic. Capturing the reversible binding, micellization, and conformational transitions remains a key area for future research.
(iii) Existing models have a limited ability to predict the combined effects of multiple environmental parameters (e.g., simultaneous changes in pH, temperature, and ionic strength), which are critical in real systems.
(iv) Experimental studies have shown that a single protein–surfactant system can yield structurally distinct complexes under different conditions.3,80 Therefore, a unified model framework capable of integrating and interpreting such variability is essential.
(v) Most models are limited to individual surfactants and are not equipped to describe mixed surfactant systems, which are increasingly employed to modulate protein unfolding, refolding, and aggregation.
(vi) These models often fail to anticipate macroscopic behaviors such as thermal gelation or phase transitions and typically do not account for time-resolved or kinetic phenomena.
6. Protein–surfactant interactions for guiding the heat-induced protein gelation
As discussed and illustrated above the protein–surfactant interactions are largely driven by hydrophobic and electrostatic interactions. Interestingly, both of these interactions also play a major role in determining different phases of protein solutions, such as denaturation, coacervation, aggregation, gelation, etc. Globular proteins are known to undergo denaturation upon heating, finally leading to gelation at elevated temperatures. The process is driven by breaking of hydrogen and disulphide bonds by heat and resultant exposure of the hydrophobic segments of protein, which are otherwise buried inside the core. Such exposure of the hydrophobic patches towards the aqueous medium induces hydrophobic attraction, which in turn causes the formation of small protein aggregates. These aggregates then assemble to form the 3-dimensional network structure, leading to gelation of the protein at high temperatures. Effects of enhanced hydrophobicity on protein gelation kinetics have been probed in several studies.129,130 As an example, thermosensitive hydrogels of polypeptides were developed using PEG and poly(L-glutamate) copolymers with varying hydrophobic side groups. Polypeptides with methyl and ethyl groups showed lower critical gelation temperatures compared to n-propyl and n-butyl derivatives. The sol–gel transition was attributed to PEG dehydration and increased β-sheet formation in the polypeptides, highlighting the decisive role of hydrophobic side groups on gelation behavior.131 Also, addition of the hydrophobic moiety has been shown to modulate the textural deterioration of protein gels at high temperature.132 In another interesting work, pathways to control the hydrogelation of β-hairpin peptides via utilizing hydrophobic amino acid substitutions have been illustrated.133On the other hand, charge on the protein causes a counter balancing effect on the hydrophobic attraction by generating electrostatic repulsion between protein molecules. It has been recently shown that the heat-induced unfolding and gelation of proteins can be effectively inhibited by multivalent counterions like Zr4+.105 It is believed that the excess condensation of Zr4+ ions generates strong hydration interactions due to increased surface dipoles, which suppress the exposure of hydrophobic patches on the protein surface. This mechanism prevents protein aggregation and gelation, offering a potential strategy to control protein behavior under thermal stress. The suppression of the electrostatic barrier by utilizing physicochemical parameters such as pH, ionic strength etc. promotes the protein gelation and even causes the gelation at significantly much lower temperatures, via process, in general, known as cold-set protein gelation.134,135
Furthermore, it has been seen that the addition of the artificial denaturant reduces the gelation temperature and assists the gel formation at lower temperatures. In this context, the surfactant can play a crucial role in directing the heat-induced protein gelation or achieving cold-set gelation due to their capability of modifying both of these interactions (electrostatic and hydrophobic), which largely govern the protein gelation upon heating.136 We first discuss some of our recent studies showing the potential of the surfactants in tuning heat-indued protein gelation, in terms of underlying interactions. However, the later part of this section will also provide an account of the literature, discussing surfactant driven modifications in protein gelation.
Fig. 6 shows the physical state of the 4 wt% BSA protein solutions (pH ∼ 5), heated at 80 °C, in the absence and presence of anionic, cationic and nonionic surfactants. All the three surfactants have the same length of the hydrophobic carbon (NC = 12) tail, but differ in their head groups and charge on them. As is obvious, protein solution becomes gel upon heating; however, remarkably different situations are observed in the presence of surfactants. It is expected that being a protein denaturant, the presence of SDS may support the protein gelation. However, in contrast to such understanding, it is observed that SDS may also provide a protective effect on BSA against its thermal gelation.9,48 The protein–SDS dispersion does not undergo gelation even after heating for longer times at elevated temperatures. Contrary to this, the cationic surfactant DTAB (C12TAB) promotes gelation, leading to significant reduction in the gelation temperature.9 On the other hand, the nonionic surfactant C12E10 also supports protein gelation.48 As discussed in the sections above, nonionic surfactants do not interact strongly with the proteins. Therefore, the role of nonionic surfactants in modifying protein gelation is also little beyond the common understanding. It has been further shown that the presence of surfactants significantly affects the rheological properties of the heat-induced protein gels.9,48
 | ||
| Fig. 6 (a) Physical states of the BSA solutions when heated in the presence of different surfactants (SDS, DTAB, and SDS + C12E10) and (b) phase behavior of the BSA–DTAB system as a function of DTAB (C12TAB) concentration and temperature. The experiments were conducted at pH 5 with 4 wt% BSA and varying surfactant concentrations. The results show that BSA–DTAB mixtures undergo thermally induced gelation at lower DTAB concentrations, where DTAB facilitates gelation. However, at higher DTAB concentrations, this gelation is completely suppressed. In contrast, SDS, a similarly charged anionic surfactant, inhibits BSA gelation at significantly lower concentrations. Notably, this suppression by SDS can be reversed through the addition of a nonionic surfactant (C12E10), highlighting the tunable nature of protein gelation through surfactant combinations. Reproduced from ref. 9 with permission from the Royal Society of Chemistry, copyright (2024) and ref. 48 with permission from the American Physical Society, copyright 2023. | ||
In order to understand the opposite role of SDS and DTAB, small-angle neutron scattering measurements were carried out on 4 wt% BSA in the absence and presence of 5 mM SDS or 5 mM DTAB [Fig. 7(a)]. The concentration of the surfactants (SDS/DTAB) is chosen in the specific binding region, where no significant changes in the protein structure are expected. One can observe a broad correlation peak appearing at about (Qp ∼ 0.035 Å−1) in pristine BSA solution, reflecting the presence of interacting charged molecules. Upon addition of SDS, the scattering data show a further decrease in the scattering intensity, particularly in the low Q region, suggesting evolution of the repulsive interaction among protein molecules. This can be understood in terms of specific binding of the surfactant monomers on the oppositely charged patches of the protein, thereby enhancing the overall electrostatic repulsion on the protein molecules. On the other hand, the scattering intensity increases in the low Q region for the BSA–DTAB system, indicating the enhanced attraction among BSA–DTAB complexes. Similar argument may here as well be used to understand the attractive nature of the system. The oppositely charged DTAB molecules strongly bind to the protein, thereby reducing the overall charge on the protein molecules. Such suppression of the charge results in a relatively attractive system compared to pure BSA solution. As a result, BSA–DTAB and BSA–SDS systems do and do not undergo gelation upon heating due to attractive and repulsive electrostatic interactions, respectively [Fig. 7(b)].9,134 This hypothesis has been verified by modelling the data and extracting overall potential among protein–surfactant complexes [Fig. 7(b)] as well as by measuring the effective zeta potential of the system.9
 | ||
| Fig. 7 (a) Small-angle neutron scattering (SANS) profiles of 4 wt% BSA solution at pH 5 and 25 °C, in the absence and presence of 5 mM surfactants, anionic (SDS) and cationic (DTAB). (b) Corresponding fitted interaction potentials derived from the SANS data. The data reveal that SDS and DTAB modulate inter-protein interactions in distinct ways. SDS enhances repulsive interaction among BSA molecules, while DTAB drives attractive interaction. The fitted potentials suggest that DTAB causes a more pronounced alteration in the total interaction potential compared to SDS, which is attributed to its stronger binding affinity toward BSA. Reproduced from ref. 9 with permission from the Royal Society of Chemistry, copyright 2024. | ||
Such tendency of promoting and suppressing the heat-induced gelation of protein solutions by the surfactant could also be reversed by different means. In the case of the BSA–DTAB system, the gel formation is completely inhibited upon increasing the surfactant concentration [Fig. 6(a)], substantially in the cooperative binding region (beyond the specific binding regime). In cooperative binding (Section 2.1), the surfactants are able to completely unfold the protein and form protein-decorated surfactant micelles along the unfolded protein chain (Section 5). In fact gelation of the protein (BSA)–surfactant (DTAB) system could be observed only for a limited concentaration range of DTAB at elevated temperatures [Fig. 6(b)].
It is interesting to note here that in spite the complete unfolding of the protein at higher DTAB concentrations, the gelation is inhibited. In general, the presence of denaturants is expected to promote the protein gelation, via increasing the exposure of the hydrophobic segments. However, counter-intuitive results here may be attributed to following two factors: (i) Enhanced inter-micellar electrostatic repulsion between micelles formed at different protein–surfactant complexes: such electrostatic repulsion usually stabilizes the pristine micellar solution, where increase in the temperature does not cause significant alterations in the micellar morphologies. (ii) Suppression of the hydrophobic attraction, by hiding the hydrophobic segments of the protein due to formation of micelle-like clusters of the surfactants (as discussed in Section 5): if we compare the role of SDS vs. DTAB, the suppression of the protein gelation is observed at much lower concentrations of SDS, suggesting a prominent role of electrostatic repulsion, along with hydrophobic. Nnyigide et al. have utilized molecular dynamics simulations to reveal that the water density around the protein is substantially reduced in the presence of SDS, leading to the inhibition of the protein gelation. Such understanding can also be correlated to the reduced hydrophobic attraction, responsible for the protein gelation.137 The authors have attributed the stabilizing effect of SDS to bridging of the non-covalent interactions between polar amino acid and non-polar residues in the protein. The contrasting effect of surfactant concentration at low and high concentrations on protein gelation has also been observed in the case of SDS, where the suppression of the protein gelation in the presence of lower concentrations of SDS is reversed at higher SDS concentrations.137
As we have seen in Section 4.2.1, the ionic surfactant driven unfolding of protein can be reversed by addition of a nonionic surfactant, along with an ionic surfactant. The same strategy can also be adopted in reversing the effect of ionic surfactants on protein gelation. It has been shown that the presence of nonionic C12E10 surfactant along with SDS reverses SDS-driven inhibition of gelation and the solution state of the BSA–SDS system undergoes gelation [Fig. 6(a)]. However, it is noted that the amount of nonionic surfactant required to neutralize the impact of SDS is at least three times more than the SDS.48 The role of non-ionic surfactant in suppressing the effect of SDS on protein gelation is similar to its role in protein refolding.
Effect of surfactants on gelation of fibrous protein has also been explored in a number of studies.138–141 The interaction of surfactant with silk fibroin protein has been utilized to accelerate sol to gel transition in an aqueous solution of silk fibroin (SF), which otherwise takes several days to complete. It has been shown that the SDS can effectively modulate the SF gelation at an optimal SDS-to-silk fibroin concentration. Beyond this concentration, gelation slows down. SDS interacts specifically with tyrosine- and valine-rich segments of SF, driving the aggregation of interspersed hydrophobic segments. However, at higher SDS concentrations, the micellar clusters formed along the SF chain are spaced further apart, hindering the proximity of hydrophobic segments necessary for β-sheet structure formation. Consequently, a reduction in β-sheet content is observed at elevated SDS levels. This interaction mechanism between SF and SDS differs notably from SDS interactions with other proteins.140 Another interesting study introduces a new approach of integrating hydrophobic interactions as sacrificial bonds to develop silk fibroin-based hydrophobic-association hydrogels within an alginate ionic network.142 This design significantly enhances the hydrogels' mechanical extensibility, strength, toughness and grants self-recovery as well as self-healing capabilities through reversible hydrophobic interactions at room temperature without external stimuli. The hydrophobic interaction system utilizes stearyl methacrylate (C18M) and amphiphilic regenerated silk fibroin (RSF). Mechanical tests and rheometry demonstrate that hydrophobic interactions act as sacrificial bonds, breaking preferentially under stress and dissipating energy effectively, thereby boosting the hydrogels' overall mechanical performance. Similarly, interactions with surfactants can also be utilized to modify the gel formation and other rheological properties of other fibrous proteins like collagen, elastin etc.143,144 Overall, this section shows the profound ability of surfactants to modify the protein gelation for desired sol–gel transitions as well as rheological properties as desired for different applications.
7. Applications of protein–surfactant complexes
Protein–surfactant complexes possess profound applications in a wide variety of fields including but not limited to pharmaceuticals, food industry, cosmetics and biotechnology (Fig. 8). This section provides a glimpse of these applications and some recent updates based on recent examples from the literature. We start the discussion with the food related applications, where the protein forms an integral part of the food while surfactants contribute in the form of different phases of food materials such as dispersions, emulsions, foams, gels, etc.145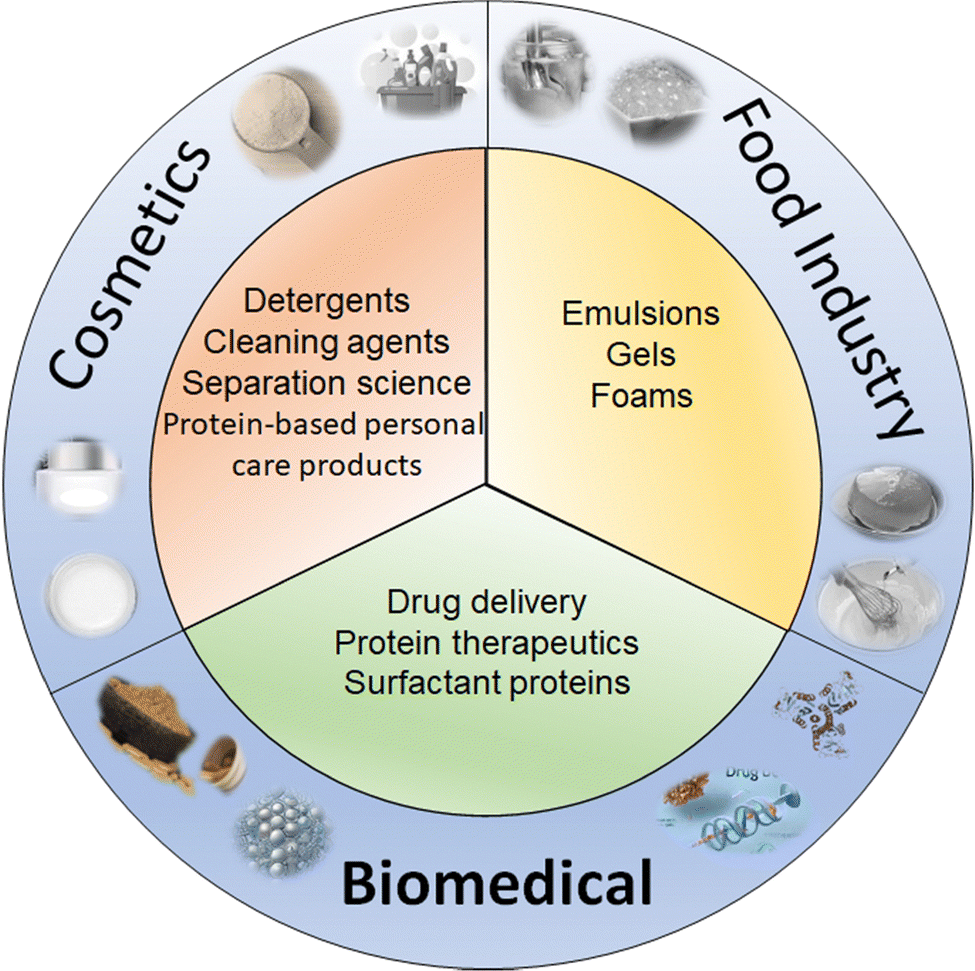 | ||
| Fig. 8 Schematic representation illustrating key application areas of protein–surfactant complexes. The diagram highlights three major sectors where these complexes exhibit significant functional relevance. | ||
7.1. Food industry
The emulsions, such as oil-in-water, formulate an esssential ingredient of many food items, such as sauces, soups, milk, butter, cream, mayonnaise, beverages etc. Surfactants play a critical role in stabilizing these food emulsions, preventing undesirable effects like phase separation, coalescence, and ripening during production or storage. However, protein–surfactant complexes have been identified to be even more useful as such emulsifiers. The most familiar examples can be combination of lecithin surfactant, naturally present in egg yolk, with various milk proteins to obtain various food products, e.g., mayonnaise, cream dressings, deserts, etc.The emulsions stabilized by combination of soy protein as well as tea saponin surfactant have been shown to display synergistic effects like improved oxidative stability of the emulsion and prevention of release of free fatty acids during digestion.146 These complexes have also been identified as a possible candidate for enhancing the nutritional values of the food. As an example, pea protein isolate (PPI) and different surfactants (rhamnolipid (Rha), tea saponin (TS) and ethyl lauroyl arginate hydrochloride (ELA)) have been prepared to encapsulate curcumin, which is known for its antioxidant, anti-inflammatory, antibacterial, and anti-diabetic properties, however, rarely used in commercial food and/or beverages owing to its poor stability and low water solubility.147–150
Similarly, pea protein isolate-high methoxyl pectin-rhamnolipid (PPI-HMP-Rha) complexes have been developed to incorporate curcumin and resveratrol, enhancing their stability and controlled release. Resveratrol, alongside curcumin, offers antioxidant, anti-inflammatory, and cardiovascular benefits. The complexes, with high rhamnolipid content, improve the stability of these nutraceuticals and delay their release during in vitro digestion, offering potential for advanced delivery systems.147
7.2. Medicine
Another key area, which should be specifically discussed for applications of protein–surfactant complexes, is medicine. One of the important aspects is the protein therapeutics, where aggregation and surface-induced denaturation pose major challenges in therapeutic protein production, formulation, and administration. Surfactant interaction with proteins is being widely used in processing and formulation, mitigating these issues by coating interfaces and forming protein–surfactant associations, effectively reducing adsorption loss and aggregation, with their efficacy depending on their mechanisms and protein properties.17,151,152 Another intriguing area of research involves studying surfactant proteins and their interactions with other proteins, lipids, and biomolecules. For instance, the pulmonary surfactant, a complex blend of surface-active lipids and proteins in the alveolar lining, plays a vital role in gas exchange. While its functions are well-recognized, the molecular-scale interactions of its components remain poorly understood. Experimental and simulation-based studies are actively exploring these interactions, offering insights into their significance in lung immunity and homeostasis.153 Protein–surfactant complexes have also been utilized as drug delivery vehicles, due to biocompatibility and acceptability of the proteins in biological milieu and drug encapsulation ability of the surfactant.154 In fact, the pulmonary surfactant, having inherent association of protein and surfactant-like properties, has also been shown to be a potential drug delivery vehicle.155–157 Protein interaction with surfactants has been utilized to enhance encapsulation of proteins in calcium alginate beads and their controlled release in the intestinal environment.158 It has been further shown that the ternary nanocarriers combining proteins, biosurfactants, and polysaccharides enhance cargo loading capacities and carrier properties. Systems like zein-propylene glycol alginate-rhamnolipid carriers demonstrate stability across varying pH and ionic conditions, improving encapsulation and bioaccessibility of antioxidants such as curcumin, resveratrol, and coenzyme Q10.159–162 Similarly, carriers made with pea protein isolate and high methoxyl pectin, modified by pH adjustments, efficiently encapsulate these compounds when paired with surfactants like rhamnolipid, tea saponin, or ethyl lauroyl arginate hydrochloride. Rhamnolipids outperform others in encapsulation efficiency, thermal resistance, and cargo protection, making them preferred surfactants for these systems.7.3. Cosmetics
Soy protein interacts with rhamnolipids primarily via hydrophobic forces, resulting in a more flexible protein conformation. This complex enhances foaming properties, foam stability, surface tension reduction, and ordered assembly, making it an effective natural foaming agent for body wash and cosmetic applications.163 Surface active complexes between keratin polypeptides and ionic surfactants have been prepared and studied. It has been shown that SDS aids in reducing disulfide bond formation and improving keratin solubility, while purification ensures complete SDS removal. The formation of keratin–surfactant complexes significantly reduced surface tension, indicating their potential in delivering keratin via surface-active formulations.164In summary, this section highlights a few recent examples to illustrate the versatility of protein–surfactant complexes in driving advancements in food, medicine, cosmetics, and separation science. Their ability to leverage hydrophobic and electrostatic interactions makes them indispensable for innovative and sustainable solutions across industries.
8. Conclusions, emerging trends and future scope
The interaction between proteins and surfactants remains a crucial area of research due to its broad applications in food science, cosmetics, and biomedicine. This review highlights the fundamental mechanisms by which surfactants influence protein structure. It outlines the binding energy curve, the key interactions governing surfactant-induced protein unfolding, and their dependence on various physicochemical parameters. Additionally, this review discusses how a combination of ionic and nonionic surfactants can facilitate protein refolding. The preferential binding of the ionic surfactant to the nonionic surfactant leads to the detachment of surfactants from the protein, ultimately restoring its folded state.Furthermore, this article explores the ability of surfactants to modify heat-induced protein gelation by altering the underlying interactions that drive the gelation process. Such insights not only enhance our understanding of protein–surfactant interactions but also create opportunities to design protein-based systems with improved stability, functionality, and performance. Toward the end, recent advances in protein–surfactant applications, ranging from the food industry to advanced drug delivery systems, are reviewed. This article also presents a roadmap for developing innovative strategies to further advance this field. Table 1 summarizes the key issues highlighted in this review article.
| Key issue | Sub-issue | Proteins under investigation | Surfactants under investigation | Relevant ref. | Common techniques used to probe the issue |
|---|---|---|---|---|---|
| Surfactant binding to the protein | Specific binding non-cooperative cooperative binding saturation | BSA, bovine acyl-coenzyme-A-binding protein (ACBP), ubiquitin | SDS, CnTAB, | 3, 13, 15, 24, 35 and 112 | SANS, SAXS, isothermal titration calorimetry (ITC), dynamic light scattering (DLS), capillary electrophoresis (CE), circular dichroism (CD) spectroscopy simulations |
| Major interactions between proteins and surfactants | Hydrophobic interaction | Human serum albumin (HSA), BSA, bovine α-lactalbumin (α-LA) | Surfactant–cobalt(III) complexes, CnTAB, | 16, 17, 25 and 26 | Simulations, circular dichroism (CD), fluorescence spectroscopy, ITC, nuclear magnetic resonance (NMR), SAXS, UV-spectroscopy, conductivity, surface tension |
| Electrostatic interaction | Lysozyme | Octyl sulfate | 21 | ||
| Hydrogen bonding | BSA | Sodium N-lauroylsarcosinate sodium N-lauroylglycinate, SDS, CTAB, octyl glucoside (OG), 3-[hexadecyl(dimethyl)ammonio]-1-propanesulfonate (HPS), | 28 and 29 | ||
| Non-covalent interactions | BSA | Sodium dodecyl benzene sulfonate (SDBS) | 30 | ||
| Parameters affecting protein–surfactant interactions | pH of solution | BSA, HSA | SDS, gemini surfactants, ionic liquids | 27, 54 and 84 | Simulations, circular dichroism (CD), fluorescence spectroscopy, ITC, nuclear magnetic resonance (NMR), SAXS, UV-spectroscopy, conductivity, surface tension |
| Ionic strength of solution | BSA | SDS | 76 | ||
| Type of surfactant | BSA, HSA, casein | SDS, CnTAB, C12E10, gemini surfactants, biosurfactants (rhamnolipid [RL] and lactone sophorolipid [SL]) | 11, 12, 29, 32, 53 and 165 | ||
| Type of protein | Globular protein, fibroin protein, membrane protein | SDS, octyl glucose neopentyl glycol (OGNG), lauryl maltose neopentyl glycol (LMNG), | 11, 12, 50, 140 and 166 | ||
| Temperature | BSA | Gemini surfactants | 54 | ||
| Chain length of surfactant | BSA, bovine α-lactalbumin (α-LA) | CnTAB | 12 and 26 | ||
| Presence of cosolvents | BSA | Cetyl pyridinium chloride (CPC), SDBS | 30 and 31 | ||
| Unfolding of protein | — | BSA, ubiquitin, α-synuclein, HSA | SDS, CnTAB, ionic liquids | 11, 12, 22, 77, 79 and 84 | UV-Vis spectroscopt, CD spectroscopy, SANS, SAXS, fluorescence spectroscopy, high-performance liquid chromatography (HPLC) |
| Refolding of protein | Refolding by mixed micelles | BSA, HAS, human serum transferrin (Fe-hTF) | SDS, CnTAB, C12E10, tri-block copolymers (F127 and P123), bile salts (sodium cholate and sodium deoxycholate) | 11, 12, 87, 88 and 167 | |
| Refolding by reverse micelles | bovine pancreatic ribonuclease | AOT | 91–93 | ||
| Refolding by dilution | BSA, carbonic anhydrase, and beta-galactosidase | SDS, C16TAB | 94 and 95 | ||
| Refolding by dialysis | Human interferon-γ (rhIFN-γ), human AFP (rhAFP) | Tween 20 | 99 and 100 | ||
| Models of protein surfactant complexes | Beads-on-a-string | BSA | SDS | 11 and 15 | |
| Protein-decorated micelles | BSA, cellulase Cel7b | Mixed micelles of SDS and DTAB, SDS, | 80 and 120 | ||
| Flexible capped helical cylindrical micelle | Water soluble protein, membrane protein | SDS | 168 | ||
| Effect of surfactant on protein gelation | Promoting protein gelation | BSA, silk fibroin | DTAB, C12E10, SDS | 9, 48, 138, 140 and 166 | Rheology, SANS, DLS, simulations |
| Suppressing protein gelation | BSA | SDS, DTAB | 9, 48, 137 and 169 | ||
| Application of protein–surfactant complexes | Food industry | Soy protein, pea protein | Tea saponin, rhamnolipid (Rha), ethyl lauroyl arginate hydrochloride (ELA)) | 146 and 147 | |
| Medical industry | Surfactant proteins, BSA | Pulmonary surfactants, SDS | 154, 155, 157 and 158 | ||
| Cosmetics | Soy protein, keratin | Rhamnolipid, SDS | 163 and 164 |
Despite significant progress in understanding protein–surfactant interactions, several open questions persist, driving emerging trends in this evolving research field. One area of growing interest is the controlled refolding of proteins, which holds great promise for recovering bioactivity from denatured proteins. This approach is particularly relevant to therapeutic protein formulation and stabilization. An intriguing direction involves exploring whether proteins denatured by agents such as urea or heat can also be effectively refolded using surfactant-mediated strategies.
The development of time-resolved, in situ experimental techniques (e.g., small-angle scattering spectroscopy) is enabling researchers to monitor unfolding, binding, and refolding dynamics in real time, offering insights into intermediates and transient structural states.170 Complementarily, the integration of artificial intelligence (AI) and machine learning (ML) into computational modeling is opening new avenues for predicting protein–surfactant interactions, surfactant design, and formulation optimization.171
Another exciting aspect is the development of responsive or “smart” surfactants that enable the design of protein–surfactant complexes with tunable and stimuli-responsive properties.172 Concurrently, surfactants are being investigated for their role in modulating protein aggregation and amyloid fibrillation, an area with direct implications for combating neurodegenerative diseases.173,174 The effect of surfactants on other phases of protein solutions such as gelation, coacervation, and crystallization under diverse environmental conditions could provide valuable insights for designing adaptable, high-performance systems tailored to specific industrial needs.
Looking ahead, future research is likely to focus also on mimicking naturally occurring surfactants, such as protein-based or biosurfactants, using advanced chemical synthesis and bioengineering approaches. The development of sustainable, biocompatible, and eco-friendly surfactants to replace synthetic ones will also be crucial to align with global sustainability initiatives. Moreover, integrating protein–surfactant systems with other biopolymers or nanoparticles may enable the fabrication of multifunctional materials with applications in tissue engineering, controlled drug delivery, and innovative food formulations.
Data availability
This being a review article, no new data were created or analysed during this study. Data sharing is not applicable to this article.Conflicts of interest
There are no conflicts to declare.References
- I. Fatma, V. Sharma, R. C. Thakur and A. Kumar, J. Mol. Liq., 2021, 341, 117344 CrossRef CAS
.
- O. Kirk, T. V. Borchert and C. C. Fuglsang, Curr. Opin. Biotechnol, 2002, 13, 345–351 CrossRef CAS PubMed
- D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 562–591 CrossRef CAS PubMed
- D. E. Otzen, J. N. Pedersen, H. Ø. Rasmussen and J. S. Pedersen, Adv. Colloid Interface Sci., 2022, 308, 102754 CrossRef CAS PubMed
- M. López Hernández, J. S. Pedersen and D. E. Otzen, Curr. Opin. Colloid Interface Sci., 2023, 68, 101746 CrossRef
- A. Javadi, S. Dowlati, S. Shourni, R. Miller, M. Kraume, K. Kopka and K. Eckert, Adv. Colloid Interface Sci., 2022, 301, 102601 CrossRef CAS PubMed
- N. D. Savić, D. E. Salazar Marcano, S. Swinnen, A. Mullaliu and T. N. Parac-Vogt, ACS Appl. Nano Mater., 2022, 5, 17159–17172 CrossRef
- D. Saha, S. Kumar, J. P. Mata, A. E. Whitten and V. K. Aswal, Phys. Chem. Chem. Phys., 2023, 25, 22130–22144 RSC
- S. Kumar and V. K. Aswal, Soft Matter, 2024, 20, 5553–5563 RSC
- J. N. Pedersen, J. Lyngsø, T. Zinn, D. E. Otzen and J. S. Pedersen, Chem. Sci., 2020, 11, 699–712 RSC
- S. Mehan, V. K. Aswal and J. Kohlbrecher, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2015, 92, 032713 CrossRef PubMed
- D. Saha, D. Ray, J. Kohlbrecher and V. K. Aswal, ACS Omega, 2018, 3, 8260–8270 CrossRef CAS PubMed
- M. N. Jones, Chem. Soc. Rev., 1992, 21, 127–136 RSC
- M. N. Jones, Biochem. J., 1975, 151, 109–114 CrossRef CAS PubMed
- N. J. Turro, X.-G. Lei, K. P. Ananthapadmanabhan and M. Aronson, Langmuir, 1995, 11, 2525–2533 CrossRef CAS
- J. Bergfreund, P. Bertsch and P. Fischer, Curr. Opin. Colloid Interface Sci., 2021, 56, 101509 CrossRef CAS
- T. A. Khan, H.-C. Mahler and R. S. K. Kishore, Eur. J. Pharm., 2015, 97, 60–67 CAS
- J. A. Reynolds and C. Tanford, J. Biol. Chem., 1970, 245, 5161–5165 CrossRef CAS PubMed
- J. A. Reynolds and C. Tanford, Proc. Natl. Acad. Sci. U. S. A., 1970, 66, 1002–1007 CrossRef CAS PubMed
- G. Ratkeviciute, B. F. Cooper and T. J. Knowles, Biochem. Soc. Trans., 2021, 49, 1763–1777 CrossRef CAS PubMed
- A. Stenstam, D. Topgaard and H. Wennerström, J. Phys. Chem. B, 2003, 107, 7987–7992 CrossRef CAS
- G. F. Schneider, B. F. Shaw, A. Lee, E. Carillho and G. M. Whitesides, J. Am. Chem. Soc., 2008, 130, 17384–17393 CrossRef CAS PubMed
- B. F. Shaw, G. F. Schneider, H. Arthanari, M. Narovlyansky, D. Moustakas, A. Durazo, G. Wagner and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 17681–17695 CrossRef CAS PubMed
- H. G. Mortensen, D. E. Otzen and J. S. Pedersen, J. Colloid Interface Sci., 2021, 596, 233–244 CrossRef CAS PubMed
- S. Veeralakshmi, G. Sabapathi, S. Nehru, P. Venuvanalingam and S. Arunachalam, Colloids Surf., B, 2017, 153, 85–94 CrossRef CAS PubMed
- Y. Sun, P. L. Oseliero Filho, Y. Song, Z. Wang, H. Ji and C. L. P. Oliveira, Colloids Surf., B, 2023, 230, 113490 CrossRef CAS PubMed
- R. Srivastava and Md. S. Alam, Int. J. Biol. Macromol., 2019, 122, 72–81 CrossRef CAS PubMed
- S. Ghosh and J. Dey, J. Phys. Chem. B, 2015, 119, 7804–7815 CrossRef CAS PubMed
- O. S. Nnyigide, S.-G. Lee and K. Hyun, Sci. Rep., 2019, 9, 10643 CrossRef PubMed
- V. Sharma, O. Yañez, M. Alegría-Arcos, A. Kumar, R. C. Thakur and P. Cantero-López, J. Chem. Thermodyn., 2020, 142, 106022 CrossRef CAS
- V. Sharma, O. Yañez, C. Zúñiga, A. Kumar, G. Singh and P. Cantero-López, Chem. Phys. Lett., 2020, 747, 137349 CrossRef CAS
- Q. Tian, L. Lai, Z. Zhou, P. Mei, Q. Lu, Y. Wang, D. Xiang and Y. Liu, J. Agric. Food Chem., 2019, 67, 6336–6349 CrossRef CAS PubMed
- S. Denzinger, Adv. Mater., 1996, 8, 367 CrossRef
-
C. Tanford, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, 1970, vol. 24, pp. 1–95 Search PubMed
- K. K. Andersen, C. L. Oliveira, K. L. Larsen, F. M. Poulsen, T. H. Callisen, P. Westh, J. S. Pedersen and D. Otzen, J. Mol. Biol., 2009, 391, 207–226 CrossRef CAS PubMed
- A. Malhotra and J. N. Coupland, Food Hydrocolloids, 2004, 18, 101–108 CrossRef CAS
- J. M. Rodríguez Patino, Ma. R. Rodríguez Niño and C. Carrera Sánchez, Curr. Opin. Colloid Interface Sci., 2007, 12, 187–195 CrossRef
- R. Miller, V. B. Fainerman, M. E. Leser and M. Michel, Curr. Opin. Colloid Interface Sci., 2004, 9, 350–356 CrossRef CAS
- J. Chayen, Cell Biochem. Funct., 1996, 14, 75 CrossRef CAS
- M. Bogunia and M. Makowski, J. Phys. Chem. B, 2020, 124, 10326–10336 CrossRef CAS PubMed
- A. Bartosik, M. Wiśniewska and M. Makowski, J. Phys. Org. Chem., 2015, 28, 10–16 CrossRef CAS
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef
- B. A. Fong, W.-Y. Wu and D. W. Wood, Protein Expression Purif., 2009, 66, 198–202 CrossRef CAS PubMed
- H. A. Scheraga, J. Biomol. Struct. Dyn., 1998, 16, 447–460 CrossRef CAS PubMed
- E. Sobolewski, M. Makowski, C. Czaplewski, A. Liwo, S. Ołdziej and H. A. Scheraga, J. Phys. Chem. B, 2007, 111, 10765–10774 CrossRef CAS PubMed
- F. W. F. Wong, A. B. Ariff and D. C. Stuckey, Crit. Rev. Biotechnol., 2018, 38, 31–46 CrossRef CAS PubMed
-
M. J. Rosen and R. J. Kunjappu, Surfactants and Interfacial Phenomena, 2012, pp. 123–201 Search PubMed
- S. Kumar, D. Saha and V. K. Aswal, Phys. Rev. Mater., 2023, 7, 015601 CrossRef CAS
-
K. Holmberg, B. Jönsson, B. Kronberg and B. Lindman, Surfactants and Polymers in Aqueous Solution, John Wiley & Sons, Ltd, Chichester, UK, 2002, pp. 305–315 Search PubMed
- H. J. Lee, H. S. Lee, T. Youn, B. Byrne and P. S. Chae, Chem, 2022, 8, 980–1013 CAS
- S. Duangjit, B. Pamornpathomkul, P. Opanasopit, T. Rojanarata, Y. Obata, K. Takayama and T. Ngawhirunpat, Int. J. Nanomed., 2014, 9, 2005–2017 CrossRef PubMed
- H. Liu, Y. Chang, Y. Li, C. Cao and R. Li, Molecules, 2024, 29, 619 CrossRef CAS PubMed
- S. Tardioli, A. Bonincontro, C. L. Mesa and R. Muzzalupo, J. Colloid Interface Sci., 2010, 347, 96–101 CrossRef CAS PubMed
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, Biomacromolecules, 2009, 10, 2508–2514 CrossRef CAS PubMed
- T. Yin, M. Qin and W. Shen, Colloids Surf., A, 2014, 461, 22–29 CrossRef CAS
- L. Guerrero-Hernández, H. I. Meléndez-Ortiz, G. Y. Cortez-Mazatan, S. Vaillant-Sánchez and R. D. Peralta-Rodríguez, Int. J. Mol. Sci., 2022, 23, 1798 CrossRef PubMed
- W. Gospodarczyk and M. Kozak, Colloid Polym. Sci., 2015, 293, 2855–2866 CrossRef CAS PubMed
- S. Halder, R. Aggrawal, S. Jana and S. K. Saha, J. Photochem. Photobiol., B, 2021, 225, 112351 CrossRef CAS PubMed
- V. Vagenende, M. G. S. Yap and B. L. Trout, Biochemistry, 2009, 48, 11084–11096 CrossRef CAS PubMed
- M. Hirai, S. Ajito, M. Sugiyama, H. Iwase, S. Takata, N. Shimizu, N. Igarashi, A. Martel and L. Porcar, Biophys. J., 2018, 115, 313–327 CrossRef CAS PubMed
- X. Chen, H. Zhang, Y. Hemar, N. Li and P. Zhou, Food Chem., 2020, 308, 125596 CrossRef CAS PubMed
-
W. Kauzmann, in Advances in Protein Chemistry, ed. C. B. Anfinsen, M. L. Anson, K. Bailey and J. T. Edsall, Academic Press, 1959, vol. 14, pp. 1–63 Search PubMed
- L. Hua, R. Zhou, D. Thirumalai and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16928–16933 CrossRef CAS PubMed
- R. Zangi, R. Zhou and B. J. Berne, J. Am. Chem. Soc., 2009, 131, 1535–1541 CrossRef CAS PubMed
- A. P. Singh Raman, A. A. Muhammad, H. Singh, T. Singh, Z. Mkhize, P. Jain, S. K. Singh, I. Bahadur and P. Singh, ACS Omega, 2022, 7, 47471–47489 CrossRef CAS PubMed
- C. Bernhard, S. J. Roeters, J. Franz, T. Weidner, M. Bonn and G. Gonella, Phys. Chem. Chem. Phys., 2017, 19, 28182–28188 RSC
- M. Martínez-Negro, D. Russo, S. Prévost, J. Teixeira, S. Morsbach and K. Landfester, Biomacromolecules, 2022, 23, 4282–4288 CrossRef PubMed
- R. Rajan, S. Ahmed, N. Sharma, N. Kumar, A. Debas and K. Matsumura, Mater. Adv., 2021, 2, 1139–1176 RSC
- S. De, A. Girigoswami and S. Das, J. Colloid Interface Sci., 2005, 285, 562–573 CrossRef CAS PubMed
- A. Valstar, M. Vasilescu, C. Vigouroux, P. Stilbs and M. Almgren, Langmuir, 2001, 17, 3208–3215 CrossRef CAS
- P. N. Moore, S. Puvvada and D. Blankschtein, Langmuir, 2003, 19, 1009–1016 CrossRef CAS
- M. Bayliss, J. Infect. Dis., 1936, 59, 131–136 CrossRef CAS
- A. Yonath, A. Sielecki, J. Moult, A. Podjarny and W. Traub, Biochemistry, 1977, 16, 1413–1417 CrossRef CAS PubMed
- A. K. Bordbar, A. A. Saboury, M. R. Housaindokht and A. A. Moosavi-Movahedi, J. Colloid Interface Sci., 1997, 192, 415–419 CrossRef CAS PubMed
- F. Mallamace, C. Corsaro, D. Mallamace, S. Vasi, C. Vasi, P. Baglioni, S. V. Buldyrev, S.-H. Chen and H. E. Stanley, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3159–3163 CrossRef CAS PubMed
- B. Shweitzer, D. Zanette and R. Itri, J. Colloid Interface Sci., 2004, 277, 285–291 CrossRef CAS PubMed
- S. F. Santos, D. Zanette, H. Fischer and R. Itri, J. Colloid Interface Sci., 2003, 262, 400–408 CrossRef CAS PubMed
- J. C. Gimel and W. Brown, J. Chem. Phys., 1996, 104, 8112–8117 CrossRef CAS
- L. Giehm, C. L. P. Oliveira, G. Christiansen, J. S. Pedersen and D. E. Otzen, J. Mol. Biol., 2010, 401, 115–133 CrossRef CAS PubMed
- D. Saha, D. Ray, S. Kumar, J. Kohlbrecher and V. K. Aswal, Soft Matter, 2021, 17, 6972–6984 RSC
- R.-C. Lu, A.-N. Cao, L.-H. Lai and J.-X. Xiao, Colloids Surf., B, 2008, 64, 98–103 CrossRef CAS PubMed
- D. E. Otzen, Biophys. J., 2002, 83, 2219–2230 CrossRef CAS PubMed
- N. El Kadi, N. Taulier, J. Y. Le Huérou, M. Gindre, W. Urbach, I. Nwigwe, P. C. Kahn and M. Waks, Biophys. J., 2006, 91, 3397–3404 CrossRef CAS PubMed
- R. Ravikanth Reddy, D. Saha, A. Pan, V. K. Aswal, S. S. Mati, S. P. Moulik and B. V. N. Phani Kumar, Langmuir, 2023, 39, 3729–3741 CrossRef CAS PubMed
- F. E. Cohen and J. W. Kelly, Nature, 2003, 426, 905–909 CrossRef CAS PubMed
- F. U. Hartl, Annu. Rev. Biochem., 2017, 86, 21–26 CrossRef CAS PubMed
- R. Yadav, A. Nandy, A. Bisoi and S. Mukherjee, Langmuir, 2024, 40, 6172–6186 CrossRef CAS PubMed
- R. Mondal, N. Ghosh, B. K. Paul and S. Mukherjee, Langmuir, 2018, 34, 896–903 CrossRef CAS PubMed
- R. Modi, L. Khamari, A. Nandy and S. Mukherjee, Spectrochim. Acta, Part A, 2019, 216, 52–60 CrossRef CAS PubMed
- X. Liu, Y. Liu, Z. Zhang, F. Huang, Q. Tao, R. Ma, Y. An and L. Shi, Chem. – Eur. J., 2013, 19, 7437–7442 CrossRef CAS PubMed
- A. J. Hagen, T. A. Hatton and D. I. C. Wang, Biotechnol. Bioeng., 1990, 35, 955–965 CrossRef CAS PubMed
- A. J. Hagen, T. A. Hatton and D. I. C. Wang, Biotechnol. Bioeng., 1990, 35, 966–975 CrossRef CAS PubMed
- M. Goto, Y. Hashimoto, T. Fujita, T. Ono and S. Furusaki, Biotechnol. Prog., 2000, 16, 1079–1085 CrossRef CAS PubMed
- U. Anand, C. Jash and S. Mukherjee, Phys. Chem. Chem. Phys., 2011, 13, 20418–20426 RSC
- G. Azadi, A. Chauhan and A. Tripathi, Protein Sci., 2013, 22, 1258–1265 CrossRef CAS PubMed
- S. Yamaguchi, E. Yamamoto, T. Mannen, T. Nagamune and T. Nagamune, Biotechnol. J., 2013, 8, 17–31 CrossRef CAS PubMed
- D. L. Daugherty, D. Rozema, P. E. Hanson and S. H. Gellman, J. Biol. Chem., 1998, 273, 33961–33971 CrossRef CAS PubMed
- H. Yamaguchi, M. Miyazaki, M. P. Briones-Nagata and H. Maeda, J. Biochem., 2010, 147, 895–903 CrossRef CAS PubMed
- S. M. Kelly and N. C. Price, Biochem. J., 1991, 275, 745–749 CrossRef CAS PubMed
- S. D. Webb, J. L. Cleland, J. F. Carpenter and T. W. Randolph, J. Pharm. Sci., 2002, 91, 543–558 CrossRef CAS PubMed
- S. S. J. Leong and A. P. J. Middelberg, Food Bioprod. Process., 2006, 84, 9–17 CrossRef CAS
- G. Stempfer, B. Höll-Neugebauer and R. Rudolph, Nat. Biotechnol., 1996, 14, 329–334 CrossRef CAS PubMed
- Z. Gu, Z. Su and J.-C. Janson, J. Chromatogr. A, 2001, 918, 311–318 CrossRef CAS PubMed
- S. Kumar, I. Yadav, D. Ray, S. Abbas, D. Saha, V. K. Aswal and J. Kohlbrecher, Biomacromolecules, 2019, 20, 2123–2134 CrossRef CAS PubMed
- S. Kumar, D. Saha, D. Ray, S. Abbas and V. K. Aswal, Phys. Rev. E, 2021, 104, L012603 CrossRef CAS PubMed
- J.-H. Seo, R. Matsuno, Y. Lee, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 4859–4867 CrossRef CAS PubMed
- R. Rajan and K. Matsumura, Sci. Rep., 2017, 7, 45777 CrossRef CAS PubMed
- D. Saha, S. Kumar, D. Ray, J. P. Mata, A. E. Whitten and V. K. Aswal, Soft Matter, 2022, 18, 434–445 RSC
- Y. Sun, P. L. O. Filho, J. C. Bozelli, J. Carvalho, S. Schreier and C. L. P. Oliveira, Soft Matter, 2015, 11, 7769–7777 RSC
- D. E. Otzen, P. Sehgal and P. Westh, J. Colloid Interface Sci., 2009, 329, 273–283 CrossRef CAS PubMed
- J. Neumann, N. Klein, D. E. Otzen and D. Schneider, Arch. Biochem. Biophys., 2014, 564, 281–296 CrossRef CAS PubMed
- D. Winogradoff, S. John and A. Aksimentiev, Nanoscale, 2020, 12, 5422–5434 RSC
- S.-H. Chen and J. Teixeira, Phys. Rev. Lett., 1986, 57, 2583–2586 CrossRef CAS PubMed
- X. H. Guo, N. M. Zhao, S. H. Chen and J. Teixeira, Biopolymers, 1990, 29, 335–346 CrossRef CAS PubMed
- M. Jones and A. Wilkinson, Biochem. J., 1976, 153, 713–718 CrossRef CAS PubMed
- U. Anand and S. Mukherjee, Serum Albumin, 2013, 1830, 5394–5404 CAS
- T. K. Das, S. Mazumdar and S. Mitra, Eur. J. Biochem., 1998, 254, 662–670 CrossRef CAS PubMed
- E. L. Gelamo, C. H. T. P. Silva, H. Imasato and M. Tabak, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1594, 84–99 CrossRef CAS PubMed
- S. Chodankar, V. K. Aswal, P. A. Hassan and A. G. Wagh, Phys. B, 2007, 398, 112–117 CrossRef CAS
- H. Ø. Rasmussen, J. J. Enghild, D. E. Otzen and J. S. Pedersen, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129434 CrossRef CAS PubMed
- W. Burchard, K. Kajiwara and D. H. Whiffen, Proc. R. Soc. London, Ser. A, 1997, 316, 185–199 Search PubMed
- J. N. Pedersen, H. K. S. Frislev, J. S. Pedersen and D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140505 Search PubMed
- K. K. Andersen and D. E. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 2338–2345 CrossRef CAS PubMed
- K. K. Andersen, B. S. Vad, S. Roelants, I. N. A. van Bogaert and D. E. Otzen, Front. Microbiol., 2016, 7, 1711 Search PubMed
- S. Mondal, A. Banerjee and B. Das, J. Mol. Liq., 2020, 301, 112450 Search PubMed
- R.-C. Lu, A.-N. Cao, L.-H. Lai, B.-Y. Zhu, G.-X. Zhao and J.-X. Xiao, Colloids Surf., B, 2005, 41, 139–143 CrossRef CAS PubMed
- S. Kumar, D. Saha, S. Takata, V. K. Aswal and H. Seto, Appl. Phys. Lett., 2021, 118, 153701 CrossRef CAS
- S. K. Chaturvedi, J. M. Khan, M. K. Siddiqi, P. Alam and R. H. Khan, Int. J. Biol. Macromol., 2016, 83, 315–325 CrossRef CAS PubMed
- C. Camilloni, D. Bonetti, A. Morrone, R. Giri, C. M. Dobson, M. Brunori, S. Gianni and M. Vendruscolo, Sci. Rep., 2016, 6, 28285 CrossRef CAS PubMed
- M. Morand, A. Dekkari, F. Guyomarc’h and M.-H. Famelart, Int. Dairy J., 2012, 25, 103–111 CrossRef CAS
- Y. Cheng, C. He, C. Xiao, J. Ding, X. Zhuang, Y. Huang and X. Chen, Biomacromolecules, 2012, 13, 2053–2059 CrossRef CAS PubMed
- B. Chen, J. Guo, Y. Xie, K. Zhou, P. Li and B. Xu, Food Chem., 2021, 341, 128274 CrossRef CAS PubMed
- C. Chen, Y. Gu, L. Deng, S. Han, X. Sun, Y. Chen, J. R. Lu and H. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 14360–14368 CrossRef CAS PubMed
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS PubMed
- Y. Mao, M. Huang, J. Bi, D. Sun, H. Li and H. Yang, Food Hydrocolloids, 2023, 134, 108031 CrossRef CAS
- J. Du, Y. You, R. L. Reis, S. C. Kundu and J. Li, Adv. Colloid Interface Sci., 2023, 318, 102950 CrossRef CAS PubMed
- O. S. Nnyigide and K. Hyun, Food Hydrocolloids, 2020, 103, 105656 CrossRef CAS
- J. H. Park, M. H. Kim, L. Jeong, D. Cho, O. H. Kwon and W. H. Park, J. Sol-Gel Sci. Technol., 2014, 71, 364–371 CrossRef CAS
- N. Chantong, S. Damrongsakkul and J. Ratanavaraporn, J. Surfactants Deterg., 2019, 22, 1395–1407 CrossRef CAS
- S. Hirlekar, D. Ray, V. K. Aswal, A. Prabhune, A. Nisal and S. Ravindranathan, Langmuir, 2019, 35, 14870–14878 CrossRef CAS PubMed
- Y. I. González and E. W. Kaler, Langmuir, 2005, 21, 7191–7199 CrossRef PubMed
- L. Meng, C. Shao, C. Cui, F. Xu, J. Lei and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 1628–1639 CrossRef CAS PubMed
- S. Jeong, G. S. Schultz and D. J. Gibson, Int. Wound J., 2017, 14, 786–790 CrossRef PubMed
- C. Ma, B. Li, B. Shao, B. Wu, D. Chen, J. Su, H. Zhang and K. Liu, Angew. Chem., Int. Ed., 2020, 59, 21481–21487 CrossRef CAS PubMed
- I. Kralova and J. Sjöblom, J. Dispersion Sci. Technol., 2009, 30, 1363–1383 CrossRef CAS
- S. Yan, J. Xu, G. Liu, X. Du, M. Hu, S. Zhang, L. Jiang, H. Zhu, B. Qi and Y. Li, Food Chem., 2022, 387, 132891 CrossRef CAS PubMed
- Q. Guo, I. Bayram, W. Zhang, J. Su, X. Shu, F. Yuan, L. Mao and Y. Gao, Food Hydrocolloids, 2021, 111, 106214 CrossRef CAS
- B. T. Sather and B. S. Goldstein, J. Exp. Agric. Int., 2016, 11, 1–9 CAS
- M. G. Semenova, L. E. Belyakova, Y. N. Polikarpov, M. M. Il’in, T. A. Istarova, M. S. Anokhina and E. N. Tsapkina, Biomacromolecules, 2006, 7, 101–113 CrossRef CAS PubMed
- S. Moghbeli, S. M. Jafari, Y. Maghsoudlou and D. Dehnad, J. Food Eng., 2019, 242, 124–132 CrossRef CAS
- H. J. Lee, A. McAuley, K. F. Schilke and J. McGuire, Formul. Biomol. Mech. Insights Mol. Interact., 2011, 63, 1160–1171 CAS
- H. L. Kim, A. Mcauley and J. Mcguire, J. Pharm. Sci., 2014, 103, 1337–1345 CrossRef CAS PubMed
- J. Liekkinen, A. Olżyńska, L. Cwiklik, J. Bernardino de la Serna, I. Vattulainen and M. Javanainen, Langmuir, 2023, 39, 4338–4350 CrossRef CAS PubMed
- P. Kaur, T. Garg, G. Rath, R. S. R. Murthy and A. K. Goyal, Drug Delivery, 2016, 23, 717–728 CrossRef CAS PubMed
- C. García-Mouton, A. Hidalgo, R. Arroyo, M. Echaide, A. Cruz and J. Pérez-Gil, Front. Bioeng. Biotechnol., 2021, 8, 613276 CrossRef PubMed
- A. Hidalgo, C. Garcia-Mouton, C. Autilio, P. Carravilla, G. Orellana, M. N. Islam, J. Bhattacharya, S. Bhattacharya, A. Cruz and J. Pérez-Gil, J. Controlled Release, 2021, 329, 205–222 CrossRef CAS PubMed
- L. Herman, S. C. De Smedt and K. Raemdonck, J. Controlled Release, 2022, 342, 170–188 CrossRef CAS PubMed
- N. Kahya and F. B. Erim, Carbohydr. Polym., 2019, 224, 115165 CrossRef CAS PubMed
- Y. Wei, Z. Yu, K. Lin, C. Sun, L. Dai, S. Yang, L. Mao, F. Yuan and Y. Gao, Food Hydrocolloids, 2019, 95, 336–348 CrossRef CAS
- Y. Wei, L. Zhang, Z. Yu, K. Lin, S. Yang, L. Dai, J. Liu, L. Mao, F. Yuan and Y. Gao, Food Hydrocolloids, 2019, 94, 333–344 CrossRef CAS
- Y. Wei, S. Yang, L. Zhang, L. Dai, K. Tai, J. Liu, L. Mao, F. Yuan, Y. Gao and A. Mackie, Food Hydrocolloids, 2020, 105, 105791 CrossRef
- L. Dai, Y. Wei, C. Sun, L. Mao, D. J. McClements and Y. Gao, Food Hydrocolloids, 2018, 85, 75–85 CrossRef CAS
- Q.-J. Ruan, M.-P. Wang, Y. Zou, C. Lin, D.-C. Cai and J.-M. Wang, Ind. Crops Prod., 2020, 153, 112587 CrossRef CAS
- F. Pan, Z. Lu, I. Tucker, S. Hosking, J. Petkov and J. R. Lu, J. Colloid Interface Sci., 2016, 484, 125–134 CrossRef CAS PubMed
- J. Aslam, I. H. Lone, N. R. E. Radwan, M. F. Siddiqui, S. Parveen, R. B. Alnoman and R. Aslam, ACS Omega, 2019, 4, 22152–22160 CrossRef CAS PubMed
- P. Dubey, S. Kumar, V. K. Aswal, S. Ravindranathan, P. R. Rajamohanan, A. Prabhune and A. Nisal, Biomacromolecules, 2016, 17, 3318–3327 CrossRef CAS PubMed
- J. D. Kaspersen, A. Søndergaard, D. J. Madsen, D. E. Otzen and J. S. Pedersen, Biophys. J., 2017, 112, 1609–1620 CrossRef CAS PubMed
- P. Lundahl, E. Greijer, M. Sandberg, S. Cardell and K.-O. Eriksson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1986, 873, 20–26 CrossRef CAS
- Y. Moriyama, Y. Kawasaka and K. Takeda, J. Colloid Interface Sci., 2003, 257, 41–46 CrossRef CAS PubMed
- R. Chen, Y. Song, Z. Wang, H. Ji, Z. Du, Q. Ma, Y. Yang, X. Liu, N. Li and Y. Sun, Int. J. Biol. Macromol., 2023, 251, 126288 CrossRef CAS PubMed
- A. Dhakal, C. McKay, J. J. Tanner and J. Cheng, Briefings Bioinf., 2022, 23, 1–23 CAS
- P. Brown, A. M. Khan, J. P. K. Armstrong, A. W. Perriman, C. P. Butts and J. Eastoe, Adv. Mater., 2012, 24, 6244–6247 CrossRef CAS PubMed
- X.-J. Ma, Y.-J. Zhang and C.-M. Zeng, Biochemistry, 2018, 83, 60–68 CAS
- Y. Wang, B. Jia, M. You, H. Fan, S. Cao, H. Li, W. Zhang and G. Ma, J. Phys. Chem. B, 2019, 123, 6200–6211 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |