
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural interplay in block copolymer-bile salt complexes: from globules to ribbons†
Suelen Gauna
Trindade
ab,
Guanqun
Du‡
a,
Luciano
Galantini
 c,
Lennart
Piculell
a,
Watson
Loh
*b and
Karin
Schillén
*a
c,
Lennart
Piculell
a,
Watson
Loh
*b and
Karin
Schillén
*a
aDivision of Physical Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: karin.schillen@fkem1.lu.se
bInstitute of Chemistry, University of Campinas (UNICAMP), P.O. Box 6154, 13083-970, Campinas, São Paulo, Brazil. E-mail: wloh@unicamp.br
cDepartment of Chemistry, Sapienza University of Rome, P.O. Box 34-Roma 62, Piazzale A. Moro 5, 00185 Roma, Italy
First published on 22nd April 2025
Abstract
The supramolecular structures resulting from the complexation between the neutral–cationic block copolymer poly(ethylene oxide)-block-poly(2-(trimethylammonium)ethyl methacrylate iodide) (PEO114-b-PTMAEMAI95) and the bile salt sodium deoxycholate (NaDC) were investigated by dynamic light scattering, small-angle X-ray scattering, cryogenic transmission electron microscopy and proton NMR techniques. Complexes were produced using different preparation protocols: the direct mixing of the pure solutions of block copolymer and bile salt, containing their respective simple counterions, and the dispersion in water of a freeze-dried complex salt, free of simple counterions. While the direct mixing protocol produced a mixture of ordered ribbon-like aggregates and globular particles with disordered cores, the complex salt protocol yielded exclusively ordered “ribbons”. The globular particles resembled classical spherical “complex coacervate core micelles” with a core of anionic deoxycholate micelles complexed with cationic PTMAEMA(+) blocks, the core radius being limited by the PTMAEMA contour length, and a shell composed of neutral PEO blocks. The drastically different ribbon morphology was found to result from (1) the organization of DC anions into hexagonally packed helices in the core and (2) the limitations on the ribbon thickness imposed by the lengths of the copolymer blocks. By varying temperature and sample treatments, it was found that the ordered ribbon morphology represents the equilibrium structure at 25 °C, while the globular morphology is favored at 50 °C. The results suggest strategies to design the morphology and tune the dimensions of aqueous block copolymer–bile salt aggregates.
1. Introduction
Complexes of hydrophilic neutral-ionic block copolymers and oppositely charged polymers or surfactants have been extensively explored in the last few decades. In aqueous dispersions, these complexes are typically organized as core–shell nanoparticles, with a concentrated core composed of the charged block of the copolymer electrostatically complexed with the oppositely charged species, and a shell composed of the neutral hydrophilic block.1 These co-assembled structures are often called complex coacervate core micelles (C3Ms).2,3Even though the most abundant C3M systems reported consist of core–shell spheres, other morphologies can appear depending on the molecular structure of the components.4,5 It is well known that the block length ratio of the copolymer has a determining effect on the C3Ms shape. Relatively long neutral hydrophilic blocks generally lead to the formation of spheres, whereas shorter shell-forming blocks often give rise to lower-curvature aggregates such as elongated micelles or vesicles.6–8 Additionally, not only the morphology, but also the size, internal structure and colloidal stability of the aggregates can be sensitive to the preparation protocol and factors such as concentration, ionic strength, and temperature.9–13 When surfactant ions are used as complexing agents, surfactant liquid crystalline structures may form in the core, giving rise to various angular particle shapes that reflect the underlying core structure.4,12,14–17 We note, however, that also for these systems, core–shell spheres with disordered cores typically form as a result of conventional preparation protocols where solutions of the respective components are mixed. We will return to this point below.
Although not extensively explored, the use of biosurfactants such as bile salts can confer unique characteristics to oppositely charged polymer–surfactant systems. Unlike conventional surfactants with a single hydrophilic head and a hydrophobic tail, bile salts display a rigid chiral steroidal skeleton with a complex distribution of hydrophobic and hydrophilic regions.18 Due to the complexity of their structure, the aggregation of the bile salts often follows a stepwise process in a broad concentration interval, generating aggregate morphologies that cannot be rationalized by the geometric rules of conventional surfactant packing parameters.19,20
Due to their special self-assembly behavior as described above, bile salts and their derivatives are also capable of forming intriguing structures when complexed with cationic polymers or multivalent cations,21,22 and similar structures have recently been found in C3Ms containing bile salts in their cores.21,23 Thus, Du et al.21 demonstrated that the complexation between the anionic bile salt sodium deoxycholate (NaDC), and cationic block copolymers with a neutral hydrophilic block of either poly(N-isopropylacrylamide) (PNIPAM) or poly(ethylene glycol) (MPEG) and a ionic block of poly(3-acrylamidopropyl)trimethylammonium (PAMPTMA(+)) (PNIPAMm-block-PAMPTMA(+)30 (m = 65 or 48) or MPEG45-block-PAMPTMA(+)21) promoted the arrangement of DC helices into hexagonally packed bundles and toroids. Cryogenic transmission electron microscopy (cryo-TEM) and cryogenic electron tomography (cryo-ET) analysis provided crucial structural information, enabling the development of a model to describe these assemblies: the DC helices are wrapped together in ordered hexagonally packed bundles by the cationic block of the copolymer while the neutral hydrophilic block composes the shell, providing colloidal stability to the assemblies.
In an earlier study, where a block copolymer with a longer PNIPAM block was used (PNIPAM120-b-PAMPTMA(+)30) in mixtures with NaDC, globular particles with disordered cores were obtained in addition to the internally structured bundles.23 If the bile salt was chemically modified, only supramolecular twisted ribbons were obtained.24 Those same systems were also explored by varying the temperature.24,25 Due to the presence of the thermoresponsive block of PNIPAM, which displays a lower critical solution temperature (LCST) typically around 32 °C, it was observed that the complexes can display aggregation or a morphological transformation upon heating.
The previous studies highlight the versatility of block copolymer–bile salt complexes in forming diverse supramolecular morphologies. However, the factors governing the formation and stability (kinetic or thermodynamic) of each possible morphology, as well as their dimensions, remain to be understood. In the present contribution we therefore explore the supramolecular structures formed by the complexation between DC and the neutral–cationic block copolymer poly(ethylene oxide)114-block-poly(2-(trimethylammonium)ethyl methacrylate)95 (PEO114-b-PTMAEMA(+)95). Considering that the previous studies exclusively used PAMPTMA(+) as the positively charged block we sought in this study to analyze, by using a copolymer with a PTMAEMA(+) block, if the formation of DC helices and bundles of such helices in aggregates with the block copolymer critically depends on the detailed chemistry of the cationic block. Furthermore, the use of PEO as the neutral hydrophilic block allows us to explore the influence of higher temperatures on the assemblies without compromising their colloidal stability in aqueous media. The LCST of the PEO homopolymer in water is above 100 °C.26–28 Lastly, here we use a neutral–cationic diblock copolymer where both blocks are longer than in the block copolymer–NaDC mixtures studied previously. This could play an important role for the resulting morphology and internal structure of the co-assemblies.
Dynamic light scattering (DLS), small-angle X-ray scattering (SAXS), cryo-TEM, and proton NMR techniques are employed to elucidate the morphology, size and structure of the complexes obtained. Importantly, we also employ different protocols to prepare and treat the complexes, to establish what may be their equilibrium morphology under various conditions. Our main focus is on conditions where the electrostatic attraction between the cationic block and the bile salt are at a maximum, that is, a stoichiometric charge ratio and no added salt. Occasional comparisons are made using samples with a twofold excess of bile salt.
2. Experimental
2.1 Materials
Poly(ethylene oxide)-block-poly(2-(dimethylamino)ethyl methacrylate) (PEO114-b-PDMAEMA95) block copolymer (Mn = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Mw/Mn = 1.4) was purchased from Polymer Source. Inc. (Quebec, Canada). The contour lengths of the two blocks are estimated to 24 nm for PDMAEMA95 and 41 nm for PEO114 (see below). Prior to use, the copolymer was reacted with methyl iodide, following the procedure described by Scandalis et al.,29 to promote the complete quaternization of the amine groups of the PDMAEMA block, resulting in the block copolymer poly(ethylene oxide)-block-poly(2-(trimethylammonium)ethyl methacrylate iodide) (PEO114-b-PTMAEMAI95) (Mn = 33484 g mol−1). The complete quaternization of the copolymer was confirmed by 1H NMR measurement, as shown in Fig. S1, ESI.† Sodium deoxycholate (NaDC, assay ≥99%), deoxycholic acid (HDC, assay ≥99%), sodium cholate (NaC, assay ≥99%), and sodium iodide (NaI, assay ≥99.5%) were purchased from Sigma Aldrich and used as received. AmberTec™ UP550 OH anion exchange resin was obtained from Sigma Aldrich and used as received. Water used for the sample preparation was purified using a Milli-Q system (Millipore Corporation, Bedford, MA, U.S.).
000 g mol−1, Mw/Mn = 1.4) was purchased from Polymer Source. Inc. (Quebec, Canada). The contour lengths of the two blocks are estimated to 24 nm for PDMAEMA95 and 41 nm for PEO114 (see below). Prior to use, the copolymer was reacted with methyl iodide, following the procedure described by Scandalis et al.,29 to promote the complete quaternization of the amine groups of the PDMAEMA block, resulting in the block copolymer poly(ethylene oxide)-block-poly(2-(trimethylammonium)ethyl methacrylate iodide) (PEO114-b-PTMAEMAI95) (Mn = 33484 g mol−1). The complete quaternization of the copolymer was confirmed by 1H NMR measurement, as shown in Fig. S1, ESI.† Sodium deoxycholate (NaDC, assay ≥99%), deoxycholic acid (HDC, assay ≥99%), sodium cholate (NaC, assay ≥99%), and sodium iodide (NaI, assay ≥99.5%) were purchased from Sigma Aldrich and used as received. AmberTec™ UP550 OH anion exchange resin was obtained from Sigma Aldrich and used as received. Water used for the sample preparation was purified using a Milli-Q system (Millipore Corporation, Bedford, MA, U.S.).
2.2 Sample preparation
Collectively, the dispersions prepared by this preparation protocol are referred to in this paper as PEODC (CR = x), where PEO refers to the PEO114-b-PTMAEMA(+)95 block copolymer, DC refers to deoxycholate, and CR denotes the negative-to-positive charge ratio of the complex as defined above.
:1 stoichiometry of charges at a final pH of 7.4. The mixture was left to react under magnetic stirring for several hours to guarantee complete homogeneity. Then, the dispersion was freeze-dried, and the complex salt was isolated as a white powder. During the ion exchange step, the quaternary amine group of PTMAEMA(+) block inevitably undergoes a Hoffman elimination reaction under alkaline conditions, generating trimethylamine and poly(vinyl methacrylate) chains as subproducts, as confirmed by 1H NMR (Fig. S2, ESI†). From the pH titration curves of the alkaline copolymer solution after removal from the anion exchange resin, the second inflection point corresponding to the weak base trimethylamine indicated a 5 mol% content of Hoffman elimination products. However, since this reaction progresses over time, the extent may be higher in the complex salt, due to the delay between the titration and the addition and complete dissolution of deoxycholic acid to neutralize the copolymer solution.
The samples to be investigated were prepared by dispersing appropriate amounts of complex salt in pure water under vigorous magnetic stirring. Dispersions obtained by employing this preparation protocol will be referred to as PEODC (CS), where the terms PEO and DC are defined above, and CS refers to the complex salt protocol employed.
2.3 Methods of characterization
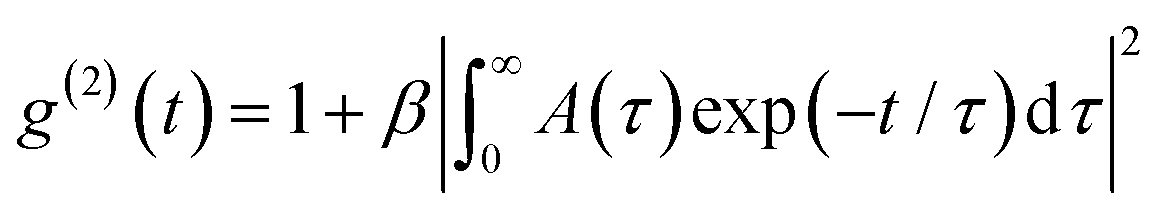 , where τ is the relaxation time and β(≤1) takes into account deviations from ideal correlation. The obtained relaxation time distributions are presented in an equal area representation as τA(τ) versus log (τ/μs). Furthermore, the apparent diffusion coefficient Dapp of the scattering particles undergoing translational motion corresponds to the mutual diffusion coefficient in the low q regime and at finite concentrations and is given by:
, where τ is the relaxation time and β(≤1) takes into account deviations from ideal correlation. The obtained relaxation time distributions are presented in an equal area representation as τA(τ) versus log (τ/μs). Furthermore, the apparent diffusion coefficient Dapp of the scattering particles undergoing translational motion corresponds to the mutual diffusion coefficient in the low q regime and at finite concentrations and is given by:  , where Γ is the relaxation frequency (Γ ≡ τ−1) and q is the magnitude of the scattering vector defined as q = 4πn
, where Γ is the relaxation frequency (Γ ≡ τ−1) and q is the magnitude of the scattering vector defined as q = 4πn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin(θ/2)/λ, where n is the refractive index of the solvent (here water). Here, in the case of the globular complexes, the measured relaxation rates were linearly dependent upon q2 through the origin thus confirming that the intensity correlation function is associated with a translational diffusion process. The DLS measurements on the globular complexes at 25 and 50 °C were thereafter only performed at θ = 90°. The Z-averaged apparent hydrodynamic radius (RH,app) of the globular complexes was calculated using the Stokes–Einstein equation: RH,app = kT/6πη0Dapp in which k is the Boltzmann constant and η0 is the viscosity of the solvent (here water) at the absolute temperature T.
sin(θ/2)/λ, where n is the refractive index of the solvent (here water). Here, in the case of the globular complexes, the measured relaxation rates were linearly dependent upon q2 through the origin thus confirming that the intensity correlation function is associated with a translational diffusion process. The DLS measurements on the globular complexes at 25 and 50 °C were thereafter only performed at θ = 90°. The Z-averaged apparent hydrodynamic radius (RH,app) of the globular complexes was calculated using the Stokes–Einstein equation: RH,app = kT/6πη0Dapp in which k is the Boltzmann constant and η0 is the viscosity of the solvent (here water) at the absolute temperature T.
Temperature dependent DLS measurements were performed using a Zetasizer Nano ZS instrument (Malvern Panalytical, Malvern, U.K.) equipped with a 4 mW He–Ne laser source (λ = 632.8 nm) and with a backscattering geometry at a scattering angle of θ = 173°. Samples were placed in square glass cuvettes and measured at 25 and 50 °C. The temperature was controlled by using the Peltier-thermostatted sample holder of the instrument (accuracy: ±0.1 °C). The obtained time autocorrelation functions of the scattered intensity were evaluated by the non-linear least square procedure provided by the instrument software. The latter analysis expresses the results as apparent hydrodynamic radii distributions and the quoted RH,app values are obtained from the main mode in the distributions.
sin(θ/2)/λ). The measured intensities were corrected using background scattering and sample transmission. Data were brought to absolute scale by using the absolute scattering intensity of water at 20 °C. Complementary SAXS and WAXS experiments were conducted at the Institute of Chemistry, University of Campinas (UNICAMP, Campinas, Brazil), using the laboratory SAXSPoint 5.0 equipment (Anton Paar, Graz, Austria). The wavelength of the radiation was 1.54 Å (Cu Kα). Liquid samples were loaded into 1.5 mm quartz capillaries, while gel-like samples were mounted between two mica foils. Experiments were performed at two sample-to-detector distances, covering the q-range of 7.0 to 28 nm−1 for WAXS measurements and 0.09 to 5 nm−1 for SAXS measurements. Background subtraction was performed by subtracting a water-filled capillary measurement from the liquid sample data, and a measurement of two mica foils was subtracted from the gel samples. The experimental data were fitted by Indirect Fourier Transform (IFT) analysis by using the WIFT program,32 to obtain the pair distance distribution functions.
3. Results and discussion
3.1 Samples obtained after direct mixing
 | ||
| Fig. 1 Cryo-TEM images of the PEODC dispersions at CR = 1 (a), (b) and CR = 2 (c), (d) at 0.1 wt% of copolymer. | ||
The latter are similar in appearance as the “bundle” or “tape-like” structures found in the PNIPAMm-b-PAMPTMA(+)20/NaDC21 (m = 65 or 48) and PNIPAM120-b-PAMPTMA(+)30/NaDC23 systems. The globular particles appearing in Fig. 1 present disordered cores and are similar to those previously reported for complexes formed by PNIPAM120-b-PAMPTMA(+)30 copolymer and NaDC.23 In the globular particles, the core is composed of anionic deoxycholate micelles complexed with cationic PTMAEMA(+) blocks, while the shell is composed by the neutral hydrophilic PEO blocks, which provide steric stability to the assemblies against further aggregation. The shell is invisible in the cryo-TEM images due to low electronic density contrast. The average core radius (Rc) determined from the images of the globular particles is 32 ± 8 nm and 30 ± 8 nm for CR = 1 and CR = 2, respectively (see size distribution in Fig. S3, ESI†). This insignificant variation in core size indicates that the excess bile salt added does not enter the core of the globules to a significant extent. The apparent hydrodynamic radii obtained from DLS measurements at the scattering angle 90° were also determined using RILT analysis (see Section 2.3.2 in Methods of characterization), again giving very similar values of RH,app = 60 nm for CR = 1 and RH,app = 57 nm for CR = 2. The electrophoretic mobility values were μ = 0.7 × 10−8 m2 s−1 V−1 for CR = 1 and −0.2 × 10−8 m2 s−1 V−1 for CR = 2.
Regarding the ribbon-like structures observed in Fig. 1, it is possible to discern darker stripes within the ribbons with a periodic average distance of 5.0 nm (see Fig. S4, ESI†) for both CR = 1 and CR = 2 samples. The ribbons exhibit a distribution of widths, averaging 44 ± 21 nm for CR = 1 and 54 ± 34 nm for CR = 2 (Fig. S5, ESI†), indicating a slight dependence on the charge ratio. The first model to describe these striped structures, proposed in ref. 23, was later revised in ref. 21, identifying them as supramolecular DC helices condensed into hexagonally ordered bundles by the association with the cationic blocks of several copolymers chains. The periodic distance of 5.0 nm between the stripes is interpreted as the inter-plane distance within the structure and the value obtained is close to what is observed in the PNIPAMm-b-PAMPTMA(+)n/NaDC systems21,23 and it also consistent with that of the hexagonal liquid-crystalline phase in the NaDC/water system at high concentration.33 As in the globular particles, the PEO block protrudes out from the ribbons, providing colloidal stability in aqueous media.
The locations of the two polymer blocks and the DC molecules in the aggregates were confirmed by 1H NMR analyses of the dispersions (Fig. 2). While solutions of the pure block copolymer and pure bile salt show several sharp peaks (Fig. 2(a) and (b)), most of these peaks are broadened beyond detection in the spectra of the dispersions at CR = 1 and CR = 2. The latter spectra show only three peaks: from the block copolymer, a sharp PEO peak and a broader peak corresponding to the N-methyl protons of the cationic block, and from the bile salt a non-resolved, very broad peak in the region 0–2.5 ppm. Due to the preserved mobility of the PEO chains protruding from the aggregate surfaces, their signal remains narrow and intense in the 1H NMR spectrum (analogous effects were found in the PNIPAM48-b-PAMPTMA(+)20/NaDC system).21 In the cationic block, the N-methyl groups of the (trimethylammonium)ethyl side chains remain relatively mobile, while the signals from the other protons have disappeared. As expected, the bile salt peak increases in intensity for the CR = 2 sample, which contains excess NaDC. However, no high-resolution signal is seen from the excess bile salt, indicating a rapid exchange (on the NMR timescale) of the excess DC anion in solution with DC anions residing in the aggregates.
 | ||
| Fig. 2 1H NMR spectra in D2O of the pure components: 14.18 mM of NaDC (a), 0.5 wt% of PEO114-b-PTMAEMAI95 (b), and of the dispersion of PEODC (CR = 1) (c) containing 14.18 mM of NaDC dispersed at 0.5 wt% of copolymer. (d) Superimposed spectra from panels a, b, and c. (e) 1H NMR spectra of the PEODC (CR = 2) dispersion, containing 28.36 mM of NaDC and 0.5 w% of copolymer, together with the spectra from NaDC and PEO114-b-PTMAEMAI95 pure solutions with the same concentrations as in the dispersion. For comparison of the peak intensities, all the superimposed spectra are on the same scale in (d) and (e). NaDC peaks were assigned according to ref. 34. | ||
For a quantitative evaluation of the intensities of the visible signals, the latter signals were integrated and compared to the corresponding signals from the pure reference solutions at the same concentration. Due to the presence of broad and overlapping peaks, the spectra of the PEODC complexes were deconvoluted using Lorentzian fitting to enable peak integration. Table 1 shows the relative peak area ratios for the complexes. The PEO peak displays an area unchanged compared to that of the pure copolymer solution, confirming that the PEO chains reside fully exposed on the aggregate surfaces, where they are highly mobile. By contrast, the signals from DC ions and PTMAEMA(+) chains, which are electrostatically complexed in the ribbons’ skeleton or the particles cores, are attenuated. The apparent relative intensity of the DC signal is higher at CR = 2 than at CR = 1, which is to be expected; the excess free DC ions in solution should contribute at full strength to the observed intensity. Note that, also under conditions of charge stoichiometry (CR = 1), a small fraction of DC ions are dissociated from the aggregate surfaces, as indicated by the positive surface charge seen in the electrophoretic mobility measurements. By analogy with NMR results from simpler block copolymer/surfactant aggregates,35 we conclude that the resulting NMR peak shape and intensity are the result of a surface fraction of DC ions in rapid (on the NMR timescale) exchange with free DC ions. Both at CR = 1 and at CR = 2, the reduction in intensity could be due to DC ions residing deep into the aggregates, which do not participate in the rapid exchange.
| Sample | PEO | N-Methyl PTMAEMA(+) | DC |
|---|---|---|---|
| PEODC (CR = 1) | 0.95 | 0.68 | 0.50 |
| PEODC (CR = 2) | 1.03 | 0.78 | 0.81 |
The coexistence of globular particles and ribbons, two structures with such drastic morphological differences, turns their characterization by scattering techniques challenging. The expected scattering features of both structures, such as a globular form factor and a sharp correlation peak related to the periodic distribution of the deoxycholate helices in the ribbons were not discerned in the SAXS scattering profile of the dispersions, as can be seen from Fig. S6 (ESI†). This effect in the resulting scattering can be attributed to the polydispersity and polymorphism of the samples.
 | ||
| Fig. 3 Cryo-TEM images of the top phase (a) and the redispersed bottom phase (b) collected after centrifugation of a PEODC dispersion at CR = 1. The copolymer concentration was 0.1 wt%. | ||
The separation of the globular particles from the ribbons was also confirmed by SAXS, which revealed a scattering profile characteristic of globular particles (Fig. 4(a)). The SAXS data was fitted using the Indirect Fourier Transform (IFT) method.32 The pair distance distribution function (p(r)) (Fig. 4(a), inset), resulting from the fit, displays a symmetric bell shape curve with a maximum distance of 72 nm. This distribution profile is characteristic of spherical particles.10,38 The radius of gyration (Rg) calculated from the fit is 25.6 ± 0.3 nm.
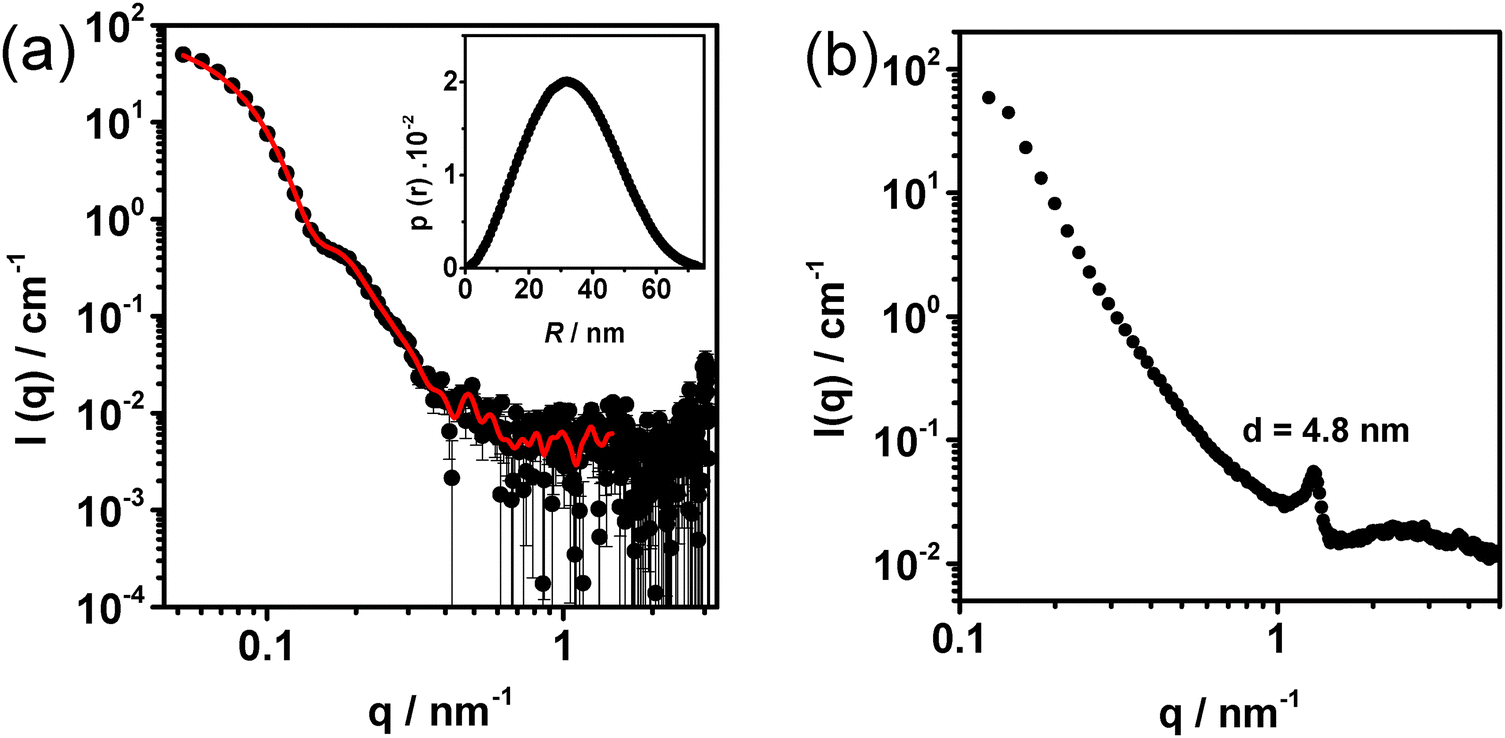 | ||
| Fig. 4 SAXS patterns (black symbols) for the top phase (a) and bottom phase (b) obtained after centrifugation of a PEODC dispersion at CR = 1 with a copolymer concentration of 0.5 wt%. Panel (a) also displays the curve fitting (red line) obtained by IFT analysis and the corresponding pair distance distribution function (p(r)) (inset). | ||
A similar result was obtained by filtration of PEODC (CR = 1) sample with a 0.45 μm pore size filter. After filtration, only globular particles remained in the sample, indicating that the ribbons were retained in the filter or transformed to globules upon shearing (see cryo-TEM image in Fig. S8a, ESI†). The Rg value obtained from the IFT analysis of the SAXS curve (Fig. S8b, ESI†) was Rg = 26.7 ± 0.4 nm. These Rg values are in good agreement with the average core radius obtained by cryo-TEM (Rc = 32 ± 8 nm and 30 ± 8 nm for CR = 1 and CR = 2, respectively, Fig. S3, ESI†). In these systems, we can infer that the Rg value obtained by SAXS refers to the core radius of the particles, since the PEO chains that compose the shell are highly hydrated and do not present a sufficient electronic density contrast to be discerned from the background.
The concentrated bottom phase of the centrifuged sample was found to primarily contain the ribbon-like structures as confirmed by the presence of a first order Bragg peak at q = 1.3 nm−1 in the SAXS curve (Fig. 4(b)) from which a mean inter-plane distance, indicating the hexagonal liquid crystalline structure of the ribbons, can be calculated using d = 2π/q. The d-spacing obtained is 4.8 nm, which is in excellent agreement with that obtained in ref. 21, and in close agreement with the mean distance of 5.0 nm estimated from the cryo-TEM images (Fig. S4, ESI†). This result demonstrates that the formation and structure of the DC helices are not critically dependent on the chemistry of the cationic block, as the use of PAMPTMA(+) and PTMAEMA(+) yielded complexes with indistinguishable internal structure characteristics.
The bottom phase was redispersed in water and further analyzed by cryo-TEM confirming that, after concentration, the ribbons can be redispersed, preserving their structure, as depicted in Fig. 3(b). The ribbon-rich bottom phase was also characterized by WAXS, which revealed the presence of Bragg diffraction peaks characteristic of the helical arrangement of the deoxycholate molecules inside the ribbons (see Fig. S9a, ESI†). This result is in good agreement with the X-ray pattern obtained for dry helical fibers of pure NaDC39,40 and the results previously reported for similar block copolymer/DC systems.21
Furthermore, as can be seen in the cryo-TEM images, especially in Fig. 1, the ribbons are relatively transparent, so that it is possible to observe one ribbon through the other in the microscopy grid. Additionally, the images in Fig. 1(b) and (d) show that the PEODC ribbons are quite flexible, in such a way that they can twist, fold and form rolled up assemblies. Overall, these features indicate that the ribbons are composed of only a few layers of long bile salt helices. No thicker bundles or toroidal structures, as reported by Du et al.21 in some systems, were observed in the present study.
 | ||
| Fig. 5 (a) Cryo-TEM image of the collected top phase of PEODC (CR = 1) dispersion 15 days after centrifugation. (b) Cryo-TEM image obtained from a PEODC (CR = 1) dispersion recorded the day after filtration with a 0.45 μm pore size filter. Both images show ribbons coexisting with globular particles. The copolymer concentration was 0.1 wt%. | ||
Comparing the image in Fig. 5(a) with that in Fig. 3(a) (recorded the same day as the centrifugation process), it is clear that globular particles evolve, given sufficient time, into the ribbon morphology. The same behavior was observed for the dispersion analyzed one day after filtration (Fig. 5(b)), where short ribbons were already detected. Note, however, that both Fig. 5(a) and (b) still show the presence of globular particles coexisting with the formed ribbons; the transition is not complete. Nevertheless, the fact that the globular particles transition to ribbons over time at room temperature strongly suggests that the ribbon-like structure represents a lower free energy state for the PEODC complexes.
3.2 Changing the preparation protocol
 | ||
| Fig. 6 Cryo-TEM images of PEODC complex salt dispersions at 0.1 wt% of copolymer without (a) and with the addition of NaI (b) at an equivalent amount to that in the PEODC (CR = 1) prepared by direct mixing protocol. (c) SAXS curves for the same dispersions at 0.5 wt% of copolymer. Gray symbols: pure PEODC (CS) dispersion; blue symbols: PEODC (CS) dispersion + NaI. | ||
This result suggests that, in the complex salt dispersion, the components are involved exclusively in ribbons. The larger proportion of ribbons was also reflected in the appearance of a correlation peak in the SAXS curve referring to the periodicity of the NaDC helices in the ribbons (inter-plane distance), as depicted in Fig. 6(c). This peak was not visible in the direct mixing sample at the same copolymer concentration (see Fig. S6, ESI†). An analysis of the cryo-TEM images revealed that the ribbons in the PEODC (CS) sample had a larger average width compared to those in the PEODC (CR = 1) sample, with values of 65 ± 24 nm (Fig. S10, ESI†) and 44 ± 21 nm (Fig. S5a, ESI†), respectively. However, the ribbons retained the same helical arrangement of the deoxycholate molecules, as confirmed by the Bragg diffraction peaks observed in the WAXS data presented in Fig. S9b (ESI†).
To check whether the presence of counterions per se plays a critical role in the formation of globular particles, a sample of PEODC complex salt was dispersed in NaI solution, yielding a dispersion with the same composition as the direct mixing sample. Analyses of the former sample using cryo-TEM and SAXS (see Fig. 6(b) and (c)) showed no presence of globular particles. The only structural effect detected was that the ribbons were narrower (W = 53 ± 24 nm) in the presence of NaI, compared to those in the salt-free PEODC complex salt (W = 65 ± 24 nm) (see Fig. S10, ESI†). This result suggests that the presence of small amounts of simple salt is not a critical factor for the formation of globules.
As the PEODC (CS) sample only contained ribbon-like aggregates, their 1H NMR peak intensities were analyzed and compared with PEODC (CR = 1), which contained both ribbons and globules, and the PEODC (CR = 1) top phase sample, which contained essentially only globules. As shown in Table S1 (ESI†), the relative DC integrals reveal that the DC signal intensity is more attenuated for the PEODC (CS) sample compared to the other samples, especially compared to the PEODC (CR = 1) top phase sample, indicating that intensity loss is significantly larger for ribbon-like aggregates than for globules. This aligns with the expected greater mobility of DC anions in the disordered core of the globules compared to the tightly packed DC anions in the helical structures within the ribbons. A smaller fraction of DC ions in the core should also be involved in rapid molecular exchange with free DC ions in the bulk when the DC molecules are assembled into crystalline helical rods.
The above findings indicate that the formation of the ribbons is facilitated when there is a locally high concentration of bile salt and block copolymer during mixing. This effect is even more pronounced when these components are associated in powder form before dispersion in water, as is the case with the complex salt and the freeze-dried directly mixed samples.
3.3 Reversible melting of ribbons to globules
In contrast to PNIPAM, PEO is known not to undergo phase transition in water upon heating below the normal boiling point.46,47 To evaluate the impact of temperature on the stability of the two competing structures, the PEODC (CS) sample was characterized by SAXS and DLS at 25 and 50 °C. Fig. 7(a) compares the SAXS curves of the PEODC (CS) dispersion measured at 25 °C, 50 °C and then again at 25 °C. | ||
| Fig. 7 (a) SAXS curves for a PEODC (CS) dispersion with a copolymer concentration of 0.5 wt% measured at different temperatures: at 25 °C (black curve); heated and equilibrated at 50 °C (yellow curve); cooled and equilibrated at 25 °C again (red curve). (b) SAXS pattern of the sample at 50 °C (same black curve as in panel a) and curve fitting (red line) obtained by IFT analysis and the corresponding pair distance distribution function (p(r)) (inset). | ||
The scattering profile shows that, after heating to 50 °C, the correlation peak corresponding to the periodic distance between the deoxycholate helices in the hexagonal array (inter-plane distance) in the ribbons disappears, indicating that the ribbons disintegrate upon heating. The increase in temperature disrupts the helical packing of the bile salt in the ribbon structure.19,48,49 This leads to a structural rearrangement to a globular morphology, as revealed by the bell shape of the pair distance distribution function (p(r)) shown in Fig. 7(b) (inset), resulting from the fit of the SAXS profile at 50 °C. The Rg obtained from the fit for these globular particles was 23.1 ± 0.4 nm, in close agreement with the globules obtained by direct mixing. After the sample was cooled back to 25 °C, the ribbons gradually reformed, and the correlation peak became apparent again, evidencing the reversibility of the temperature-induced morphological transition. The melting transition was also studied and confirmed by DLS measurements (Fig. S13, ESI†).
3.4 Shape and dimensions of ribbons
From the above observations, we can conclude that the core organization of deoxycholate anions into hexagonally packed helices, and the resulting formation of ribbon-like aggregates, represent a lowering of the free energy of the PEODC aggregates compared to the disordered globules. How do we understand the shapes of the ribbon-like aggregates? The elongated structure is a natural consequence of the hexagonal packing of the helices combined with the restrictions imposed by the neutral chains of the block copolymer, the latter preventing formation of an infinite hexagonal array. For a more detailed understanding, we need to consider the lengths of the two blocks in the copolymer.From the cryo-TEM images, we observe that the aggregates are not only elongated along the long-axis of the DC helices but also tend to lie flat on the grid displaying a width significantly greater than their thickness. The flat ribbon shape differs from the most intuitive shape expected for a rod with a 2D hexagonal internal structure, which would possess a regular hexagonal cross section with equal lengths of all sides. The deviation must ultimately be due to the repulsion between the PEO chains located on the surfaces of the aggregates. A hypothetical regular hexagonal rod, with a single cross-sectional dimension limited only by the PTMAEMA(+) contour length, would possess a significantly lower curvature than the globule with the corresponding diameter and, consequently, a stronger repulsion between the PEO chains in the shell. To decrease this repulsion, a thinner rod would be required, with a higher curvature and, also, a larger area per copolymer molecule at the surface of the core (since the volume per cationic block in the core must be conserved). However, the thinner rod would also reduce the number of stabilizing interactions per unit volume in the core. A way to make the cross-sectional area expand is to allow the aggregate to grow in one lateral dimension, into a ribbon, and we propose that this is the reason for the anisometric shape of the cross section. On the other hand, the ribbon shape results in a further decrease in curvature of the interface, compared to the hypothetical hexagonal rod. This should lead to a further reduction of the thickness of the ribbon, because of the increased repulsion between the PEO chains in the shell.
The resulting morphology is a ribbon-like aggregate with a thickness (as is clear especially from the occasional twisted ribbons found in the cryo-TEM images) much thinner than twice the contour length of the PTMAEMA(+) block. However, the length of the latter block is still important for the thickness of the ribbon, since a longer block would allow the thickness of the ribbon to increase at a preserved repulsion between the PEO chains on the surface. Conversely, a shorter neutral block would decrease the headgroup repulsion and, at a constant cationic block length, allow the ribbon to grow in thickness. This balance explains why thicker hexagonally packed bundles and toroids are obtained when copolymers with a shorter neutral hydrophilic block (PNIPAMm-b-PAMPTMA(+)20 (m = 65 or 48) and MPEG45-b-PAMPTMA(+)21) are complexed with NaDC.21 In good agreement with this hypothesis, the formation of toroidal assemblies was not observed in the system containing NaDC and the block copolymer containing a long PNIPAM block with 120 repeating units.23
The edges of the ribbon are highly curved, which serves to decrease the average repulsion between PEO chains. This effect should influence the equilibrium width of the ribbon. However, our results clearly show that the ribbon width is at least partly determined by kinetic effects. Ribbons obtained from the complex salt preparation protocol are, on average, wider than those obtained from the direct mixing protocol (see Fig. S5 and S10, ESI†).
3.5 Understanding the globular aggregates
To further investigate the preferred aggregation behavior in the absence of bile salt helix formation, we studied a mixture of PEO114-b-PTMAEMAI95 with sodium cholate (PEOC complex), prepared by the direct mixing protocol at CR = 1. Trihydroxy bile salts, such as cholate, are known to self-assemble into smaller aggregates and are less prone to form helical arrangements.50–52 A cryo-TEM image of the PEOC complex (Fig. 8(a)) displays only globular particles with cores lacking internal order and an average Rc = 36 ± 7 nm (Fig. S14, ESI†). These particles presented a mean RH,app value of 70 nm and a slight positive electrophoretic mobility value of μ = 0.96 × 10−8 m2 s−1 V−1. Similarly to the PEODC (CR = 1) globules, the globular structures of PEOC (CR = 1) were preserved at 50 °C, maintaining an RH,app unchanged at 70 nm after heating, as illustrated by the relaxation time distributions shown in Fig. 8(b).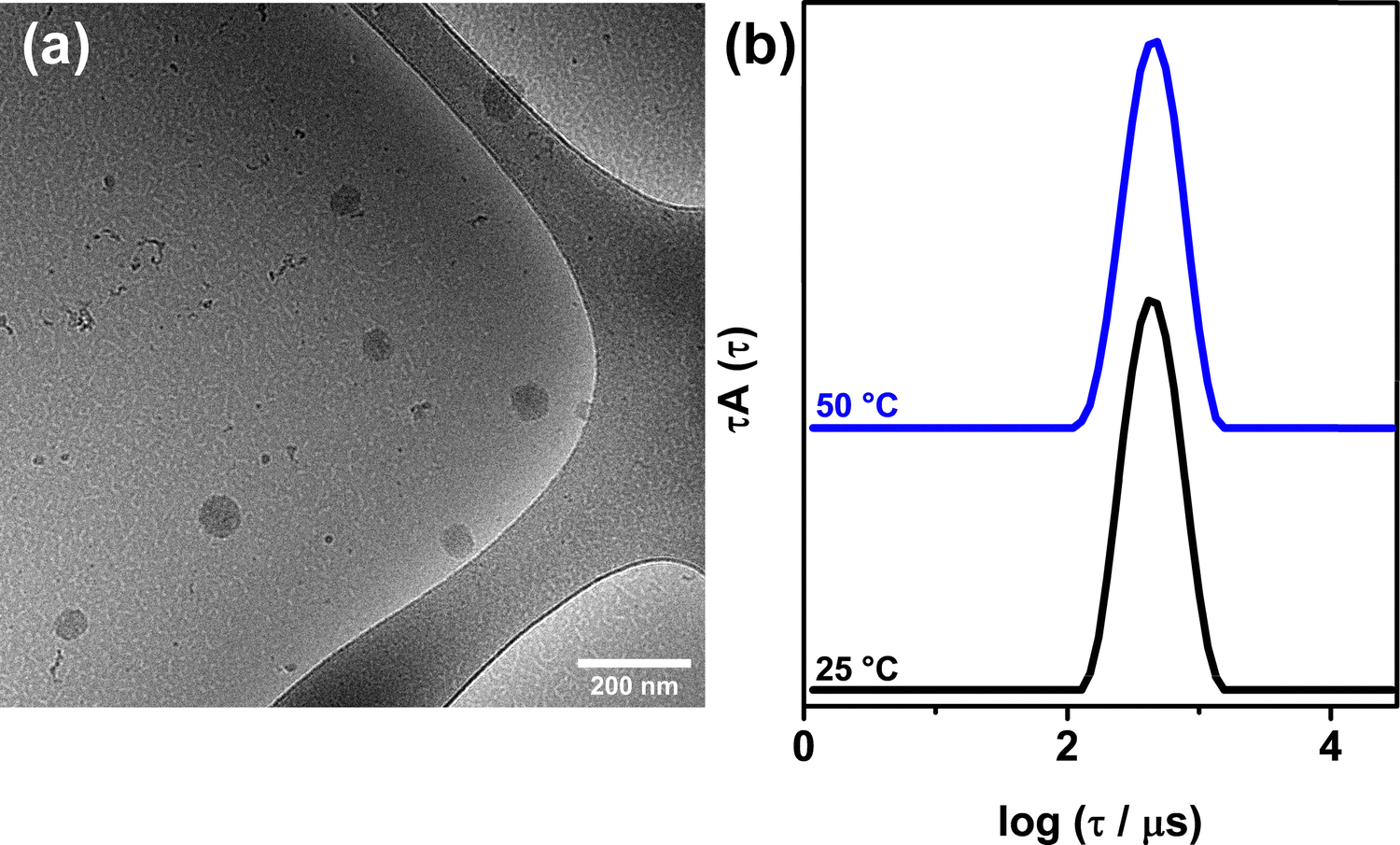 | ||
| Fig. 8 (a) Cryo-TEM image of a PEO-cholate (CR = 1) aqueous dispersion at 0.1 wt% and (b) relaxation times distributions obtained by non-linear least squares fit of the intensity autocorrelation functions from DLS at 25 °C and 50 °C (both corresponding to RH,app = 70 nm) for the same sample diluted to 0.05 wt%. The x-axis is corrected for the change in absolute temperature and solvent viscosity relative to 25 °C by multiplying a correction factor of (T/η0)/(298/η298). The distributions were normalized to their maximum height and shifted along the y-axis to enhance visualization. The scattering angle was 173°. | ||
In directly mixed complexes of neutral-ionic diblock copolymers with oppositely charged single-chain surfactants, the resulting assemblies are typically core–shell aggregates where the core radius is limited in at least one dimension by the length of the ionic block.12,53,54 In PEO114-b-PTMAEMA(+)95, the contour lengths, i.e., the maximum end-to-end distance in all-trans conformation, Rmax, of the two blocks are estimated to 24 nm for PTMAEMA(+) and 41 nm for PEO, using Rmax,PTMAEMA = 0.252 NPTMAEMA (in nm)23 and Rmax,PEO = 0.358 NPEO,55,56 respectively, where N is the number of repeat units in each case. Considering the block length asymmetry of PEO114-b-PTMAEMAI95 – the neutral PEO block is almost twice as long as the PTMAEMA(+) block – and the strong steric repulsion between the long PEO chains on the particles shell, high curvature aggregates, such as globules, are likely to form in the absence of core ordering, as found in the present study. Similar globular particles were also obtained in the PNIPAM120-b-PAMPTMA(+)30/NaDC system reported in ref. 23. Table 2 summarizes the measured core radii of the globular aggregates in the present work and in ref. 23, and shows that the core radius is comparable in length to Rmax of the cationic block. Cases where the core radius exceeds this delimitation can be explained by the inevitable polydispersity of the ionic block length.57–59 A fraction of long polyions can extend further inside the core, resulting in particles with larger average core radii. Additionally, slight deviations from a spherical shape, such as oblate and prolate shapes,12 can also contribute to generate particles with core radii exceeding the ionic block average length.
| Samples | R max/nm (cationic block) | R c/nm (cryo-TEM) |
|---|---|---|
| PEODC (CR = 1) | 24 | 32 |
| PEODC (CR = 2) | 24 | 30 |
| PEOC (CR = 1) | 24 | 36 |
| PNIPAM120-b-PAMPTMA(+)30/NaDC CR = 123 | 8 | 10 |
3.6 When and why do we find metastable globules?
Our results show that globular particles represent an initial aggregation state when dilute solutions of NaDC and PEO114-b-PTMAEMAI95 are directly mixed, and that the globules evolve over time into ribbons or bundles. Why, then, are globules not always observed in systems where NaDC and oppositely charged block copolymers are directly mixed at charge stoichiometry? Complexes formed from shorter block copolymers, as reported in ref. 21, feature only hexagonally packed bundles and toroids; globules are not detected. Possibly, in these systems, the comparatively lower repulsion between the shorter neutral hydrophilic chains reduces the energy barrier for the transition from globules to bundles of helices. As a result, the initially formed globules rapidly evolve into bundles, rendering them undetectable. An alternative explanation is that the preferred shape of an initially formed aggregate with a disordered core is not a globule but, rather, an elongated aggregate, owing to the relatively short length of the neutral block. This already elongated shape would facilitate the rapid ordering of the core into an extended hexagonally packed ribbon.For dispersions produced by dispersing a freeze-dried complex, DC helices likely exist already in the solid state. Accordingly, no globular intermediates were seen in the complex salt dispersions. Similarly, the liquid-crystalline core order clearly increased in a directly mixed sample after freeze-drying and redispersion (Fig. S11, ESI†). We note in passing that the initial states, before (re)dispersion, were not identical in the two freeze-dried samples. Only one combination of ions can form in the solid state on freeze-drying a complex salt, while three additional combinations are possible when freeze-drying a solution containing also the “original” simple counterions, sodium and iodide.
Metastable core–shell aggregates with disordered cores form also in situations when neutral-ionic diblock copolymers are directly mixed with common single-tail surfactants of opposite charge, and where the equilibrium surfactant ion-polyion core features a liquid-crystalline order. Specifically, systematic studies on stoichiometric complexes between alkyltrimethylammonium surfactants and poly(acrylamide)-block-poly(acrylate) copolymers of a range of block lengths (PAAmx-b-CnTAPAy complexes, for short) have shown that small core–shell aggregates with disordered cores result from a direct mixing protocol, whereas larger aggregates with liquid crystalline cores are formed when using the complex salt protocol.1,12,16 The liquid crystalline structures found in the block copolymer complex salts were the same as those observed in hydrated complex salts formed by the same surfactant ions with the polyacrylate homopolymer.17 A study of very concentrated (up to 80 wt%) mixtures of the PAAmx-b-CnTAPAy complex salts in water showed that their equilibrium states were, in fact, hierarchically ordered structures, featuring one liquid crystalline order on the block copolymer length scale and another one on a much shorter surfactant length scale inside the surfactant ion-polyion domains.60 All systems studied in ref. 60 showed a finite swelling in water, indicating that the concentrated hierarchically ordered phases represent the equilibrium states of the complexes, also in the presence of excess water.
Notably, no results obtained so far in PAAmx-b-CnTAPAy systems have indicated that the small core–shell spheres formed on direct mixing eventually transform into larger aggregates with ordered surfactant domains. Thus, there are striking differences in behavior between the block copolymer–deoxycholate systems studied here and in ref. 23, on the one hand, and the previously studied PAAmx-b-CnTAPAy systems: although both types of system feature (1) an initial formation of metastable core–shell globules with amorphous cores on direct mixing and (2) larger structures with ordered cores at equilibrium, aggregate growth and core ordering occurs relatively rapidly in deoxycholate based systems, whereas no such transition with time has been observed in the PAAmx-b-CnTAPAy systems. From available experimental data, the origin of this interesting and practically important difference is not immediately obvious. The copolymer block lengths and length ratios do not offer any explanation. Including the results from ref. 23 we find mixtures of ribbons and globules in two NaDC-based systems featuring block copolymers that differ significantly in the block length ratio (4 and 1.7), in the length of the charged block (30 or 95 repeat units) and in the chemistries of both blocks. For comparison, the studied PAAmx-b-CnTAPAy systems include the PAAm133-b-PA(−)49 block copolymer, which is comparable to PNIPAM120-b-PAMPTMA(+)30 and PEO114-b-PTMAEMA(+)95 regarding the block lengths, and intermediate between them regarding the length ratio.
The additional electrostatic stabilization of the dispersed particles could potentially be important for the rate of aggregate growth, but in the present study we saw little effect of changing the surface charge (CR = 2 vs. CR = 1; Fig. 1 and Table 2) on the aggregates obtained in directly mixed systems. Similarly, for the PAAm420-b-C12TAPA70 system, Berret et al. saw no strong variation of the size of the (metastable) globules over a range of charge ratios around unity, where the surface charge is expected to change sign.1
In making the above comparisons we have tacitly assumed that aggregate growth is a necessary first step in the development of a core structure with long-range order. For PAAmx-b-CnTAPAy systems, this assumption is supported by the finding that a partial ordering into a micellar cubic structure could indeed develop after direct mixing in a chemically quite similar system, PEO705-b-PMAA476 complexed with N-dodecylpyridinium cloride, but where the very long block copolymer resulted in globules with exceptionally large cores.14
4. Conclusions and outlook
Two radically different types of aggregate in diblock copolymer–bile salt complexes, sometimes coexisting, have been found and studied in this work: either ordered elongated ribbon-like structures, or globules with disordered cores, both featuring a concentrated core of the PTMAEMA–bile salt complex and a shell of fully hydrated PEO chains. The equilibrium structure depends on two key factors: the propensity of the bile salt to assemble into supramolecular helices, and the lengths of the two blocks of the copolymer. At room temperature, deoxycholate, which exhibits a strong tendency to pack into helices, prefers to form ribbons of hexagonally packed bile salt helices in its complexes with PEO114-b-PTMAEMA(+)95. Under conditions where helix formation is not favored, such as for deoxycholate at 50 °C or for cholate already at room temperature, the aggregate shape is dominated by the geometrical characteristics of the block copolymer leading, for the copolymer used here, to the formation of classical globular particles with disordered cores. When dispersions are produced at room temperature by the direct mixing of stock solutions of NaDC and PEO114-b-PTMAEMAI95, metastable globular aggregates are formed in addition to the ribbons, but with time, the globules disappear in favor of the ribbons.Previous studies on complexes between NaDC and cationic–neutral block copolymers with other block lengths have demonstrated internally ordered aggregates with shapes other than ribbons, such as thicker bundles or toroids. In one case, where the block copolymer had relatively long blocks, globular particles, analogous to those found here, were also formed. It is thus quite clear that the block copolymer lengths and length ratios control, in complexes with NaDC, both the occurrence of long-lived metastable globules with disordered cores, and the shapes and dimensions of the thermodynamically preferred elongated aggregates with ordered deoxycholate helices in their cores. Collectively, the present and previous results also show that especially the width of the ribbon-like aggregates can also be varied by choice of the preparation conditions and the charge ratio between bile salt and block copolymer.
Clearly, we have only begun to explore the possibilities to vary the rich morphology of aqueous block copolymer–bile salt aggregates in a controlled way. In addition to varying the copolymer block lengths and the nature of the bile salt – possibilities that have not nearly been exhausted – our results suggest strategies to use deliberate polymer mixtures. For instance, replacing some of the block copolymer with a homopolymer of the polycation would inflate the aggregate core, and using bidisperse block copolymers (two lengths of either the neutral or the cationic block, or both) would affect the curvature, and thus the shape, of the elongated aggregate. A more detailed investigation of how the life-time of metastable aggregates with disordered cores depend on the block copolymer block lengths is warranted, but also other variables, such as the content of simple salt in the dispersion, may be important.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Center for High Resolution Electron Microscopy (nCHREM) at Lund University is acknowledged for providing the access to the cryo-TEM facility. We are grateful to Crispin Hetherington for assisting with the cryo-TEM experiments at nCHREM. We thank Viveka Alfredsson for discussions. We would like to thank LNNano/CNPEM for the access to the cryo-TEM facility and technical support, in the execution of the proposal CRYO-EM-C2-20210263. Laura Caetano Escobar da Silva is acknowledged for her help with the cryo-TEM experiments. We are thankful to the São Paulo Research Foundation (FAPESP) for the financial support (grant numbers 2019/15956-9 and 2021/12071-6). S. G. T. thanks the Brazilian Agency CAPES (Finance Code 001) for the PhD scholarship and to INCT-Catálise/FAPESC/CNPq/CAPES, Brazil, for supporting her visit to Lund University, and W. L. thanks the Brazilian Agency CNPq for the senior researcher grant (grant number 306398/2018-4). We also acknowledge financial support from the Lund University/FAPESP (SPRINT) collaboration mobility project (to K. S.), The Swedish Research Council (K. S., grant number 2022-02860) and Magnus Bergwalls Stiftelse (K. S., grant number 2017-02225). G. D. acknowledges the China Scholarship Council (CSC) for the PhD scholarship (grant number 201706870015).References
- J. F. Berret, G. Cristobal, P. Hervé, J. Oberdisse and I. Grillo, Eur. Phys. J. E, 2002, 9, 301–311 CrossRef CAS.
- S. Van Der Burgh, A. De Keizer and M. A. C. Stuart, Langmuir, 2004, 20, 1073–1084 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS PubMed.
- S. G. Trindade, L. Piculell and W. Loh, Langmuir, 2022, 38, 2906–2918 CrossRef CAS.
- L. Shen, Y. Li, Q. Lu, X. Qi, X. Wu, Z. Zhou and J. Shen, Chem. Commun., 2020, 56, 2411–2414 RSC.
- S. Bayati, K. E. Bergquist, K. Zhu, B. Nyström, J. Skov Pedersen, L. Galantini and K. Schillén, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1457–1470 CrossRef CAS.
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953 CrossRef CAS.
- Y. Anraku, A. Kishimura, M. Oba, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 1631–1636 CrossRef CAS PubMed.
- H. M. Van Der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. Van Der Gucht, Langmuir, 2012, 28, 14180–14191 CrossRef CAS PubMed.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, Langmuir, 2024, 40, 2015–2027 CrossRef CAS PubMed.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, J. Colloid Interface Sci., 2025, 678, 1012–1021 CrossRef CAS PubMed.
- G. A. Ferreira, M. Ram-On, Y. Talmon, K. Schillén, L. Piculell and W. Loh, Langmuir, 2023, 39, 4113–4124 CrossRef CAS PubMed.
- I. A. Van Hees, P. J. M. Swinkels, R. G. Fokkink, A. H. Velders, I. K. Voets, J. Van Der Gucht and M. Kamperman, Polym. Chem., 2019, 10, 3127–3134 RSC.
- M. Uchman, M. Štěpánek, S. Prévost, B. Angelov, J. Bednár, M. S. Appavou, M. Gradzielski and K. Procházka, Macromolecules, 2012, 45, 6471–6480 CrossRef CAS.
- M. Uchman, M. Gradzielski, B. Angelov, J. Oh and T. Chang, Macromolecules, 2013, 46, 2172–2181 CrossRef CAS.
- L. Vitorazi, J. F. Berret and W. Loh, Langmuir, 2013, 29, 14024–14033 CrossRef CAS PubMed.
- G. A. Ferreira, L. Piculell and W. Loh, ACS Omega, 2016, 1, 1104–1113 CrossRef CAS PubMed.
- L. Galantini, M. C. di Gregorio, M. Gubitosi, L. Travaglini, J. V. Tato, A. Jover, F. Meijide, V. H. Soto Tellini and N. V. Pavel, Curr. Opin. Colloid Interface Sci., 2015, 20, 170–182 CrossRef CAS.
- D. Madenci and S. U. Egelhaaf, Curr. Opin. Colloid Interface Sci., 2010, 15, 109–115 CrossRef CAS.
- M. C. Di Gregorio, L. Travaglini, A. Del Giudice, J. Cautela, N. V. Pavel and L. Galantini, Langmuir, 2019, 35, 6803–6821 CrossRef CAS PubMed.
- G. Du, D. Belić, A. Del Giudice, V. Alfredsson, A. M. Carnerup, K. Zhu, B. Nyström, Y. Wang, L. Galantini and K. Schillén, Angew. Chem., Int. Ed., 2022, 134, 1–8 Search PubMed.
- Y. Qiao, Y. Lin, Y. Wang, Z. Yang, J. Liu, J. Zhou, Y. Yan and J. Huang, Nano Lett., 2009, 9, 4500–4504 CrossRef CAS PubMed.
- K. Schillén, L. Galantini, G. Du, A. Del Giudice, V. Alfredsson, A. M. Carnerup, N. V. Pavel, G. Masci and B. Nyström, Phys. Chem. Chem. Phys., 2019, 21, 12518–12529 RSC.
- M. C. Di Gregorio, M. Gubitosi, L. Travaglini, N. V. Pavel, A. Jover, F. Meijide, J. Vázquez Tato, S. Sennato, K. Schillén, F. Tranchini, S. De Santis, G. Masci and L. Galantini, Phys. Chem. Chem. Phys., 2017, 19, 1504–1515 RSC.
- G. Du, A. Del Giudice, V. Alfredsson, A. M. Carnerup, N. V. Pavel, W. Loh, G. Masci, B. Nyström, L. Galantini and K. Schillén, Polymer, 2020, 206, 122871 CrossRef CAS.
- S. Saeki, N. Kuwahara, M. Nakata and M. Kaneko, Polymer, 1976, 17, 685–689 CrossRef CAS.
- G. Karlström, J. Phys. Chem., 1985, 89, 4962–4964 CrossRef.
- Y. C. Bae, S. M. Lambert, D. S. Soane and J. M. Prausnitz, Macromolecules, 1991, 24, 4403–4407 CrossRef CAS.
- A. Skandalis and S. Pispas, Polym. Chem., 2017, 8, 4538–4547 RSC.
- A. Svensson, L. Piculell, B. Cabane and P. Ilekti, J. Phys. Chem. B, 2002, 106, 1013–1018 CrossRef CAS.
- J. Janiak, S. Bayati, L. Galantini, N. V. Pavel and K. Schillén, Langmuir, 2012, 28, 16536–16546 CrossRef CAS.
- C. L. P. Oliveira, M. A. Behrens, J. S. Pedersen, K. Erlacher, D. Otzen and J. S. Pedersen, J. Mol. Biol., 2009, 387, 147–161 CrossRef CAS.
- H. Amenitsch, H. Edlund, A. Khan, E. F. Marques and C. La Mesa, Colloids Surf., A, 2003, 213, 79–92 CrossRef CAS.
- O. B. Ijare, B. S. Somashekar, Y. Jadegoud and G. A. N. Gowda, Lipids, 2005, 40, 1031–1041 CrossRef CAS.
- G. A. Ferreira, W. Loh, D. Topgaard, O. Söderman and L. Piculell, Polymers, 2021, 13, 3265 CrossRef CAS.
- V. Sharma, K. Park and M. Srinivasarao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4981–4985 CrossRef CAS.
- M. Boksebeld, N. P. Blanchard, A. Jaffal, Y. Chevolot and V. Monnier, Gold Bull., 2017, 50, 69–76 CrossRef CAS.
- C. L. Pinto Oliveira, Current Trends in X-Ray Crystallography, 2011 Search PubMed.
- A. Rich and D. Blow, Nature, 1958, 182, 423–426 CrossRef CAS PubMed.
- A. A. D’Archivio, L. Galantini, E. Giglio and A. Jover, Langmuir, 1998, 14, 4776–4781 CrossRef.
- A. Naderi and P. M. Claesson, J. Dispers. Sci. Technol., 2005, 26, 329–340 CrossRef CAS.
- A. Naderi, P. M. Claesson, M. Bergström and A. Dedinaite, Colloids Surf., A, 2005, 253, 83–93 CrossRef CAS.
- A. Mezei, R. Mészáros, I. Varga and T. Gilányi, Langmuir, 2007, 23, 4237–4247 CrossRef CAS PubMed.
- E. Guzmán, L. Fernández-Peña, F. Ortega and R. G. Rubio, Curr. Opin. Colloid Interface Sci., 2020, 48, 91–108 CrossRef.
- R. A. Campbell, A. Angus-Smyth, M. Yanez Arteta, K. Tonigold, T. Nylander and I. Varga, J. Phys. Chem. Lett., 2010, 1, 3021–3026 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- S. Saeki, N. Kuwahara, M. Nakata and M. Kaneko, Polymer, 1977, 18, 1027–1031 CrossRef CAS.
- J. Zhang, H. Wang, X. Li, S. Song, A. Song and J. Hao, J. Phys. Chem. B, 2016, 120, 6812–6818 CrossRef CAS PubMed.
- A. Jover, J. Troncoso, M. C. di Gregorio and F. Fraga López, J. Mol. Liq., 2022, 361, 119621 CrossRef CAS.
- A. A. D’Archivio, L. Galantini and E. Tettamanti, J. Phys. Chem. B, 2000, 104, 9255–9259 CrossRef.
- H. Sugioka, K. Matsuoka and Y. Moroi, J. Colloid Interface Sci., 2003, 259, 156–162 CrossRef CAS PubMed.
- C. Leggio, L. Galantini, E. Zaccarelli and N. V. Pavel, J. Phys. Chem. B, 2005, 109, 23857–23869 CrossRef CAS PubMed.
- J. F. Berret, B. Vigolo, R. Eng, P. Hervé, I. Grillo and L. Yang, Macromolecules, 2004, 37, 4922–4930 CrossRef CAS.
- J. F. Berret, J. Chem. Phys., 2005, 123, 164703 CrossRef PubMed.
- F. Oesterhelt, M. Rief and H. E. Gaub, New J. Phys., 1999, 1, 6.1 CrossRef.
- H. J. Kreuzer, R. L. C. Wang and M. Grunzez, New J. Phys., 1999, 1, 21.1 CrossRef.
- D. Nguyen, X. F. Zhong, C. E. Williams and A. Eisenberg, Macromolecules, 1994, 27, 5173–5181 CrossRef CAS.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- M. Asad Ayoubi, K. Zhu, B. Nyström, U. Olsson, K. Almdal, A. R. Khokhlov and L. Piculell, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1657–1671 CrossRef CAS.
- G. A. Ferreira, L. Piculell and W. Loh, Macromolecules, 2018, 51, 9915–9924 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00097a |
| ‡ Present address: Physical and Chemical Analysis Center at Suzhou Institute for Advanced Research, University of Science and Technology of China, Suzhou 215123, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2025 |