
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting and parameterizing the glass transition temperature of atmospheric organic aerosol components via molecular dynamics simulations†
Panagiota
Siachouli
ab,
Vlasis G.
Mavrantzas
 *abc and
Spyros N.
Pandis
*ab
*abc and
Spyros N.
Pandis
*ab
aDepartment of Chemical Engineering, University of Patras, Patras, GR 26504, Greece. E-mail: vlasis@chemeng.upatras.gr; spyros@chemeng.upatras.gr
bInstitute of Chemical Engineering Sciences (ICE-HT/FORTH), Patras, GR 26504, Greece
cParticle Technology Laboratory, Department of Mechanical and Process Engineering, ETH Zürich, CH-8092 Zürich, Switzerland
First published on 7th April 2025
Abstract
Atmospheric aerosols contain thousands of organic compounds that exhibit an array of functionalities, structures and characteristics. Quantifying the role of these organic aerosols in climate and air quality requires an understanding of their physical properties. A key property determining their behavior is the glass transition temperature (Tg). Tg defines the phase state of aerosols, which in turn influences crucial aerosol processes. Molecular Dynamics (MD) simulations were implemented to predict Tg of a range of atmospheric organic compounds. The predictions were used to develop a Tg parameterization. The predictions and the parameterization link Tg with molecular characteristics such as the type and number of functional groups present in the molecule, its architecture, as well its carbon and oxygen content. The MD simulations suggest that Tg is sensitive to the functional groups in the organic molecule with the following order: –COOH > –OH > –CO. This trend is maintained even when more than one of these functional groups is present in a molecule. Molecular structure was also found to play a significant role. Cyclic structures exhibited consistently higher predicted Tg values compared to linear counterparts. Tg, as expected, increased as the number of carbon atoms increased. The parameterization was evaluated using a leave-one-out approach, providing insights into the contributions of various molecular features.
1. Introduction
Secondary organic aerosol consists of thousands of complex organic compounds, most of which remain un-investigated or even unidentified.1 These organic compounds exhibit an array of functionalities including alcohols, acids, ketones, etc. and also often feature a combination of such functional groups. Quantifying the physicochemical properties of these compounds is a scientifically challenging task. Organic aerosols (OA) are not always liquid, as was previously assumed;2 instead, they can also be glassy,2,3 or semi-solid.4,5 Deciphering the phase state of organic compounds is crucial for gaining insight into a variety of atmospheric processes. For example, the presence of a solid or glassy phase state can reduce the rate of heterogeneous chemical reactions, preclude water uptake, change the atmospheric lifetime of particles as well as influence their long-range transport.The glass transition temperature (Tg) is a key property for the determination of the ambient aerosol phase state. Over the years, various definitions of Tg have been suggested, with the one widely accepted connecting it to viscosity. Specifically, Tg is defined as the temperature at which the zero shear rate viscosity reaches the value of 1012 Pa s.6–8Tg can be determined by several experimental approaches. However, the discrepancies in these approaches can be significant.9 Common approaches for measuring Tg involve monitoring properties such as the thermal heat capacity over a relevant temperature range and identifying an abrupt change in the property. However, synthesis and purification of atmospheric organic compounds remain highly laborious tasks, which underlines the essential role of computational approaches for Tg prediction.
Over the years, efforts have been made to predict the glass transition temperature of atmospheric organic compounds. These efforts range from semi-empirical equations such as the Boyer–Kauzmann rule (often referred to also as the Boyer–Beaman rule),10–12 to Tg parameterizations,13,14 and more recently, to the use of machine learning algorithms.15,16Tg is sensitive to the thermal history of the system under study and the approach used to analyse the measurements. If these two factors are unknown, it is difficult to verify and reproduce results.9,10 Furthermore, the development of Tg parameterizations and machine learning algorithms requires an extensive dataset of Tg values of relevant organic compounds with the range of functionalities and complex structures encountered in the atmosphere. However, the compounds which have been studied experimentally are often limited to small molecules, such as alcohols, a few carboxylic acids and some hydroxy acids. The complexity of the atmospheric organic compounds far exceeds that of these simple compounds, with a lot of the OA components having multiple different functional groups and exhibiting intricate structural architectures.17,18 Therefore, the need for more robust datasets to support predictive approaches is becoming increasingly apparent.
The first atmospherically relevant Tg parameterization was developed by Shiraiwa et al. (2017)13 based on the compound's molecular weight and O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C. A refinement of the method was introduced by DeRieux et al. (2018)14 incorporating atomic and bond contributions, but neglecting structural and functional group effects. The influence of functional group on Tg has been widely documented.19–21 Moreover, functional groups have been shown to significantly affect the viscosity of organic compounds,19,22 a property closely associated with Tg. The structure of a compound is known to affect Tg.21,23 However, existing efforts do not include the structure as a feature in the available parameterizations.
:C. A refinement of the method was introduced by DeRieux et al. (2018)14 incorporating atomic and bond contributions, but neglecting structural and functional group effects. The influence of functional group on Tg has been widely documented.19–21 Moreover, functional groups have been shown to significantly affect the viscosity of organic compounds,19,22 a property closely associated with Tg. The structure of a compound is known to affect Tg.21,23 However, existing efforts do not include the structure as a feature in the available parameterizations.
In this work, we use molecular dynamics (MD) simulations to create a dataset of glass transition temperatures of atmospherically relevant organic compounds and to develop a new parameterization of Tg. MD simulations can bypass some of the difficulties encountered in experimental studies. Simultaneously, they offer insights at the molecular level, thus they represent a valuable tool for understanding physicochemical OA properties.21,24–26 MD simulations have been widely used in polymer science to predict the Tg of a variety of polymers.27–30 However, similar efforts to apply MD simulations to investigate the Tg of atmospherically relevant organic compounds are rare; one of them was reported recently by our team.21 That preliminary effort is extended here to include more organic compounds with diverse and multiple functional groups, and different structural architectures.
The compounds selected for this study were chosen based on their atmospheric relevance and the availability of experimental data, enabling the comparison of the MD predictions as well as the evaluation of the predictive performance of the developed parameterization. Our efforts focus on expanding our understanding of how functional groups, their interactions, carbon chain length, molecular architecture, O:C ratio and molecular weight influence Tg. The Tg parameterization derived from our atomistic MD simulations can eventually improve the accuracy of atmospheric model predictions. It aims to fill an important gap in the existing literature and pave the way for a deeper understanding of a key physicochemical property of organic compounds and its role in atmospheric processes.
2. Methodology
2.1 Simulation details
The materials and processes simulations platform (MAPS)31 was used to build all the organic molecules and initial configurations for the systems of interest. The systems were composed of 2000 molecules placed in a cubic box, with the initial box size estimated according to the density of each organic species at ambient temperature and pressure. The latest version of all-atom OPLS (optimized potentials for liquid simulations) force field was implemented (OPLS/2020).32,33 The LigParGen platform34,35 was used as a generator of “personalized” atomic charges for each compound under study. The systems were subjected to potential energy minimization to avoid atom overlaps present in the initial configuration. Following this, the systems were equilibrated at a relatively high temperature, well above each compound's melting point. Equilibration was confirmed by monitoring changes in density and the time autocorrelation function of the unit end-to-end distance vector for each compound. All simulations were performed with periodic boundary conditions applied in all spatial dimensions. The isothermal–isobaric (NpT) statistical ensemble was implemented, utilizing the Nosé–Hoover thermostat–barostat36 for pressure and temperature control and the velocity-Verlet algorithm37 for the integration of the microscopic equations of motion with a 1 fs timestep. The simulations were conducted using the open-source large-scale atomic/molecular massively parallel simulator (LAMMPS).38A common approach for determining Tg is to monitor volumetric, mechanical and structural properties during cooling simulations.39–41 We selected a temperature range based on each compound's melting point to perform stepwise cooling simulations. Each simulation step lasted 2 ns at each temperature, followed by stepwise reduction of 20 K before the next simulation step. The properties monitored were the density and the energy due to non-bonded interactions. Only the final nanosecond was used in post-processing. The properties of interest were analysed as a function of temperature to identify any abrupt changes in slope, and a bilinear fit was used to identify Tg. To ensure the reproducibility of our results, we conducted each simulation three times, starting from different initial configurations. The Tg determination protocol used in this work is detailed in Siachouli et al. (2024).21
2.2 Systems examined
The examined organic compounds had different architecture (linear/non-linear), length of carbon chain and type, number and co-existence of functional groups present. In the group of linearly structured compounds we extended our previous study21 by considering alcohols and carboxylic acids with a longer carbon chain length. Specifically, we examined linear alcohols with nine carbon atoms in their main backbone, including 1-nonanol (1 hydroxyl group), 1,2-nonanediol (2 hydroxyl groups), and 1,2,9-nonanetriol (3 hydroxyl groups). We then investigated linear alcohols with twelve carbon atoms in the backbone, namely 1-dodecanol (1 hydroxyl group) and 1,2-dodecanediol (2 hydroxyl groups). Similarly, we examined carboxylic acids with nine carbon atoms in the backbone, including nonanoic acid (1 carboxyl group) and azelaic acid (2 carboxyl groups). For the case of the twelve-carbon atom chain length, we examined dodecanoic (1 carboxyl group) and dodecanedioic acid (2 carboxyl groups).Cyclic compounds were selected based on the same reasoning for alcohols and carboxylic acids. The groups of cyclo-alcohols investigated had six and nine carbon atom rings. For the six-carbon ring case, we examined cyclohexanol (1 hydroxyl group), 1,2-cyclohexanediol (2 hydroxyl groups) and 1,2,3-cyclohexanetriol (3 hydroxyl groups). For the nine-carbon ring compounds, we investigated only cyclononanol (1 hydroxyl group) due to the lack of available experimental melting points for other compounds in this category. In addition to the compounds already examined in our previous work, we also considered two monocarboxylic acids with ring-like structures: cyclopentanecarboxylic acid and cycloheptanecarboxylic acid.
Linear and cyclic compounds with carbonyl functional groups were included in the study. For ketones, the investigation focused on linear compounds such as 2-propanone (1 carbonyl group – 3 carbon atoms), 2-hexanone (1 carbonyl group – 6 carbon atoms) and 2-nonanone (1 carbonyl group – 9 carbon atoms). The corresponding cyclic ketones studied were cyclopropanone (1 carbonyl group – 3 carbon atoms), cyclohexanone (1 carbonyl group – 6 carbon atoms) and cyclononanone (1 carbonyl group – 9 carbon atoms). The case of diacetyl (2 carbonyl groups – 4 carbon atoms) was also investigated. For aldehydes, propanal (1 carbonyl group – 3 carbon atoms), hexanal (1 carbonyl group – 6 carbon atoms) and nonanal (1 carbonyl group – 9 carbon atoms) were studied.
Finally, we examined compounds containing multiple functional groups, combining the carboxyl, hydroxyl and carbonyl groups already considered. For hydroxyl/carboxyl groups we considered lactic acid (1 hydroxyl, 1 carboxyl and 3 carbon atoms) and tartronic acid (1 hydroxyl, 2 carboxyl and 3 carbon atoms). For the hydroxyl/carbonyl group combination, we considered hydroxy acetone (1 hydroxyl, 1 carbonyl and 3 carbon atoms) and dihydroxy acetone (2 hydroxyl groups, 1 carbonyl group and 3 carbon atoms). Lastly, the combination of carboxyl/carbonyl groups was considered by investigating linear compounds with an increasing carbon chain length, namely, pyruvic acid (1 carboxyl, 1 carbonyl and 3 carbon atoms), 5-oxohexanoic acid (1 carboxyl, 1 carbonyl and 6 carbon atoms) and 6-oxononanoic acid (1 carboxyl, 1 carbonyl and 9 carbon atoms). The same compounds with the addition of one carboxyl group to their structure were also considered, i.e., oxomalonic acid (2 carboxyl, 1 carbonyl and 3 carbon atoms) and 2-oxoadipic acid (2 carboxyl, 1 carbonyl and 6 carbon atoms). The study of multifunctional organic compounds aims to develop an understanding of the combined effect of the coexistence of multiple functional groups and their interactions.
2.3 T g parameterization
The Tg parameterization developed in this work aims to link the structure of an organic compound to its glass transition temperature. A property of interest for a given compound can be predicted based on contributions of its structural fragments, which can be systematically determined.42 This approach is commonly known as the group contribution (GC) method and has been widely used to predict properties of organic compounds such as viscosity,43–45 boiling point,19,46 liquid vapor pressure and enthalpy of vaporization.47 We developed a parameterization for predicting Tg based on contributions of carbon atoms, oxygen atoms, functional groups and molecular structure. The proposed parameterization is formulated as follows:| Tg = A + ncCc + noCo + nfuncCfunc + narchCarch | (1) |
:C) ratio are indirectly included. The influence of both parameters has been examined in past studies.13,14,48
3. Results
3.1 Insights from the MD simulations
Ketones/aldehydes. The Tg of aldehydes increases as their carbon chain length increases (Fig. 1). Furthermore, aldehydes have consistently lower predicted Tg values compared to the corresponding ketones. Aldehydes, due to the presence of a hydrogen atom attached to the carbonyl functional group, tend to have lower steric hindrance compared to ketones, thereby allowing for more flexibility. Additionally, the lack of that hydrogen in the carbonyl group of ketones could possibly allow them to form more tight molecular packing compared to aldehydes which ultimately could contribute to a higher Tg. Moreover, the case of the di-ketone (diacetyl) shows that the existence of a second carbonyl group can lead to higher predicted Tg even for small molecules that contain only four carbon atoms.
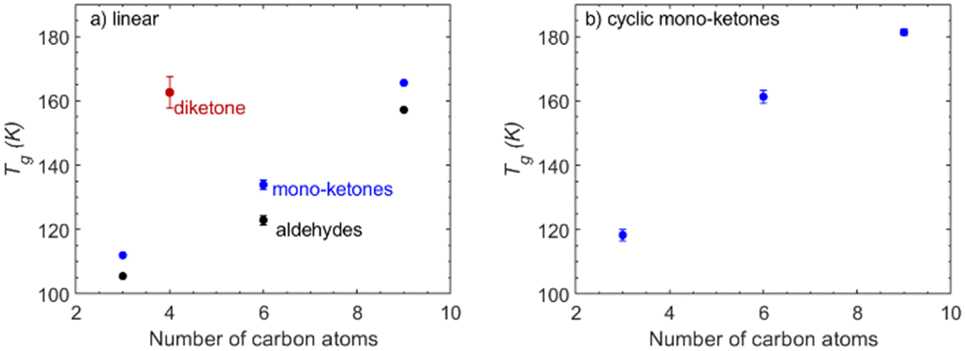 | ||
| Fig. 1 T g of ketones and aldehydes as a function of the number of carbon atoms present in the molecule, from the MD-based density predictions: (a) linear compounds and (b) cyclic compounds. | ||
Alcohols. Addition of carbon atoms along the main backbone increases the Tg of alcohols (Fig. 2). The increasing trend of Tg with carbon chain length is smooth for mono-hydroxyl compounds. The addition of a second hydroxyl group results in an increase in Tg, but with a non-linear dependence of Tg on the number of carbon atoms. Interestingly, there appears to be a plateau in the predicted Tg for diols and triols after six carbons. The compounds containing three hydroxyl groups consistently show a higher predicted Tg compared to compounds of the same carbon chain length but with fewer hydroxyl groups. Overall, the primary factor driving the increase in Tg is the presence of additional hydroxyl groups. While a longer carbon chain further increases Tg, this effect seems to saturate at long enough chains, suggesting that there may be a limit to the impact of carbon chain length on Tg for alcohols.
 | ||
| Fig. 2 T g of alcohols as a function of the number of carbon atoms present in the molecule, from the MD-based density predictions: (a) linear compounds and (b) cyclic compounds. | ||
Carboxylic acids. The mono-carboxylic acids show a steady increase in Tg that – similarly to the case of alcohols – reaches a plateau for longer carbon chains (Fig. 3). The smaller mono-carboxylic acid (propionic acid) has a Tg of approximately 160 K whereas dodecanoic acid has a Tg of 200 K, indicating a moderate increase with the addition of carbon atoms. Di-carboxylic acids exhibit a significantly higher Tg compared to mono-carboxylic acids, indicating the impact of the addition of a carboxyl group. The smallest investigated di-carboxylic acid (malonic acid) has a predicted Tg equal to 275.3 K and dodecanedioic acid a predicted Tg of 316.3 K. The addition of carbon atoms above the sixth does not lead to a significant increase in Tg. Finally, the tri-carboxylic acids have the highest predicted Tg, further highlighting the importance of the number of functional groups compared to the addition of carbon atoms.
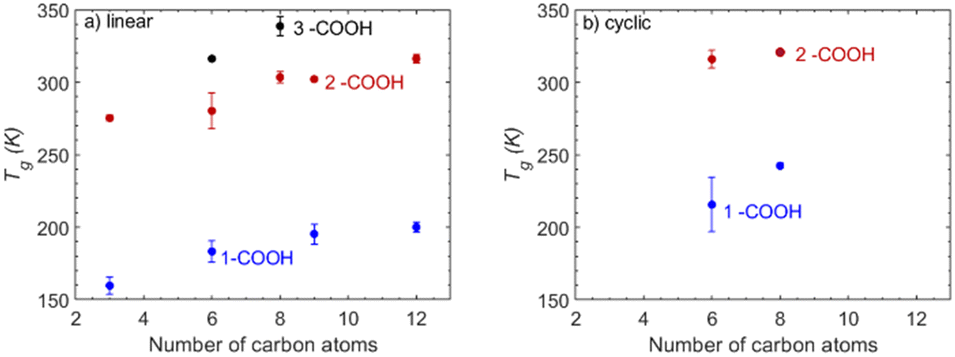 | ||
| Fig. 3 T g of carboxylic acids as a function of the number of carbon atoms present in the molecule, from the MD-based density predictions: (a) linear compounds and (b) cyclic compounds. | ||
Overall, Tg seems to be primarily sensitive to the type and number of functional groups and secondarily to the length of the carbon chain. Carbonyls consistently lead to lower predicted Tg than hydroxyls, regardless of the size of the carbon chain length. Furthermore, the addition of carboxyl groups has a higher impact on Tg compared to the addition of hydroxyl groups. The sensitivity trend (–COOH) > (–OH) > (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) can be attributed to the enhanced ability of carboxyl groups to form hydrogen bonds compared to hydroxyls, and of hydroxyls compared to carbonyls. Carboxyl groups, with both a hydroxyl and a carbonyl, form stronger hydrogen bonding networks, resulting in a more significant impact on Tg. Hydroxyl groups, while capable of hydrogen bonding, form weaker networks compared to carboxyls. Carbonyl groups, with their lower hydrogen bonding potential, seem to have the least influence on Tg compared to the other two functional groups.
O) can be attributed to the enhanced ability of carboxyl groups to form hydrogen bonds compared to hydroxyls, and of hydroxyls compared to carbonyls. Carboxyl groups, with both a hydroxyl and a carbonyl, form stronger hydrogen bonding networks, resulting in a more significant impact on Tg. Hydroxyl groups, while capable of hydrogen bonding, form weaker networks compared to carboxyls. Carbonyl groups, with their lower hydrogen bonding potential, seem to have the least influence on Tg compared to the other two functional groups.
Ketones/aldehydes. The compounds considered in this section are divided into two groups, linear ketones and cyclic ketones. In both groups, Tg increases with the addition of carbon atoms. The range of Tg values for linear ketones increases from approximately 110 K for 2-propanone up to almost 180 K for 2-nonanone. Cyclic ketones have a higher predicted Tg than their linear counterparts. Their Tg is characterized by a more stable, increasing trend, while linear ketones exhibit a steeper increase in their Tg with the addition of carbon atoms (Fig. 1). Although linear ketones show a steeper increase in Tg, cyclic ketones have consistently higher predicted Tg values. This can be attributed to the fact that linear ketones have more conformational flexibility due to their open-chain structure compared to the rigid ring-like structure of cyclic ketones. The rigid ring structure can also facilitate close-packing and intermolecular interactions that enhance the hydrogen bonding potential for cyclic ketones compared to linear ones.
Alcohols. Similarly to ketones and cycloketones, alcohols with a cyclic structure have a higher predicted Tg than their linear counterparts with the same number of hydroxyl groups and carbon atoms. Again, the rigid ring-like structure enables a closer packing and an enhanced hydrogen bond network which, combined, can lead to higher Tg. For both groups, the number of hydroxyl groups plays a dominant role in determining Tg, with more hydroxyl groups leading to higher Tg. Regarding the longest chains (those with nine carbon atoms), both linear and cyclic alcohols seem to converge toward similar Tg values, indicating that at higher chain lengths, the influence of hydroxyl groups dominates Tg.
Carboxylic acids. Based on their structure, the investigated carboxylic acids can be divided into linear and cyclic. The linear mono-carboxylic acids exhibit the lower predicted Tg (Fig. 3). Comparing hexanoic acid (a linear mono-carboxylic acid) to its cyclic counterpart (cyclopentanecarboxylic acid) reveals a significant difference in Tg of approximately 30 K. The trend is similarly seen in the case of linear/cyclic di-carboxylic acids, with cyclic structures having consistently higher Tg.
The structural influence on Tg is particularly pronounced for the case of carboxylic acids. Unlike carbonyl or hydroxyl compounds where structural differences between linear and cyclic forms have a notable but nevertheless small effect, the carboxyl group influences significantly the compound's Tg when considering cyclic structures. This observation emphasizes the pronounced sensitivity of the carboxyls to changes in molecular conformation.
Co-existence of carbonyl and carboxyl functional groups. The compounds containing one carbonyl and one carboxyl group have a slightly higher predicted Tg compared to mono-carboxylic acids (Fig. 4). The difference in Tg remains more or less constant regardless of the carbon chain length, suggesting that carbonyls have a slight and yet consistent contribution to that of the carboxyl group. The difference between the di-carboxylic acids and their corresponding compounds with an extra carbonyl group is similar to that seen in mono-carboxylic acids and compounds containing one carbonyl and one carboxyl group. The overall increase of the Tg by the addition of a carbonyl group in a compound with an existing carboxylic acid is small. The tri-carboxylic acids remain the compounds with the higher predicted Tg regardless of the carbon chain length.
 | ||
| Fig. 4 T g predictions for multifunctional organic compounds containing carboxyl and carbonyl groups, as a function of the number of carbon atoms along the main carbon chain. Compounds with only carboxyl groups are also included for comparison. | ||
Co-existence of carbonyl–hydroxyl and carboxyl–hydroxyl functional groups. Hydroxyacetone is predicted to have a Tg similar to that of 1,2-propanediol, showing that the addition of a carbonyl group in an alcohol can lead to a significant increase in Tg. However, di-hydroxyacetone exhibits a lower Tg than 1,2,3-propanetriol, indicating that the effect of additional hydroxyl groups is more impactful than that of carbonyls. This supports the conclusion that carbonyls contribute less than hydroxyls to the increase of Tg.
Lactic acid contains one hydroxyl and one carboxyl group; however, it has a slightly higher predicted Tg compared to 1,2,3-propanetriol (Fig. 5). This observation suggests that the combination of a hydroxyl and a carboxyl group may yield a synergistic effect that elevates Tg. Furthermore, tartronic acid, with its two carboxyl groups, exhibits the highest predicted Tg. The addition of a second carboxyl group appears to lead to significant structural changes that can possibly enhance intermolecular interactions which in turn cause an increase in the Tg.
 | ||
| Fig. 5 T g predictions of multifunctional organic compounds containing hydroxyl–carbonyl groups and hydroxyl–carboxyl, as a function of the number of functional groups. Compounds with only hydroxyl groups are also included for comparison. | ||
These findings emphasize the importance of functional group interactions in determining Tg. The trends that have been observed in compounds comprised solely of carbonyls, hydroxyls and carboxyls remain similar in the multifunctional compounds.
 | ||
| Fig. 6 MD-predicted Tg as a function of the molecular weight for all the compounds considered in this study. | ||
We have analyzed our results to detect whether correlations between Tg and molecular mass M proposed in the literature for molecular (non-polymeric) and polymeric glass formers49 apply also to the small organic compounds studied here. Namely, Novikov and Rössler (2013)49 proposed a correlation between Tg and molecular mass M of the form Tg ∝ Mα, with α = 0.51 ± 0.02 for molecular and short-chain length (i.e., low molecular weight) polymers (also called oligomers). Novikov and Rössler have also found that subclasses of molecular glasses with homologous chemical structure exhibit a similar universal correlation but with significantly lower scatter. Motivated by this study, we considered the entire dataset as well as subcategories based on the type of functional group (Fig. S5–S9, ESI†) and analyzed the dependence of the MD-predicted Tg values with M. The alcohols and carboxylic acids were found to agree well with the correlation proposed by Novikov and Rössler for α = 0.50 and α = 0.51, respectively. For carbonyls and multifunctional compounds, on the other hand, the best correlation was found for α = 0.42 and α = 0.43, respectively. If one considers the entire dataset, then the best correlation requires α = 0.62 indicating a stronger dependence of the glass transition temperature on molecular weight across the dataset. Interestingly the multifunctional compounds exhibit a lower value of α, suggesting a more complex interplay between molar mass and glass transition temperature when multiple functional groups coexist in the molecule.
The oxygen to carbon ratio (O:C) is another parameter commonly used as an indicator of Tg. The O:C ratio indicates the degree of oxygenation of a compound, and a higher O:C ratio tends to lead to a higher Tg. In our study (Fig. 7) the correlation between O:C ratio and Tg is weak (R2 ≈ 0.18). While Tg generally increases with an increasing O:C ratio (Fig. 7), there are several instances where compounds with a high O:C ratio exhibit a lower Tg compared to others with lower O:C. A characteristic example is malonic acid (O:C ≈ 1.33, Tg = 275.3 K) compared to tricarballylic acid (O:C ≈ 0.75, Tg = 338.7 K). A detailed analysis of the O:C ratio for each compound category can be found in Section S2 of the ESI.†
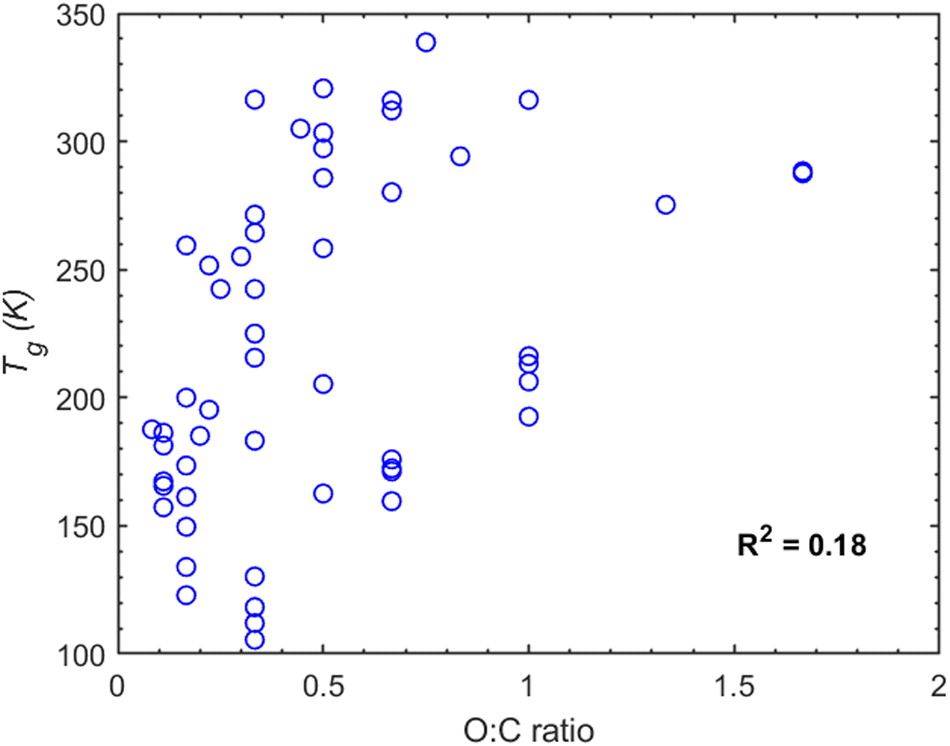 | ||
| Fig. 7 MD-predicted Tg values as a function of the O:C ratio for all the organic compounds considered in this study. | ||
3.2 Evaluation of the parameterization
Most parameterization predictions are within 10% of the MD values, with only a few of them exceeding this range (Fig. 8). Compounds exceeding the 10% include 1-propanol, 1,2-hexanediol, cyclohexanediol, tricarballylic acid, 2,2-dimethylsuccinic acid, diacetyl and pinonaldehyde. Therefore, the proposed Tg parameterization generalizes the results obtained from the MD simulations. Moreover, it could be used to extend the results of the MD simulations to a broader range of compounds within the interval of those included in the training dataset.
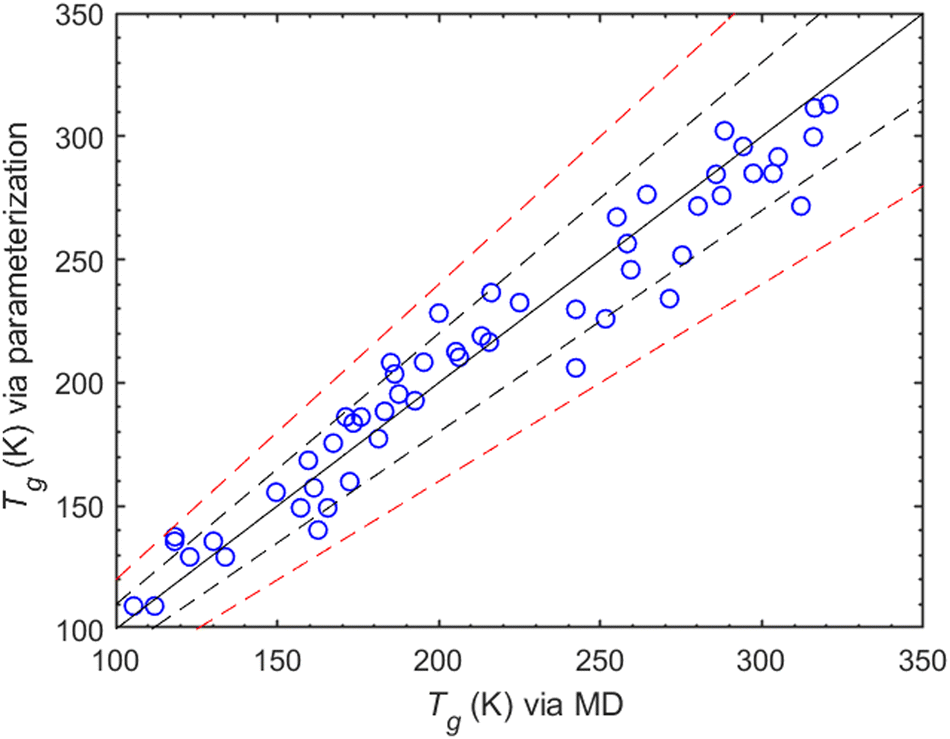 | ||
| Fig. 8 Comparison of the Tg values predicted from the MD simulations with those from the proposed parameterization. The solid line is the 1:1 curve while the dashed black line indicates the ±10%, and the dashed red line represents the ±20% deviations from 1:1. | ||
Robustness of the parameterization and limitations. The robustness of the Tg parameterization was evaluated using a leave-one-out approach, where each compound was removed and then the contributions were re-evaluated. The procedure was repeated for the removal of all the compounds considered in the dataset. Deviations of each parameter's contribution (e.g., carbons, carboxyls, hydroxyls, etc.) were monitored using a tolerance threshold of 5% from the contribution value extracted upon the training of the parameterization with the full dataset. The tolerance threshold was implemented to point out which compounds stand out and drive the contributions considered (Fig. S10–S16, ESI†).
Removal of triols increased the contribution of hydroxyl groups. The Tg of triols with longer carbon chain length was found to reach a plateau, meaning that the removal of such compounds reduces this effect. The carbonyl contribution increased upon the removal of hydroxyl and carboxyl functional groups, but decreased when multifunctional compounds containing carbonyl groups, or compounds composed solely of carbonyl groups, were removed. Finally, the contribution of cyclic structures increased with the removal of linear compounds and decreased when cyclic compounds with a high Tg were removed.
Overall, the compounds that were under-represented in the developed dataset had a notable impact on the variation of each contribution parameter upon their removal. For example, diacetyl, as the only diketone in the dataset, altered the carbonyl contribution's variation by more than 5% compared to its value in the full dataset. Carbonyl compounds were under-represented in our dataset due to limited experimental input. Moreover, most multifunctional compounds contain carbonyl groups alongside hydroxyl or carboxyl groups, making the carbonyl contribution's behaviour closely linked to that of other functional groups, and therefore difficult to interpret independently. To improve the dataset, measurements such as melting and boiling points are needed for compounds containing only carbonyl groups and for compounds with complex structures and functional groups similar to aldehydes or cis-pinonic acid.
MD simulations compared to measurements. All MD predictions are within 20% of the measured values (Fig. 9). Most of the compounds included in the comparison are small alcohols, with average Tg values taken from Rothfuss et al. (2017). MD predictions for Tg perform relatively well, with a normalized mean error (NME) of 12.4%, a normalized mean bias (NMB) of 12.4% and R2 = 0.79.
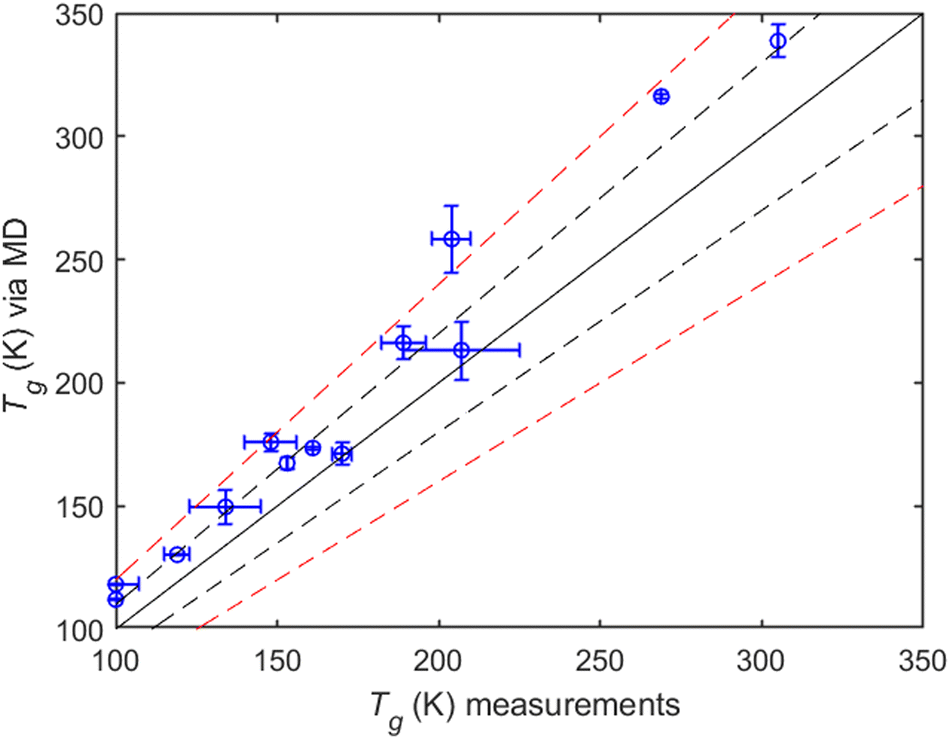 | ||
| Fig. 9 MD-predicted Tg values against available experimental data. The solid line depicts the 1:1 curve while the dashed black line indicates the ±10%, and the dashed red line represents the ±20% deviations from 1:1. | ||
It should be noted that although available measurements of experimental Tg's are rather limited (Table S1, ESI†), one can still indirectly estimate them from viscosity data (given that such data are available). An example is provided by the relatively recent work of Fu et al. (2021)50 for 1-dodecanol and 1,12-dodecanediol. We fitted the reported viscosity data in that work to the Vogel–Fulcher–Tammann (VFT) equation:
 | (2) |
T g parameterization compared to measurements. The parameterization developed in this work was compared to the available Tg measurements (Fig. 10(a)). There is a moderately good agreement (NME ≈ 19.6%, NMB ≈ 19.6%, R2 ≈ 0.52) with a slight but consistent overprediction of Tg. This overprediction is mainly due to the fact that the MD simulations themselves exhibit an at least 10% overprediction of the measured values. One reason for this is the rather fast cooling rates implemented in the MD simulations due to computational constraints. In an effort to correct for this bias we implement a 0.9 scaling factor to account for the combined overprediction of both the MD simulations and the parameterization. The parameterization takes now the following form:
| Tg = 0.9(A + ncCc + noCo + nOHCOH + nCOOHCCOOH + nCO + nringCring) | (3) |
| Tg = 0.9(65.15 + nc6.57 + no29.98 + nOH20.68 + nCOOH23.58 − nCO5.93 + nring28.48) | (4) |
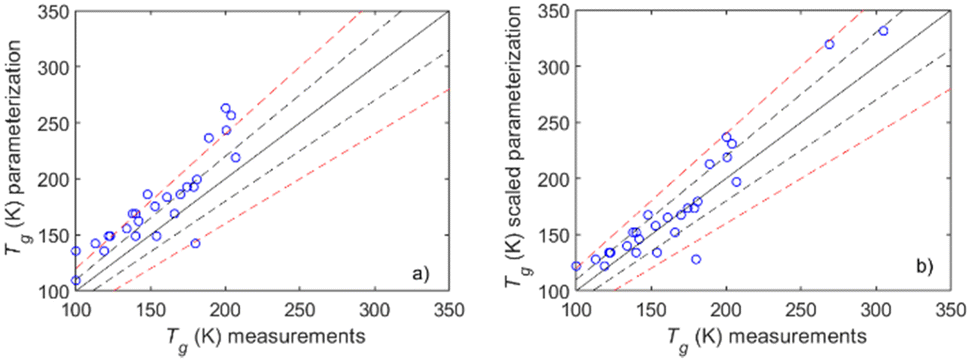 | ||
| Fig. 10 Comparison of the Tg values predicted from the proposed parameterization, with (a) original version and (b) the 0.9 scaling factor, with measurements. The solid line is the 1:1 curve while the dashed black line indicates the ±10%, and the dashed red line represents the ±20% deviations from 1:1. | ||
The scaling factor as expected reduces the overprediction (Fig. 10(b)), particularly at lower Tg values, which are mostly associated with alcohols – compounds that exhibited the highest deviations when comparing MD simulations with measurements. The implementation of the scaling factor improves the performance of the parameterization (NME ≈ 8.9%, NMB ≈ 7.6%, R2 ≈ 0.87), making it more reliable for predicting Tg – especially in the case of low Tg compounds.
The effect of cooling rate. A major impediment in comparing experimental studies with molecular simulations regarding properties such as Tg is the cooling rate implemented in the latter, which is several orders of magnitude larger than the typical one characterizing experimental protocols.30 The higher Tg values predicted via MD simulations are therefore qualitatively consistent with the expectation that measurements at a high cooling rate should lead to a higher Tg value. Bridging the gap between measured and simulated Tg values requires calculating the shift of Tg due to a high cooling rate. A system specific equation30,60 based on the Williams–Landel–Ferry (WFL)61 equation is
 | (5) |
 being the ratio of experimental and simulated cooling rates, R the ideal gas constant and Δh the apparent activation energy associated with structural relaxation. We calculated the Tg shift for 1-propanol using the average reported experimental value19 of Tg = 100 K and the value of Δh = 97487.2 J mol−1.62 The resulting dTg was found to be 21.72 K. Based on this, the expected Tg (MD) would be 121.7 K, which is very close to the (average) value of 118.2 K that came out from our simulations (Table S1, ESI†).
:C ratio to predict Tg for compounds with molar mass up to 450 g mol−1. A refinement of the Shiraiwa et al. (2017) parameterization was later introduced by DeRieux et al. (2018),14 extending the previous work to include higher molar mass compounds, up to 1100 g mol−1. The predicted Tg values from the DeRieux et al. parameterization are in good agreement with the experimental Tg values. The predictions are falling within ±10% of the measurements and none exceeding the ±20% error (Fig. S19, Section S5, ESI†). However, the experimental data used in this comparison were already part of the training set for the DeRieux et al. (2018) parameterization, therefore good agreement was to be expected.
being the ratio of experimental and simulated cooling rates, R the ideal gas constant and Δh the apparent activation energy associated with structural relaxation. We calculated the Tg shift for 1-propanol using the average reported experimental value19 of Tg = 100 K and the value of Δh = 97487.2 J mol−1.62 The resulting dTg was found to be 21.72 K. Based on this, the expected Tg (MD) would be 121.7 K, which is very close to the (average) value of 118.2 K that came out from our simulations (Table S1, ESI†).
:C ratio to predict Tg for compounds with molar mass up to 450 g mol−1. A refinement of the Shiraiwa et al. (2017) parameterization was later introduced by DeRieux et al. (2018),14 extending the previous work to include higher molar mass compounds, up to 1100 g mol−1. The predicted Tg values from the DeRieux et al. parameterization are in good agreement with the experimental Tg values. The predictions are falling within ±10% of the measurements and none exceeding the ±20% error (Fig. S19, Section S5, ESI†). However, the experimental data used in this comparison were already part of the training set for the DeRieux et al. (2018) parameterization, therefore good agreement was to be expected.
The limitations of Tg parameterizations such as those by Shiraiwa et al. (2017) and DeRieux et al. (2018) are due to the lack of available experimental data to train them. The datasets used for both parameterizations primarily consist of experimental Tg data for small compounds, such as alcohols or small acids,19 and do not include the effect of different functional groups or that of molecular architecture. DeRieux's parameterization generally agrees mostly within 10% with our parameterization for compounds at low temperatures. However, for compounds with a predicted Tg above 250 K, the two parameterizations exceed the 10% difference (Fig. 11). The compounds that have a higher predicted Tg than 250K are mostly multifunctional or non-linear organic compounds that have not been experimentally investigated and therefore have not been used as input in the past parameterizations. The increasing difference between the two parameterizations at higher temperatures underlines the importance of enriching the current datasets. To refine Tg parameterizations and include more complex compounds, methods such as MD simulations, as implemented in this work, are useful.
 | ||
| Fig. 11 Comparison of the developed parameterization with that by DeRieux et al. (2018). The compounds shown are divided in two categories: (i) blue for the compounds that are experimentally tested, and (ii) black for untested compounds both experimentally and DeRieux's parameterization. The solid line is the 1:1 curve while the dashed black line indicates the ±10%, and the dashed red line represents the ±20% deviations from 1:1. | ||
4. Conclusions
MD simulations were carried out to investigate the glass transition temperature of atmospherically relevant organic compounds. Two properties were monitored to predict Tg, density and non-bonded potential energy. The two methods agree well within 7% with each other. The MD predictions are within 20% of available experimental measurements while the parameterization is following the MD within 10% of agreement. The scaled parameterization agrees mostly within 10% of the available measurements.The Tg of aldehydes increases smoothly as the carbon chain length increases; however, their Tg is consistently lower compared to those of ketones with the same carbon atom number. This can be attributed to the flexibility of aldehydes due to an attached hydrogen to their carbonyl group, while ketones can exhibit comparatively tighter packing at the molecular level, contributing to a higher Tg.
Alcohols’ Tg shows a linear increase for mono-hydroxyl compounds upon the addition of carbon atoms. However, in the case of additional hydroxyl groups the increase of Tg seems no longer to be linear. On longer carbon chain, a plateau is reached in the increase of Tg. This plateau of Tg is observed in alcohols with nine carbon atoms or more. The addition of hydroxyl groups has a much more significant effect on Tg than the carbon chain length, marking hydroxyl functionality the primary factor of the two influencing Tg.
In the case of carboxylic acids, the compounds with the most carboxyls show a higher Tg compared to mono- or di-carboxylic acids, which highlights the dominant role of the carboxyl group. The plateau effect of Tg for longer carbon chains that was seen in alcohols is also present in carboxylic acids. The functional group hierarchy for Tg sensitivity is (–COOH) > (–OH) > (–CO). A key factor for this sensitivity can be the enhanced ability of carboxyl groups to form stronger hydrogen bonds, which ultimately increases Tg. Overall, the number and type of functional groups, particularly carboxyl and hydroxyl groups, tend to have a stronger effect on Tg than the length of the carbon chain.
Cyclic compounds have higher predicted Tg compared to their linear counterparts, a fact that can be attributed to the rigidity imparted by a non-aromatic carbon ring. A similar behaviour is encountered in the case of cyclic alcohols. The structural effect is particularly important in the case of carboxyl groups.
When both carboxyl and hydroxyl groups are present within a compound, the Tg tends to be higher than for the respective compounds with only one type of functional group. This can be attributed to the synergistic effect of the hydrogen bonding networks formed between the carboxyl and hydroxyl groups. Conversely, the combination of carbonyl with a hydroxyl or carboxyl group results in a relatively lower increase in Tg. Such interactions underline the importance of functional group synergy, where the presence of multiple functional groups can enhance or moderate the sensitivity of Tg to the addition of more functional groups.
The molecular weight was found to have a clear, positive correlation with Tg. The O:C ratio was found to have no significant influence, given the several examples of compounds (e.g., malonic acid, oxomalonic, lactic acid, etc.) with high O:C ratio but comparatively low predicted Tg value.
Overall, the limitations primarily involve compounds with complex structures and multiple functional groups. Compounds such as diketones with cyclic structure and multifunctionality were under-represented in the dataset and are needed to improve the parameterization. Experimental data of melting and boiling point for such compounds are needed to guide the MD simulations. Using a lower stepwise cooling rate could help account for the scaling factor.
Author contributions
PS contributed to the design of the study, conducted the simulations, analysed the results, developed the parameterization, and wrote the paper. VGM contributed to the design of the study, the analysis of the results and the writing of the paper. SNP was responsible for the design and coordination of the study and the synthesis of the results.Data availability
All software programs used in this study are referenced throughout the Methodology and Results sections of this work. Data files for all simulated systems are available in the Zenodo repository, accessible through the C-STACC community records, https://zenodo.org/communities/c-stacc_group/records?q=&l=list&p=1&s=10&sort=newest.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Atmospheric Nanoparticles, Air Quality, and Human Health (NANOSOMs) research project co-funded by the Stavros Niarchos Foundation (SNF) and the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the 5th Call of “Science and Society” Action Always strive for excellence – Theodoros Papazoglou” (Project Number: 11504). The authors acknowledge the computational time granted by the Greek Research & Technology Network (GRNET) in the national HPC facility ARIS under project GlaSOA III (pr015029).References
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirilä, J. Leskinen, J. M. Mäkelä, J. K. Holopainen, U. Pöschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS PubMed.
- B. Zobrist, C. Marcolli, D. A. Pedernera and T. Koop, Atmos. Chem. Phys., 2008, 8, 5221–5244 CrossRef CAS.
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS PubMed.
- M. Song, P. F. Liu, S. J. Hanna, Y. J. Li, S. T. Martin and A. K. Bertram, Atmos. Chem. Phys., 2015, 15, 5145–5159 CrossRef CAS.
- C. D. Cappa, E. R. Lovejoy and R. Ravishankara, Direct, 2008, 105, 18687–18691 CAS.
- C. Marcolli, B. Luo and T. Peter, J. Phys. Chem. A, 2004, 108, 2216–2224 CrossRef CAS.
- Y. Cheng, H. Su, T. Koop, E. Mikhailov and U. Pöschl, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- O. Mazurin and Yu. V. Gankin, Glass transition temperature: problems of measurements and analysis of the existing data, Strasbourg, France, 2007 Search PubMed.
- O. V. Mazurin, Glass Physics and Chemistry, 2007, vol. 33, pp. 22–36 Search PubMed.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259 CrossRef CAS PubMed.
- D. Champion, M. Le Meste and D. Simatos, Trends Food Sci. Technol., 2000, 11, 41–55 CrossRef CAS.
- M. Shiraiwa, Y. Li, A. P. Tsimpidi, V. A. Karydis, T. Berkemeier, S. N. Pandis, J. Lelieveld, T. Koop and U. Pöschl, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- W. S. W. DeRieux, Y. Li, P. Lin, J. Laskin, A. Laskin, A. K. Bertram, S. A. Nizkorodov and M. Shiraiwa, Atmos. Chem. Phys., 2018, 18, 6331–6351 CrossRef.
- T. Galeazzo and M. Shiraiwa, Environ. Sci. Atmos., 2022, 2, 362–374 CAS.
- G. Armeli, J. H. Peters and T. Koop, ACS Omega, 2023, 8, 12298–12309 CrossRef CAS PubMed.
- A. Kołodziejczyk, P. Pyrcz, K. Błaziak, A. Pobudkowska, K. Sarang and R. Szmigielski, ACS Omega, 2020, 5, 7919–7927 CrossRef PubMed.
- A. Kołodziejczyk, P. Pyrcz, A. Pobudkowska, K. Błaziak and R. Szmigielski, J. Phys. Chem. B, 2019, 123, 8261–8267 CrossRef PubMed.
- N. E. Rothfuss and M. D. Petters, Environ. Sci. Technol., 2017, 51, 271–279 CrossRef CAS PubMed.
- M. Nakanishi and R. Nozaki, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 3–7 CrossRef PubMed.
- P. Siachouli, K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Soft Matter, 2024, 20, 4783–4794 RSC.
- J. W. Grayson, E. Evoy, M. Song, Y. Chu, A. Maclean, A. Nguyen, M. A. Upshur, M. Ebrahimi, C. K. Chan, F. M. Geiger, R. J. Thomson and A. K. Bertram, Atmos. Chem. Phys., 2017, 17, 8509–8524 CrossRef CAS.
- K. Li, G. Jiang, F. Zhou, L. Li, Z. Zhang, Z. Hu, N. Zhou and X. Zhu, Polym. Chem., 2017, 8, 2686–2692 RSC.
- K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Atmos. Chem. Phys., 2019, 19, 5571–5587 CrossRef CAS.
- K. S. Karadima, V. G. Mavrantzas and S. N. Pandis, Phys. Chem. Chem. Phys., 2017, 19, 16681–16692 RSC.
- P. G. Mermigkis, K. S. Karadima, S. N. Pandis and V. G. Mavrantzas, ACS Omega, 2023, 8, 33481–33492 CrossRef CAS PubMed.
- S. Napolitano, E. Glynos and N. B. Tito, Reports Prog. Phys., 2017, 80, 036602 CrossRef PubMed.
- G. Tsolou, V. A. Harmandaris and V. G. Mavrantzas, J. Nonnewton. Fluid Mech., 2008, 152, 184–194 CrossRef CAS.
- O. Alexiadis, V. G. Mavrantzas, R. Khare, J. Beckers and A. R. C. Baijon, Macromolecules, 2008, 41, 987–996 CrossRef CAS.
- N. J. Soni, P. H. Lin and R. Khare, Polymer, 2012, 53, 1015–1019 CrossRef CAS.
- Scienomics SARL, version 3.4.2, MAPS platform, France, 2015 Search PubMed.
- M. M. Ghahremanpour, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2022, 126, 5896–5907 CrossRef CAS PubMed.
- W. L. Jorgensen, M. M. Ghahremanpour, A. Saar and J. Tirado-Rives, J. Phys. Chem. B, 2024, 128, 250–262 CrossRef CAS PubMed.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2017, 121, 3864–3870 CrossRef CAS PubMed.
- L. S. Dodda, I. C. De Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- William G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulation of liquids, Oxford University Press, New York, 1991 Search PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- Q. Yang, X. Chen, Z. He, F. Lan and H. Liu, RSC Adv., 2016, 6, 12053–12060 RSC.
- K. K. Bejagam, C. N. Iverson, B. L. Marrone and G. Pilania, Phys. Chem. Chem. Phys., 2020, 22, 17880–17889 RSC.
- C. Wen, B. Liu, J. Wolfgang, T. E. Long, R. Odle and S. Cheng, J. Polym. Sci., 2020, 58, 1521–1534 CrossRef CAS.
- R. Gani, Curr. Opin. Chem. Eng., 2019, 23, 184–196 CrossRef.
- S. R. S. Sastri and K. K. Rao, Chem. Eng. J., 1992, 50, 9–25 CrossRef CAS.
- J. Marrero-Morejón and E. Pardillo-Fontdevila, Chem. Eng. J., 2000, 79, 69–72 CrossRef.
- N. R. Gervasi, D. O. Topping and A. Zuend, Atmos. Chem. Phys., 2020, 20, 2987–3008 CrossRef CAS.
- S. E. Stein and R. L. Brown, J. Chem. Inf. Comput. Sci., 1994, 34, 581–587 CrossRef CAS.
- J. F. Pankow and W. E. Asher, Atmos. Chem. Phys., 2008, 8, 2773–2796 CrossRef CAS.
- T. Koop, J. Bookhold, M. Shiraiwa and U. Pöschl, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC.
- V. N. Novikov and E. A. Rössler, Polymer, 2013, 54, 6987–6991 CrossRef CAS.
- Y. Fu, X. Meng, X. Liang and J. Wu, J. Chem. Eng. Data, 2021, 66, 712–721 CrossRef CAS.
- Y. S. Elmatad, D. Chandler and J. P. Garrahan, Glass, 2009, 2, 5563–5567 Search PubMed.
- Y. S. Elmatad, D. Chandler and J. P. Garrahan, J. Phys. Chem. B, 2010, 114, 17113–17119 CrossRef CAS PubMed.
- E. A. Di Marzio and A. J. M. Yang, J. Res. Natl. Inst. Stand. Technol., 1997, 102, 135–157 CrossRef CAS PubMed.
- V. V. Ginzburg, Soft Matter, 2020, 16, 810–825 RSC.
- J. C. Mauro, Y. Yue, A. J. Ellison, P. K. Gupta and D. C. Allan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19780–19784 CrossRef CAS PubMed.
- J. Zhao, S. L. Simon and G. B. McKenna, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- G. B. McKenna and J. Zhao, J. Non. Cryst. Solids, 2015, 407, 3–13 CrossRef CAS.
- H. Yoon and G. B. McKenna, Sci. Adv., 2018, 4, 1–8 CAS.
- O. V. Laukkanen and H. H. Winter, J. Non. Cryst. Solids, 2018, 499, 289–299 CrossRef CAS.
- C. T. Moynihan, A. J. Easteal, J. Wilder and J. Tucker, J. Phys. Chem., 1974, 78, 2673–2677 CrossRef CAS.
- M. L. Williams, R. F. Landel and J. D. Ferry, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- S. B. Dake and R. V. Chaudhari, J. Mol. Catal., 1986, 35, 119–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01533a |
| This journal is © The Royal Society of Chemistry 2025 |