
A novel microscopic origin of co-nonsolvency†
Xingye
Li
a,
Zhiyuan
Wang
a,
Zheng
Wang
a,
Yuhua
Yin
a,
Run
Jiang
a,
Pengfei
Zhang
 *b and
Baohui
Li
*a
*b and
Baohui
Li
*a
aKey Laboratory of Weak-Light Nonlinear Photonics, Ministry of Education, School of Physics, Nankai University, Tianjin 300071, China. E-mail: baohui@nankai.edu.cn
bState Key Laboratory for Advanced Fiber Materials, Center for Advanced Low-Dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: pfzhangphy@dhu.edu.cn
First published on 13th May 2025
Abstract
Co-nonsolvency presents a fundamental paradox in polymer physics where macromolecules undergo collapse or precipitation in mixed good solvents. Through investigations combining simulations of various binary good solvent systems of polymers, including single-chain and multi-chain of homopolymers and block copolymers, and ternary Flory–Huggins theoretical validation, we reveal that the competition between the enthalpy of the system and the mixing entropy of binary solvents results in the liquid–liquid phase separation (LLPS) of the better solvent (S-solvent) and the co-nonsolvency phenomenon. To reduce the enthalpy, the polymer and S-solvent tend to mix together to maximize their contact, which, however, is entropically unfavorable due to the localization of the S-solvent in the polymer domain. The LLPS of the S-solvent, where different chain segments share the localized S-solvent molecules, simultaneously lowers the enthalpy and reduces the loss of the mixing entropy. This sharing leads the chain in single-chain systems to be in a locally folding conformation with a size being much smaller than that of the ideal chain. In multi-chain systems, however, the sharing can be among segments from different chains, which causes chain condensation and hence an average chain size larger than its ideal value. Our study provides a novel mechanism for co-nonsolvency and may provide insights into the LLPS in other soft matter systems.
1. Introduction
The ability of stimuli-responsive polymers to change their structure, function, and stability in response to external stimuli makes them excellent candidates as high-performance multifunctional soft materials.1–3 In this context, polymers in solution belong to such a class of smart materials since their conformation may change significantly with the solvent quality. In contrast to the single solvent whose quality can be varied with temperature, mixed solvents provide a convenient way to regulate the solubility and conformation of polymers by changing the solvent composition at a fixed temperature, and thus, they are widely used in various polymeric applications such as in asymmetric membrane fabrication and fiber wet spinning.4,5 Some polymers in certain mixed solvents exhibit much richer phase behaviors than in a single solvent, and thus, have received great attention in the past few decades. One of the most intriguing phenomena is called co-nonsolvency, where a polymer perfectly soluble in each solvent may become insoluble in mixtures of both.3,6 As a well-known example of co-nonsolvency, solutions of poly(N-isopropylacrylamide) (PNIPAM) in water–methanol mixtures have been investigated extensively. At room temperature, PNIPAM readily dissolves in both pure water and pure methanol and forms optically transparent solutions. However, mixing these solutions at certain proportions leads to the appearance of turbidity, indicating the occurrence of phase separation.2,3,6,7 Furthermore, a re-entrant coil-to-globule-to-coil transition was observed with varying the solvent composition for the first time for a single PNIPAM chain in water–methanol mixtures by Zhang and Wu using static and dynamic laser light scattering (SDLLS).8 That is, a long flexible PNIPAM homopolymer chain can collapse into a thermodynamically stable globule in a suitable water–methanol mixture though both pure water and pure methanol are good solvents for PNIPAM. Moreover, co-nonsolvency was observed in several other polymer binary solvent solutions, such as poly(N,N-dimethylacrylamides) in water–ethanol mixtures,9 polystyrene in cyclohexane-N,N-dimethylformamide mixtures,10 and other polymer binary solvent systems as reviewed recently.6Co-nonsolvency has also demonstrated applications in block copolymer assembly. For example, for diblock copolymers consisting of a PNIPAM block and a co-nonsolvency inactive block with both soluble in either pure water or pure methanol, co-nonsolvency leads to the formation of micelles with PNIPAM as micelle cores and the other block as swollen coronas in water–methanol mixtures at a low polymer concentration.11 When the polymer concentration is high, well ordered mesoscopic lattice structures such as lamellar and cylindrical morphologies form due to co-nonsolvency of PNIPAM blocks in certain solvent compositions.12 On the other hand, if the co-nonsolvency inactive block is insoluble in pure water, it forms the core of the micelle and the PNIPAM block constitutes the micelle corona in pure water and in solvent mixtures with a higher water ratio. The thickness of the micelle corona can be adjusted by varying the solvent composition due to co-nonsolvency.13,14 Moreover, Kyriakos et al. found that these spherical micelles can form large aggregates and further demonstrated that the aggregation process of diblock copolymer micelles exhibits rather different features from that of homopolymers.15 Besides block copolymer solutions, co-nonsolvency was also employed to regulate the physical properties of many other polymeric systems such as thin films,16,17 brushes,18,19 and gels.20–24
The microscopic origin of co-nonsolvency, however, remains controversial despite numerous efforts made.3,8,9,25–46 Currently, there are three main classes of viewpoints. The first class considers that the chain collapse in solvent mixtures arises from the preferential adsorption of one type of solvent over the other.25–28,41 Tanaka and coworkers attributed the coil-to-globule-to-coil transition of a PNIPAM chain in mixed solvents of water–methanol to the preference in the cooperative hydrogen bond formation between methanol molecules and monomers over that between water molecules and monomers.25 This viewpoint was supported by the later fluorescence correlation spectroscopy experiment by Wang et al., where they observed a re-entrant change of the scaling index for a single PNIPAM chain in a water–ethanol mixture.26 Subsequently, by performing molecular dynamics simulations based on the coarse-grained Kremer–Grest model, Mukherji et al. found that, to lower the system enthalpy, the better solvent (called cosolvent) molecules bridge different monomers together and thus drive the chain collapse.27,28 They stressed that the co-nonsolvency is a generic phenomenon driven by the preferential adsorption of monomers to the cosolvent over to the other solvent. Moreover, by combining the preferential attraction concept with the Alexander–de Gennes brush theory, Sommer found that the preferential attraction can induce a swelling-to-collapse-to-swelling transition with varying the solvent composition for polymer brush in mixtures of two good solvents,41 which was latter verified by experiments and computer simulations by Sommer and coworkers.44 By contrast, the second class of viewpoints attributes the chain collapse to the formation of solvent complexes or the attraction between the two types of solvents.29,35,42 By performing the SDLLS experiments, Zhang and Wu postulated that the PNIPAM chain collapse in a water–methanol mixture arises from the formation of different water/methanol complexes which are poor to PNIPAM.35 Recently, by using the neutron total scattering technique and all-atom molecular dynamic simulations, Zuo et al. suggested that the collapse of poly(N-diethylacrylamide) in a water–ethanol mixture results from the strong attraction between water and ethanol molecules.42 Moreover, the third class of viewpoint proposed by van der Vegt and coworkers rationalizes the co-nonsolvency by viewing the cosolvent as a surfactant.33,47,48 Specifically, by classifying the conformation space of a polymer into a coil state and a globule state, van der Vegt et al. performed the coarse-grained molecular dynamics simulation to estimate the free energy of the polymer in each of these two states both in a pure solvent and in mixtures of solvent and cosolvent.33 They found that the free energy of a globule polymer decreases more rapidly than that of a coil polymer with the addition of a small amount of cosolvent, and thus, attributed the chain collapse to the smaller polymer–solvent excluded volume of the globule polymer than that of the coil polymer. Besides, other viewpoints, such as the composition fluctuation of the two solvents38 and the cosolvent induced geometric frustration,49 have also been proposed to address the mechanism of co-nonsolvency.
On the other hand, the differences in the co-nonsolvency-induced chain conformation transition between single-chain and multi-chain systems, as well as those between homopolymer and block copolymer systems, remain much less understood. This is mainly because most of the previous studies concerning chain conformation focused on single-chain homopolymer systems, and only few have examined it in multi-chain solution systems50 and brush systems.41,44 Clarifying these differences may be helpful not only in revealing the microscopic origin of co-nonsolvency but also in providing valuable guidance for the rational design of co-nonsolvency—based smart materials. In addition, previous studies on diblock copolymer dilute solutions in binary good solvent mixtures primarily reported the spherical micelles;11,13,14 however, it is expected that other aggregates such as vesicles and micelles of rich morphologies, obtained in amphiphilic diblock copolymer dilute solutions,51 should also be induced by co-nonsolvency.
In this paper, we report an extensive investigation on the microscopic origin of co-nonsolvency, based on our systematic studies of chain conformation and phase behaviors of both single-chain and multi-chain systems of homopolymers and diblock copolymers in binary good solvent mixtures. In the simulations, we observe co-nonsolvency-induced chain conformation changes in all the studied systems, and chain condensation in multi-chain systems where it adopts the forms of vesicles and micelles of rich morphologies in block copolymer systems. Furthermore, we find that the better solvent (S-solvent) undergoes liquid–liquid phase separation, resulting in the coexistence of an S-solvent-concentrated phase and an S-solvent-dilute matrix in both single-chain and multi-chain systems. Moreover, we find that in multi-chain systems, each chain has an average size larger than the corresponding ideal chain size over the entire solvent composition range, rather different from that in the corresponding single-chain systems. Based on our systematic quantitative analysis, we propose a novel microscopic origin of co-nonsolvency and further verify it using the ternary Flory–Huggins theory.
2. Model and methods
We use a simulated annealing technique52 based on the single-site bond fluctuation model53 in our simulations. The model and the method have been demonstrated to be efficient for studying the self-assembly and elucidating the underlying mechanisms of block copolymer solutions in our previous studies where details of the model and the method can be found.54,55 Specifically, our simulation system consists of block copolymer or homopolymer chain(s) and binary solvents. The system is embedded in a simple cubic lattice with periodic boundary conditions in all three directions. Each segment or solvent molecule occupies one lattice site, and the polymers are self- and mutually avoiding. The copolymer consists of a co-nonsolvency inactive block (A-block) and a co-nonsolvency active block (B-block). The homopolymer is co-nonsolvency active. The polymer concentration ϕP is defined as the fraction of chain segments in the system, ϕP = nN/V where n is the total number of chains with each having N segments in a system of volume V. The binary solvents are named O- and S-solvents, and the solvent composition CS is defined as the fraction of the S-solvent, i.e., CS = nS/(nO + nS), where nO and nS are the numbers of the O-solvent and S-solvent molecules, respectively. Our lattice model assumes that the ternary mixtures are incompressible and thus, it cannot address the effect of pressure. Nevertheless, we think the essential physics of the co-nonsolvency effect has been captured by this model and further, the simplicity of the model allows us to systematically examine the chain conformation and phase behaviors in the huge parameter space. The interactions are modeled by assigning an energy Eij = εijkBTref to each nearest-neighboring pair of components i and j, where εij is the reduced interaction energy, εij = εji, with i, j = A, B (or H), O, and S, representing the A-segment, B-(or homopolymer) segment, O-solvent, and S-solvent, respectively; kB is the Boltzmann constant and Tref is a reference temperature. We concentrate on systems where any two species are miscible except the two blocks in the copolymer system, and further assume that the S-solvent is better than the O-solvent to the B- or H-segment. For comparison, we assume that the nature of homopolymer segments is the same as that of the B-block in the copolymer system. For simplicity, we first set all the interactions to be zero except εAB and εBS (εHS), i.e., εij = 0 with i, j = A, B (or H), O and S, except that εAB = 1.0 and εBS = εHS < 0. We also carry out simulations with εOS < 0 or εOB < 0. This parameter setting is consistent with the preferential adsorption concept mentioned in the Introduction section. The effect of other parameters such as the interaction between solvents O and S will be examined in future. The initial state of a simulated annealing simulation is a random distribution of polymer(s) and solvents at a given set of ϕP and CS. Starting from this initial state, we perform a sequence of Monte Carlo simulations with each at a decreasing temperature. A final temperature is obtained when the difference of the ensemble-averaged quantities obtained at two consecutive temperatures is less than or comparable to the statistical error. Snapshots of the morphologies of the simulation system are then extracted and various quantities are calculated by the ensemble average. Specifically, we calculate the radial density profiles, the average nearest neighbor contact number for each polymer segment, and the chain and segment mean-square radius of gyration for both single-chain and multi-chain homopolymer and copolymer systems.We also calculate the difference in the Helmholtz mixing free energy density between the phase-separated system and the homogeneous system for homopolymer solutions, as well as the energetic contribution and the entropic contribution using the ternary Flory–Huggins theory. The details are included in the ESI.†
3. Results and discussion
In this section, we present our results along with discussions and comparisons with related results in the literature. We first exhibit co-nonsolvency-induced aggregates in terms of morphological phase diagrams and radial density profiles of typical morphologies. We then show the solvent composition CS dependence of the averaged nearest neighbor contact number for each polymer segment to manifest its local environments. We also show the CS dependence of the chain mean-square radius of gyration in each system to characterize the chain conformation. Based on the systematic quantitative analysis on simulation results, we propose a view on the microscopic origin of co-nonsolvency and further verify it using the ternary Flory–Huggins theory. Finally, we present the CS dependence of the segment mean-square radius of gyration to elucidate the different behaviors of the chain mean-square radius of gyration between the single-chain and multi-chain systems. In our simulations, we primarily use the chain length N = 10. We also study the cases with N = 30 for comparison. Our results show that the calculated quantities depend on N; however, the variation trend of all calculated quantities with CS does not depend on N, and thus we mainly present results for N = 10 unless otherwise specified.3.1 Co-nonsolvency-induced aggregates
Aggregates are observed in various studied systems. Fig. 1 shows the morphological phase diagram and the radial density profiles of typical morphologies in multi-chain diblock copolymer solutions with εBS = −2.0 and εAB = 1.0, and all other interactions being zero. In Fig. 1a, we note that the morphologies of aggregates change with varying the volume fraction of block copolymer fA (= NA/N, where NA and N are the lengths of A-block and the copolymer chain, respectively) and the solvent composition CS. Dispersed chains (DC) are observed at the two limiting cases of CS = 0 and CS close to 1. For CS in between, the copolymers aggregate into micelles or vesicles. With increasing CS, a total of six morphological sequences are observed: QV(quasi-vesicles) → V(vesicles) → TQV(tubular quasi-vesicles) → CM(compound micelles), DM(disk-micelles) → V → LM(lamellar micelles) → CM, DM → LM → CM, SM(spherical micelles) → DM → CM → SLM(spherical-like micelles), SM → HM(hamburger micelles) → SLM, and HM → SLM, at fA = 0.2, 0.3, 0.4, 0.5, 0.6 and 0.7–0.8, respectively. It is noted that in these micelles and vesicles, the B-blocks form the micellar cores and the vesicle shells while the A-blocks form the corona. On the other hand, the variation trend from vesicles to micelles with increasing fA is similar to that observed in amphiphilic diblock copolymer dilute solutions.51 The CS window to form the DC phase near CS = 1.0 enlarges with increasing fA, due to the decrease in the length of the co-nonsolvency active B-block. As both types of solvents are good to the two blocks, the only force driving the formation of aggregates should be the co-nonsolvency of B-blocks. Furthermore, we also observe co-nonsolvency-induced micelles and vesicles in multi-chain copolymer solutions with different combinations of ϕP, CS, and εBS. As examples, Fig. S1a and b (ESI†) show the morphological phase diagrams in the CS − ϕP plane at εBS = −2 and in the CS − εBS plane at ϕP = 0.01, respectively. We notice that the CS window of a specific aggregate in each diagram depends on ϕP, CS, and εBS. Specifically, the CS window of vesicles increases with decreasing εBS (i.e., increasing |εBS|) at ϕP = 0.01 and the window of tubular quasi-vesicles increases with increasing ϕP, as shown in Fig. S1a and b (ESI†), respectively. The formation of vesicles and micelles of rich morphologies in our case is similar to the cases of amphiphilic diblock copolymer dilute solutions as reviewed by Mai and Eisenberg,51 with the only difference being in the driving force for micelle formation. In theirs, it is the solvophobic interaction of the core-forming block, while in ours, it is the co-nonsolvency effect of the core-forming block. | ||
| Fig. 1 Typical aggregates obtained in multi-chain diblock copolymer systems with εBS = −2.0, εAB = 1.0, and all other interactions being zero. (a) Morphological phase diagram as a function of the solvent composition CS and the volume fraction of the A-block fA at ϕP = 0.02. (b) and (c) Snapshots (in the front and cross-sectional views) and the corresponding radial density profiles ρi for aggregates obtained with fA = 0.2 and CS = 0.2, where i = A, B, O or S, in (b) ϕP = 0.005 and (c) ϕP = 0.02. Color in the snapshots: A(green) and B(red). | ||
Co-nonsolvency-induced micelles were observed experimentally in block copolymers consisting of a co-nonsolvency active block (PNIPAM) and a co-nonsolvency inactive block, in water–methanol mixtures.11 However, only nearly spherical micelles have been reported so far. Our results here demonstrate that much richer morphologies of aggregates can be induced by co-nonsolvency of one block in block copolymer solutions and they will serve as a valuable guidance for further experimental studies.
On the other hand, we also observe co-nonsolvency-induced aggregates in multi-chain homopolymer solutions. Taking the case with ϕP = 0.02 and εHS = −2 as an example, Fig. S1c (ESI†) shows that the spherical aggregate forms when 0.1 < CS < 0.7 and this window increases slightly with decreasing εHS. In Fig. S1d (ESI†), we notice that the specific heat curve presents a peak at εHS ≈ −0.41 where the chains start to aggregate, and when |εHS| > 0.52 the chains in the system are all in the aggregated state for the system with N = 10 and CS = 0.1. For single-chain homopolymer and copolymer systems, we also observe similar aggregates as those shown in Fig. S1c (ESI†) at intermediate CS, but the aggregate size is much smaller and the CS window of these aggregates depends on εHS (or εBS) (data not shown). All the above conclusions hold true for systems with εOS < 0 or εOB < 0, as long as |εOS| ≪ |εBS| and |εOB| ≪ |εBS|, that is, for systems dominated by εBS. For clarity, in the following we only present results for systems with εBS = εHS = −2.0, εAB = 1.0, and all other interactions being zero.
To quantitatively characterize the composition distributions in each system, in Fig. 1b and c we show the radial density profiles ρi of species i (= A, B, O, and S), for typical snapshots of the spherical micelle and vesicle obtained in multi-chain diblock copolymer solutions, where each ρi curve is plotted along the radial direction r of the aggregate with r = 0 denoting its center of mass of polymer segments. Specifically, Fig. 1b suggests that for a spherical micelle, the core is mainly composed of B-blocks and the S-solvent, and the corona is composed of A-blocks as reflected by the shallow peak of ρA; outside the micelle, the solution is a mixture of O- and S-solvents. Likewise, Fig. 1c indicates that the shell of a vesicle is also mainly composed of B-blocks and the S-solvent with the A-blocks being distributed around the two surfaces of the shell. One notable feature of Fig. 1b and c is that in the micelle core (or vesicle shell), the S-solvent concentration (>70%) is much higher than that of polymer, indicating that the micelle core (or vesicle shell) is in a liquid state. Another notable feature from Fig. 1b and c is that the S-solvent concentration in the micelle core (or vesicle shell) is much higher than that outside it, which indicates that the S-solvent is under the liquid–liquid phase separation (LLPS) state where the B-block domain is in the S-solvent-concentrated phase and the O-solvent in the S-solvent-dilute phase (forming matrix). Moreover, the LLPS of the S-solvent is also observed in the single-chain copolymer system, as well as in the single-chain and multi-chain homopolymer systems (Fig. S2a and b, ESI†), where the B-block (or the homopolymer) domain is in the S-solvent-concentrated phase and the O-solvent in the S-solvent-dilute phase (forming matrix). Hereafter, we refer to the S-solvent-concentrated phase, including the dissolved B-block (or homopolymer-) domain, as the droplet.
It was reported that a number of other multi-chain systems, such as proteins in the intracellular environments,56 oppositely charged polyelectrolytes in solutions,57 and amphiphilic copolymers in mixtures of a good organic solvent and a selective solvent (water),58,59 can undergo LLPS under some conditions, forming a polymer-rich liquid phase coexisting with a polymer-poor phase. Electrostatic interactions are important to the LLPS in the former two systems,56,57 while the hydrophobic interaction drives the LLPS in the third system.58,59 However, there are neither electrostatic nor hydrophobic interactions in our systems. Furthermore, different from the above-mentioned systems,56–59 the LLPS in our systems takes place in both single-chain and multi-chain systems of the copolymer and homopolymer.
The radial density profiles in Fig. 1b, c and Fig. S2 (ESI†) show that there is a large number of S-solvent molecules inside each droplet. To characterize the local environments of the chain segments, we show the averaged nearest neighbor contact number for each B- or H-segment with each of all species, CNBi, or CNHk (i = A, B, O, S and k = H, O, S) in Fig. S3a–d (ESI†). It is noted that there are on average 15–16 O-solvent and 15–16 S-solvent molecules in contact with each B- (or H-) segment at CS = 0 and CS > 0, respectively, rather close to the theoretical maximum value of 16.125 or 16.2 for the copolymer and homopolymer systems, respectively in our lattice model. These features indicate that each B (or H)- segment is nearly fully surrounded by solvents over the entire CS range for all the studied systems. This finding is consistent with previous observation or prediction that there is a large portion of solvent molecules inside the “collapsed” polymer domain in single-chain systems8,28 and inside the micelle cores for the copolymer solution systems.11 However, it is in stark contrast to the collapsed globule formed by the homopolymer chain(s) in a pure poor solvent, where each segment is mainly surrounded by other segments instead of solvents. This comparison reflects a fundamental difference in the globule formed in pure poor solvents and in the globule/aggregates formed in mixtures of two good solvents. The enrichment of the S-solvent (such as methanol) in the co-nonsolvency-induced globule/aggregates can be experimentally verified by the small angle neutron scattering or spectroscopic techniques in future.
3.2 Chain conformation
To characterize the evolution of the chain conformation along CS, in Fig. 2a–c we show the CS dependence of the mean-square radius of gyration of the chain/block 〈RgJ2〉 (J = A, B, AB, and H) in both single-chain and multi-chain systems. To reflect the deviation of the chain size relative to the ideal chain, we normalize each curve by the mean-square radius of gyration of the corresponding ideal chain given by RgJ-ideal2 = (N2 − 1)b2/6N, where b = (5/3)1/2 ≈ 1.291 is the statistical segment length in the unit of lattice spacing for our single-site bond fluctuation model; specifically, RgB-ideal2 = 2.188 for B-blocks with NB = 8 and RgH-ideal2 = 2.750 for homopolymer chains with NH = 10 used here. Fig. 2a shows that in single-chain systems, both 〈RgH2〉 and 〈RgB2〉 first decrease at very small CS and then increase with increasing CS. Furthermore, the normalized 〈RgH2〉 (or 〈RgB2〉) is smaller than 1 when 0 < CS < 34% (or 25%), indicating that the chain seems “collapsed”. This CS window increases with the chain length N, and it is 0 < CS < 45% for the homopolymer with N = 30 (Fig. S4, ESI†). Hence, with increasing CS, both 〈RgH2〉 and 〈RgB2〉 manifest the so-called reentrant coil-to-globule-to-coil transition. The variation trend of 〈RgH2〉 and 〈RgB2〉 shown in Fig. 2a and Fig. S4 (ESI†) for single-chain systems is consistent with that in previous observations.8,27,31,34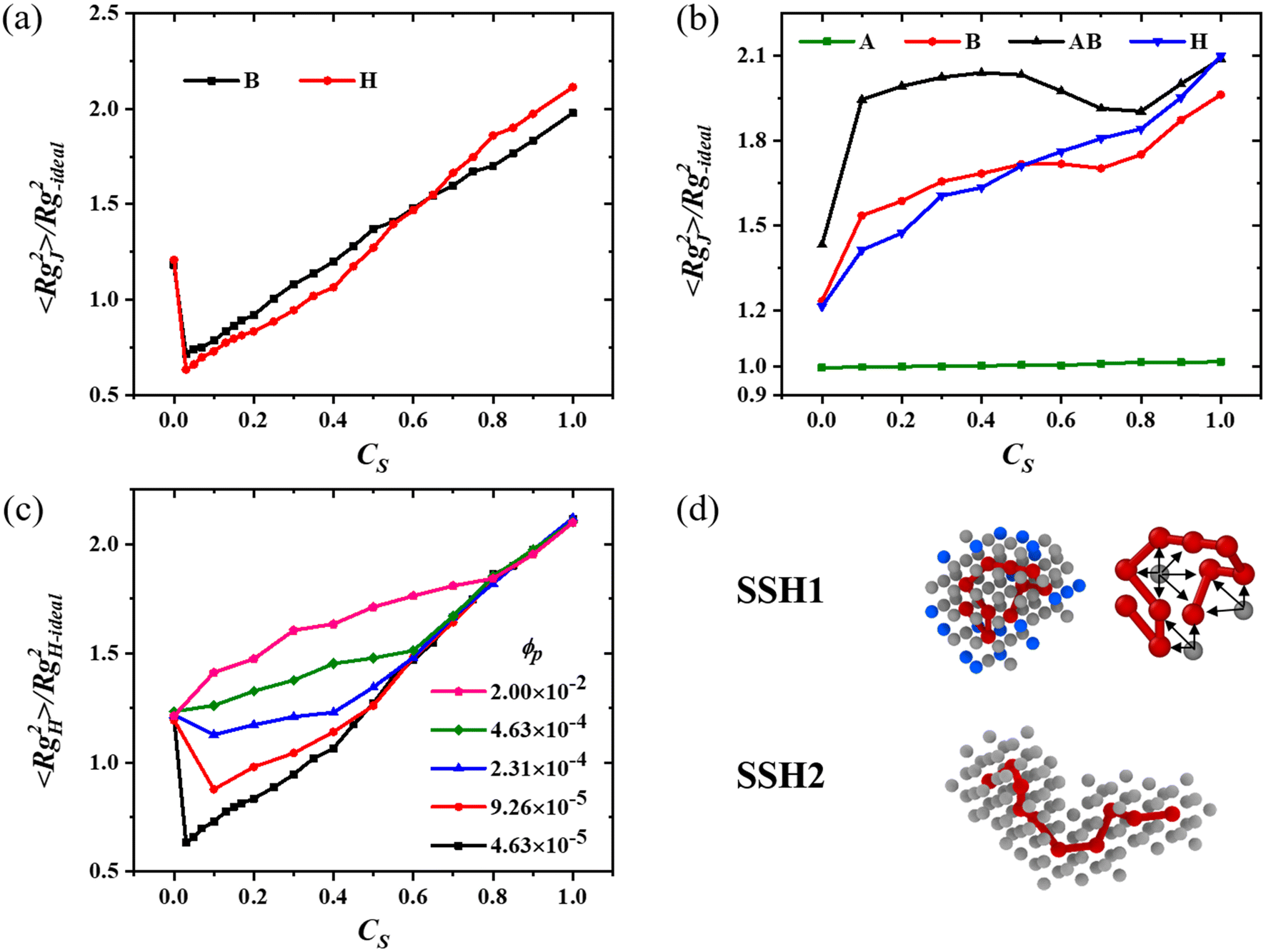 | ||
| Fig. 2 The CS dependence of the normalized chain mean-square radius of gyration for both copolymer (with fA = 0.2) and homopolymer systems (a) and (b), and of the homopolymer systems with various ϕP (c). (d) Snapshots of the homopolymer chain with its nearby solvent molecules, where both chain segments and the bonds (connecting two consecutive segments) are shown in red, the S-solvent in grey, and the O-solvent in blue, and CS = 0.2 and 1.0 for SSH1 and SSH2, respectively. (a) and (d) Single-chain systems. (b) Multi-chain systems with ϕP = 0.02. | ||
On the other hand, for multi-chain systems with ϕP = 0.02, it is surprising that all 〈RgJ2〉 curves do not exhibit the so-called reentrant coil-to-globule-to-coil transition as shown in Fig. 2b. Instead, in the entire CS range, both 〈RgB2〉 and 〈RgH2〉 increase almost monotonically with increasing CS; this is reasonable considering that the attraction of the S-solvent/B-block is stronger than that of the O-solvent/B-block so that the B-blocks and the homopolymer chains become increasingly more stretched with increasing CS. Furthermore, the normalized values of 〈RgB2〉 and 〈RgH2〉 are all much larger than 1 in Fig. 2b, indicating that the B-block and the entire homopolymer or copolymer chain are not “collapsed” in the whole CS range. On the other hand, 〈RgA2〉 remains roughly unchanged, mainly because the A-blocks are co-nonsolvency inactive. The above simulation results on the chain mean-square radius of gyration for multi-chain systems (Fig. 2b) are different from those by Mohammadyarloo and Sommer for the polymer solution system.50 This may be due to the different treatments of the two solvents: they modeled the solvent implicitly and the cosolvent explicitly, while we treat both the solvent and cosolvent in an explicit way.
The above results suggest that over a certain CS range, the chain conformation in multi-chain systems with ϕP = 0.02 is rather different from that in the corresponding single-chain system. To elucidate how the polymer concentration induces this difference, in Fig. 2c we show the CS dependence of the normalized mean-square radius of gyration for homopolymer systems with various ϕP. It is noted that each 〈RgH2〉 increases almost monotonically with increasing CS when ϕP ≥ 4.63 × 10−4, while the so-called reentrant coil-to-globule-to-coil transition is observed only when ϕP < 2.31 × 10−4 (corresponds to only 5 chains in our system), i.e., the reentrant transition occurs only in single-chain or few-chain systems. We also note that in Fig. 2c, 〈RgH2〉 at each ϕP manifests a linear increase with CS and 〈RgH2〉 at different ϕP overlaps in the large CS range. However, in the small CS range, 〈RgH2〉 is slightly larger than that expected from the linear increase with CS for few-chain and multi-chain systems. With an increasing ϕP, this deviation becomes more pronounced and the related CS range enlarges. Furthermore, such a deviation also exists in the 〈RgB2〉 curve as shown in Fig. 2b. We will explain this deviation later.
3.3 Microscopic origin of co-nonsolvency
To elucidate the microscopic origin of co-nonsolvency, we first examine the typical snapshots of the homopolymer chain and its nearby solvent molecules for the single-chain systems with various CS. The snapshot SSH1 shown in Fig. 2d indicates that, at CS = 0.2 the chain is in a locally folding conformation inside the approximately spherical droplet. This chain conformation is in stark contrast to the expanded coil state in the pure S-solvent as shown in the snapshot SSH2 of Fig. 2d. Obviously, it is the local folding of the chain that results in a much smaller chain size than the ideal value in the so-called “collapsed” CS range mentioned above.In Fig. 1b, c and Fig. S2 (ESI†), we have shown that the droplet is composed of the S-solvent and B-block (or homopolymer) with negligible O-solvent. This partitioning is energetically favorable since more polymer/S-solvent (P/S) contact can be achieved to lower the system enthalpy. However, it is entropically unfavorable since the localization of S-solvent molecules in the droplet will lead to a loss in the mixing entropy of the binary solvents. On one hand, the enthalpy is lowered by increasing the P/S contact. On the other hand, the loss in the mixing entropy of the binary solvents is reduced by decreasing the number of S-solvent molecules in the droplet. The former can only be realized by forming a phase composed of S-solvent and polymer without O-solvent, while the latter limits the number of S-solvent molecules in that phase. Therefore, the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents results in the LLPS of the S-solvent and the formation of a droplet with a specific size. The occurrence of LLPS indicates that the free energy of the system is lowered when the loss in the mixing entropy is surpassed by the gain in the enthalpy. In single-chain systems, when CS is very small, the number of S-solvent molecules in the droplet is very small (due to the small droplet size and for reducing the loss in the mixing entropy) where the P/S contact can be maximized only when different segments share the small number of the S-solvent in the droplet. This sharing leads to the chain in a locally folding conformation; see the snapshot SSH1 shown in Fig. 2d, where arrows are included to indicate the sharing of each S-solvent molecule by several segments. In short, the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents leads to the chain in a locally folding conformation that manifests in the form of chain collapse. This is the origin of co-nonsolvency. With increasing CS, the osmotic pressure and the exchange chemical potential of the S-solvent in the matrix (outside the droplet) increase gradually.34 To maintain the chemical and mechanical equilibria between the droplet and the matrix, a certain amount of the S-solvent must enter into the droplet, which further causes a gradual increase of the droplet size and hence a weakening in the degree of the chain folding. Accordingly, the chain radius of gyration increases gradually with CS. While the increase of the chain size starts from a rather small CS, it is worthwhile noting that there is a relatively wide CS window over which the chain size is smaller than the ideal value; see Fig. 2a.
Fig. 2c further shows that, at a fixed CS ∈ (0, 0.4), 〈RgH2〉 increases with ϕP when ϕP is small; this is mainly because more chains and S-solvents are included in the droplet. In the few-chain systems, segments from different chains start to share the S-solvent molecules to lower the enthalpy, and simultaneously to lower the loss in the mixing entropy, the fraction of S-solvents shared by segments from different chains increases gradually with ϕP, which weakens the chain folding degree for each individual chain and thus leads to an increase of 〈RgH2〉. When ϕP ≥ 2.31 × 10−4, 〈RgH2〉 is comparable to or larger than the size of the corresponding ideal chain RgH-ideal2 even at very small CS, which means that 〈RgH2〉/RgH-ideal2 ≥ 1 for all CS and thus the chain collapse does not occur any more. With further increasing ϕP, the sharing of S-solvents by segments from different chains dominates and chains are condensed together; accordingly, 〈RgH2〉 becomes independent of ϕP. In our homopolymer case, 〈RgH2〉 no longer depends on ϕP when ϕP ≥ 0.005. In short, in multi-chain systems, chains are condensed together to share the S-solvent molecules and these S-solvent molecules act as bridges among different chains to lower the enthalpy. On the other hand, the loss in the mixing entropy of the binary solvents is reduced by decreasing the number of S-solvent molecules in the droplet, the same as that in the single-chain system. Therefore, it is also the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents that results in the condensation of chains in multi-chain systems. Different from the chain collapse in single-chain or few-chain systems, co-nonsolvency in multi-chain systems manifests as chain condensation. Furthermore, to further lower the polymer/solvent enthalpy, each condensed chain in the droplet is more stretched than the uncondensed chain in the O-solvent. With increasing ϕP, the fraction of S-solvents shared by segments from different chains increases, and thus, the deviation of 〈RgH2〉 from the expected linear increase becomes more pronounced and the related CS range enlarges, as observed in Fig. 2c.
One may argue that our view of the microscopic origin of co-nonsolvency seems the same as the preferential-adsorption mechanism proposed previously.25–28,41 This preferential-adsorption mechanism attributes the chain collapse in mixed solvents to the preferential-adsorption of one type of solvent over the other to lower the enthalpy, and thus emphasizes the importance of enthalpy. Our studies here show that co-nonsolvency is indeed triggered by the stronger P/S attraction. However, the mixing entropy of the binary solvents is equally important in inducing co-nonsolvency. If there were no loss of the mixing entropy of the binary solvents, the droplet formed by polymer and S-solvent molecules could be large enough (as there are enough S-solvent molecules in each system with LLPS, as shown in Fig. 1b and c) so that the P/S contact could be maximized without the chain local folding in single-chain systems and without chain condensation in multi-chain systems. If these were the case, there would be no observable reentrant coil-to-globule-to-coil transition in the single-chain systems and phase separation in the multi-chain solutions. Our results elucidate the importance of the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents in inducing co-nonsolvency, which is totally different from previous views. Moreover, our results specifically highlight mainly the competitive role of the polymer–S-solvent attractive enthalpy and mixing entropy of the O-solvent and S-solvent; this is rather different from the conventional enthalpy–entropy competition.45,60
3.4 Helmholtz mixing free energy calculation
To quantitatively verify the above view of the microscopic origin of the co-nonsolvency effect, we need to calculate the differences in the energetic contribution and in entropic contribution, especially the mixing entropic contribution purely due to the binary solvents, to the Helmholtz mixing free energy between the phase-separated system and the homogeneous system. However, calculating the free energy and entropy is a challenging task in Monte Carlo-based simulations and we thus resort to the ternary Flory–Huggins theory.6,45,61Using this theory, we calculate the difference in the Helmholtz mixing free energy density between the phase-separated system and the homogeneous system Δf = xf(ϕIP, ϕIS) + (1 − x)f(ϕIIP, ϕIIS) − f(ϕP, ϕS), where f(ϕP, ϕS) is the free energy density in the homogeneous system with ϕS = (1 − ϕP) × CS being the concentration of the S-solvent, f(ϕIP, ϕIS) and f(ϕIIP, ϕIIS) are, respectively, the free energy density in the S-solvent-dilute phase (phase I with superscript I) and the S-solvent-concentrated phase (phase II with superscript II) with ϕIP, ϕIS and ϕIIP, ϕIIS being the corresponding concentrations of polymers and S-solvent in the two coexisting phases; moreover, x is the volume fraction of the S-solvent-dilute phase. In this treatment, we ignore the contribution of the interface between the coexisting phases. Moreover, the Flory–Huggins theory treats the concentrations of various species in a mean-field way, and thus, ignores the concentration fluctuations that are usually important in the critical regime; the chain conformation entropy is also not considered. However, as demonstrated in the previous work,45,46 the main features of the co-nonsolvency should be qualitatively captured in the Flory–Huggins theory. In this theory, Δf can be decomposed into an energetic contribution Δu and an entropic contribution Δs via Δf = Δu − TΔs, with similar definitions for Δu and Δs. We refer readers to the ESI† (details of the ternary Flory–Huggins theory, ESI†) for the explicit expressions of f, u, s as well as the methodology to calculate ϕIP, ϕIS, ϕIIP, ϕIIS, and x.
We use χij = (z − 2)Eij/kBT = (z − 2)εij/(T/Tref) to relate the ε parameter in the simulations to the χ parameter in the Flory–Huggins theory, where the lattice coordination number is z = 18 in our model. For the simulation system with εHS = −2, εHO = εOS = 0, and T/Tref = 1.6, we can transform the simulation parameters to χPS = −20, χPO = χOS = 0 in our Flory–Huggins calculations. Based on these parameters, we can calculate the related quantities. Taking the multi-chain homopolymer system with ϕP = 0.02 as an example, we plot the CS dependence of βΔf, βΔu, and Δs/kB obtained from the Flory–Huggins theory in Fig. 3a. We note that Δf < 0 when 0 < CS < 0.88, indicating that the phase-separated system is thermodynamically stable in this CS window; this is qualitatively consistent with that indicated from the morphologies obtained from the simulations (Fig. S1c, ESI†). Moreover, both Δu and Δs are negative in this CS window, suggesting that both the enthalpy and the entropy decrease after phase separation. However, the magnitude of |βΔu| is larger than |Δs/kB|, indicating that it is the lowering of the enthalpy that drives the phase separation, at the expense of the decrease of the mixing entropy. Since the only non-zero interaction in our homopolymer system is the P/S attraction, the lowering of the enthalpy is thus achieved by increasing the P/S contact. On the other hand, since the mixing entropy density Δs/kB consists of the contributions from polymers and from the binary solvents, we further introduce Δs′/kB to denote the contribution purely due to the binary solvents. In Fig. 3a, we note that Δs′/kB is rather close to Δs/kB in the entire CS range and that they exhibit a similar variation trend, indicating that the mixing entropy of the binary solvents is indeed the dominant factor governing the behaviors of Δs/kB. Therefore, these analyses based on the Flory–Huggins theory support our view from the simulations: it is the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents that results in the LLPS and co-nonsolvency effect.
 | ||
| Fig. 3 The CS dependence of (a) the differences in the free energy density βΔf, the energetic and entropic contributions βΔu, Δs/kB, as well as the entropic contribution purely due to the binary solvents Δs′/kB, between the phase-separated system and the homogeneous system calculated from the Flory–Huggins theory, and of (b) and (c) the concentrations of various components in the coexisting phases (phases I and II denote the S-solvent-dilute and S-solvent-concentrated phases, respectively) obtained from (b) the Flory–Huggins theory and (c) simulations. | ||
To further verify the consistency between our simulations and Flory–Huggins theoretical calculations in treating the thermodynamics of homopolymers in binary solvents, we calculate the concentrations of the polymer and S-solvent in the S-solvent-dilute phase, ϕIP, ϕIS, and those in the S-solvent-concentrated phase, ϕIIP and ϕIIS; the concentrations of the O-solvent in each phase are then calculated as ϕO = 1 − ϕP − ϕS. In Fig. 3b and c, we show the CS dependence of ϕIP, ϕIS, ϕIO, ϕIIP, ϕIIS, and ϕIIO obtained from theoretical calculations and simulations, respectively, for the above-mentioned example of the multi-chain homopolymer system with ϕP = 0.02 and T/Tref = 1.6. We find that the simulation results are in good agreement with the theoretical calculations, suggesting that the ternary Flory–Huggins theory can qualitatively describe the main features of our simulation systems. This agreement is mainly due to the fact that the droplet formed at this parameter set in the simulation is sufficiently large such that the interfacial contribution is small. The interfacial contribution is in general important when the droplet is small and the Flory–Huggins theory is usually not valid for the finite-sized aggregates or droplet. Besides, the good agreement also suggests that the chain conformation entropy in the multi-chain system is small in comparison with the polymer–solvent enthalpy and mixing entropy of the two solvents; this is consistent with the recent study for single chain systems using Monte Carlo simulations and Flory-type mean-field theory.34
3.5 Chain segment distribution
To reflect the distribution of chain segments in multi-chain systems, we further compute the mean-square radius of gyration of all segments for the block copolymer 〈Rgs-AB2〉, that for A- and B-blocks 〈Rgs-A2〉 and 〈Rgs-B2〉, and that for homopolymers 〈Rgs-H2〉. Different from 〈Rg2〉 shown in Fig. 2, 〈Rgs-J2〉 (J = A, B, AB, and H) here includes both intra-chain and inter-chain contributions. In Fig. 4, we plot the normalized (by that in the respective athermal system) 〈Rgs-J2〉 as a function of CS for the multi-chain copolymers (with fA = 0.2) and homopolymers in systems with ϕP = 0.02. It is interesting to note that with increasing CS, all four curves in Fig. 4 manifest the pronounced feature of the re-entrant transition, similar to the re-entrant coil-to-globule-to-coil transition of 〈RgB2〉 or 〈RgH2〉 in single-chain systems (Fig. 2a) but in stark contrast to the corresponding 〈RgJ2〉 in the same multi-chain system (Fig. 2b). The large value of 〈Rgs-H2〉 at both CS = 0 and CS = 1 limits indicates that the chains are well dispersed in the system, while the small value of 〈Rgs-H2〉 at intermediate CS is a reflection of the condensation of chains into a droplet; this feature in the intermediate CS range is consistent with that shown in Fig. 3c where a droplet is formed. The behaviors of 〈Rgs-B2〉 for B-blocks of block copolymers are similar. On the other hand, since the A-blocks are co-nonsolvency inactive, the relatively smaller value of 〈Rgs-A2〉 at intermediate CS is certainly not due to the co-nonsolvency. Instead, since each A-block is linked to a B-block, the condensation of B-blocks must lead to a localization of A-blocks. As condensation of chains can be regarded as localization of chains, we may conclude that localization of homopolymer chains or copolymer blocks causes the relatively smaller values of the corresponding 〈Rgs-J2〉 at intermediate CS for all curves shown in Fig. 4. Based on these, we further conclude that localization of the chain in the single-chain systems will definitely cause the chain “collapse’’ since 〈Rgs-J2〉 = 〈RgJ2〉 in all the single-chain systems. By contrast, in multi-chain systems, localization of chains may not necessarily lead to the chain “collapse’’ since 〈Rgs-J2〉 ≠ 〈RgJ2〉. This may explain the different behaviors of 〈RgJ2〉 between the single-chain and multi-chain systems shown in Fig. 2. | ||
| Fig. 4 The CS dependence of the normalized segment mean-square radius of gyration for the multi-chain copolymers (fA = 0.2), as well as that of A-blocks and of B-blocks, and that of homopolymers in the systems with εBS = εHS = −2.0 and ϕP = 0.02. 〈Rgs-02〉 is the segment mean-square radius of gyration of the corresponding system in the athermal state. | ||
4. Conclusion
Using a simulated annealing technique, we have systematically investigated the chain conformation and phase behaviors of both single-chain and multi-chain of homopolymers and A-b-B diblock copolymers in binary solvent mixtures. The binary solvents, named S-solvent and O-solvent, are completely miscible and are good for all the studied polymer species. The S-solvent is better than the O-solvent for the co-nonsolvency active polymer (i.e., the homopolymer or the B-block of the copolymer). Our simulation results show that the competition between the enthalpy of polymer/solvent interactions and the mixing entropy of binary solvents leads to the LLPS of the S-solvent and further to co-nonsolvency, and we have verified this conclusion using the ternary Flory–Huggins theory.To the best of our knowledge, the following results are all obtained for the first time. First, the competition between the mixing entropy of the binary solvents and the enthalpy of the polymer/solvent interactions is the microscopic origin of co-nonsolvency. Second, both single-chain and multi-chain systems can undergo LLPS. Third, under the occurrence of co-nonsolvency, the chain conformation in multi-chain systems is rather different from that in single-chain systems. In multi-chain systems, co-nonsolvency manifests as chain condensation with each chain remaining swollen, while in single-chain systems it manifests as a locally folding conformation. Fourth, in multi-chain copolymer systems, each co-nonsolvency-induced droplet manifests as a vesicle or micelle, and micelles of various morphologies are observed. Our findings would be helpful for a better understanding of co-nonsolvency, and further for the rational design of co-nonsolvency-based smart materials. Furthermore, although the above discussions are restricted to the co-nonsolvency effect, our results may also provide insight into the LLPS occurring in biological systems when the S-solvent is interpreted as proteins in cellular milieu.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. Li acknowledges the financial support provided by the National Natural Science Foundation of China (NSFC) (22173051, 21829301, and 21774066), by the PCSIRT (IRT1257), and by the 111 Project (B16027). P. Zhang acknowledges the financial support provided by the NSFC (22473024 and 22073016) and the award of the Shanghai Dongfang Scholar.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- D. Mukherji, C. M. Marques and K. Kremer, J. Phys.: Condens. Matter, 2018, 30, 024002 CrossRef PubMed.
- R. W. Baker, Membrane Technology and Applications, Wiley, 2012 Search PubMed.
- M. Müller and V. Abetz, Chem. Rev., 2021, 121, 14189–14231 CrossRef PubMed.
- S. Bharadwaj, B.-J. Niebuur, K. Nothdurft, W. Richtering, N. F. A. van der Vegt and C. M. Papadakis, Soft Matter, 2022, 18, 2884–2909 RSC.
- I. Bischofberger, D. C. E. Calzolari and V. Trappe, Soft Matter, 2014, 10, 8288–8295 RSC.
- G. Zhang and C. Wu, Phys. Rev. Lett., 2001, 86, 822–825 CrossRef CAS PubMed.
- D. Jia, T. Zuo, S. Rogers, H. Cheng, B. Hammouda and C. C. Han, Macromolecules, 2016, 49, 5152–5159 CrossRef CAS.
- B. A. Wolf and M. M. Willms, Die Makromol. Chem., 1978, 179, 2265–2277 CrossRef CAS.
- J. Rao, J. Xu, S. Luo and S. Liu, Langmuir, 2007, 23, 11857–11865 CrossRef CAS PubMed.
- Y. Higaki, T. Masuda, M. Nakamura and M. Takahashi, Macromolecules, 2023, 56, 6208–6216 CrossRef CAS.
- C.-H. Ko, C. Henschel, G. P. Meledam, M. A. Schroer, R. Guo, L. Gaetani, P. Müller-Buschbaum, A. Laschewsky and C. M. Papadakis, Macromolecules, 2021, 54, 5825–5837 CrossRef CAS.
- C.-H. Ko, C. Henschel, G. P. Meledam, M. A. Schroer, R. Guo, P. Müller-Buschbaum, A. Laschewsky and C. M. Papadakis, Macromolecules, 2023, 56, 8980–8992 CrossRef CAS.
- K. Kyriakos, M. Philipp, J. Adelsberger, S. Jaksch, A. V. Berezkin, D. M. Lugo, W. Richtering, I. Grillo, A. Miasnikova, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromolecules, 2014, 47, 6867–6879 CrossRef CAS.
- L. P. Kreuzer, C. Lindenmeir, C. Geiger, T. Widmann, V. Hildebrand, A. Laschewsky, C. M. Papadakis and P. Müller-Buschbaum, Macromolecules, 2021, 54, 1548–1556 CrossRef CAS.
- C. Geiger, J. Reitenbach, L. P. Kreuzer, T. Widmann, P. Wang, R. Cubitt, C. Henschel, A. Laschewsky, C. M. Papadakis and P. Müller-Buschbaum, Macromolecules, 2021, 54, 3517–3530 CrossRef CAS.
- H. Yong, B. Molcrette, M. Sperling, F. Montel and J.-U. Sommer, Macromolecules, 2021, 54, 4432–4442 CrossRef CAS.
- Y. Yu, R. A. Lopez de la Cruz, B. D. Kieviet, H. Gojzewski, A. Pons, G. Julius Vancso and S. de Beer, Nanoscale, 2017, 9, 1670–1675 RSC.
- K. Nothdurft, D. H. Müller, S. D. Mürtz, A. A. Meyer, L. P. B. Guerzoni, A. Jans, A. J. C. Kühne, L. De Laporte, T. Brands, A. Bardow and W. Richtering, J. Phys. Chem. B, 2021, 125, 1503–1512 CrossRef CAS PubMed.
- B. Liu, J. Wang, G. Ru, C. Liu and J. Feng, Macromolecules, 2015, 48, 1126–1133 CrossRef CAS.
- C. Scherzinger, A. Schwarz, A. Bardow, K. Leonhard and W. Richtering, Curr. Opin. Colloid Interface Sci., 2014, 19, 84–94 CrossRef CAS.
- C. Scherzinger, P. Lindner, M. Keerl and W. Richtering, Macromolecules, 2010, 43, 6829–6833 CrossRef CAS.
- C. H. Hofmann, F. A. Plamper, C. Scherzinger, S. Hietala and W. Richtering, Macromolecules, 2013, 46, 523–532 CrossRef CAS.
- F. Tanaka, T. Koga and F. M. Winnik, Phys. Rev. Lett., 2008, 101, 028302 CrossRef PubMed.
- F. Wang, Y. Shi, S. Luo, Y. Chen and J. Zhao, Macromolecules, 2012, 45, 9196–9204 CrossRef CAS.
- D. Mukherji, C. M. Marques and K. Kremer, Nat. Commun., 2014, 5, 4882 CrossRef PubMed.
- D. Mukherji, C. M. Marques, T. Stuehn and K. Kremer, J. Chem. Phys., 2015, 142, 114903 CrossRef PubMed.
- J. Dudowicz, K. F. Freed and J. F. Douglas, J. Chem. Phys., 2015, 143, 131101 CrossRef PubMed.
- F. Rodríguez-Ropero, T. Hajari and N. F. A. van der Vegt, J. Phys. Chem. B, 2015, 119, 15780–15788 CrossRef PubMed.
- S. Bharadwaj and N. F. A. van der Vegt, Macromolecules, 2019, 52, 4131–4138 CrossRef CAS.
- X. Zhang, J. Zong and D. Meng, Soft Matter, 2020, 16, 7789–7796 RSC.
- S. Bharadwaj, D. Nayar, C. Dalgicdir and N. F. A. van der Vegt, Commun. Chem., 2020, 3, 165 CrossRef CAS PubMed.
- P. Zhang, Z. Wang and Z.-G. Wang, Macromolecules, 2023, 56, 153–165 CrossRef CAS.
- G. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, M. Garcia-Garibay and N. J. Turro, Macromolecules, 1992, 25, 6007–6017 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, W. Pan, M. Garcia-Garibay and N. J. Turro, Macromolecules, 1993, 26, 4577–4585 CrossRef CAS.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- M. J. A. Hore, B. Hammouda, Y. Li and H. Cheng, Macromolecules, 2013, 46, 7894–7901 CrossRef CAS.
- D. Jia, M. Muthukumar, H. Cheng, C. C. Han and B. Hammouda, Macromolecules, 2017, 50, 7291–7298 CrossRef CAS.
- J.-U. Sommer, Macromolecules, 2017, 50, 2219–2228 CrossRef CAS.
- T. Zuo, C. Ma, G. Jiao, Z. Han, S. Xiao, H. Liang, L. Hong, D. Bowron, A. Soper, C. C. Han and H. Cheng, Macromolecules, 2019, 52, 457–464 CrossRef CAS.
- C. E. Mills, E. Ding and B. D. Olsen, Biomacromolecules, 2019, 20, 2167–2173 Search PubMed.
- H. Yong and J.-U. Sommer, Macromolecules, 2022, 55, 11034–11050 CrossRef CAS.
- P. Zhang, Macromolecules, 2024, 57, 4298–4311 CrossRef CAS.
- X. Zhang, J. Zong and D. Meng, Macromolecules, 2024, 57, 8632–8642 CrossRef CAS PubMed.
- S. Bharadwaj, D. Nayar, C. Dalgicdir and N. F. A. van der Vegt, J. Chem. Phys., 2021, 154, 134903 CrossRef CAS PubMed.
- N. F. A. van der Vegt, J. Phys. Chem. B, 2021, 125, 5191–5199 CrossRef CAS PubMed.
- C. Dalgicdir, F. Rodríguez-Ropero and N. F. A. van der Vegt, J. Phys. Chem. B, 2017, 121, 7741–7748 CrossRef CAS PubMed.
- Z. Mohammadyarloo and J.-U. Sommer, Macromolecules, 2022, 55, 8975–8986 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- S. Kirkpatrick, D. Gelatt Jr and M. Vecchi, Science, 1983, 220, 671–680 CrossRef CAS PubMed.
- R. Larson, J. Chem. Phys., 1989, 91, 2479–2488 CrossRef CAS.
- W. Kong, B. Li, Q. Jin, D. Ding and A.-C. Shi, J. Am. Chem. Soc., 2009, 131, 8503–8512 CrossRef CAS PubMed.
- J. Wu, Z. Wang, Y. Yin, R. Jiang, B. Li and A.-C. Shi, Macromolecules, 2015, 48, 8897–8906 CrossRef CAS.
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef PubMed.
- S. Chen and Z.-G. Wang, Phys. Rev. Lett., 2023, 131, 218201 CrossRef CAS PubMed.
- G. Cristobal, J.-F. Berret, C. Chevallier, R. Talingting-Pabalan, M. Joanicot and I. Grillo, Macromolecules, 2008, 41, 1872–1880 CrossRef CAS.
- A. Ianiro, H. Wu, M. M. J. van Rijt, M. P. Vena, A. D. A. Keizer, A. C. C. Esteves, R. Tuinier, H. Friedrich, N. A. J. M. Sommerdijk and J. P. Patterson, Nat. Chem., 2019, 11, 320–328 CrossRef CAS PubMed.
- H. Yong, H. Merlitz, A. Fery and J.-U. Sommer, Macromolecules, 2020, 53, 2323–2335 CrossRef CAS.
- J. Dudowicz, K. F. Freed and J. F. Douglas, J. Phys. Chem. B, 2016, 120, 5753–5758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00164a |
| This journal is © The Royal Society of Chemistry 2025 |