
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of alpha-synuclein and poly(N-isopropylacrylamide) complex formation through detailed atomistic simulations†
Sisem
Ektirici
a and
Vagelis
Harmandaris
 *abc
*abc
aComputation-based Science and Technology Research Center, The Cyprus Institute, 2121 Nicosia, Cyprus. E-mail: v.harmandaris@cyi.ac.cy
bDepartment of Mathematics and Applied Mathematics, University of Crete, Heraklion, GR-71110, Greece
cInstitute of Applied and Computational Mathematics, Foundation for Research and Technology – Hellas, Heraklion, GR-71110 Crete, Greece
First published on 21st January 2025
Abstract
This work presents an investigation of the influence of poly(N-isopropylacrylamide) (PNIPAM) polymer on the structural dynamics of intrinsically disordered alpha-synuclein (α-syn) protein, exploring the formation and intricate features of the resulting α-syn/PNIPAM complexes. Using atomistic molecular dynamics (MD) simulations, our study analyzes the impact of initial configuration, polymer molecular weight, and protein mutations on the α-syn and the α-syn/PNIPAM complex. Atomistic simulations, of a few μs, of the protein/polymer complex reveal crucial insights into molecular interactions within the complex, emphasizing a delicate balance of forces governing its stability and structural evolution. Our findings indicate that PNIPAM polymer engages in significant non-polar interactions with the non-amyloid component (NAC) region of α-syn, which plays a crucial role in fibril formation, under various conditions such as the mutations in the protein structure and polymer chain length. Especially the PNIPAM polymer with a 40mer monomer exhibits a stabilizing effect on the structural properties of the protein, reducing intramolecular interactions that contribute to misfolding. These findings, which delve into protein/polymer interactions, hold promise as potential guidance for therapeutic strategies in various neurodegenerative disorders.
1. Introduction
Alpha-synuclein (α-syn) is a 14 kDa protein found in the human brain that regulates the release of neurotransmitters and is synthesized by the SNCA (synuclein alpha) gene. Although this protein has been investigated in detail in numerous studies, its exact function is still unknown. A mutation in the gene encoding α-syn was associated with family cases of early-onset Parkinson's disease (PD) in 1997,1 and its aggregates were found to be the main components of Lewy bodies that are the hallmark of PD. The α-syn protein is associated not only with Parkinson's disease, but also with other neurodegenerative diseases such as Alzheimer's disease (AD)2 and dementia.3 Because it is an intrinsically disordered protein (IDP), it has been the subject of research for years to clarify the deformations in its structure and explain the causes of the mentioned diseases. IDPs are characterized by the absence of a stable, well-defined three-dimensional structure under native functional conditions, allowing them to adopt multiple conformations. Although the reason why the structure and pathways of α-syn cause these diseases are not yet fully understood, intensive studies suggest that these reasons are due to protein misfolding and defects in binding to cell membranes.4–6 The study of these protein misfolding is of great importance, as studies have shown that protein misfolding is associated not only with neurodegenerative diseases, but also with cancer,7 heart disease,8 and type II diabetes.9Using the protein regulators10,11 and pharmacological chaperones12,13 are the most common methods to prevent protein misfolding. Pharmacological chaperones focus on secondary interactions such as hydrogen bonds and van der Waals bonds between proteins and molecules with specific properties designed to prevent misfolding. Polymer chains,14 complex micelles,15 hydrogels,16 and nanoparticles17 can be mentioned as examples of pharmacological chaperones specifically designed for proteins to prevent interactions and degradation processes that lead to protein misfolding. In particular, polymers that can change their properties in response to light, pH, ionic strength, and temperature are frequently used in the diagnosis and treatment of diseases.18–20
One of the extensively studied ‘smart’ polymer is poly(N-isopropylacrylamide) (PNIPAM), known for its unique thermoresponsive behavior and wide range of applications for biological systems.21–23 PNIPAM undergoes a phase transition at its lower critical solution temperature (LCST), which is close to physiological temperature (∼32 °C) making it ideal for biomedical applications.24–28 Below LCST, PNIPAM is hydrophilic and water-soluble but above this temperature, it becomes hydrophobic and collapses into a globular conformation due to dehydration of isopropyl groups.29,30 Furthermore, polymer's hydrophobic/hydrophilic transition allows it to from stabilizing interactions with proteins, making it valuable tool for drug delivery systems.31–34 The stereo regularity of PNIPAM chains has also been shown to its influence thermoresponsive properties, with isotactic PNIPAMs adopting more ordered conformations.35,36
Experimental techniques to study and prevent protein misfolding encompass a diverse array of approaches aimed at understanding and mitigating this phenomenon. An example involves the use of molecular chaperones, specialized proteins that aid in proper protein folding by interacting with unfolded or misfolded intermediates, thus preventing aggregation and facilitating correct folding.33,34,37 Chemical chaperones,38 including osmolytes39 and small molecules,40,41 are also used to stabilize protein structures under denaturing conditions, thus reducing the probability of misfolding. Site-directed mutagenesis, another powerful technique, allows the engineering of proteins with greater stability against misfolding by introducing specific amino acid substitutions that strengthen intramolecular interactions crucial to maintaining proper folding.42 Advanced biophysical techniques, such as nuclear magnetic resonance (NMR) spectroscopy43–45 and X-ray crystallography,38 provide valuable information on protein folding dynamics and structural stability, aiding in the design of strategies to prevent misfolding. On the other hand, protein/polymer complexes play a crucial role in various biological processes46 and have significant implications in biomedical applications, including drug delivery,47,48 biomaterial development,49,50 and therapeutic strategies51 for neurodegenerative disorders. Understanding their formation kinetics and stability at the molecular level is essential to advance our knowledge in these fields and for the development of effective treatments.
In addition to the above experiments, computational approaches and, in particular, molecular simulations, such as atomistic molecular dynamics (MD) and free energy computation methods, have been widely used in biomedical applications in recent years to elucidate the structures and interactions of biomolecules.52–55 Molecular simulations can be used in drug design and disease treatment by providing insights into protein structures and protein–ligand interactions. By controlling parameters such as temperature, pH, and ionic strength, these simulations help to illuminate how these factors influence molecular behavior.56–59 Therefore, such techniques have also been used to study the structure and formation of amyloid-based biomolecules. For example, in the study of Chewook Lee et al. MD simulations were used to examine misfolding of the amyloid beta protein, intramolecular hydrogen bonds, and the hydrophobic surface region.57 The results of group computations investigating regions conducive to amyloid fibril formation were found to coincide with the findings obtained by NMR. Several MD studies examine the causes of protein misfolding, leading to real-time experiments with a detailed examination of protein structures.56,58
Molecular simulations using atomistic and/or coarse-grained models have been particularly instrumental in elucidating the conformational dynamics of α-syn60 and its interactions with membranes61 and other cellular components.62 These simulations have revealed the propensity of α-syn to adopt various structural states, including helical conformations and β-sheet-rich aggregates, which sheds light on the mechanisms underlying its aggregation and misfolding. Furthermore, simulation studies have explored the effects of post-translational modifications63–65 and environmental factors66,67 on α-syn structure and behavior of α-syn, offering crucial mechanistic insights into the pathological processes associated with α-syn aggregation in neurodegenerative diseases.
Despite the extensive research conducted, several issues remain unresolved in relation to the formation of α-syn and its complexes with polymers. Specifically, the interactions between polymer chains, such as PNIPAM, and different regions of α-syn, and how these interactions influence the structure of the protein, are not fully understood. Additionally, the structure of the dependence of the protein/polymer complex on parameters such as the molecular weight of the polymer chain of the polymer chain remains unclear. To address these questions, a detailed atomic-level investigation of the α-syn/polymer complex formation and structure is essential.
The goal of this work is to provide a detailed investigation about the intramolecular interactions of α-syn that leads to the aggregation of the protein and the changes in these interactions in the presence of the thermoresponsive polymer PNIPAM via all-atom MD simulations. By examining the structural changes and behavioral variations of α-syn in the presence of PNIPAM, this study aims to elucidate the intricate factors within the polymer–protein complex. This exploration is intended to enhance our understanding of different folding pathways influenced by the polymer and applications in biomedicine and materials science. Initially, native α-syn and four distinct mutant forms related to Parkinson's disease (A30P, A53E, A53T, A53V, and E46K)68,69 and their complexes with PNIPAM were subjected to simulations of 0.5 μs. This part of the study provides a detailed benchmark for folding parameters and interaction dynamics at the atomic level, which was crucial for establishing a framework for understanding how mutations affect protein behavior and polymer interactions. Subsequently, the study changed to examining the native α-syn protein to offer a baseline to compare the effects of mutations and clarify deviations from the natural properties of the wild-type protein. Following this, the importance of accurately sampling in molecular simulations the complex configuration space of multicomponent protein/polymer systems was investigated by examining different initial configurations, in which the PNIPAM molecules are close to one of the three key regions of α-syn (see Fig. 1), that is the N-terminal involved in membrane binding, the central hydrophobic nonamyloid component (NAC) crucial for aggregation, and the C-terminal that regulates protein–protein and protein-small molecule interactions. Therefore, the mutant proteins and initial configuration assessments are described in detail in the ESI† section, to ensure that the methodology and rationale are transparent and reproducible.
 | ||
| Fig. 1 (a) Monomeric α-syn structure with the polar N- and C-terminal regions depicted in red and the apolar NAC region depicted in blue, highlighting the charged sites. (b) Structure of the 40mer PNIPAM polymer. | ||
In the following section, details of MD simulations, atomistic models for α-syn and PNIPAM, as well as generation and equilibration of their initial configurations, are provided, followed by presentation of the simulation results.
2. Model and simulations
Our work focuses on a detailed investigation of the interactions between the C-terminal, N-terminal, and NAC regions of the protein and the polymer (PNIPAM) chains, addressing the problem of misfolding of α-syn. As part of the benchmarking section of the study, the configurations of the mutated protein and the mutated protein/PNIPAM complexes after simulations are shown in Fig. S2 and S3 (ESI†), respectively.The initial structures of α-syn and a 40mer PNIPAM chain are shown in Fig. 1, while in Table 1 the different regions of α-syn are denoted. To provide a more detailed representation of the properties of the amino acid sequence properties, which is very important in the study presented, Fig. S1 (ESI†) shows the polarity chart of the amino acids within the structure of α-syn. Crystal configuration of the α-syn protein obtained from the Protein Data Bank (PDB) with the protein code 1XQ8,70–73 has been widely used for understanding the structural parameters for the protein.64–66
| N-terminal | NAC region | C-terminal | |
|---|---|---|---|
| Residue number | 1–60 | 61–95 | 96–140 |
| Atom number | 1–891 | 892–1351 | 1352–2016 |
The protein's N-terminal is capped with a methyl group, while the C-terminal is capped with a hydrogen atom. To achieve charge neutrality in the α-syn protein, 9 Na+ ions were incorporated into the simulation box in all systems.
In this context, the α-syn protein was initially subjected to a 1.5 μs simulation in water, followed by an examination of the effect with different parameters. A detailed investigation of the influence of the polymer was carried out by subjecting the PNIPAM polymer and α-syn to a simulation of 1.5 μs at 298 K. We also investigate the impact of polymer chain length on the formation and characteristics of α-syn/PNIPAM complexes, by examining various polymer lengths: 10mer, 40mer, 60mer, and 100mer PNIPAM. All PNIPAM polymer chains are generated using the CHARMM-GUI74,75 interface and adhere to the CHARMM parameters. The PNIPAM polymers were chosen in their isotactic form due to its well-defined stereochemistry, which allows for a clearer understanding of the impact of polymer structure on the stability and conformation of α-syn.
Table S1 (ESI†) provides more details of the simulations.
All simulations were performed using the GROMACS76 simulation package, using the CHARMM2777 force field which is particularly suited for examining both the fibril formation78 of α-syn and the structural properties of the monomeric form of the protein.79,80 The TIP3P model81 was used to represent water molecules in MD simulations. The simulations were conducted in a simulation box of size 8 × 16 × 8 nm. The leap frog integrator was used for a total simulation time of 1.5 μs in the study of the α-syn/40mer PNIPAM complex. Subsequent simulations, including examination of the initial configuration and investigation of the effect of molecular weight were carried out over a duration of 1.5 μs, employing identical simulation parameters. The short-range electrostatic and van der Waals cut-offs were both set to 1.0 nm. Long-range electrostatics were treated with the Particle Mesh Ewald (PME)82,83 method using cubic interpolation of order 4 and a Fourier grid spacing of 0.16 nm. For the temperature coupling, we used the modified Berendsen Thermostat84 with a time constant of 0.1 ps and a reference temperature of 298 K. For pressure coupling, we used the Berendsen method84 with isotropic scaling and a time constant of 2.0 ps, referring to a pressure of 1.0 bar. All visual analyzes of the simulations were performed using visual molecular dynamics (VMD).85 The total number of atoms, and water atoms, PNIPAM polymer atoms used in all simulations is provided in Table S1 (ESI†).
The simulation results were analyzed to calculate binding free energy (ΔGbinding) and its components, non-bonded interaction energies, as well as to probe changes in the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), asphericity, prolateness, pair radial distribution function (RDF), variations in hydrogen bonds (HBs), and DSSP (secondary structure assignments). The methods used to calculate these quantities are provided in the equations below. After benchmarking studies with different protein mutants and changes in the initial polymer position were performed, a detailed examination of the interactions in the polymer–protein complex was performed. By sequentially exploring the effects of molecular weight our work aims to provide a comprehensive understanding of the multifaceted factors that influence the behavior of polymer–protein complexes.
Calculation of binding free energy with gmx_MMPBSA Tool:
The binding free energy ΔGbinding was calculated using the MM-PBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) method, which estimates the free energy difference between the bound complex and its unbound components (α-syn and PNIPAM):
| ΔGbinding = ΔGcomplex − (ΔGα-syn + ΔGPNIPAM) | (1) |
The total free energy (ΔG) for each system was decomposed into gas-phase molecular mechanics (MM) energy and solvation free energy (ΔGsolvation):
| ΔG = ΔGMM + ΔGsolvation | (2) |
Here, the gas-phase energy ΔGMM accounts for van der Waals ΔEVDW and electrostatic ΔECol contributions:
| ΔGMM = ΔEVDW + ΔECol | (3) |
The solvation free energy ΔGsolvation consists of a polar component ΔGpolar computed using the Generalized Born (GB) method and a nonpolar component ΔGnonpolar estimated from the solvent-accessible surface area (SASA):
| ΔGsolvation = ΔGpolar + ΔGnonpolar | (4) |
Thus, the final binding free energy incorporates both the gas-phase molecular mechanics contributions and the solvation free energy differences between the complex, α-syn, and PNIPAM.
2.1. Minimum distances, number of contacts, RDF
The minimum distances between hydrophobic and hydrophilic residues (a), the number of contacts within a threshold of 0.6 nm (b), and the RDF (c) data for the NAC region are presented in Fig. S8 (ESI†). Hydrophobic and hydrophilic groups were identified for both α-syn and PNIPAM to distinguish polar and non-polar interactions. For the NAC region of α-syn (residues 61–95), hydrophobic residues were selected based on their apolar nature, including alanine (Ala), valine (Val), leucine (Leu), isoleucine (Ile), phenylalanine (Phe), methionine (Met), proline (Pro), and tryptophan (Trp). Hydrophilic residues were defined as polar residues, comprising aspartic acid (Asp), glutamic acid (Glu), asparagine (Asn), glutamine (Gln), serine (Ser), threonine (Thr), histidine (His), tyrosine (Tyr), lysine (Lys), and arginine (Arg). For PNIPAM, hydrophilic groups were identified as the amide nitrogen and oxygen atoms, while the hydrophobic groups included the methyl (–CH3) and methylene (–CH2) groups.2.2. RMSD
The RMSD measures the average deviation of atomic positions with respect to a reference structure over time. Higher RMSD values often correlate with increased flexibility or structural fluctuations in a protein. In the context of α-syn, flexibility is a key characteristic. The polymer might induce changes in the conformational dynamics and flexibility of α-syn. The RMSD is calculated using the following relation: | (5) |
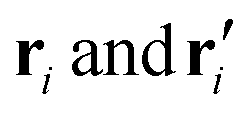 are the three-dimensional coordinates of the i-th atom in the reference (initial configuration of α-syn) and target structures (post-simulation configuration of α-syn), respectively.
are the three-dimensional coordinates of the i-th atom in the reference (initial configuration of α-syn) and target structures (post-simulation configuration of α-syn), respectively.
2.3. RMSF
In order to investigate the structure of each residue of the α-syn in the α-syn/PNIPAM complex we compute RMSF which quantifies the degree of mobility (movement) exhibited by atoms relative to their average positions, providing valuable insight into the local flexibility of specific regions within a protein or complex. This is particularly important because different parts of a protein may exhibit varying degrees of flexibility, and regions that interact with ligands may display unique patterns of fluctuation. Ligand binding often induces conformational changes in the protein, especially in regions directly involved in binding. RMSF analysis helps identify allosteric effects by revealing changes in flexibility in regions that do not directly interact with the ligand, contributing to a comprehensive understanding of ligand-induced alterations. The RMSF is computed using the following equation: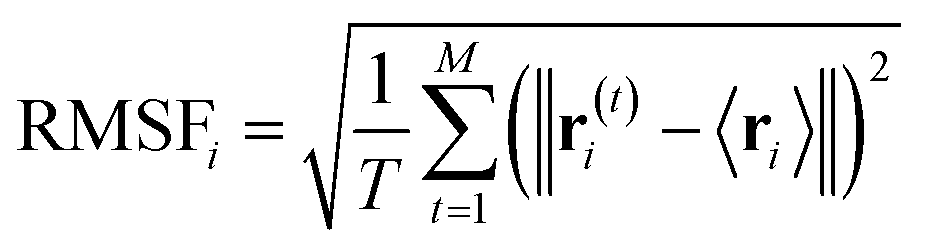 | (6) |
2.4. R g, asphericity and prolateness
To demonstrate the impact of protein–polymer interactions on the alteration of the protein's shape we compute the radius of gyration, Rg, as well as the aspericity and the prolateness of the α-syn within the complex. Rg is computed as follows: | (7) |
To further examine the impact of the polymer on the structural conformations and the overall shape of the protein, we calculate the asphericity, A, and prolateness, P, of the α-syn defined by the following equations:
 | (8) |
 | (9) |
The time evolution of Rg for both the isolated protein and the polymer complex is provided in Fig. S9 (ESI†), while the profiles for aspericity and prolateness are presented in Fig. S10 and S11 (ESI†), respectively. Table 4 shows the average Rg, over the last 0.5 μs of the simulation.
2.5. DSSP
We used the default settings for the DSSP tool within GROMACS76 to calculate the hydrogen bonding interactions and to identify the secondary structure elements. These default parameters include the criteria for identifying hydrogen bonds, such as the distance between donor and acceptor atoms and the angle between the donor-hydrogen-acceptor atoms. By using these standard parameters, we were able to ensure that the secondary structure analysis was conducted.3. Result and discussion
3.1. [α-Syn/40mer PNIPAM]1 complex
As mentioned above, the water solubility and biocompatibility of the PNIPAM polymer, along with its potential hydrogen bonding regions (–NH2, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), suggest its potential interaction with amino acids in the protein structure. To investigate the indirect impact of these interactions on the misfolding of α-syn protein, we first examined the changes in the non-bonded energies during the formation of the protein–polymer complex. For this, non-bonded interaction energies (Fig. S7, ESI†), total binding free energy (ΔGTotal) and its van der Waals (ΔVDW) and electrostatic contributions (ΔCol) have calculated by using eqn (1). Subsequently, because of the significant interactions of both structures with water, we analyzed changes in hydrogen bonds throughout the simulation, focusing on the average hydrogen bonds in protein–water, protein–protein, protein–polymer and polymer–water interactions. We also examined the pair radial distribution function, RDF, add between hydrophobic and hydrophilic groups in the NAC region to quantify the spatial arrangement and distribution of atoms within the system, which provides insights into the structural organization and interactions between the protein and polymer. Following these analyzes, which offer detailed information on the formation and stabilization of the protein–polymer complex, we evaluated the effects of the PNIPAM polymer on the structural and shape characteristics using RMSD, RMSF, Rg, asphericity, and prolateness.
O), suggest its potential interaction with amino acids in the protein structure. To investigate the indirect impact of these interactions on the misfolding of α-syn protein, we first examined the changes in the non-bonded energies during the formation of the protein–polymer complex. For this, non-bonded interaction energies (Fig. S7, ESI†), total binding free energy (ΔGTotal) and its van der Waals (ΔVDW) and electrostatic contributions (ΔCol) have calculated by using eqn (1). Subsequently, because of the significant interactions of both structures with water, we analyzed changes in hydrogen bonds throughout the simulation, focusing on the average hydrogen bonds in protein–water, protein–protein, protein–polymer and polymer–water interactions. We also examined the pair radial distribution function, RDF, add between hydrophobic and hydrophilic groups in the NAC region to quantify the spatial arrangement and distribution of atoms within the system, which provides insights into the structural organization and interactions between the protein and polymer. Following these analyzes, which offer detailed information on the formation and stabilization of the protein–polymer complex, we evaluated the effects of the PNIPAM polymer on the structural and shape characteristics using RMSD, RMSF, Rg, asphericity, and prolateness.
| Energy component | Average (kJ mol−1) |
|---|---|
| ΔVDW | −76.1 ± 0.1 |
| ΔCol | −59.1 ± 0.2 |
| ΔGTotal | −53.2 ± 0.3 |
To better understand the driving forces behind the interactions between the two structures, minimum distance and contact analyses, along with RDF calculations for the hydrophilic and hydrophobic residues of both structures, were conducted. The methodology for this analysis is described in the Methods and Simulation section. Fig. S8 (ESI†) shows that the hydrophobic groups of PNIPAM consistently maintained a distance below 0.6 nm from the NAC hydrophobic residues, indicating close and persistent non-polar interactions. In contrast, the hydrophilic groups exhibited larger distances (>1 nm) and greater fluctuations, reflecting weaker and less stable interactions. Hydrophobic contacts between PNIPAM and the NAC hydrophobic residues were significantly higher, more persistent, and stabilized after approximately 0.7 μs, indicating sustained non-polar interactions. The RDF for hydrophobic interactions exhibited a pronounced peak at ∼0.5 nm, indicating significant clustering of PNIPAM hydrophobic atoms around NAC hydrophobic residues, while the RDF for hydrophilic interactions was less prominent, reflecting weaker clustering of PNIPAM hydrophilic atoms. The findings from the contact analysis, minimum distance measurements, and RDF calculations strongly support the conclusion that PNIPAM stabilizes its interaction with the NAC region of α-syn primarily through non-polar (hydrophobic) interactions. These results align with the binding free energy decomposition (Table 2) and total potential energy analysis (Fig. S7, ESI†), which highlight van der Waals forces as the dominant contributor to the stabilization of the [α-syn/40mer PNIPAM]1 complex after approximately 1 μs.
The initial configurations are specified in Fig. S4 (ESI†), which outlines the different initial positions of the PNIPAM polymer relative to the α-syn molecule. These configurations were systematically tested to assess their impact on the interaction between the molecules Data in Table S2 (ESI†) indicate that the initial position of PNIPAM slightly influences its interactions with the protein, with similar values observed across all complexes except for the [α-syn/40mer-PNIPAM]3 complex. Notably, van der Waals interactions are more pronounced in all complexes. The interactions between the bulky isopropyl groups of the PNIPAM polymer and the apolar regions of the protein structure are particularly dominant compared to Coulombic interactions. In particular, the [α-syn/40mer-PNIPAM]3 complex exhibits significantly higher van der Waals interactions compared to other structures. As illustrated in Fig. S5 (ESI†), this is due to the polymer being placed in the protein region where the apolar groups are most densely located, resulting in the highest degree of apolar–apolar interactions within this complex.
In the context of mutant proteins, the non-bonded energies presented in Table S2 (ESI†) support the observation that the intermolecular α-syn/PNIPAM interactions are mainly of apolar–apolar type. This trend becomes pronounced in the mutants of α-syn when amino acids change from polar to apolar residues. For instance, the E46K mutation, characterized by the substitution of a negatively charged glutamic acid with a positively charged lysine, introduces a longer aliphatic side chain and alters the local electrostatic environment. This change facilitates stronger van der Waals interactions with PNIPAM, as evidenced by the highest total non-bonded interaction energy recorded for this mutant Table S2 (ESI†), suggesting that the increased hydrophobic contact area between lysine and the polymer's isopropyl groups enhances stability. In contrast, mutations like A53E, which replace a small apolar alanine with a negatively charged glutamic acid, reduce hydrophobic interactions and disrupt local binding in the NAC region, as reflected in the comparatively weaker interaction energies.
From the data presented Fig. S7 and Table S2 (ESI†), it is also observed that the α-syn/PNIPAM complex achieves an energetic equilibrium on a time scale of around 1 μs. Based on these findings in the analysis below, we compute the average equilibrium properties that characterize the α-syn/PNIPAM complex for the period between 1 μs and 1.5 μs.
Hydrogen bonds. Following the analysis of the free binding energy and non-bonded interaction energies, we examined hydrogen bonds (h-bonds) to provide a more detailed understanding of the specific intermolecular forces that contribute to the stabilization and structural integrity of the α-syn/PNIPAM complexes. The formation or disruption of specific h-bonds becomes a molecular signature that guides the design of ligands targeting the misfolded state. To investigate the molecular-level changes in the structure of the protein in the presence of the polymer, we calculated the average number of h-bonds during the simulation for protein–water, protein–protein, protein–polymer, polymer–water and polymer–polymer interactions. Hydrogen bonds between structures were identified using a donor–acceptor distance cutoff of 0.35 nm and a hydrogen bond angle of up to 30 degrees, for the simulation time from 1 to 1.5 μs. The average number of HBs across all temperatures in the last 0.5 μs of simulation is shown in Table 3.
| System | [α–W] | [α–α] | [α–P] | [P–W] | [P–P] |
|---|---|---|---|---|---|
| α-Syn in water | 363.6 ± 0.2 | 100.2 ± 0.1 | — | — | — |
| [α-Syn/40mer PNIPAM]1 | 336.2 ± 0.2 | 90.9 ± 0.1 | 2.8 ± 0.02 | 68.4 ± 0.07 | 3.3 ± 0.02 |
| PNIPAM in water | — | — | — | 76.0 ± 0.19 | 3.9 ± 0.06 |
The observed alterations in the hydrogen bonding patterns, particularly the decrease in the average number of hydrogen bonds between the protein and water in the presence of PNIPAM, and the concurrent establishment of hydrogen bonds between the polymer and both the protein and water, can be attributed to a combination of intricate molecular interactions and environmental effects. The decrease in [α–α] h-bonds in the presence of PNIPAM indicates a weaker association between protein molecules, potentially driven by the altered dynamics induced by the polymer. Similarly, the observed decrease in [P–P] hydrogen bonds is attributed to changes induced by the protein itself. The limited number of h-bonds between the polymer and the protein suggests a specific and nuanced interaction pattern at this interface. Furthermore, the average number of h-bonds between the polymer and water points to the pronounced affinity of PNIPAM for water molecules, likely facilitated by its inherently hydrophilic nature. This affinity may result in competing interactions with water molecules for hydrogen bonding sites on the protein surface, leading to a reduction in protein–water hydrogen bonds. The establishment of hydrogen bonds between the polymer and water may signify the displacement of water molecules in specific regions by the polymer. The observed decrease in polymer–water hydrogen bonds in the presence of the protein further supports the establishment of polymer–protein hydrogen bonding, indicating that the polymer interacts directly with the protein, displacing water molecules at specific regions of the interface. Furthermore, the interaction with PNIPAM might induce conformational changes in the protein, exposing or concealing residues involved in hydrogen bonding with water.
 | ||
| Fig. 2 Conformational changes in the structure of the α-syn and [α-syn/40mer PNIPAM]1 complex. (a) and (c) Show the initial conformation of the α-syn and [α-syn/40mer PNIPAM]1 complex, respectively, before the MD simulations. (b) and (d) Show the altered conformation of the α-syn and [α-syn/40mer PNIPAM]1 complex at the end (1.5 μs) of the simulation. | ||
Data on the RMSD of alpha carbons of α-syn in water and in the [α-syn/40mer-PNIPAM]1 complex are shown in Fig. 3 while the mean RMSD values of the protein polymer complexes are shown in Table S3 (ESI†) for the last 0.5 μs part of the simulation. As seen in the RMSD graph, there is a sharp increase within the first few steps, indicating very fast kinetics and a rapid structural reorganization. Following this initial phase, the RMSD values stabilize and remain relatively constant, suggesting that the system has reached a stable conformation. In contrast, the [α-syn/40mer PNIPAM]1 complex exhibits a much smaller initial increase and stabilizes at a lower RMSD value, reflecting the restrictive effect of PNIPAM on the protein's structural flexibility. It is clear that the presence of a 40mer polymer chain decreases the RMSD of α-syn; for the other out of five initial configurations the decrease is substantial whereas in the other two is rather weak. This adjustment in α-syn's structure may lead to more stable configurations with PNIPAM, resulting in lower RMSD values and potentially establishing an energetically favorable state for the complex. The structural adaptation of α-syn becomes evident as PNIPAM induces molecular changes to accommodate the polymer. These adaptations likely involve alterations in amino acid residue orientation or stabilization of specific secondary structures, which contribute to the observed decrease in RMSD. By stabilizing the secondary or tertiary structure of α-syn, PNIPAM can mitigate conformational fluctuations, leading to a lower RMSD.
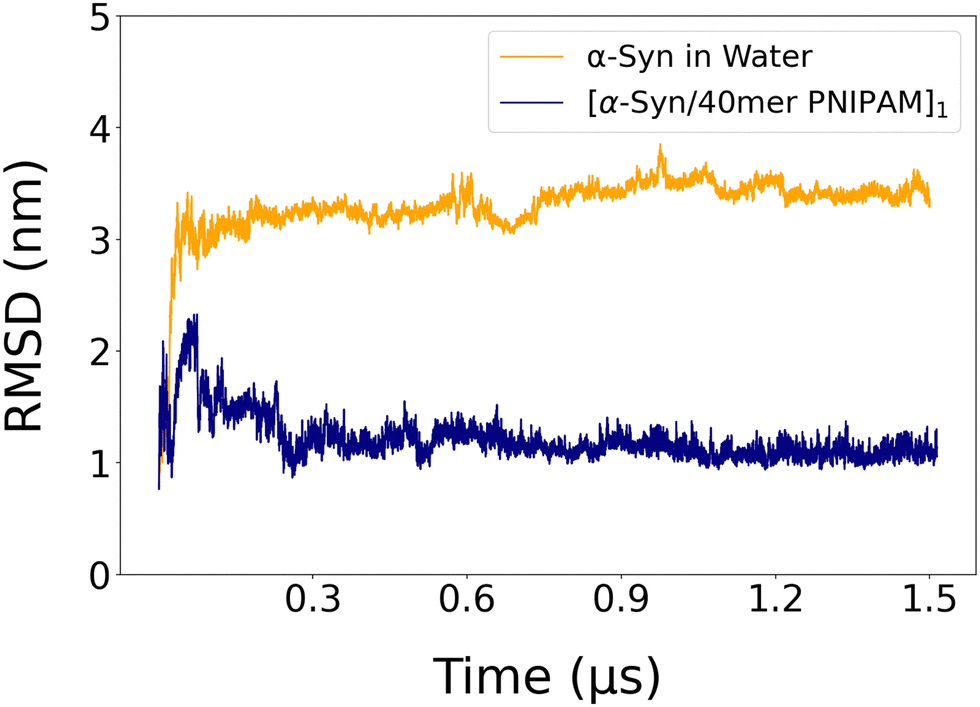 | ||
| Fig. 3 Time evolution of the RMSD of alpha carbons of α-syn in water and in α-syn/PNIPAM complex. | ||
As shown in Fig. 4, a general decrease in RMSF values is observed in the presence of the PNIPAM polymer. This reduction, seen in almost every region of the protein, can be explained by the stabilization effect of the polymer, as observed in the RMSD values. This leads to a decrease in the overall flexibility of the protein and can be attributed to a reduction in conformational entropy, which occurs when the binding of a ligand reduces the conformational entropy of the protein by limiting the range of accessible conformational states.
 | ||
| Fig. 4 Comparative analysis of the RMSF of the α-syn protein in water and within the [α-syn/40mer-PNIPAM]1 complex in the last 0.5 μs of the simulation. The red dashed lines indicate the distinction between the regions of α-syn. | ||
When examining this change at the amino acid level, it is important to note that the amino acids within the N-terminal region (residue numbers 1–60) are known as the membrane-binding region of the protein.86,87 In this region, which contains predominantly polar amino acid groups (Fig. S1, ESI†), the polymer improves the stabilization of these amino acids and the RMSF behavior mirrors the trend observed in the native state of the protein. In the NAC region, where the interaction between the two structures is most pronounced, the polymer again leads to a decrease in the RMSF values, particularly affecting amino acids with residue numbers 80 to 94. This effect can be attributed to the predominant hydrophobic interactions between the isopropyl groups and the apolar amino acid groups in this region. In particular, amino acids with residue numbers 95 and 96, phenylalanine and valine, respectively, exhibit the same RMSF values in both native α-syn and its complex with PNIPAM. This phenomenon can be primarily explained by the close proximity of the two structures and the hydrophobic interactions between PNIPAM's isopropyl groups and the side chains of these residues, creating a microenvironment where the flexibility of both residues is similarly restricted. In the C-terminal region, where the native protein exhibits the highest mobility, the generally lower RMSF values observed in the presence of PNIPAM are the result of the general stabilization effect. Hydrophobic amino acid–polymer interactions observed in the NAC region are also observed in the amino acids at residues 112 and 113 (isoleucine and leucine, respectively) in this region. In summary, the PNIPAM polymer reduces RMSF values throughout the entire α-syn protein, reflecting a general decrease in flexibility and enhanced stabilization in all regions, including membrane binding and NAC regions, due to polar and hydrophobic interactions with the polymer.
Radius of gyration, aspericity and prolateness. Next, we provide a more comprehensive understanding of the conformational changes and stability of the complex by investigating the shape of α-syn. In the initial configuration of the complex, the PNIPAM polymer is approximately 8 Å away from the N-terminal of the protein, as illustrated in Fig. 2c.
The comparison of the protein after simulation in the absence of polymer (Fig. 2b) with its configuration after simulation in the presence of polymer (Fig. 2d) is of great importance at this point. Although the protein is observed to be folded in water, a configuration very close to its native structure is observed after simulation in the presence of a polymer. It has been observed that the polymer, particularly interacting significantly with the NAC region of the protein during the simulation, greatly reduces the intramolecular interactions of the protein throughout the simulation in the [α-syn/40mer PNIPAM]1 complex.
As with other IDPs, previous experimental and computational studies on α-syn have demonstrated variations in its Rg.88 These works revealed a nuanced representation of α-syn's Rg value, showing fluctuations within the range of 2.4–4.1 nm under neutral pH and room temperature conditions. Computational investigations have also observed differences in the Rg of α-syn depending on the force field and the number of water molecules around α-syn.89–91 Although the inherent structural instability of the protein prevents a precise determination of the mean radius of gyration, the impact of the presence of other molecules under the same conditions provides insights into the structural alterations. In this study, the average Rg of α-syn was found to be around 2.5 nm in the aqueous environment after a 1.5 μs simulation. Notably, as shown in Table 4, the presence of the PNIPAM polymer resulted in an increase in the average Rg value for α-syn, which was measured at 4.8 nm. It is important to emphasize that in all cases, independently of the initial configurations, the Rg value of the protein within the α-syn/PNIPAM complex increases compared to the one of the pure α-syn. However, there is considerable variation in these Rg values in different configurations, reflecting the diverse conformational responses of α-syn to the presence of the polymer. Flexible and dynamic configurations allow the system to explore a larger conformational space within its existing degrees of freedom, enabling α-syn to adopt a more extended conformation compared to its intrinsically disordered state in the aqueous environment. Furthermore, the change in Rg may reflect the influence on α-syn's intramolecular interactions. The introduced complexation could lead to alterations in the secondary and tertiary structures, affecting its overall compactness. It is important to consider the potential role of water molecules in mediating these interactions. In an aqueous environment, water molecules may act as mediators of protein–polymer interactions, influencing the overall compactness. The dynamic interaction between water, the PNIPAM polymer, and α-syn could lead to nuanced conformational changes, influencing the observed Rg values.
| System | R g (nm) | Aspericity | Prolateness |
|---|---|---|---|
| α-Syn in water | 2.5 ± 0.08 | 0.9 ± 0.0003 | 0.2 ± 0.004 |
| α-Syn in complex | 4.8 ± 0.05 | 1.9 ± 0.0004 | 0.3 ± 0.001 |
Asphericity is a measure of irregularity or roughness of a surface, quantified by the ratio of the surface area of an object to that of a sphere with equivalent volume; a lower asphericity value indicates a more spherical or compact shape, while a higher value indicates a more elongated or irregular shape. In the aqueous environment, α-syn exhibits a lower asphericity value (0.9), indicating a globular or spherical conformation. This suggests that, in the absence of the PNIPAM polymer, the protein tends to adopt a compact and rounded structure. In Fig. S10 (ESI†), the probability density of protein's asphericity, depicted by the orange plot, exhibits a density ranging from 0.8 to 1.2, which is proportional to the average asphericity data. In the presence of the PNIPAM polymer, it shows a more elongated density range (1.8–2.0). The favorable interactions among its constituent amino acids contribute to a more cohesive and tightly packed protein state. In contrast, when embedded in the 40mer PNIPAM complex, α-syn shows an increase in asphericity, suggesting a departure from the globular shape towards a more elongated or irregular structure. The substantial influence of the polymer on the amino acid residues of the protein is consistent with the observations of the RMSF graph, indicating a complementary relationship between these results. The presence of the polymer results in a less compact and more elongated protein structure.
Prolateness is a measure of an object's deviation from spherical symmetry, defined in terms of the principal moments of inertia. It quantifies the extent to which the shape is elongated or anisotropic, with values closer to zero indicating a more spherical shape and higher positive values reflecting greater elongation along the principal axis. In the context of proteins, prolateness specifically refers to the elongation or oblateness of the molecular shape. In the aqueous environment, α-syn exhibits a prolateness value of 0.2. However, after interaction with the 40mer PNIPAM polymer, α-syn undergoes a change in prolateness, with a value of 0.3. The observation in Fig. S11 (ESI†) where the probability of prolateness, is significantly dense around 0.30–0.32 in the presence of the PNIPAM polymer suggests that the polymer maintains the protein in a more elongated structure. This considerable increase in prolateness correlates with the higher asphericity observed in the complex, indicating a transition toward a more elongated structure. Collectively, these changes denote a pronounced deviation from the native compact conformation of α-syn, suggesting a considerable impact of the PNIPAM polymer on structural dynamics.
DSSP analysis. Next, we investigate how different regions of the protein undergo significant structural changes upon binding to PNIPAM. For this, we perform a detailed DSSP analysis to identify binding regions, characterizing structural alterations, and understanding interaction dynamics. This step is essential for our analysis, since α-syn is an IDP, meaning that it lacks a stable or fixed secondary structure. DSSP analysis helps us to observe how PNIPAM affects the secondary structure of α-syn, such as inducing the formation of alpha helices, beta sheets, or turns in regions that are otherwise disordered. These structural alterations can provide information on how PNIPAM influences the misfolding pathways of α-syn.
As shown in Fig. 5, the DSSP analysis of α-syn and its complex with the 40mer PNIPAM polymer reveals that PNIPAM generally increases the alpha-helix content of the protein. Specifically, residues 35–40 initially exhibit a 5-helix and turn structure up to 0.7 μs before transitioning to an alpha-helix, but in the presence of PNIPAM, they convert to an alpha-helix at 0.1 μs. This early transition is likely due to the fact that PNIPAM stabilizes the alpha helix by interacting with the peptide backbone and side chains, reducing the energy barrier for helix formation. Residues 40–45, which predominantly show coil and bend characteristics without PNIPAM, transition from bend structures to turn structures after 1.1 μs with PNIPAM. PNIPAM likely induces structural constraints that favor the formation of turn structures over bends by stabilizing specific turn conformations through interactions with hydrophobic and hydrophilic regions. Residues 45–50, which normally exhibit bend and coil features, adopt an alpha-helix structure throughout the simulation with PNIPAM. This stabilization of the helical conformation is likely due to hydrophobic interactions and backbone hydrogen bonding facilitated by PNIPAM, which reduces the entropy of this segment and promotes a more ordered alpha-helix structure. Similarly, residues 65–75 transition to an alpha-helix in the presence of PNIPAM. The polymer stabilizes the helical structure by interacting with the side chains and the backbone, reducing conformational freedom and promoting helical stability. For residues 85–95, a transition from the alpha helix to the turn structure occurs at 0.7 μs without PNIPAM, while with PNIPAM, turn structures predominate until 0.9 μs, followed by a transition to the alpha helix. PNIPAM probably initially stabilizes turn structures through specific interactions, but eventually promotes alpha-helix formation as the simulation progresses, facilitated by the polymer's dynamic interactions. In particular, residues 100–105 exhibit a β-bridge structure without PNIPAM, which is completely absent in the presence of PNIPAM. The polymer likely disrupts beta-sheet interactions by sterically hindering β-bridge formation and altering the local solvent environment, favoring other secondary structures or disordered states by interfering with the necessary backbone hydrogen bonding pattern.
 | ||
| Fig. 5 DSSP analysis of α-syn (a) and α-syn in [α-syn/40mer PNIPAM]1 complex (b), for 1.5 μs. | ||
Overall, it is clear that the presence of PNIPAM promotes alpha-helix formation and alters secondary structure transitions in α-syn by stabilizing specific conformations through hydrophobic interactions, stabilization of hydrogen bonds, steric effects, and changes in the local solvent environment. These interactions reduce conformational entropy and promote the formation of more ordered structures, such as alpha helices, while destabilizing others, such as β-bridges, demonstrating the significant impact of PNIPAM on protein structural dynamics.
3.2. Impact of the length of the PNIPAM polymeric chain on the structural dynamics of α-syn and the protein/polymer complex
To explore the impact of the length of the PNIPAM polymeric chain on the structure of the α-syn protein, we performed additional MD simulations, up to 1.5 μs, of α-syn/PNIPAM systems with varying polymer chain lengths of 10mer, 40mer, 60mer, and 100mer. Fig. S12 (ESI†) illustrates the post-simulation configurations for these complexes. In all cases, the structure of the α-syn/PNIPAM complex was stabilized within time scales of approximately one 0.5 μs.The average RMSD data of α-syn in all complexes with different molecular weights of PNIPAM are shown in Table S3 (ESI†). It is clear from the data shown in Table S3 (ESI†) for the α-syn/10mer PNIPAM complex, there is a subtle decrease in RMSD, hinting at localized structural changes induced by polymer interactions. The limited spatial reach of the 10mer PNIPAM could result in less effective shielding or stabilization of critical regions within the protein, leading to a slightly lower average RMSD. However, the stabilization observed with the 40mer PNIPAM, reflected in a remarkably low RMSD, prompts a deeper exploration into the specific amino acid residues involved. This intriguing finding suggests that the intermediate chain length of 40mer PNIPAM may have an optimal length for interactions with specific regions of α-syn, inducing a pronounced effect on its structural dynamics. Furthermore, the impact of 40mer PNIPAM on protein structural fluctuations and conformational changes underscores the unique and impactful role played by this specific chain length in the complex, highlighting the intricate relationship between polymer chain length and its influence on protein flexibility and structure.
Moreover, in complexes containing 60mer and 100mer PNIPAM polymers, a decrease in the average RMSD values is again observed, whereas no clear and distinct difference is observed in the complex with 40mer PNIPAM polymer. The 60mer polymer chain can introduce steric hindrance or crowding effects that limit its ability to interact effectively with the protein, resulting in a moderate decrease in RMSD compared to the 40mer PNIPAM chain. The increased flexibility observed in the 100mer complex may be attributed to the longer polymer chain providing more binding sites for extensive hydrogen bond formation, potentially influencing the protein's structural fluctuations and local flexibility. The extended structure of these polymers could introduce an increased conformational variability, potentially leading to clashes or steric hindrances with specific protein residues. Furthermore, longer polymers (60mer and 100mer) could exhibit greater conformational rigidity, limiting their ability to dynamically adapt and interact with the flexible regions of α-syn. The intermolecular dynamics at the atomic level may involve intricate yet disruptive interactions, preventing the establishment of a stabilizing influence observed in the 40mer complex.
It is also of interest to examine the modification of the shape of α-syn within the different [α-syn/PNIPAM] complexes. Table S3 (ESI†) shows the calculated average Rg values of α-syn with different polymer chain length complexes. The [α-syn/10mer PNIPAM] complex exhibits an average Rg value close to that of the polymer-free simulation. In contrast, the average Rg values of α-syn increase in the 40 and 60mer PNIPAM complexes, while a decrease is observed in the 100mer PNIPAM complex. As highlighted in the RMSD analysis, the 10mer PNIPAM complex maintains an average Rg value that resembles the isolated protein structure because of its limited interaction region with the polymer. For the 40mer and 60mer complexes, the average Rg values align with the Rg range reported in the literature for α-syn.88
In particular, the 40mer complex exhibits the highest average Rg value (4.47 nm), indicating a configuration relatively similar to the native structure of the protein, as previously discussed. On the contrary, the 60mer complex exhibits a more moderate increase in Rg compared to the 40mer complex, indicating that the intermediate-sized polymer induces structural changes without reaching the extent observed with the larger 40mer complex. The nuanced impact could stem from a balance between long-range polymer-induced unfolding and local stabilization of certain protein regions, possibly through specific secondary structure alterations. The observed decrease in the average Rg in the 100mer polymer complex suggests extensive interactions between the polymer and all regions of the protein. This phenomenon could be attributed to the extensive coverage of the protein surface by the polymer, promoting stabilized interactions across various protein regions. The large polymer can effectively act as a structural scaffold, providing a myriad of interaction sites that enable the protein to fold around it (see Fig. S12, ESI†). This molecular weight-dependent disruption in the native state is consistent with the findings in the literature where polymers with different degrees of hydrophobicity have been shown to induce significant conformational changes in intrinsically disordered proteins.92,93
In summary, the observed differences in average Rg across the varying molecular weights of α-syn/PNIPAM complexes can be attributed to the intricate interaction between molecular size, specific molecular interactions, and the resulting conformational dynamics. Smaller polymers may induce localized structural changes, whereas larger polymers can lead to more extensive disruptions and even a reorganization of the protein's native state. The complex with 40mer PNIPAM was selected for further study due to its optimal stabilizing effect on α-syn, as evidenced by the low RMSD and high Rg values observed in the simulations. The 40mer PNIPAM polymer strikes a balance between effective interaction with α-syn and avoiding steric hindrance or excessive flexibility seen in larger polymers like the 60mer and 100mer. This intermediate chain length provides the most pronounced and reliable stabilization, allowing for a clearer understanding of the impact of polymer size on protein structure.
4. Conclusion
We provide, to the best of our knowledge for the first time in the literature, a detailed investigation of the impact of single-molecule PNIPAM on the formation of monomer α-syn protein and the protein/polymer complexes through detailed atomistic MD simulations. The structural and energetic characteristics of these complexes were thoroughly examined, with a focus on the role of protein mutations, the initial α-syn/PNIPAM configurations, and the molecular weight of the polymer. Analyses of binding free energy and hydrogen bonds revealed a high affinity between PNIPAM and α-syn. Specifically, interactions within the NAC region showed that PNIPAM stabilizes the protein via hydrophobic interactions, as indicated by minimum distance and contact number analyses of hydrophobic and hydrophilic residues. RDF data further supported this observation, highlighting the dominance of apolar–apolar interactions as the primary stabilizing force in the complexes. Structural metrics such as RMSD, Rg, asphericity, and prolateness demonstrated that PNIPAM enhances the protein's stability by maintaining a more compact structure. These effects were observed consistently across several mutant variants of α-syn, all of which exhibited strong affinities for PNIPAM molecules. Non-bonded interaction energy calculations for the mutant α-syn structures, confirmed that van der Waals forces predominantly drive these interactions, particularly within the NAC region where the protein's apolar groups are concentrated, regardless of the presence of mutations. Notably, the E46K mutation not only enhances polymer binding but also stabilizes in the NAC region, which is critical for aggregation. This suggests that such mutations may predispose the protein–polymer complex toward conformational states that reduce aggregation-prone intramolecular contacts.Further investigations into the influence of initial configurations revealed that PNIPAM primarily localizes in the NAC region, as evidenced by visual inspection and Lennard-Jones potential analyses. These findings underscored the importance of PNIPAM in stabilizing protein structure, leading to the selection of the [α-syn/40mer PNIPAM]1 complex for detailed analysis. RMSD and RMSF data for this complex confirmed that PNIPAM restricts protein mobility and enhances stability by maintaining the protein closer to its native unfolded conformation.
The study also examined the impact of polymer chain length, revealing its critical role in complex formation. While the 10mer PNIPAM exhibited limited interaction regions, the 60mer and 100mer chains caused steric effects that disrupted protein folding. In contrast, the 40mer PNIPAM showed a balanced influence, avoiding the limitations of the shorter chain while maintaining structural integrity without the disruptive effects of the longer chains.
These findings deepen our understanding of how polymer characteristics influence the behavior of intrinsically disordered proteins like α-syn and pave the way for targeted applications. Future research will expand to explore the effects of PNIPAM on protein dimers and oligomers through both molecular dynamics simulations and experimental studies. Additionally, the polymer's influence on the membrane-binding capacity of α-syn and its interactions with Parkinson's disease drugs will be investigated. This work holds promise for advancing therapeutic strategies for intrinsically disordered proteins and neurodegenerative diseases.
Data availability
Data for this article, including simulations trajectory are available at CyI Knowledge Bank that is a local repository at The Cyprus Institute, at https://repository.cyi.ac.cy/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement No. 101034267.References
- M. G. Spillantini, M. L. Schmidt, V. M.-Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed.
- D. Twohig and H. M. Nielsen, Mol. Neurodegener., 2019, 14, 23 CrossRef PubMed.
- W. J. Schulz-Schaeffer, Acta Neuropathol., 2010, 120, 131–143 CrossRef CAS PubMed.
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 261–285 CrossRef CAS PubMed.
- E. Luna and K. C. Luk, FEBS Lett., 2015, 589, 3749–3759 CrossRef CAS PubMed.
- S. Mehra, S. Sahay and S. K. Maji, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 890–908 CrossRef CAS PubMed.
- A. Bruning and J. Juckstock, Front. Oncol., 2015, 4, 1–12 Search PubMed.
- C. Hofmann, H. A. Katus and S. Doroudgar, Circulation, 2019, 139, 2085–2088 CrossRef PubMed.
- A. Mukherjee, D. Morales-Scheihing, P. C. Butler and C. Soto, Trends Mol. Med., 2015, 21, 439–449 CrossRef CAS PubMed.
- A. C. Muntau, J. Leandro, M. Staudigl, F. Mayer and S. W. Gersting, J. Inherited Metab. Dis., 2014, 37, 505–523 CrossRef CAS PubMed.
- T. Nakamura, D.-H. Cho and S. A. Lipton, Exp. Neurol., 2012, 238, 12–21 CrossRef CAS PubMed.
- M. B. Tropak, J. E. Blanchard, S. G. Withers, E. D. Brown and D. Mahuran, Chem. Biol., 2007, 14, 153–164 CrossRef CAS PubMed.
- A. Jorge-Finnigan, S. Brasil, J. Underhaug, P. Ruiz-Sala, B. Merinero, R. Banerjee, L. R. Desviat, M. Ugarte, A. Martinez and B. Perez, Hum. Mol. Genet., 2013, 22, 3680–3689 CrossRef CAS PubMed.
- Z. Huang and S. S. J. Leong, J. Biotechnol., 2009, 142, 157–163 CrossRef CAS PubMed.
- Y. Liu, H. J. Busscher, B. Zhao, Y. Li, Z. Zhang, H. C. van der Mei, Y. Ren and L. Shi, ACS Nano, 2016, 10, 4779–4789 Search PubMed.
- L. W. Simpson, G. L. Szeto, H. Boukari, T. A. Good and J. B. Leach, Acta Biomater., 2020, 112, 164–173 Search PubMed.
- M. Lundqvist, I. Sethson and B.-H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- C. M. Wells, M. Harris, L. Choi, V. P. Murali, F. D. Guerra and J. A. Jennings, J. Funct. Biomater., 2019, 10, 34 CrossRef CAS PubMed.
- Y. H. Lim, K. M. Tiemann, D. A. Hunstad, M. Elsabahy and K. L. Wooley, WIREs Nanomed. Nanobiotechnol., 2016, 8, 842–871 CrossRef CAS PubMed.
- B. S. Swami Vetha, A. G. Adam and A. Aileru, Int. J. Mol. Sci., 2021, 22, 5607 CrossRef PubMed.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef PubMed.
- M. J. Ansari, R. R. Rajendran, S. Mohanto, U. Agarwal, K. Panda, K. Dhotre, R. Manne, A. Deepak, A. Zafar, M. Yasir and S. Pramanik, Gels, 2022, 8, 454 CrossRef CAS PubMed.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS PubMed.
- G. Graziano, Int. J. Biol. Macromol., 2000, 27, 89–97 CrossRef CAS PubMed.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- Y. Okada and F. Tanaka, Macromolecules, 2005, 38, 4465–4471 CrossRef CAS.
- H. A. Pérez-Ramírez, C. Haro-Pérez, E. Vázquez-Contreras, J. Klapp, G. Bautista-Carbajal and G. Odriozola, Phys. Chem. Chem. Phys., 2019, 21, 5106–5116 Search PubMed.
- L. Tavagnacco, E. Chiessi and E. Zaccarelli, Phys. Chem. Chem. Phys., 2021, 23, 5984–5991 RSC.
- Y. Yuan, K. Raheja, N. B. Milbrandt, S. Beilharz, S. Tene, S. Oshabaheebwa, U. A. Gurkan, A. C. S. Samia and M. Karayilan, RSC Appl. Polym., 2023, 1, 158–189 RSC.
- G. Pasparakis and C. Tsitsilianis, Polymer, 2020, 211, 123146 CrossRef CAS.
- M. Chernykh, D. Zavalny, V. Sokolova, S. Ponomarenko, S. Prylutska, Y. Kuziv, V. Chumachenko, A. Marynin, N. Kutsevol, M. Epple, U. Ritter, J. Piosik and Y. Prylutskyy, Materials, 2021, 14, 3517 CrossRef CAS PubMed.
- R. Hoogenboom, Smart Polymers and their Applications, Elsevier, 2014, pp. 15–44 Search PubMed.
- F. U. Hartl, A. Bracher and M. Hayer-Hartl, Nature, 2011, 475, 324–332 CrossRef CAS PubMed.
- S. A. Broadley and F. U. Hartl, FEBS Lett., 2009, 583, 2647–2653 CrossRef CAS PubMed.
- G. Paradossi and E. Chiessi, Gels, 2017, 3, 13 CrossRef PubMed.
- C. S. Biswas, K. Mitra, S. Singh, K. Ramesh, N. Misra, B. Maiti, A. K. Panda, P. Maiti, M. Kamigaito, Y. Okamoto and B. Ray, Colloid Polym. Sci., 2015, 293, 1749–1757 CrossRef CAS.
- N. L. Wankhede, M. B. Kale, A. B. Upaganlawar, B. G. Taksande, M. J. Umekar, T. Behl, A. A. H. Abdellatif, P. M. Bhaskaran, S. R. Dachani, A. Sehgal, S. Singh, N. Sharma, H. A. Makeen, M. Albratty, H. G. Dailah, S. Bhatia, A. Al-Harrasi and S. Bungau, Biomed. Pharmacother., 2022, 147, 112647 CrossRef CAS PubMed.
- L. Cortez and V. Sim, Prion, 2014, 8, 197–202 CrossRef CAS PubMed.
- N. Kushwah, V. Jain and D. Yadav, Biomolecules, 2020, 10, 132 CrossRef CAS PubMed.
- F. Chiti and J. W. Kelly, Curr. Opin. Struct. Biol., 2022, 72, 267–278 CrossRef CAS PubMed.
- W. Tu, C. Zheng, Y. Zheng, Z. Feng, H. Lin, Y. Jiang, W. Chen, Y. Chen, Y. Lee, J. Su and W. Zheng, Int. J. Biol. Macromol., 2023, 249, 125702 CrossRef CAS PubMed.
- A. Baruah and P. Biswas, Phys. Chem. Chem. Phys., 2014, 16, 13964–13973 RSC.
- P. C. A. Van Der Wel, Solid State Nucl. Magn. Reson., 2017, 88, 1–14 CrossRef CAS PubMed.
- T. R. Alderson and L. E. Kay, Curr. Opin. Struct. Biol., 2020, 60, 39–49 CrossRef CAS PubMed.
- K. Wu, S. Horowitz and J. C. A. Bardwell, in Biophysics of Molecular Chaperones, ed. S. Hiller, M. Liu and L. He, Royal Society of Chemistry, 2023, pp. 199–216 Search PubMed.
- S. S. Stadmiller and G. J. Pielak, Curr. Opin. Struct. Biol., 2021, 66, 183–192 CrossRef CAS PubMed.
- C. A. Stevens, K. Kaur and H.-A. Klok, Adv. Drug Delivery Rev., 2021, 174, 447–460 Search PubMed.
- A. K. Umar, N. Wathoni, J. H. Zothantluanga, S. Das and J. A. Luckanagul, Heliyon, 2022, 8, e08934 CrossRef PubMed.
- Y. K. Sung, D. R. Lee and D. J. Chung, Biomater. Res., 2021, 25, 37 Search PubMed.
- S. Acosta, L. Quintanilla-Sierra, L. Mbundi, V. Reboto and J. C. Rodríguez-Cabello, Adv. Funct. Mater., 2020, 30, 1909050 Search PubMed.
- H. Wu, H. Yao, C. He, Y. Jia, Z. Zhu, S. Xu, D. Li and J. Xu, Acta Pharm. Sin. B, 2022, 12, 3548–3566 Search PubMed.
- S. A. Hollingsworth and R. O. Dror, Neuron, 2018, 99, 1129–1143 CrossRef CAS PubMed.
- Y. Zhuang, H. R. Bureau, S. Quirk and R. Hernandez, Mol. Simul., 2021, 47, 408–419 CrossRef CAS.
- M. Arnittali, A. N. Rissanou and V. Harmandaris, Procedia Comput. Sci., 2019, 156, 69–78 CrossRef.
- R. Ramis, J. Ortega-Castro, B. Vilanova, M. Adrover and J. Frau, Int. J. Biol. Macromol., 2021, 169, 251–263 CrossRef CAS PubMed.
- H. Wille, L. Dorosh, S. Amidian, G. Schmitt-Ulms and M. Stepanova, Advances in Protein Chemistry and Structural Biology, Elsevier, 2019, vol. 118, pp. 33–110 Search PubMed.
- C. Lee and S. Ham, J. Comput. Chem., 2011, 32, 349–355 CrossRef CAS PubMed.
- A. Rahman, B. Saikia, C. R. Gogoi and A. Baruah, Prog. Biophys. Mol. Biol., 2022, 175, 31–48 CrossRef CAS PubMed.
- M. W. van der Kamp and V. Daggett, in Prion Proteins, ed. J. Tatzelt, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, vol. 305, pp. 169–197 Search PubMed.
- Z. Khatooni, K. Akhtari and H. L. Wilson, Sci. Rep., 2023, 13, 19020 Search PubMed.
- C. Kang and R. Sun, J. Phys. Chem. B, 2021, 125, 1036–1048 CrossRef CAS PubMed.
- P. Robustelli, A. Ibanez-de-Opakua, C. Campbell-Bezat, F. Giordanetto, S. Becker, M. Zweckstetter, A. C. Pan and D. E. Shaw, J. Am. Chem. Soc., 2022, 144, 2501–2510 CrossRef CAS PubMed.
- B. Bu, X. Tong, D. Li, Y. Hu, W. He, C. Zhao, R. Hu, X. Li, Y. Shao, C. Liu, Q. Zhao, B. Ji and J. Diao, ACS Chem. Neurosci., 2017, 8, 2145–2151 Search PubMed.
- F. Meshkini, A. Moradi and S. Hosseinkhani, Int. J. Biol. Macromol., 2023, 230, 123216 CrossRef CAS PubMed.
- A. B. Uceda, J. Frau, B. Vilanova and M. Adrover, Int. J. Biol. Macromol., 2023, 229, 92–104 CrossRef CAS PubMed.
- O. K. Mankoo, A. Kaur, D. Goyal and B. Goyal, Phys. Chem. Chem. Phys., 2023, 25, 8128–8143 RSC.
- A. M. Barclay, D. D. Dhavale, J. M. Courtney, P. T. Kotzbauer and C. M. Rienstra, Biomol. NMR Assignments, 2018, 12, 195–199 CrossRef CAS PubMed.
- A. B. Johnson, N. Modi, K. N. Kamath, R. Rajeshkumar and V. R. Palanimuthu, Alpha-Synuclein Aggregation and Spread: Implications for the Pathology and Progression of Neurodegenerative Diseases, 2023, preprint DOI:10.20944/preprints202312.0569.v1.
- R. Guerrero-Ferreira, L. Kovacik, D. Ni and H. Stahlberg, Curr. Opin. Neurobiol., 2020, 61, 89–95 Search PubMed.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595–9603 CrossRef CAS PubMed.
- E. Jakova, M. T. Moutaoufik, J. S. Lee, M. Babu and F. S. Cayabyab, Transl. Neurodegener., 2022, 11, 9 Search PubMed.
- A. N. R. Da Silva, G. R. C. Pereira, L. F. S. Bonet, T. F. Outeiro and J. F. De Mesquita, J. Cell. Biochem., 2024, 125, e30523 CrossRef CAS PubMed.
- G. Bagree, T. Srivastava, S. Mahasivam, M. Sinha, V. Bansal, R. Ramanathan, S. Priya and S. K. Sharma, J. Biochem., 2023, 173, 107–114 CrossRef CAS PubMed.
- Y. K. Choi, S.-J. Park, S. Park, S. Kim, N. R. Kern, J. Lee and W. Im, J. Chem. Theory Comput., 2021, 17, 2431–2443 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 Search PubMed.
- L. Xu, B. Ma, R. Nussinov and D. Thompson, ACS Chem. Neurosci., 2017, 8, 837–849 CrossRef CAS PubMed.
- T. Zhang, Y. Tian, Z. Li, S. Liu, X. Hu, Z. Yang, X. Ling, S. Liu and J. Zhang, J. Chem. Inf. Model., 2017, 57, 2281–2293 CrossRef CAS PubMed.
- H. Alıcı, J. Mol. Model., 2020, 26, 132 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. Burré, M. Sharma and T. C. Südhof, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4274–83 CrossRef PubMed.
- T. Bartels, L. S. Ahlstrom, A. Leftin, F. Kamp, C. Haass, M. F. Brown and K. Beyer, Biophys. J., 2010, 99, 2116–2124 CrossRef CAS PubMed.
- D. Dibenedetto, G. Rossetti, R. Caliandro and P. Carloni, Biochemistry, 2013, 52, 6672–6683 CrossRef CAS PubMed.
- R. Ramis, J. Ortega-Castro, R. Casasnovas, L. Mariño, B. Vilanova, M. Adrover and J. Frau, J. Chem. Inf. Model., 2019, 59, 1458–1471 CrossRef CAS PubMed.
- K. B. Pedersen, J. C. Flores-Canales and B. Schiøtt, Proteins, 2023, 91, 47–61 CrossRef CAS PubMed.
- R. Klement, T. Graen, A. Grupi, E. Haas and H. Grubmüller, Biophys. J., 2016, 110, 551a CrossRef.
- L. Breydo, A. E. Sales, T. Frege, M. C. Howell, B. Y. Zaslavsky and V. N. Uversky, Biochemistry, 2015, 54, 2957–2966 Search PubMed.
- H. Zhao, E. Ibarboure, V. Ibrahimova, Y. Xiao, E. Garanger and S. Lecommandoux, Adv. Sci., 2021, 8, 2102508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01395f |
| This journal is © The Royal Society of Chemistry 2025 |