
Recent advances in atomically dispersed M–N–C coupled Pt-based oxygen reduction catalysts
Zigang
Zhao
ab,
Lezhi
Zhan
b,
Pan
Guo
 b,
Yunkun
Dai
b,
Lixiao
Shen
*c,
Yunlong
Zhang
*b,
Guiling
Wang
*a,
Zhenbo
Wang
*bc and
Lei
Zhao
*b
b,
Yunkun
Dai
b,
Lixiao
Shen
*c,
Yunlong
Zhang
*b,
Guiling
Wang
*a,
Zhenbo
Wang
*bc and
Lei
Zhao
*b
aKey Laboratory of Superlight Materials and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, Heilongjiang, China
bState Key Laboratory of Space Power-Sources, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China
cShenzhen Key Laboratory of Special Functional Materials, Shenzhen Engineering Laboratory for Advance Technology of Ceramics, Guangdong Research Center for Interfacial Engineering of Functional Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, P. R. China
First published on 19th November 2024
Abstract
Proton exchange membrane fuel cells have garnered significant attention as a sustainable energy conversion technology amidst the escalating consumption of fossil fuels. Although Pt-based catalysts are effective in oxygen reduction reactions, their limited availability and high Pt content pose challenges to the wide adoption of PEMFCs. Improving the activity and durability of Pt-based catalysts is essential for lowering Pt consumption, cutting costs, and increasing the fuel cell's efficiency and power density. Recently, atomically dispersed metal–nitrogen–carbon (M–NC) coupled platinum-based catalysts have received attention as highly promising options due to their superior performance and stability. This review explores the advancements in M–NC coupled platinum-based catalysts, encompassing various supports, alloys, and intermetallic compounds. The optimization strategies for these catalysts, spanning preparation methods, structural composition, and catalytic efficacy, are also discussed. In addition, this review discusses the comprehensive optimization strategy of the M–NC coupled platinum-based oxygen reduction catalyst, focusing on various aspects such as the preparation process, structural composition, and catalytic performance. Additionally, we offer insights into the future advancement of M–NC coupled platinum-based oxygen reduction catalysts, emphasizing this method as a potential avenue to enhance efficiency.
 Zigang Zhao | Zigang Zhao is currently a PhD candidate at the College of Materials Science and Chemical Engineering, Harbin Engineering University. His primary research focuses on the development of noble metal catalysts for fuel cells, with an emphasis on achieving high activity and stability. |
 Lezhi Zhan | Lezhi Zhan is currently a graduate student at the Department of Chemistry, University College London (UCL). Her research focuses on the development of electrochemical acoustic transducers. Working within an interdisciplinary framework, her research aligns with innovations in the field of electrochemistry, particularly in the context of gas-phase electrolytes and advanced sensing technologies. |
 Lei Zhao | Lei Zhao is currently an associate professor/doctoral supervisor in the Department of Electrochemical Engineering at the School of Chemical Engineering and Chemistry, and was selected for the Youth Top notch Talent Selection Program at Harbin Institute of Technology in 2021. His research focuses on chemical power sources, electrocatalysis, and nanoelectrode materials. He has conducted systematic and in-depth research on key technologies for high-performance lithium/sodium ion cells, low-temperature fuel cells, metal air cells, and electrocatalytic energy conversion. |
1 Introduction
PEMFCs have received attention as an emerging clean energy source in response to the continued degradation of the environment and the exhaustion of conventional fossil fuels.1–3 PEMFCs are widely valued for their efficient energy conversion, high power density, reliability, and eco-friendliness.4–6 The core structure of PEMFCs comprises an anode material, electrolyte material, and cathode material.7 The anode facilitates the HOR of H2, while the cathode enables the ORR of O2. In comparison to the HOR at the anode, the high bond strength of O–O within the oxygen molecule (494 kJ mol−1) leads to the sluggish kinetics of the ORR, as the oxygen-containing species formed during the reaction are not easily removed from the active site.8,9 The slow kinetics of the ORR, exacerbated by the substantial cathode overpotential and intricate reaction mechanisms, is a primary impediment to the advancement of fuel cell technology. Pt-based catalysts are crucial in PEMFCs, impacting the cost-effectiveness, energy conversion effectiveness, and durability of fuel cells.6,10 However, the sluggish ORR kinetics hampers the overall efficiency of fuel cell batteries.11 Moreover, the exorbitant cost and poor endurance of cathode catalyst Pt/C have significantly constrained the progress and utilization of fuel cells. Thus, the imperative task of developing ORR catalysts with enhanced performance is essential to enhance cathode ORR efficacy and accelerate the commercialization of PEMFCs.12–14ORR catalysts are divided into non-metallic catalysts, non-precious metal nitrogen–carbon, and Pt-based catalysts. Non-metallic catalysts typically consist of carbon materials doped with heteroatoms.15 To enhance the activity of non-metallic catalysts, N and S are usually introduced to increase the number of active sites, thereby obtaining high-performance ORR catalysts.16,17 Lv et al.18 reported a hydrogen-substituted graphene non-metallic catalyst doped with pyridinic nitrogen, which exhibits oxygen reduction performance comparable to that of Pt-based catalysts in acidic media. Non-precious catalysts are extensively used because of their comparatively lower cost and different valuations.19 The primary focus for ORR catalysts is on non-precious metals M–NC (M = Fe, Co, Ni, Mn, etc.). Research has shown that M–NC catalysts exhibit commendable catalytic performance, being a promising replacement for Pt.20,21 Yuan et al.22 constructed a Cyan-Fe–N–C catalyst with pyrrolic Fe–N4 as the constitutive active center using axial Fe4C atomic clusters. The catalyst exhibits a half-wave potential of up to 0.836 V (vs. RHE, the same below) in acidic environments, which is close to that of Pt/C. Nonetheless, M–NC materials encounter challenges such as metal leaching, anion binding to the active site, and protonation under acidic conditions.23 A Pt-based catalyst consists of Pt or a Pt alloy as the active component, offering benefits such as high activity, robust anti-poisoning capability, low-temperature and low-pressure resistance, and high efficiency.24,25 The straightforward adsorption and dissociation mechanisms of Pt make it the most efficient ORR catalyst. The increased cost and poor durability of Pt catalysts pose major challenges to the commercial advancement of PEMFCs. Therefore, the current research direction mainly focuses on reducing the amount of platinum and improving the catalyst performance. Using M–NC (M = Fe,14 Co,26–29 Ni,30etc.) materials as the support instead of ordinary carbon is a reliable approach to enhance the ORR performance of Pt catalysts. Incorporating a transition metal into the support material to form M–NC with a unique structure allows the M atoms to electronically regulate Pt, reducing the binding energy between Pt and oxygen intermediates, and thus improving the catalyst's ORR performance.31 Additionally, incorporating M atoms into Pt forms a Pt–M alloy structure, including disordered solid solution alloy and ordered intermetallic compound structures. The various effects (ligand effect, strain effect, geometric effect, etc.) generated by the Pt-M and M–NC combination can enhance both the performance and durability of the catalyst.32–35
We conducted a comprehensive review of the progress of M–NC coupled Pt-based oxygen reduction catalysts, emphasizing the principles for enhancing catalyst ORR activity and stability. As shown in Fig. 1, we summarized the catalysts of M–NC-supported Pt, Pt–M alloys, and Pt–M intermetallic compounds, and provided a detailed overview of strategies for optimizing M–NC coupled Pt-based ORR catalysts from various perspectives. Additionally, we outlined the current challenges and future prospects for M–NC coupled Pt-based oxygen reduction catalysts, aiming to contribute to the development of innovative research methodologies in this field.
 | ||
| Fig. 1 Schematic diagram of argument structure. | ||
2 ORR theory and limitations of Pt-based catalysts
The ORR is a complex multi-electron process within basic electrochemical reactions. In comparison to the HOR, the ORR results in the generation of various oxygen-containing intermediates, increasing the complexity of the electron transfer process. The sluggish kinetics and high energy barrier of the ORR pose a substantial challenge, necessitating the use of numerous highly active catalysts to achieve a satisfactory reaction rate. Therefore, a comprehensive understanding of the ORR mechanism, key steps determining reaction rates, and the structure–activity relationship of catalysts are crucial for the development of ORR catalysts. This knowledge will help us better optimize the structure and composition of catalysts, thereby enhancing their ORR performance.2.1 ORR theory
In the cathode of PEMFCs, the electrochemical reduction of oxygen occurs via ORR electrocatalysis. This primarily involves the four-electron reduction pathway that converts O2 into H2O and the two-electron path that produces H2O2.36–38 The four-electron path is more efficient for completely reducing O2 to H2O, resulting in higher output voltage, making it the preferred reaction route for the ORR. Key intermediates in this electrocatalytic process include O*, OH*, and OOH*, whose interconversion adds complexity to the ORR.39 Thus, an effective catalyst is crucial for optimizing the ORR.As shown in Fig. 2a and b, under acidic conditions, the adsorbed O2 molecule acquires an electron from the solution, resulting in its protonation and the creation of OOH*(1). Subsequently, two potential reaction pathways may occur.40
 | ||
| Fig. 2 The ORR: 4e-processes (a) and 2e-processes (b) (reproduced from ref. 40 with permission from Elsevier, copyright 2023). The Pt/C catalyst: aggregation, abscission of platinum particles, and carbon corrosion (c, and d). The PtM alloy: M-atom dissolution (e) (reproduced from ref. 41 with permission from Elsevier, copyright 2024). | ||
(A) In the first mechanism, a 4-electron reduction reaction takes place where H+ interacts with oxygen atoms, breaking the OOH* bond and forming OH* and H2O (2), which then receives an electron to become H2O.
(B) Alternatively, in the 2-electron reduction reaction mechanism, OOH* captures an electron combined with H+ to produce H2O2, which can either desorb directly from the catalyst and dissolve in the solution or undergo a 2-electron process to form water H2O, resembling a (2 + 2) electron reaction process (4).
2.2 Limitations of Pt-based catalysts
In the initial phases of fuel cell evolution, Pt NPs served as the exclusive catalysts for the ORR without any supporting materials. These Pt NPs showed a tendency to agglomerate, adversely affecting their catalytic properties. Consequently, substantial amounts of platinum were necessary to fulfill the ORR requirements of fuel cells.42 Researchers determined that increasing platinum loading could significantly enhance catalytic performance, yet efficiency remained critically dependent on the platinum quantity employed; only catalysts with adequate platinum loading demonstrated optimal properties.43Currently, only platinum–carbon meets the criteria for an ideal ORR process.44 Despite extensive research and refinements enhancing its catalytic activity, the Pt/C catalyst continues to encounter challenges, including platinum particle decomposition and agglomeration, Ostwald ripening, and the corrosion of the support material (Fig. 2c and d).28,45 To mitigate these issues, researchers focus on producing finer, uniform platinum nanoparticles and enhancing strong metal–support interactions (SMSI) through carbon support treatments, ensuring stable platinum anchoring. Introducing nitrogen (N), phosphorus (P), and sulfur (S) into carbon materials helps to anchor Pt.46–48 For example, Qiao et al.49 demonstrated the influence of N doping on the interaction between Pt and the carbon support through DFT calculations. The results indicate that bridge sites near the N dopant are more attractive to Pt atoms than the original graphene layer. This finding proves that N doping in carbon supports can improve the stability of Pt/C catalysts by strengthening the binding of Pt particles to the support. Niu et al.50 designed a catalyst with Pt supported on a carbon material co-doped with N and P (PNC). In this structure, the doping of P atoms further regulates the electronic environment around Pt, allowing Pt NPs to anchor more stably on N atoms, thus suppressing the leaching or agglomeration of Pt particles and enhancing the stability of the catalyst. Additionally, Chen et al.51 synthesized a Pt NW/SGs catalyst by loading Pt onto a S-doped graphene material. The S atoms in the supporting material provide strong anchoring points for Pt, enhance the interaction between the support and the metal, and improve the stability of the catalyst. Pretreating the carbon support to enrich it with oxidative functional groups enhances platinum dispersion during the support process. However, one of the key obstacles to the commercial use of platinum–carbon catalysts is the limited availability and high expense of platinum resources, which hinder mass production efforts.52–55 Consequently, minimizing platinum usage while enhancing catalyst performance is crucial.56 Developing Pt-based alloy catalysts by combining platinum with non-precious metals may address this issue. However, the facile leaching of non-precious metal atoms from platinum alloys under acidic conditions represents a major challenge. Therefore, further research and optimization of platinum alloys are essential (Fig. 2e).41 For example, Zhang et al.12 found that the D-Pt3Mn/rGO-HF catalyst sample was immersed in a 0.1 M HClO4 solution for testing. The leaching amount of Mn in acidic electrolytes was determined by ICP-AES, and after 10 days, the Mn leaching loss of the D-Pt3Mn/rGO-HF catalyst was 32.2%. This indicates that Pt alloy catalysts need further research and optimization. In summary, although Pt/C catalysts exhibit excellent ORR activity, their catalytic stability and the scarcity of platinum limit their widespread application.11 Therefore, reducing costs and boosting ORR performance have emerged as critical objectives in the commercial advancement of Pt-based catalysts.
3 M–NC@Pt catalysts
With the prevalent use of platinum carbon in PEMFCs, extensive studies aimed at reducing platinum consumption in PEMFCs have been conducted to boost utilization efficiency.57 A remarkable advancement involved successfully minimizing platinum loading several times by distributing ultra-fine platinum nanoparticles onto a high-surface-area carbon support, decreasing it from 4 mg cm−2 to 0.3 mg cm−2.58 However, decreasing particle size complicates synthesis, leading to increased production costs. Ongoing efforts focus on reducing platinum loading in Pt-based catalysts on the cathode while maintaining ORR efficiency.59 Enhancing the ORR activity of the carbon support presents an effective method to improve catalyst performance. Nevertheless, traditional carbon supports mainly facilitate the dispersion of platinum nanoparticles and act as electron conduction pathways.60 Commonly utilized commercial carbon materials like XC-72 exhibit minimal ORR catalytic activity in acidic environments.61,623.1 Synergistic effect
Recent studies reveal that by meticulously altering the surface characteristics of carbon and doping with nitrogen along with a small number of transition metals, specifically M–NC, one can achieve significant ORR performance akin to that of platinum on carbon. For example, Liu et al.63 proposed a method for customizing iron-doped zeolite imidazolate framework-8 (ZIF-8) at the atomic scale to construct multidimensional concave surface Fe@NC catalyst structure. The electrochemical evaluation shows that the E1/2 of the Fe@NC catalyst is very similar to that of Pt/C in acidic environments, exhibiting comparable catalytic performance. Based on the better ORR performance of Fe–N–C, some researchers substituted conventional carbon supports with an atomically distributed Fe–N–C structure to host platinum nanoparticles, resulting in a novel Pt/Fe–N–C catalyst configuration. This support, abundant in iron–nitrogen (Fe–Nx) sites, facilitates synergistic catalysis alongside platinum nanoparticles.64,65 For instance, Park et al.66 utilized 50/500 nm silica beads and phthalocyanines as doping agents and carbon sources. They employed a template approach to create bimodal porous crystalline structures that served as catalyst supports, followed by the even deposition of platinum nanoparticles on the FeNC framework utilizing electron beam radiation, yielding a Pt catalyst (5-Pt/FeNC) with a 5 wt% loading capacity. The FeNC structure influences the electronic properties of platinum through electron transfer effects. Furthermore, the Fe–N4 sites in the FeNC enhance catalytic activity by generating additional active sites, leading to improved ORR performance. In electrochemical assessments, the 5-Pt/FeNC showed an ORR mass activity of 2.58 A mgPt−1, surpassing that of 20% Pt/C catalysts. Additionally, the mass activity of Pt/FeNC reached 18.76 A mgPt−1 at 0.6 V, again exceeding that of commercial Pt/C. Moreover, Fe–NC can exhibit a synergistic catalytic effect with Pt single-atom sites,67 thus increasing the ORR performance of the catalyst. For instance, Sha et al.68 introduced a Pt1.1FeNC catalyst featuring distributed active sites from both Pt and Fe atoms for the first time. This catalyst, synthesized through a two-step process, first involved combining Fe ions with Zn ions to create an Fe-doped ZIF precursor, followed by heat treatment to form the dispersed active sites of FeNC under a nitrogen atmosphere. The FeNC was subsequently utilized to adsorb Pt ions, successfully synthesizing the Pt1.1FeNC catalyst. The atomic dispersion of Pt on the Fe-doped ZIF was verified using HAADF-STEM. Due to the interaction between Pt and Fe, the atomically dispersed Pt1.1FeNC catalyst exhibited superior catalytic performance and durability. In a 0.1 M HClO4 environment, the catalyst achieved an E1/2 of 0.85 V and an H2O2 yield below 1%. This research offers a prospective orientation to the innovation of high-activity bimetallic catalysts, addressing the inherent challenges associated with Pt and Fe-based ORR catalysts.Research has found that combining M with ordinary carbon carriers can improve the ORR activity of ordinary carbon supports (such as graphene), and has good adaptability of ordinary carbon carriers to Pt NPs.69 Gao et al.70 developed Fe–N-HG porous graphene supports by etching graphite oxide with H2O2 and doping with Fe/N. They subsequently deposited Pt nanoparticles on these supports using ethylene glycol reduction, resulting in Pt/Fe–N-HG (Fig. 3a). The incorporation of N and Fe atoms in Fe–N-HG introduces additional defects and active sites, which act as nucleation sites for Pt effectively securing the Pt nanoparticles, and ensuring a uniform distribution (Fig. 3b). Additionally, Fe–N–C increases the interaction between Pt and the support, optimizes electron transfer, and offers extra active sites for the ORR, thus increasing the catalytic efficiency of Pt/Fe–N-HG. The special porous structure of graphene in Pt/Fe–N-HG increases the overall surface area and facilitates O2 transport in the catalytic layer. The synergistic interactions among Pt, Fe, and porous graphene supports greatly enhance the performance of the Pt/Fe–N-HG. The onset and E1/2 for Pt/Fe–N-HG are recorded at 1.01 V and 0.88 V, respectively, surpassing those of Pt/HC (0.98 V and 0.83 V, respectively), Pt/N-graphene (1.00 V and 0.82 V, respectively), Pt/HG (0.91 V and 0.66 V, respectively), and Pt/C (0.84 V and 0.55 V, respectively).70 In single-cell evaluations, the Pt/Fe–N-HG has a current density of 280 mA cm−2 and a peak power density of 310 mW cm−2 at 0.6 V under backpressure-free conditions at 80 °C. Furthermore, the Pt/Fe–N-HG outperforms Pt/C at increased current densities. In a different study, K. Kuttiyiel et al.73 integrated Fe porphyrins into graphene, which subsequently underwent pyrolysis to yield FeNxCy. They fabricated the Pt–FeNC Janus structure catalyst by depositing Pt nanoparticles onto FeNxCy particles using pulse deposition techniques. The Pt nanoparticles and FeNC exhibit a synergistic catalytic interaction within the Janus framework, enhancing both the overall ORR activity and the longevity of the catalyst. The Pt–FeNC catalysts demonstrate a remarkable initial Pt mass activity of 0.69 A mgPt−1, nearly threefold greater than that of standard commercial Pt/C catalysts.
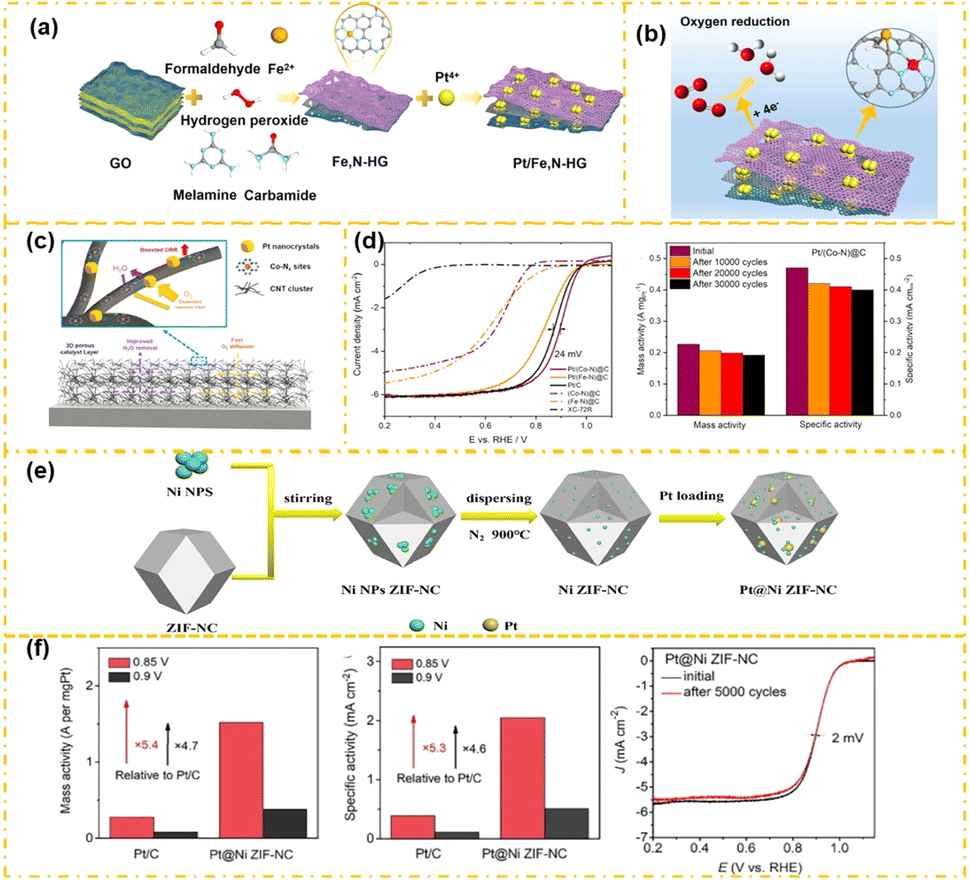 | ||
| Fig. 3 Preparation of the Pt/Fe–N-HG catalyst (a) and ORR synergistic catalytic process (b) of the Pt/Fe–N-HG catalyst (reproduced from ref. 70 with permission from ACS, copyright 2023). The ORR synergistic catalytic process (c) and ORR performance (d) of the Pt/Co–N–C catalyst (reproduced from ref. 71 with permission from Elsevier, copyright 2020). Preparation of Pt@NiNC (e) and ORR performance (f) of the Pt@NiNC catalyst (reproduced from ref. 72 with permission from Elsevier, copyright 2022). | ||
Research indicates that Co–N–C structures uniformly distributed at the atomic level demonstrate high ORR activity.74 For example, Qu et al.75 achieved dense deformation of Co–N4 sites anchored on in situ grown CNTs during the synthesis of Co–N/C catalysts by precisely adjusting the ratio of reactants in the metal–organic framework precursor. Thanks to the unique structure of the dense Co–N4 sites anchored on CNTs, the Co–N/C catalyst exhibits ORR (E1/2: 0.781 V, kinetics) performance that shows high activity. Therefore, some researchers have used atomically dispersed Co–N–C structures instead of traditional carbon materials as carriers of Pt nanoparticles.76 Bin Hu et al.71 substituted the conventional inert XC-72 carbon support with the more reactive Co–N@C. They then deposited Pt on the Co–N@C support using glycol as both a heating and reducing agent to create the Pt/Co–N@C (Fig. 3c). In this configuration, the synergistic catalysis between Pt and Co–N@C leads to exceptional ORR activity, as evidenced in the half-cell test (Fig. 3d). The mass activity was measured to be 0.227 A mgPt−1, which is double that of Pt/C at 0.105 A mgPt−1.
Additionally, in PEMFCs, the MEA of the Pt/Co–N@C cathode can achieve a power density of 1.06 W cm−2, which is 1.6 times more than that of an industrial Pt/C (JM20%) catalyst. Utilizing a microwave-assisted heating method, Guo et al.77 engineered a low-platinum hybrid catalyst characterized by small platinum nanoparticles integrated with cobalt–nitrogen (Co–Nx) groups. The Co–Nx site within Co–N–C exhibits significant catalytic properties, enabling the platinum active center to catalyze the ORR by participating actively in the ORR mechanism. Additionally, the spillover effect from Co–N–C to platinum enhances the 4-electron process, consequently improving the kinetics of the ORR. Therefore, the ORR efficiency of the Pt/Co–N–C catalyst experienced considerable enhancement.
Various research efforts have demonstrated that defect engineering can alter the bonding configuration of metal atoms within the Co–NC framework, adjust the corresponding electron redistribution during electrocatalysis, and consequently improve the catalytic capability of the catalyst. Zhang et al.78 employed an electrochemical activation technique to extract cobalt (Co) nuclei from a stable Co/C core–shell configuration. This approach could yield nitrogen-doped defective carbon that contains atomic metal Co. During activation, amorphous carbon is removed, creating channels in the graphite carbon shell that allow acidic solvents to penetrate it. Consequently, Co atoms become encapsulated within the carbon shell, leading to the gradual elimination of Co particles. Furthermore, the introduction of a platinum (Pt) counter-electrode during activation causes Pt atoms to dissolve in the electrolyte and become simultaneously trapped by the NC shell. This leads to the atomic-scale synthesis of Co/Pt-modified catalysts. A-CoPt–NC comprises a minimal quantity of Co (∼1.72 wt%) and an even lesser amount of Pt (∼0.16 wt%), and yet demonstrates outstanding ORR activity. Under alkaline conditions, the ORR mass activity surpasses that of commercial Pt/C by 267 times, while also maintaining significant ORR activity in acidic environments. DFT computations imply that the uneven electron distribution around the Pt/Co metal center, along with the bonding of metal atoms with the carbon surface's local environment—stemming from the N8V4 vacancy in the carbon shell—contributes to its high ORR activity. The existence of Pt–Co atoms on the surface of defective NC induces a synergistic catalytic effect. The atomic interactions between Pt and Co enhance the selectivity for the 4-electron pathway in the ORR, thus bolstering catalytic performance. Thus, optimizing the metal atom coordination environment through Co–NC defect engineering is crucial for boosting the electrocatalytic activity of the ORR.
In addition, some nickel–nitrogen–carbon (Ni–NC) materials are also used as carriers for Pt catalysts. For instance, Liang et al.72 effectively distributed nickel zeolitic imidazolate framework–nitrogen–carbon (Ni ZIF–NC), abundant in Ni–NC active sites, on a carbon substrate, successfully creating the Pt@Ni ZIF–NC catalyst through a dip-coating procedure (Fig. 3e). In this configuration, Ni–NC demonstrates notable catalytic capability, due to the active site of Pt@Ni ZIF–NC being raised, resulting in improved ORR catalytic performance while minimizing platinum loading. Under acidic conditions (Fig. 3f), the E1/2 potential of Pt@Ni ZIF–NC reaches 0.902 V, surpassing the 0.861 V achieved by Pt/C. Its MA attains 0.38 A mgPt−1 at 0.9 V, which is 4.7 times greater than that of Pt/C (0.08 A mgPt−1). Similarly, Wang et al.79 developed a low-loading platinum-based electrocatalyst that features platinum nanoparticles on Ni–N–C through an electrochemical substitution reaction (8.0 wt% Pt@NiNC). Due to the synergistic interactions of the nanoscale and atomic binding active sites, Pt@NiNC manifests enhanced MA compared to Pt/C. At 0.90/0.85 V, Pt@NiNC registered an initial MA of 28.0/156.0 mA mgPt−1, which is 6.5/6.2 times higher than that of Pt/C, respectively.
3.2 Strong metal–support interaction
Minimizing the cost of PEMFCs and making them suitable for diverse applications, especially by reducing platinum (Pt) consumption and improving stability, continues to be a major challenge. The primary cause of instability in Pt-based catalysts is the clustering of Pt nanoparticles and the degradation of the supporting materials. To address this, researchers have explored a range of support materials to enhance catalyst stability. Among these, modified carbon materials have demonstrated the greatest potential, owing to their large surface area, adjustable surface characteristics, customizable porous structure, and excellent electronic conductivity. Evidence indicates that Pt nanoparticles can be evenly distributed on carbon materials through modification of surface functional groups or heteroatom enhancements, leading to improved stability. For instance, Wang et al.79 described the benefits of using a N-MCF to enhance the catalytic proficiency of Pt-based catalysts in the ORR. The N-MCF, produced via ZIF pyrolysis, offers numerous edges, defects, and heteroatom doping sites that anchor Pt effectively, promoting robust Pt–metal–support interactions and optimal dispersion. At a potential of 0.90 V, electrochemical studies show that the MA and SA of Pt/N-MCF reach 0.246 A mgPt−1 and 0.276 mA cm−2, respectively.Additionally, the Pt utilization rate of Pt/N-MCF (186 mgPt kW−1) is 1.9 times greater than that of fuel cells utilizing standard Pt/C cathodes. After undergoing an ADT, the E1/2 of Pt catalysts supported by N-MCF dropped by just 8 mV, highlighting that the interaction between Pt and its supporting material is pivotal in managing catalyst degradation. Additionally, the lone pair electrons in NC strengthen the Pt–N interaction, which in turn enhances the stability. However, the limitations of functional groups in these carbon materials lead to inadequate Pt–support interactions, failing to satisfy the high stability demands of Pt-based catalysts.
To further enhance catalyst stability, numerous studies have been conducted. It has been found that doping nonprecious metal elements into an N-doped carbon carrier generates a strong metal-scaffold interaction (SMSI) with Pt. Guo et al.77 reported a Pt/Co–NC catalyst supported on Co–NC, in which the Co–Nx group is also an effective active center synergistically catalyzing with the Pt site. Specifically, in addition to being reduced to H2O at the Pt site, O2 is converted to OOH* on Co–Nx, forming O* and OH*, which then combine with H+ to generate H2O. The OOH* formed on Co–Nx in this structure is easily converted to H2O2. DFT calculations show that when Co–Nx generates H2O2, it quickly overflows to nearby Pt sites through the overflow effect, and then reduces to H2O, thereby enhancing the kinetics of the oxygen reduction reaction. In addition, Co–Nx as an electron donor can regulate the electronic structure of Pt through electronic effects, promote the downward shift of Pt's d-band center, optimize the adsorption energy between Pt and oxygen intermediates, and enhance the intrinsic activity of Pt. Meanwhile, due to the strong metal carrier interaction between Pt and the Co–NC support, it is limited. Small Pt NPs are firmly anchored and uniformly dispersed on Co–NC, greatly enhancing the activity and stability of the ORR. The performance of a catalyst with a low Pt loading can be improved while the Pt loading in a Pt-based catalyst is reduced. Xiao et al.80 successfully synthesized a Pt/Fe–N–C catalyst by depositing Pt nanoparticles onto an Fe–N–C support derived from ZIF-8. In situ ICP-MS analysis reveals that the dissolution rate of Pt/Fe–N–C was three times lower than that of Pt/C during cycling. As shown in Fig. 4a, the influence of the Fe–N–C carrier on the structure and properties of Pt is studied through DFT calculations, and compared with that of N–C and C carriers. The results show that the binding energy between Pt/Fe–N–C and the carrier is the lowest (−4.60 eV), followed by Pt/N–C (−3.86 eV) and Pt/C (−2.84 eV), indicating that Fe–N–C has the strongest supporting effect on the carrier.80 The ability to generate Pt oxides on Pt/Fe–N–C, Pt/N–C, and Pt/C was evaluated by assessing the adsorption strength of atomic oxygen (O*) on the catalyst surface. The results show that Pt/Fe–N–C has the weakest adsorption of O* (0.98 eV), followed by Pt/N–C (0.85 eV) and Pt/C (0.81 eV). In addition, the negative shift of the Pt d-band center from Pt/C and Pt/N–C to Pt/Fe–N–C weakens the interaction between Pt/Fe–N–C and the adsorbate O*. Therefore, the Fe–N–C carrier reduces the adsorption strength of O* on Pt and mitigates the formation of Pt oxide by adjusting the electronic structure of Pt, thereby improving the ORR performance of Pt/Fe–N–C. Furthermore, the Fe–N–C support exhibits minimal metal dissolution and high stability, significantly boosting the overall stability of the Pt/Fe–N–C. After 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in an acidic electrolyte (0.6–1.0 V), the Pt/Fe–N–C shows remarkable durability with an Electrocatalytic Surface Area (ECSA) retention rate of 99%. Under the same conditions, it outperformed Pt/C (78% retention) and Pt/N–C (53% retention). Extensive research has demonstrated that reinforcing the interaction between M–NC and Pt, while leveraging their combined strengths, can improve the electrocatalytic performance. For example, Liao et al.82 effectively deposited Pt nanoparticles on the synthesized Fe/NC support using a self-template solution reduction method without surfactants in situ. The robust interaction between Pt NPs and Fe/NC results in Pt–Fe/NC exhibiting superior ORR activity and stability. Electrochemical assessments revealed that the MA and ORR activity of Pt–Fe/NC were 1.20 times and 2.53 times greater than those of conventional Pt/C catalysts. Furthermore, after 20000 cycles, the MA and specific activity of Pt–Fe/NC only declined by 4.5% and 1.6%. Additionally, by employing a strategy that combines the strain-induced contraction mechanism with an impregnation reduction approach, they dispersed Pt NPs onto the Fe–NC after strain-induced shrinkage and obtained an excellent Pt@Fe–NC oxygen reduction electrocatalyst.83 The hollow porous structure facilitates mass transfer and offers multiple active centers, enhancing the catalyst's high ORR performance. DFT calculations indicate that the interaction between Pt NPs and Fe–NC effectively anchors Pt while reducing the adsorption of *OH intermediates at both Pt and Fe, thereby improving the intrinsic ORR activity. The MA of Pt@Fe–NC reached 1.34 A mgPt−1, which is 6.77 times that of commercial Pt/C (0.198 A mgPt−1). Moreover, after 20000 accelerated durability tests, the mass activity degradation rate of the Pt@Fe–NC was 14.18%, notably lower than that of Pt/C.
000 cycles in an acidic electrolyte (0.6–1.0 V), the Pt/Fe–N–C shows remarkable durability with an Electrocatalytic Surface Area (ECSA) retention rate of 99%. Under the same conditions, it outperformed Pt/C (78% retention) and Pt/N–C (53% retention). Extensive research has demonstrated that reinforcing the interaction between M–NC and Pt, while leveraging their combined strengths, can improve the electrocatalytic performance. For example, Liao et al.82 effectively deposited Pt nanoparticles on the synthesized Fe/NC support using a self-template solution reduction method without surfactants in situ. The robust interaction between Pt NPs and Fe/NC results in Pt–Fe/NC exhibiting superior ORR activity and stability. Electrochemical assessments revealed that the MA and ORR activity of Pt–Fe/NC were 1.20 times and 2.53 times greater than those of conventional Pt/C catalysts. Furthermore, after 20000 cycles, the MA and specific activity of Pt–Fe/NC only declined by 4.5% and 1.6%. Additionally, by employing a strategy that combines the strain-induced contraction mechanism with an impregnation reduction approach, they dispersed Pt NPs onto the Fe–NC after strain-induced shrinkage and obtained an excellent Pt@Fe–NC oxygen reduction electrocatalyst.83 The hollow porous structure facilitates mass transfer and offers multiple active centers, enhancing the catalyst's high ORR performance. DFT calculations indicate that the interaction between Pt NPs and Fe–NC effectively anchors Pt while reducing the adsorption of *OH intermediates at both Pt and Fe, thereby improving the intrinsic ORR activity. The MA of Pt@Fe–NC reached 1.34 A mgPt−1, which is 6.77 times that of commercial Pt/C (0.198 A mgPt−1). Moreover, after 20000 accelerated durability tests, the mass activity degradation rate of the Pt@Fe–NC was 14.18%, notably lower than that of Pt/C.
 | ||
| Fig. 4 The DFT calculation models and results of Pt/Fe–NC (a) (reproduced from ref. 62 with permission from ACS, copyright 2022). Preparation process (b) and ORR performance (c) of the Pt@Fe–N-OMC catalyst (reproduced from ref. 81 with permission from Wiley, copyright 2023). | ||
Combining M–Nx with some special framework carbon substrates as carriers can effectively exert the favorable effect of M–Nx on Pt. For example, Wang et al.81 proposed a novel approach to create efficient, low platinum-loaded electrochemical catalysts for the ORR. As shown in Fig. 4b, ordered mesoporous carbon (OMC) functions as the carbon substrate via in situ polymerization of ferrous acetate and 1,10-phenanthroline. The Fe–N-OMC is modified with atomically distributed Fe–N4 sites on the OMC surface, serving as a reliable support. Subsequently, well-dispersed platinum nanoparticles, approximately 4.0 nm in size, are integrated into Fe–N-OMC using a straightforward high-temperature reduction method to yield the Pt@Fe–N-OMC catalyst. Density functional theory calculations indicate a robust electron interaction between Fe–N4 and platinum on the OMC surface in this construct. This interaction prevents the migration of platinum nanoparticles and significantly enhances the durability of Pt@Fe–N-OMC. Furthermore, the strong electron coupling between Fe–N4 and platinum results in a downturn of the band center by 0.21 eV for pure platinum. This shift in the d-band center reduces the Pt–O energy and speeds up the desorption of oxidation products, thus enhancing the ORR efficiency of the catalyst. As shown in Fig. 4c, during the half-cell evaluation, Pt@Fe–N-OMC achieves an ORR E1/2 of 0.844 V, surpassing that of Pt@OMC (0.804 V). The qualitative and specific activities of Pt@Fe–N-OMC (0.53 A mgPt−1 and 0.97 mA cm−2) are notably superior to those of Pt@OMC (0.14 A mgPt−1 and 0.52 mA cm−2) and Pt/C (0.28 A mgPt−1 and 0.40 mA cm−2). Notably, the Pt@Fe–N-OMC-2-based membrane electrode assembly (MEA) shows a mass activity of 0.25 A mgPt−1 at 0.9 V, significantly higher than that of commercial Pt/C (0.165 A mgPt−1).
In addition, the MnN4 sites integrated within the carbon matrices are more proficient at increasing the equation point of platinum solvation than traditional carbon. This enhancement improves the resistance of ultrafine Pt nanoparticles to potential-induced dissolution, effectively minimizing both Pt dissolution and diffusion from these nanoparticles. Zeng et al.84 developed Pt/Mn–N–C catalysts by depositing platinum nanoparticles onto metal-rich active sites, particularly MnN4. In this configuration, the Mn–N–C effectively traps Pt NPs. The MnN4 sites embedded within the carbon carriers enhance the immobilization of Pt particles, significantly improving the sustained stability of the Pt/Mn–N–C catalysts under hydrothermal degradation conditions. After exposing the Pt/Mn–N–C catalysts to 150000 cycles, under accelerated stress testing with H2/N2 at 80 °C and a voltage range of 0.60–0.90 V, the Pt (40 wt%)/Mn–N–C catalysts demonstrated outstanding endurance. The current density at 0.7 V declined from 1.42 A cm−2 to 1.20 A cm−2, reflecting a modest drop of 15.5%. Likewise, the current density at 0.8 V reduced from 512.6 mA cm−2 to 413.3 mA cm−2, reflecting a loss of only 19.4%.
In addition to the optimization of M–NC, the optimization of Pt particles is also a successful approach to strengthen the interaction between M–NC and Pt. For example, Li et al.85 utilized a eutectic salt-assisted semi-closed carbonation method to synthesize atomically dispersed ZnFe–NC and subsequently reduced the MAC-Pt-dispersed ZnFe–NC support through backflow glycol reduction. The resulting MAC-Pt/ZnFe–N–C catalyst, characterized by cross-linked Pt–Pt metal centers, was successfully fabricated using this reduction method. During synthesis, the H2PtCl6 precursor was initially anchored onto the ZnFe–N–C support surface, which improved the precursor's adsorption. Thereafter, the reduced platinum atoms were swiftly captured by the surface atoms on ZnFe–NC. A polarization test of PEMFCs, using a MAC-Pt/ZnFe–N–C cathode (0.035 mgPt cm−2) and a Pt/C anode (0.1 mgPt cm−2), revealed that the MAC-Pt/ZnFe–N–C cathode achieves a peak power density of 1.02 W cm−2, nearly 1.96 times greater than the 0.52 W cm−2 power density recorded for the conventional Pt/C cathode.
The key hurdles obstructing the commercialization of Pt/C involve its limited availability and high price, hindering mass production efforts. Therefore, it is essential to minimize the quantity of Pt while enhancing the catalyst's performance. While advances in developing and refining Pt M–NC catalysts have led to excellent catalytic activity and established methodologies, issues such as increased Pt demand, interactions, and insufficient stability remain. To tackle these challenges, researchers aim not only to refine the process for producing smaller, more uniform platinum nanoparticle catalysts but also to incorporate additional non-precious metal atoms into Pt structures. This strategy transforms Pt particles into Pt alloys, thereby reducing dependence on Pt within catalysts. Non-precious metals have proven effective in enhancing the characteristics of Pt catalysts by mechanisms like electron modulation and crystal strain. Therefore, creating Pt-based alloy catalysts by alloying Pt with other non-precious metals offers a hopeful remedy to the difficulties associated with the commercialization of Pt/C catalysts.
4 M–NC@PtM alloy catalyst
Pt catalysts account for 40% of the overall cost of PEMFC installations, and they remain irreplaceable as a vital component of fuel cells. Thus, developing cost-effective and efficient Pt-based catalysts is fundamental to the progress of PEMFC commercialization.86 Extensive research indicates that alloying Pt with non-precious metals on Pt/C can alter the electronic arrangement of Pt, thereby reducing its consumption. These Pt alloys, such as PtNi,87 PtCo,88–90 and PtCu91 alloys, can significantly improve their catalytic activity through strain and electronic effects. At present, the ordinary carbon-based Pt alloy catalyst is usually a low platinum catalyst, its catalytic activity can't achieve the purpose of commercialization, so the ordinary carbon-based Pt alloy catalyst still needs to make great efforts to improve its catalytic activity.Findings suggest that M–N–C not only refines the electronic behavior of Pt alloys but also supplies more ORR active sites (M–Nx).92 For example, Xiong et al.93 developed an ORR catalyst supported by a PtCo alloy on a bimetallic MOF-derived ZnCo–N–C substrate. In this configuration, the synergistic interaction between the PtCo alloy and Co–N–C enhances ORR activity while ensuring the preservation of structural coherence and performance over extended stability testing. Electrochemical measurements reveal that the E1/2 of commercially utilized Pt/C was 0.862 V, whereas the E1/2 value of PtCo/Co–NC exhibits a positive shift of 60 mV, showing a marked advancement in core activity. Compared to Pt/C, the MA of PtCo/Co–N–C increases by a factor of 6.7, attributed to the synergistic effects between the PtCo alloy and the Co–N–C matrix, alongside the advantageous incorporation of cobalt into the PtCo alloy. Research indicates that the synergistic catalytic effect of numerous active sites can effectively reduce H2O2 formation, thereby minimizing membrane and ionomer degradation. For example, Xiao et al.80 successfully developed a Pt-loaded (1.7 wt%) ORR electrocatalyst, featuring dispersed Pt, single atom Fe, and Pt–Fe alloy nanoparticles. This catalyst displays a Pt mass activity 3.7 times greater than that of Pt/C catalysts. Notably, it demonstrates remarkable durability, maintaining 97% of its activity after 100000 cycles, without any significant activity loss at 0.6 V throughout 200 h. Theoretical simulations reveal that the Pt–N1C3, Fe–N1C3, and PtFe@Pt components in the Pt–Fe–N–C catalyst act as active sites for the ORR. The catalytic efficiency is enhanced by the synergistic interactions among the various active sites. These results highlight the crucial importance of active site synergy in Pt–Fe–N–C catalysts, offering a promising approach for creating more efficient and long-lasting low-platinum catalysts.
It is important to note that the reduction potential of metal atoms in M–NC is lower than that of Pt. This allows M–NC to supply M–Nx catalytic active sites and sufficient M atoms for Pt alloys, playing a vital role in the design of low-Pt and multi-dimensional catalyst structures.94,95 For example, Guo et al.96 implemented a gas-promoted dealloying technique, starting with Pt1.5Ni1−x nanocrystals that underwent dealloying because of the persistent depletion of Ni atoms driven by ammonia and heat, thus creating a stable Pt-skin Pt1.5Ni1−x alloy. The displaced Ni atoms bond with nearby carbon substrate defects, resulting in numerous single Ni atom sites dispersed around the Pt1.5Ni1−xN alloy.
Ultimately, this leads to the formation of a compact hybrid electrocatalyst comprised of Pt-based alloy nanocrystals and densely packed monatomic Ni sites. Fig. 5a and b illustrate that during the multi-electron transfer process of the ORR, these dense monatomic Ni sites significantly facilitate the two-electron reduction of O2. The resulting intermediate (OOH*) swiftly migrates to adjacent Pt sites for further electron migration. This efficient relay catalytic effect enables Pt1.5Ni1−x/Ni–N–C catalysts to demonstrate exceptional ORR activity, achieving a mass activity of 4.10 AmgPt−1 in acid ORR testing, which surpasses that of Pt/C by a factor of 15 (Fig. 5c). Furthermore, during practical H2/O2 fuel cell evaluations, these catalysts attained a peak power density of 1.72 W cm−2 and a current density of 0.55 A cm−2 at 0.80 V. In addition, Feng et al.24 utilized pyrolysis and leaching strategies with a Ni-polymer to fabricate a nitrogen-carbon support (Ni–NC) featuring abundant mono-metal Ni@C and Ni–Nx sites. Subsequently, they deposited Pt nanoparticles onto this support. Furthermore, they synthesized a PtNi alloy from PtNi nanoparticles, using Ni@C as a Ni atom donor through alloying, resulting in 5-PtNi/NiNC catalysts containing a mere 5 wt% of Pt. The Ni–NC not only provides an adequate Ni source for the catalytic centers of the PtNi alloy but also creates Ni–Nx sites, fostering a synergistic catalytic effect with PtNi. This interaction greatly enhances the ORR performance of PtNi/NiNC. When compared to commercial PtNi ORR electrocatalysts, the MA and SA of the 5-PtNi/NiNC catalysts increased by 5.5 and 7.2 times, respectively, at 0.9 V. After subjecting the catalyst to 10000 cycles, the MA and SA activities only saw reductions of 4% and 8%, respectively.
 | ||
| Fig. 5 The ORR catalytic mechanism (a), DFT calculation results (b), and ORR performance (c) of the Pt1.5Ni1−x/Ni–N–C catalyst (reproduced from ref. 96 with permission from RCS, copyright 2022). The ORR synergistic catalytic mechanism (d) and ORR performance (e) of the PtCuCo@Co–N–C catalyst (reproduced from ref. 97 with permission from Wiley, copyright 2021). | ||
Research has shown that encapsulating a Pt alloy in porous graphite Co–N–C can better achieve synergistic catalysis of multiple active sites.87,98,99 Huang et al.97 synthesized an ORR catalyst using a porous graphite Co–N–C encapsulated PtCuCo alloy. DFT calculations reveal that the hybridization between the 3D PtCuCo alloy and graphite Co–N–C matrix produces a synergistic effect among multiple active sites (Fig. 5d). This synergistic interaction boosts the stability of the PtCuCo@Co–N–C, thereby enhancing their ORR properties. As shown in Fig. 5e, PtCuCo@Co–N–C exhibits a mass activity of 1.14 A mgPt−1 at 0.9 V, along with a peak power density of 960 mW cm−2 in hydrogen–air fuel cells, surpassing the efficiency of Pt/C (0.12 A mgPt−1 and 780 mW cm−2). Furthermore, the PtCuCo@Co–N–C catalyst exhibits remarkable electrochemical stability in the course of the ADT. While commercial Pt/C experienced a 46% decrease in activity, the mass activity of PtCuCo@Co–N–C remains nearly constant after 50000 potential cycles, showing minimal degradation relative to its initial state.
Incorporating low-cost transition metals into platinum lattice-forming alloys functions as a practical way to boost the efficiency of platinum usage while decreasing platinum content in catalysts. However, the alloying of platinum usually occurs at elevated temperatures, which results in platinum alloy particles losing their optimal activity due to agglomeration at high temperatures. Furthermore, the leaching of transition metals from platinum alloy catalysts contributes significantly to structural degradation, impacting their stability. Studies have shown that platinum catalysts are often dispersed and supported on carbon substrates, where the van der Waals forces between alloy particles and the carbon support are relatively weak. Consequently, nanoparticles can easily detach during operation, resulting in reduced stability. As such, it is crucial to strengthen the adhesion between platinum alloys and supports. Researchers have utilized carbon supports enriched with atomic M–Nx sites (M–NC) for supporting platinum alloy nanoparticles, allowing these particles to be securely anchored by the robust interactions between M–Nx sites and platinum alloys. This approach effectively mitigates catalytic deactivation stemming from dissolution and agglomeration of platinum alloy catalysts in use, subsequently enhancing catalytic stability. Utilizing a vapor deposition method,100 Yin et al.101 successfully fabricated an ultra-low Pt-loaded (0.64 wt%) ORR catalyst, which features electron coupling to the FeN4 active site of the PtFe alloy (PtFe–FeNC). In this configuration, the FeN4 sites on the FeNC substrate effectively secure the PtFe alloy, thus preventing agglomeration over extended cycles. Furthermore, these PtFe alloys can significantly inhibit the leaching of FeN4 from the FeNC substrate. Consequently, the Pt mass activity of PtFe–FeNC during the ORR at 0.9 V reaches 2.33 A mgPt−1, which is 12.9 times higher than that of commercial Pt/C. The catalyst also exhibits strong stability, with only a 9.4% reduction in mass activity following 70000 cycles. Importantly, a fuel cell with an ultra-low Pt loading of 0.012 mgPt cm−2 in the cathode exhibits a remarkable Pt mass activity of 1.75 A mgPt−1 at 0.9 V, significantly surpassing the performance of commercial MEAs, which achieve 0.25 A mgPt−1. Additionally, the PtFe–FeNC catalyst showcases exceptional durability.
Research shows that precise regulation of the morphology and structure of M–NC can lower the reaction energy threshold of its catalytic sites and enhance the interaction between M–NC and platinum sites. Chong et al.102 introduced a catalyst featuring Co–NC nanofiber-supported PtCo nanoparticles, synthesized from an electro-spun cobalt metal–organic framework. The engineering of the catalyst's topography enhances the base curvature, reducing the thermodynamic barrier at the Co–N4 site, which promotes the 4-electron path during the ORR. X-ray absorption spectroscopy reveals that the electron configurations of both the Pt site in PtCo and Co site in Co–N4 are optimized, leading to increased catalytic activity. The catalyst displays impressive performance in fuel cell tests (Fig. 6a), achieving an MA of 2.48 A mgPt−1 and maintaining 80% of this activity across 60000 AST cycles. Furthermore, research has demonstrated that topological carbon defect treatment of M–NC can endow it with topological carbon defect structure, which is of great significance for enhancing the interaction between M–NC and Pt. For instance, Chong et al.102 successfully synthesized Pt–Co alloy NPs on a Co–NC support derived from ZIF-8@ZIF-67 (Fig. 6b), which were stabilized through ammonia heat treatment leading to topological carbon defects, yielding a PtCo–NC catalyst. Experimental data and theoretical analyses reveal that the topological carbon defects on the Co–NC support substantially improve the charge transfer mechanism at the alloy–carbon interface reinforcing electron metal–support interactions between Pt–Co alloy nanoparticles and the carbon defects. This, in turn, improves the ORR properties in acidic environments. In a 0.1 M HClO4 electrolyte, the catalyst achieves an E1/2 of 0.926 V. Moreover, durability assessments conducted via the constant current method demonstrated a mere 3.6% loss after 12 hours. Additionally, DFT calculations reveal that the strong interaction between Pt–Co nanoparticles and Co–NC provides the catalyst with excellent ORR performance (Fig. 6c).
 | ||
| Fig. 6 The ORR performance (a), ORR catalytic mechanism (b), and DFT calculation results (c) of the PtCo@Pt–Co-GF catalyst (reproduced from ref. 102 with permission from Elsevier, copyright 2023). | ||
Theoretical calculations demonstrate that M–NC can alter the electronic state of platinum and redistribute its charge, resulting in better catalytic performance. For example, Zhang et al.89 fabricated an ultra-low platinum-loaded, multifunctional PtCo/NC nano-catalyst on a porous carbon substrate derived from ZIF-67 through an in situ assembly method. Remarkably, the PtCo/NC catalyst, featuring a minimal Pt loading of just 1 wt%, achieved an E1/2 of 0.877 V in an acidic environment. Additionally, in an application for Zn–air batteries, PtCo/NC demonstrated superior power density relative to Pt/C. Moreover, by tuning the fine structure and surface composition, the advantages of both M–NC and Pt alloys can be better utilized, which is beneficial for the catalyst to exhibit better ORR catalytic performance. Zhou et al.103 doped platinum onto porous carbon nanoparticles doped with transition metals and nitrogen. This approach enabled the uniform anchoring of smaller Pt-based particles on the support, maximizing the exposure of active sites and enhancing electron and proton transfer during electrochemical reactions. Consequently, this improved the electrochemical reactivity of the Pt1Ni2Co@NC and Pt1Ni2@NC. Additionally, the interactions, both electronic and synergistic, between the Pt nanoparticles and the M@NC support effectively stabilize the nanoparticles, mitigating risks of dissolution and aggregation, which in turn bolstered their stability. The mass activities for Pt1Ni2@NC (0.84 A mgPt−1) and Pt1Ni2Co@NC (1.43 A mgPt−1) surpass that of the commercial Pt/C (0.18 A mgPt−1) by factors of 4.67 and 7.94, respectively. Following accelerated durability tests, PtNiCo@NC exhibits outstanding stability for the ORR.
Combining graphene oxide with M–NC as a composite carrier for Pt alloy catalysts has proven to be an efficient strategy for improving the catalytic performance of Pt alloy catalysts. Pan et al.104 introduced an innovative method for synthesizing a PtCo alloy by using Co-ZIF as a substrate for the ORR. The precursor, Co-ZIF@GO, was obtained through in situ controlled growth of ZIF-67 on different surfaces of GO sheets at various nanoscale dimensions. Through pyrolysis and platinum loading, they developed PtCo alloy nitrogen-doped graphene (PtCo-NG) cathode catalysts with impressive dispersion (∼5 nm). The optimized PtCo-NG catalyst displayed superior properties and stability in acidic environments when compared to those of conventional Pt/C catalysts. Its E1/2 reached 0.86 V, with MA triple that of commercial Pt/C counterparts. Extensive investigations have prompted the utilization of M–NC providing a platform for PtM alloy nanoparticles, presenting a favorable alternative to traditional carbon materials. The activity and resilience of Pt alloys have significantly improved due to the presence of abundant M–Nx and PtM sites within the M–NC structure, facilitated by synergistic interactions and the robust connection between the M–NC support and PtM alloy nanoparticles. However, challenges regarding catalytic stability remain, as non-precious metal atoms within Pt alloys tend to leach under acidic conditions, compromising the integrity of PtM active sites. To mitigate this issue, researchers have optimized the structure of PtM alloys by transforming disordered PtM alloys into PtM-intermetallic compounds, which substantially enhance Pt–M interactions. This advancement bolsters the stability of PtM alloys under acidic conditions, leading to improved ORR performance of the catalysts.
5 M–NC@Pt–M intermetallic compound catalyst
A major obstacle in the use of PtM alloy catalysts is the leaching of transition metal elements during operation, which impacts their stability and effectiveness. This issue leads to structural degradation and negatively impacts catalyst stability. Lately, Pt-intermetallic compounds have emerged as a new array of ORR catalysts, showcasing better performance and durability.105,106 The well-structured atomic configuration in intermetallic structures allows Pt intermetallic compounds to exhibit enhanced strain and ligand effects. Furthermore, the expansive crystalline framework and distinct chemical composition of intermetallic phases endow them with superior chemical stability and catalytic efficacy compared to alloys with random atomic alignment.107 Nonetheless, synthesizing intermetallic compounds poses challenges. Transitioning from disordered alloys to intermetallic structures typically necessitates high-temperature annealing exceeding 500 °C. Elevated temperatures risk accelerated metal sintering, which can deform and aggregate nanostructures into larger particles, consequently diminishing their catalytic performance.108,109 This chapter reviews recent advancements in utilizing M–NC as a support to enhance the catalytic activity of Pt-based intermetallic compounds.110Research shows that M–Nx sites can strengthen the chemical interaction between PtM and the support, and reduce the bond affinity of OH on the PtM surface, hence boosting the intrinsic ORR reactivity and resilience of the catalyst.111 For example, Zeng et al.112 reported the uniform distribution of L12-Pt3Co nanoparticle catalysts on atomically dispersed carbon-rich MnN4 supports. Experimental results and DFT calculations suggest that MnN4 sites promote a strong synergistic interaction between Pt clusters and carbon supports, enhancing the catalyst's intrinsic ORR performance. As illustrated in Fig. 7a, MnSA–NC-supported L12-Pt3Co intermetallic nanoparticles demonstrated exceptional performance in both RRDE and integrated MEA assessments. The L12-Pt3Co@MnSA–NC MEA demonstrates an outstanding MA of 0.91 A mgPt−1 at a Pt loading of 0.1 mgPt cm−2 and 0.9 V, retaining 73% of its initial performance after 30000 cycles in catalyst AST. Additionally, under heavy-duty vehicle conditions, with 0.2 mgPt cm−2 and 250 kPa, the L12-Pt3Co@MnSA–NC catalyst attains a remarkable current density of 1.75 A cm−2 at 0.7 V. It shows exceptional durability as well, experiencing only an 18% reduction in current density and a 37% mass activity drop after 90000 AST cycles. As illustrated in Fig. 7b, the synergistic effect occurred between Co–N4 sites and L10-PtCo, facilitating the rapid migration of H2O2 from Co–N4 sites to L10-PtCo, where it underwent decomposition. This mechanism critically accelerates the kinetics of oxygen reduction. Lai et al.105 developed a one-dimensional atomically dispersed Co–N–PCNF embedded with intermetallic L10-PtCo through electrospinning, designed as an advanced cathode catalyst for PEMFC applications (Fig. 7c). Furthermore, compared to conventional carbon supports, the adhesion between Co–N–PCNF and L10-PtCo is substantially stronger, greatly improving catalyst stability. Fig. 7d demonstrates that L10-PtCo/Co–N–PCNF provides exceptional initial mass activity and durability in ORR tests, retaining 99% of its mass activity following 50000 cycles and 73% after 100000 cycles.
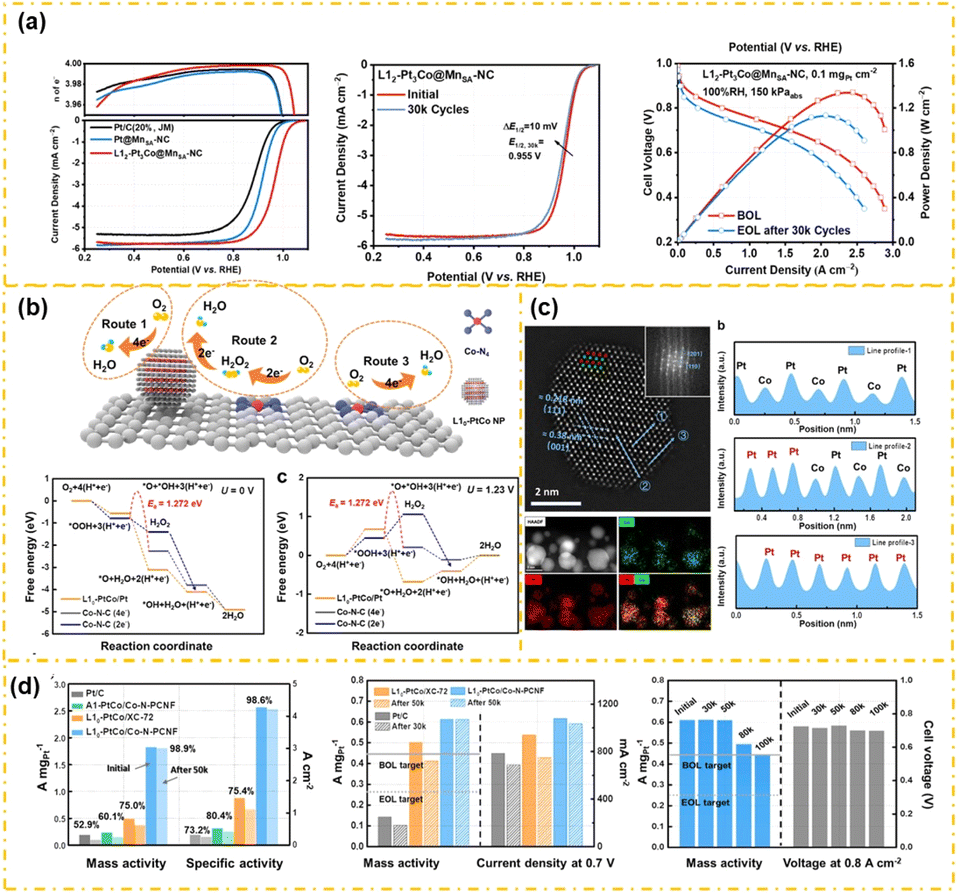 | ||
| Fig. 7 The ORR performance (a) and DFT calculation results (b) of L12-Pt3Co@MnSA–NC (reproduced from ref. 26 with permission from ACS, copyright 2023). The lattice spacing calculation (b and c), and ORR performance (d) of L10-PtCo/Co–N–PCNF (reproduced from ref. 105 with permission from ACS, copyright 2023). | ||
In addition, the strong interaction between M–NC and the Pt alloy can regulate the electronic arrangement of the Pt alloy, causing an appropriate bond energy between Pt and oxygen, which is crucial for optimizing the ORR capability of the catalyst.113 Yan et al.114 simultaneously incorporated both Pt and Co into Co–NC and developed an ORR catalyst composed of intermetallic PtCo and Co–NC sites through high-temperature reduction. This catalyst exhibits uniformly dispersed, higher-order PtCo intermetallic nanoparticles owing to the robust interaction between PtCo nanoparticles and Co–NC, along with the highly ordered L10 square structure of PtCo intermetallic compounds. Consequently, the electronic arrangement of Pt is optimized to achieve suitable binding energy with oxygen. Additionally, Co–NC successfully anchors PtCo nanoparticles, greatly enhancing the catalytic performance of PtCo/Co–NC. In the RDE test, PtCo/Co–NC exhibited impressive performance, achieving an E1/2 of 0.906 V, with a 7 mV loss after 20000 cycles and an MA of 0.63 A mgPt−1 at 0.9 V.
DFT calculations show that the synergistic interaction between the Pt alloy and adjacent M–Nx sites weakens the O2 adsorption of Pt alloy sites, reduces the activation barrier required to break the O–O bond, and increases the inherent reactivity of Pt sites,115 Qiao et al.116 developed an efficient method for synthesizing well-dispersed and highly ordered L12Pt3Co catalysts (L12Pt3Co/Fe–N–C) on FeN4–C supports. Experimental results confirmed this synergy exists between Pt3Co nanoparticles (NPs) and FeN4, resulting in reduced O2 absorption at Pt sites, thereby decreasing the activation energy for O–O bond rupture and improving the intrinsic activity of Pt3Co. Tests demonstrate that the improved performance of the ORR catalysts stemmed from the synergistic effect between Pt3Co NPs and FeN4 sites. In an MEA featuring a minimal Pt quantity at the cathode (0.1 mgPt cm−2), the MA of the L12Pt3Co/Fe–N–C catalyst achieved 0.72 A mgPt−1, maintaining 80% of its starting value following 30000 voltage cycles.
Due to the unique hierarchical pore structure and the firm metal support interaction of M–N–C, the size of the Pt alloy can be greatly limited and uniformly distributed, which holds great importance for Pt-based catalytic activity and stability.117 Mo et al.118 proposed a novel method for PtCo/Co–N–C, which is supported by nanoscale Co and N-doped carbon nanorods, thereby replacing traditional impregnation techniques of Pt sources with existing carbon materials.118 Initially, a MOF containing Co and Zn ions in rhombohedral dodecahedrons was synthesized using 2-methylimidazole. Then, the Co ions within the MOF were reduced into the Co–B–O complex via NaBH4, transforming the MOF into porous nanorods. Afterward, Pt was applied onto the Co–Zn-MOF via a displacement reaction involving PtCl6− with Co metal as well as the coordination of PtCl6− with the MOF. Following pyrolysis and an acid wash, highly dispersed PtCo/Co–N–C was synthesized. The catalyst demonstrates an MA of 0.577 A mgPt−1 and an activity of 1.4 mA cm−2 at 0.9 V. This performance is 3.6 and 3.5 times greater than that of Pt/C, underscoring its exceptional activity and stability. This innovative strategy lays the foundation for creating a strongly catalytic and stable ORR with diverse formulations to maximize active site exposure.
Researchers dispersed non-precious metal atoms on a carbon carrier and subjected it to thermal processing after loading platinum. Under high-temperature conditions, non-precious metal atoms merge into the lattice of platinum to form intermetallic catalysts. These catalysts exhibit excellent catalytic performance.119 In this regard, Zhou et al.120 designed an acidic catalysis system for the ORR, characterized by superior efficiency, low Pt loading, and strong durability. This system employed a gas-phase ordered alloying technique to design a productive synergistic catalytic architecture. This design integrates PtM intermetallic compounds and densely packed M–N4 onto NC. This method captures defects on platinum nanoparticles and NC scaffolds, effectively preventing partial aggregation through the efficient diffusion of transition metal salts in gaseous form with low boiling temperatures. Notably, the generated PtFe intermetallic compounds work synergistically with Fe–N4 sites to enhance ORR performances, yielding an E1/2 of 0.94 V, an MA of 0.51 A mgPt−1, and a mere 23.5% loss after 30000 cycles. This strategy provides a pathway for designing efficient collaborative catalytic systems by integrating platinum intermetallic compounds with isolated transition metal sites. Moreover, this synthesis methodology can adapt to other intermetallic compounds such as PtNi and PtCo. For instance, Ao et al.121 subsequently formed an intermetallic compound, Pt3Fe, through heat treatment (Fig. 8a). The Fe–Nx sites in this catalyst offer additional active sites for the ORR and exhibit strong interactions with Pt3Fe, thereby enhancing both activity and durability (Fig. 8b). Electrochemical tests showed that PtA@FeSA–N–C demonstrated remarkable reactivity and resilience, achieving an E1/2 of 0.923 V with minimal activity loss after 5000 acceleration cycles (Fig. 8c). Additionally, in single-cell tests of PEMFCs, the MEA utilizing PtA@FeSA–N–C as a cathode catalyst outperformed commercial Pt/C, delivering higher electrochemical performance (1.31 W cm−2, 2.81 A cm−2vs. 0.92 W cm−2, 2.46 A cm−2). PtA@FeSA–N–C achieves an impressive MA of 0.45 A mgPt−1 at 0.9 V, with just a 24.4% drop after 10000 cycles.
 | ||
| Fig. 8 The preparation (a), DFT calculation results (b), and ORR performance (c) of PtA@FeSA–NC (reproduced from ref. 102 with permission from ACS, copyright 2018). The DFT calculation results and ORR performance of Pt–Co/Co–NC (d) (Reproduced from ref. 121 with permission from RSC, copyright 2020). | ||
The close contact between Pt–M nanoparticles and M–N4 site surfaces can promote charge transfer and transfer of reaction intermediates, thereby accelerating the oxygen reduction reaction.122 Chong et al.123 introduced a method to create exceptionally active and steady electrocatalysts with ultra-low Pt content using cobalt or bimetallic Co-ZIFs. As shown in Fig. 8d, the DFT simulations indicate that strong bonds exist between the Pt surface and Co–N4 sites, facilitating their binding and preventing the separation of Pt–Co nanoparticles from the supports. As shown in Fig. 8d, Pt–Co/Co–NC exhibits exceptional performance in PEMFCs, driven by the synergistic catalysis of Pt–Co core–shell nanoparticles, achieving an MA of 1.77 A mgPt−1, retaining 64% and 15% of its starting activity even after 30000 voltage cycles.124 On this basis, Wang et al.125 embedded atomically dispersed Co sites into Co-ZIF carriers during the synthesis process of loading Pt nanoparticles, and then annealed the atomically dispersed cobalt and platinum nanocrystals to form ordered Pt3Co intermetallic structures. The ordered Pt3Co intermetallic catalyst structure has been optimized, and the optimized Pt3Co nanoparticles exhibit excellent activity and durability. Their E1/2 reaches 0.92 V, and after 30000 cycles in the voltage range, it only decreases by 12 mV. Atomic scale element mapping confirms that the intermetallic structure maintains a highly ordered structure after rigorous cyclic testing. In addition, Guo et al.126 loaded Pt onto Co–N–C through polyol reduction, followed by heat treatment to allow Co encased in the Co–N–C carrier to diffuse into the Pt, resulting in the formation of ultra-small PtCo NPs. The PtCo/Co–N–C catalyst, benefiting from both SMSI and alloying effects, demonstrates excellent ORR performance.
Research has found that at carbonization temperatures above 900 °C, the residual Zn in the derived carbon of ZIF-8 can still exceed 10 wt%. Therefore, using high Zn content Zn NC as a carrier, Zn NC-supported L10 PtZn intermetallic compound catalysts can be synthesized. Xia et al.127 prepared an A-Zn–NC-Ar PtZn catalyst. The Zn–Nx component in Zn–NC not only supplies zinc for the formation of L10-PtZn intermetallic compounds but also serves as a spatial barrier, preventing the development of inactive Pt–Nx and enhancing the active Pt ratio. In addition, according to DFT calculations, the combined ligand and SMSI effects between ZnN4 and PtZn enable Zn–NC-Ar-PtZn to demonstrate outstanding durability and high catalytic activity. In comparison to the leading 20%Pt/C (0.228 A mgPt−1 and 69.3%), the resultant Zn–NC-Ar-PtZn exhibits superior MA (0.557 A mgPt−1) and MA retention (74.0%). Notably, in the context of PEMFCs, the Zn–NC-Ar-PtZn demonstrates exceptional ORR catalytic performance (1.485 W cm−2 at 0.40 V). In addition, Xue et al.128 successfully synthesized highly active, structured PtZn intermetallic nanoparticles incorporated into 10% Zn NC by one-step heat treatment of the Pt doped ZIF-8-PtZn@NC catalyst. Due to the highly graphitized Zn NC framework and the specific electronic structure of intermetallic PtZn NPs, the PtZn@NC shows ideal performance.
Combining conventional low-platinum group metals (LP) with platinum group metal-free (PF) components to form LP/PF composite catalysts has become a promising strategy for enhancing the intrinsic performance of ORR catalysts.129,130 Zhou et al.131 elucidated the specific synergistic effects between LP and PF in detail in their report. As shown in Fig. 9a, both physical characterization and theoretical calculations were employed to explore the potential synergistic interaction between PtCo and the Co/N/C matrix. The findings indicated that the asymmetric electron distribution, induced by Co's presence in both the PtCo nanoparticles and the supports, significantly enhances catalyst performance. This synergistic effect modulates the d-band center of Pt, thereby easing O2 dissociation at the *OOH step and *OH desorption step. Furthermore, this interaction fosters a strong bond between PtCo NPs and Co/N/C, ensuring good stability. As shown in Fig. 9b, the MA of the Co/N/C@PtCo NPs catalyst was measured at 0.24 A mgPt−1 @ 0.9 V, surpassing that of Co/N/C@PtSAs (0.12 A mgPt−1 @ 0.9 V) and Pt/C (0.12 A mgPt−1 @ 0.9 V). Additionally, the catalyst sustained an ECSA of up to 98% after underwent DOE standard 30000 cycle ADTs. This research highlighted the role of the synergistic effect in LP/PF catalysts by using the Co/N/C@PtCo NP catalyst as a model, providing valuable insight for future advancements in oxygen reduction catalysts.
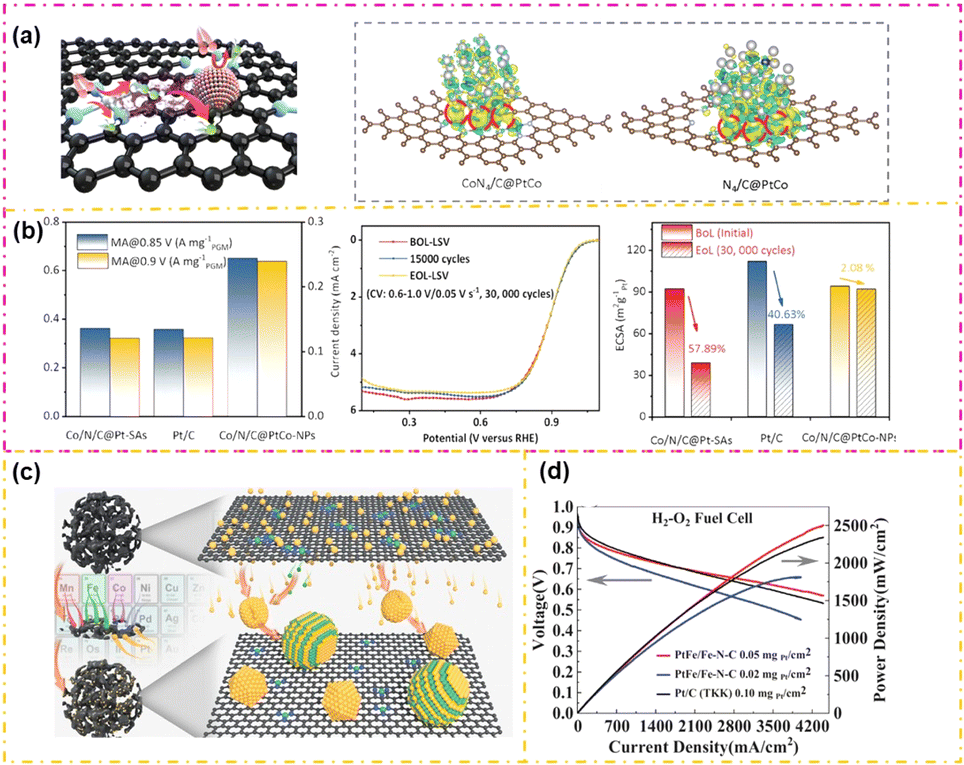 | ||
| Fig. 9 The DFT calculation results (a) and ORR performance (b) of CoN/C@PtCo (reproduced from ref. 131 with permission from Wiley, copyright 2024). The preparation (c) and ORR performance (d) of Pt–Fe/Fe–N–C (reproduced from ref. 132 with permission from Wiley, copyright 2023). | ||
Research shows that the SMSI between some alkaline earth metals M–NC and PtM alloys is more conducive to the anchoring of PtM, thereby promoting the stability of the catalyst. This presents an innovative method for the production of high-quality ultra-low platinum-loaded catalysts.133 Xu et al.134 achieved the synthesis of an atomically dispersed Mg–N–C material using alkaline earth metal Mg as a support, creating highly crystalline Pt3M intermetallic compounds as PtMg–N–C catalysts via annealing and subsequent treatment of Pt–Mg-HMT-MOF. Within this system, Mg–N–C can not only serve as a platform for anchoring Pt nanoparticles but also promote the binding of Mg and Pt face-center cubic lattices, thereby forming highly crystalline Pt3Mg nanoalloys through metal carrier interface interactions. Due to the robust interaction between Pt3Mg and the Mg–N–C, along with strain in Pt3Mg, the ORR activity of PtMg–N–C surpasses that of commercial 40% Pt/C.
Lowering the Pt content in PEMFCs raises oxygen mass transport resistance because of the restricted amounts of active sites and decreases catalyst stability as a result of Pt particle growth over prolonged operation.135 In this respect,136 Luo et al.132 developed a Pt–Fe/Fe–N–C aerogel catalyst that significantly mitigates mass transport resistance associated with oxygen and enhances long-term durability (Fig. 9d). The accompanying diagram indicated that the Fe–N–C aerogels feature layered and interconnected pores, allowing PEMFCs with an ultra-low Pt loading (50 ± 5 μgPt cm−2) to sustain low local oxygen transport resistance (0.18 s cm−1). The atomically dispersed Fe–Nx sites provide stability for the loaded Pt during synthesis at temperatures up to 1000 °C and in practical PEMFC applications. As illustrated in Fig. 9d, the PtFe/Fe–N–C catalyst shows a peak power density of 2.30 W cm−2 at 4.2 A cm−2 and a current density of 2.99 A cm−2 at 0.6 V, with a cathode loading of merely 0.02 mgPt cm−2. Following 60000 accelerated durability tests, the Pt catalyst displayed a negligible voltage loss of 8 mV at 0.80 A cm−2, while the ECSA stayed constant. This combination of stratified pores, aerogel structure, and single-atom Fe–N–C sites emphasizes their structural benefits, paving the way for advancing fuel cell catalyst development.137,138
6 Summary and future perspectives
This review summarizes the advancements made over the past few decades in atomic-scale metal–nitrogen–carbon coupled platinum-based ORR catalysts. The synthesis and modification techniques of M–NC@Pt (Pt NPs, Pt alloys, and Pt intermetallic compounds) oxygen reduction catalysts, as well as the principle of improving their properties, are introduced in detail. The interaction between M–NC and Pt (synergistic catalysis effect, electronic regulatory effect, anchoring effect, overflow effect, etc.) is emphasized. The SMSI between M–NC and Pt can enhance the performance of Pt-based catalysts.In the third part, we outline the use of a new M–NC material instead of traditional carbon as the carrier of platinum particles to improve the ORR performance of Pt/C catalysts. On the one hand, the M–Nx site contained in the M–NC structure endowed the carbon carrier with good ORR catalytic activity. M–Nx can produce a synergistic catalytic mechanism with the Pt site, thus improving the overall catalytic performance of the catalyst. On the other hand, due to the special structure of M–NC and abundant M–Nx sites, Pt and carrier have a strong interaction, which is of great significance in enhancing the catalytic activity and durability of Pt sites (Table 1).
| Types of catalysts | E 1/2 (V vs. RHE) | ECSA (m2 g−1) | MA (A mgPt−1) | Retention rate (%) | Power density (W cm−2) | Reference |
|---|---|---|---|---|---|---|
| Pt/Fe, N-HG | 0.88 | 140.89 | 0.208 | 73.52 | 0.28 | 70 |
| Pt–FeNC | — | — | 0.69 | 81 | — | 73 |
| Pt/(Co–N)–C | 0.894 | 48 | 0.227 | 84.6 | 1.06 (0.6V) | 71 |
| Pt/Co–N–C | 0.886 | 157 | 0.223 | 82 | — | 77 |
| Pt@Ni ZIF–NC | 0.902 | 74 | 0.38 | 91 | — | 74 |
| Pt–Fe/NC | — | — | 0.42 | 98.4 | 0.75 | 80 |
| Pt/Fe–N-OMC | 0.844 | 59.26 | 0.53 | — | 1.22 | 81 |
| Pt1.5Ni1-x/Ni–N–C | 0.967 | 89.88 | 4.1 | — | 1.72 | 96 |
| PtCuCo@Co–N–C | 0.95 | — | 1.14 | — | 0.96 | 94 |
| PtCo@Pt–Co-GNF | 0.93 | — | 5.48 | 86 | 0.63 | 104 |
| Pt1Ni2Co@NC | — | 54.78 | 1.43 | 85.32 | 103 | |
| L12Pt3Co@MnSA–NC | 0.915 | 125.8 | 1.8 | 87.8 | 1.66 | 112 |
| PtCo–CoNC | 0.906 | — | 0.63 | 88.97 | — | 102 |
| Pt3Co/FeN4 | 0.90 | 72.2 | 1.34 | — | 0.824 | 116 |
| PtCo/Co–N–C | 0.916 | 62.8 | 0.577 | — | — | 93 |
| Pt1Fe1 IMC | 0.94 | — | 0.51 | 76.5 | 1.44 | 116 |
| PtA@FeSA–N–C | 0.923 | — | 0.45 | 75.6 | 1.31 | 121 |
| Pt–Co NPs | 0.96 | — | 1.77 | — | — | 131 |
In the fourth part and the fifth part, we summarized the Pt-based catalysts prepared by M–NC structure loaded with a Pt alloy and Pt intermetallic compound respectively. The important role of M–NC in the preparation and catalysis of Pt-based catalysts is emphasized. On the one hand, the preparation of Pt alloys and intermetallic compounds requires high-temperature treatment, which means that the dispersion of Pt substances will be a severe test for the carrier. It is found that Pt is firmly anchored on the surface of M–NC by the strong interaction between M–NC and Pt, which can greatly limit the adverse effect of high-temperature treatment on Pt. At the same time, the anchoring effect of M–NC on Pt also greatly improves the durability. On the other hand, M–NC materials are rich in M single atoms (M–Nx), and M–Nx can provide sufficient M atoms for Pt alloys and intermetallic compounds, thereby inducing M atoms to enter the lattice of Pt through diffusion under relatively mild conditions, thus generating low-load Pt-based catalysts with excellent ORR properties.
As shown in Fig. 10, researchers have created numerous Pt-based catalysts that exhibit outstanding performance. However, in order to promote the commercial application of ORR catalysts, more efforts are needed in terms of low platinum, high efficiency, and stability. We can further optimize the Pt-based catalysts in the following directions: firstly, we can refine the synthesis methods of platinum-based catalysts by building on existing studies, thereby boosting their performance while lowering costs. Additionally, intermetallic compounds, formed by combining platinum with various elements, show increased catalytic activity, which could improve the efficacy and longevity of platinum alloy catalysts. However, current research on platinum intermetallic compounds remains limited, particularly regarding how composition and structure influence their properties. Both theoretical and experimental investigations are essential moving forward. Secondly, it is crucial to develop highly performing M–NC. Although some research exists on M–NC ORR catalysts, their performance has yet to meet commercial benchmarks, necessitating further studies to match or surpass platinum catalyst capabilities. Finally, we can also harness advanced AI technologies to expedite the design and development of ORR catalysts. Traditional catalyst development relies heavily on trial and error, resulting in significant time and economic costs along with unpredictability. Recently, the emergence of data and supercomputing has facilitated the widespread application of advanced artificial intelligence in materials research and development. These technologies offer powerful learning, data analysis, and predictive capabilities, which can significantly accelerate development and reduce costs. Leveraging advanced AI technology in ORR catalyst development promises extensive growth potential.
 | ||
| Fig. 10 The challenges and future directions of ORR catalysts. | ||
Data availability
Data will be made available on request.Author contributions
Zigang Zhao, writing – original draft; conceptualization; Lezhi Zhan, writing – original draft; Pan Guo, formal analysis; Yunkun Dai, supervision; Lixiao Shen, supervision; Yunlong Zhang, conceptualization; Guiling Wang, writing – review & editing; Zhenbo Wang, conceptualization; Lei Zhao, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22075062 and U23A20573), the High-Level Professional Team in Shenzhen (KQTD20210811090045006), the Shenzhen Science and Technology Program (20220809194504001, JCYJ20210324120400002 and SGDX20210823103803017), the Key Research and Development Program of Shandong Province (2022CXGC010305), Heilongjiang Touyan Team (Grant No. HITTY-20190033), Heilongjiang Postdoctoral Fund (LBH-Z22113), the Fundamental Research Funds for the Central Universities (Grant No. FRFCU5710051922), and Guangdong Basic and Applied Basic Research Foundation (2022B1515120001).References
- S. Arunachalam, B. Kirubasankar, D. Pan, H. Liu, C. Yan, Z. Guo and S. Angaiah, Green Energy Environ., 2020, 5, 259–273 CrossRef.
- H. Wan, W. Ma, K. Zhou, Y. Cao, X. Liu and R. Ma, Green Energy Environ., 2022, 7, 205–220 CrossRef CAS.
- Y. Pan, F.-F. Deng, Z. Fang, H.-J. Chen, Z. Long and X.-D. Hou, Chin. Chem. Lett., 2021, 32, 3440–3445 CrossRef CAS.
- K. Wang, N. Li, Y. Yang, S. Ke, Z. Zhang, M. Dou and F. Wang, Chin. Chem. Lett., 2021, 32, 3159–3163 CrossRef CAS.
- L. Chen, X. Xu, W. Yang and J. Jia, Chin. Chem. Lett., 2020, 31, 626–634 CrossRef CAS.
- Q. Xue, Z. Wang, Y. Ding, F. Li and Y. Chen, Chin. J. Catal., 2023, 45, 6–16 CrossRef CAS.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 CrossRef.
- L. Shen, F. Tu, Z. Shang, M. Ma, Y. Xia, Z. Zhao, L. Zhao, Z. Wang and G. Shao, Int. J. Hydrogen Energy, 2022, 47, 15001–15011 CrossRef CAS.
- Q. Xue, X. Y. Bai, Y. Zhao, Y. N. Li, T. J. Wang, H. Y. Sun, F. M. Li, P. Chen, P. Jin and S. B. Yin, J. Energy Chem., 2022, 65, 94–102 CrossRef CAS.
- B. Sun, Y. C. Jiang, Q. L. Hong, X. Liu, F. M. Li, D. S. Li, Y. Yang and Y. Chen, J. Energy Chem., 2023, 4, 150–162 Search PubMed.
- L. Liang, M. Li, B. Zhang, J. Liang, B. Zeng, L. Wang, Y. Tang, G. Fu and Z. Cui, Adv. Energy Mater., 2023, 13, 2203803 CrossRef CAS.
- B. Zhang, G. Fu, Y. Li, L. Liang, N. S. Grundish, Y. Tang, J. B. Goodenough and Z. Cui, Angew. Chem., Int. Ed., 2020, 59, 7857–7863 CrossRef CAS.
- W. Yan, X. Wang, M. Liu, K. Ma, L. Wang, Q. Liu, C. Wang, X. Jiang, H. Li, Y. Tang and G. Fu, Adv. Funct. Mater., 2024, 34, 2310487 CrossRef CAS.
- Y. Wu, L. Chen, S. Geng, Y. Tian, R. Chen, K. Wang, Y. Wang and S. Song, Adv. Funct. Mater., 2024, 34, 2307297 CrossRef CAS.
- M. Y. Chen, Y. Li, H. R. Wu, B. A. Lu and J. N. Zhang, Materials, 2023, 16, 2311564 Search PubMed.
- H. Kim, T. Y. Yoo, M. S. Bootharaju, J. H. Kim, D. Y. Chung and T. Hyeon, Advanced Science, 2022, 9, 2104054 CrossRef CAS.
- R. Zhao, C. Ziheng, Q. Li, W. Xuan, Y. Tang, G. Fu, H. Li, J.-M. Lee and S. Huang, Chem Catal., 2022, 2, 3590–3606 CrossRef CAS.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang, W. Deng, Y. Long, C. Huang and Y. Li, Nat. Commun., 2018, 9, 33–36 CrossRef PubMed.
- I. Jang, S. Lee, J.-H. Jang, M. Ahn and S. J. Yoo, Int. J. Energy Res., 2022, 46, 13602–13612 CrossRef CAS.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS PubMed.
- S. Ning, M. Li, X. Wang, D. Zhang, B. Zhang, C. Wang, D. Sun, Y. Tang, H. Li, K. Sun and G. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314565 CrossRef CAS PubMed.
- L.-J. Yuan, B. Liu, L.-x. Shen, Y.-K. Dai, Q. Li, C. Liu, W. Gong, X.-L. Sui and Z.-B. Wang, Adv. Mater., 2023, 35, 2305945 CrossRef CAS.
- C. Wang, C. An, C. Qin, H. Gomaa, Q. Deng, S. Wu and N. Hu, Nanomaterials, 2022, 12, 385–396 CrossRef PubMed.
- Q. Feng, X. Wang, M. Klingenhof, M. Heggen and P. Strasser, Angew. Chem., Int. Ed., 2022, 61, e202203728 CrossRef CAS.
- A. Pavlets, A. Alekseenko, E. Kozhokar, I. Pankov, D. Alekseenko and V. Guterman, Int. J. Hydrogen Energy, 2023, 48, 22379–22388 CrossRef CAS.
- Y. Zeng, J. Liang, C. Li, Z. Qiao, B. Li, S. Hwang, N. N. Kariuki, C.-W. Chang, M. Wang, M. Lyons, S. Lee, Z. Feng, G. Wang, J. Xie, D. A. Cullen, D. J. Myers and G. Wu, J. Am. Chem. Soc., 2023, 145, 17643–17655 CrossRef CAS.
- S. Hu and Z.-A. Li, Chem. Eng. J., 2023, 471, 144828 CrossRef CAS.
- L. Zhang, T. Liu, X. Liu, S. Li, X. Zhang, Q. Luo, T. Ding, T. Yao and W. Zhang, Nanoscale, 2024, 16, 221–236 Search PubMed.
- X. Li, X. Duan, S. Zhang, C. Wang, K. Hua, Z. Wang, Y. Wu, J. Li and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202400549 CrossRef CAS.
- L. Yang, J. Bai, N. Zhang, Z. Jiang, Y. Wang, M. Xiao, C. Liu, S. Zhu, Z. J. Xu, J. Ge and W. Xing, Angew. Chem., Int. Ed., 2024, 63, e202315119 CrossRef CAS.
- S. Li, J.-J. Li, C. Xu, L. Zhang, A. Li, T.-W. Song, W. Zhang, L. Tong and H.-W. Liang, ACS Mater. Lett., 2024, 6, 706–712 CrossRef CAS.
- T.-W. Song, M.-X. Chen, P. Yin, L. Tong, M. Zuo, S.-Q. Chu, P. Chen and H.-W. Liang, Small, 2022, 18, 2202916 CrossRef CAS PubMed.
- J. Lin, J. Wang, Y. Wu, P. Yang, Q. Liu, M. Li, S. Du, R. Chen and L. Tao, Chem.–Asian J., 2023, 18, e202300137 CrossRef CAS.
- A. Gunnarson, J. De Bellis, T. Imhof, N. Pfänder, M. Ledendecker and F. Schüth, Chem. Mater., 2023, 35, 2006–2015 CrossRef CAS.
- Y. Gao, N. Thakur, T. Uchiyama, W. Cao, K. Yamamoto, T. Watanabe, M. Kumar, R. Sato, T. Teranishi, H. Imai, Y. Sakurai and Y. Uchimoto, ACS Appl. Mater. Interfaces, 2023, 2311248 Search PubMed.
- C. Wang, X. Wang, H. Ren, Y. Zhang, X. Zhou, J. Wang, Q. Guan, Y. Liu and W. Li, Nat. Commun., 2023, 14, 51–58 CrossRef.
- Y. Zhao, Z. Shen, J. Huo, X. Cao, P. Ou, J. Qu, X. Nie, J. Zhang, M. Wu, G. Wang and H. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308349 CrossRef CAS PubMed.
- Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu, Y. Li and Y. Huang, Adv. Mater., 2019, 31, 1808115 CrossRef.
- J. Zhang, P. Liang, X. Xu, R. Wang, S. Liu, C. Wang, B. Liu, L. Luo, M. Jin, H. Liu, H. Yi and S.-Y. Lu, Nano Mater. Sci., 2024, 5, 12–25 Search PubMed.
- Z.-G. Zhao, P. Guo, L.-X. Shen, Y.-Y. Liu, Z.-Y. Zhang, F.-D. Tu, M. Ma, X.-W. Liu, Y.-L. Zhang, L. Zhao, G.-J. Shao and Z.-B. Wang, Appl. Surf. Sci., 2023, 609, 155302 CrossRef CAS.
- Z. Zhao, P. Guo, M. Ma, W. Ye, P. Shao, J. Liu, B. Xu, L. Shen, Y. Zhang, L. Zhao, G. Wang and Z. Wang, Int. J. Hydrogen Energy, 2024, 81, 40–46 CrossRef CAS.
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218269 CrossRef CAS PubMed.
- S. Yi, H. Jiang, X. Bao, S. Zou, J. Liao and Z. Zhang, J. Electroanal. Chem., 2019, 848, 113279 CrossRef CAS.
- C.-L. Yang, L. Wang, P. Yin, J. Liu, M. Chen, Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H.-W. Liang, Science, 2021, 374, 459–464 CrossRef CAS.
- Y.-F. Chang, C.-Y. Wu and M.-H. Chang, Int. J. Hydrogen Energy, 2024, 54, 437–445 CrossRef CAS.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen, H. Liu, Y. Li and G. Wang, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed.
- M. Janssen, P. Weber and M. Oezaslan, Curr. Opin. Electrochem., 2023, 40, 101337 CrossRef CAS.
- Z. Pei, H. Zhang, Y. Guo, D. Luan, X. Gu and X. W. Lou, Adv. Mater., 2024, 36, 2306047 CrossRef CAS PubMed.
- Z. Qiao, S. Hwang, X. Li, C. Wang, W. Samarakoon, S. Karakalos, D. Li, M. Chen, Y. He, M. Wang, Z. Liu, H. Zhou, G. Wang, Z. Feng, D. Su, J. Spendelow and G. Wu, Energy Environ. Sci., 2019, 12, 2830–2841 RSC.
- L. Niu, J. Zhao, X. Chen, G. Wang, W. Zhang and X. Wang, Mol. Catal., 2024, 557, 113997 CrossRef CAS.
- M. A. Hoque, F. M. Hassan, M.-H. Seo, J.-Y. Choi, M. Pritzker, S. Knights, S. Ye and Z. Chen, Nano Energy, 2016, 19, 27–38 CrossRef CAS.
- Q. Lenne, A. Mattiuzzi, I. Jabin, J. Hamon, Y. R. Leroux and C. Lagrost, Adv. Mater. Interfaces, 2023, 10, 2202219 CrossRef CAS.
- T. Y. Yoo, J. M. Yoo, A. K. Sinha, M. S. Bootharaju, E. Jung, H. S. Lee, B.-H. Lee, J. Kim, W. H. Antink, Y. M. Kim, J. Lee, E. Lee, D. W. Lee, S.-P. Cho, S. J. Yoo, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 14190–14200 CrossRef CAS.
- J. Wei, P. Li, J. Shi, M. Huang, W. Tian and H. Wang, Sustainable Energy Fuels, 2022, 6, 3383–3393 RSC.
- X. Wang, J. Zhang, P. Wang, L. Li, H. Wang, D. Sun, Y. Li, Y. Tang, X. F. Lu, Y. Wang and G. Fu, Energy Environ. Sci., 2023, 16, 5500–5512 RSC.
- C. Ouyang, L. Zheng, Q. Zhang and X. Wang, Adv. Mater., 2022, 34, 2205372 CrossRef CAS PubMed.
- S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2020, 11, 4302 CrossRef CAS PubMed.
- Z. Li, P. Zhou, W. Jiang, B. Zhao, X. Chen and M. Li, Catalysts, 2024, 14, 57–71 CrossRef CAS.
- A. Sarapuu, J. Lilloja, S. Akula, J. H. Zagal, S. Specchia and K. Tammeveski, ChemCatChem, 2023, 15, e202300849 CrossRef CAS.
- A. Parkash, ECS J. Solid State Sci. Technol., 2020, 9, 25–41 Search PubMed.
- Y. Zeng, J. Liang, B. Li, H. Yu, B. Zhang, K. Reeves, D. Cullen, X. Li, D. Su, G. Wang, S. Zhong, H. Xu, N. Macauley and G. Wu, ACS Catal., 2023, 13, 11871–11882 CrossRef CAS.
- F. Xiao, Y. Wang, G. L. Xu, F. Yang, S. Zhu, C. J. Sun, Y. Cui, Z. Xu, Q. Zhao, J. Jang, X. Qiu, E. Liu, W. S. Drisdell, Z. Wei, M. Gu, K. Amine and M. Shao, J. Am. Chem. Soc., 2022, 144, 20372–20384 CrossRef CAS.
- Y. Liu, F. Tu, Z. Zhang, Z. Zhao, P. Guo, L. Shen, Y. Zhang, L. Zhao, G. Shao and Z. Wang, Appl. Catal., B, 2023, 324, 122209 CrossRef CAS.
- X. b. Gao, Y. Wang, W. Xu, H. Huang, K. Zhao, H. Ye, Z.-Y. Zhou, N. Zheng and S.-G. Sun, J. Am. Chem. Soc., 2023, 145, 15528–15537 CrossRef CAS.
- D. Shin, S. Bhandari, M. F. Tesch, S. A. Bonke, F. Jaouen, S. Chabbra, C. Pratsch, A. Schnegg and A. K. Mechler, J. Energy Chem., 2022, 65, 433–438 CrossRef CAS.
- J.-Y. Park, D.-H. Kwak, K.-B. Ma, S.-B. Han, G. Chai, S.-K. Kim, D. H. Peck, C.-S. Kim, A. Kucernak and K.-W. Park, J. Catal., 2018, 359, 46–54 CrossRef CAS.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS.
- Y. Sha, J. Ji, S. Li, X. Gao, B. Zhang, M. Ling, C. Liang and Z. Lin, Eur. J. Inorg. Chem., 2020, 2020, 165–168 CrossRef CAS.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- Z. Gao, Z. Chen, X. Zhan, L. Zhou, Y. Xie, X. Yang, J. Tian, G. Zhang, S. Sun and X. Tong, ACS Appl. Nano Mater., 2023, 6, 10521–10530 CrossRef CAS.
- B. Hu, X. Deng, L. Zhou, J. Dai, G. Yang, W. Tan, W. Zhou and Z. Shao, Composites, Part B, 2020, 193, 108012 CrossRef CAS.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, J. Zhu, Z. Wang, H.-W. Li, S. Liu, D. He and S. Mu, J. Energy Chem., 2022, 65, 48–54 CrossRef CAS.
- R. Adzic, K. Kuttiyiel, K. Sasaki, G.-G. Park, M. Vukmirovic, L. Wu, Y. Zhu and J. Chen, Chem. Commun., 2017, 53, 25–41 Search PubMed.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, Z. Wang, Z.-Y. Hu, Y. Zhao, L. Hai-Wen, D. He and S. Mu, Nano Energy, 2021, 88, 106221 CrossRef CAS.
- X.-M. Qu, S.-H. Yin, Y.-N. Yan, J. Yang, Y.-R. Li, X.-Y. Cheng, F. Lu, C.-T. Wang, Y.-X. Jiang and S.-G. Sun, Chem. Eng. J., 2023, 461, 142054 CrossRef CAS.
- J. Shan, J. Liao, C. Ye, J. Dong, Y. Zheng and S.-Z. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202213412 CrossRef CAS.
- P. Guo, B. Liu, Y.-K. Dai, X.-F. Gong, Y.-F. Xia, Y.-L. Zhang, B. Liu, L. Zhao, X.-L. Sui and Z.-B. Wang, J. Colloid Interface Sci., 2022, 613, 276–284 CrossRef CAS.
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed.
- X. Wang, S. Yang, Y. Yu, M. Dou, Z. Zhang and F. Wang, Catal. Sci. Technol., 2019, 10, 35–41 Search PubMed.
- F. Xiao, G.-L. Xu, C.-J. Sun, I. Hwang, M. Xu, H.-w. Wu, Z. Wei, X. Pan, K. Amine and M. Shao, Nano Energy, 2020, 77, 105192 CrossRef CAS.
- K. Wang, H. Yang, Q. Wang, J. Yu, Y. He, Y. Wang, S. Song and Y. Wang, Adv. Energy Mater., 2023, 13, 2204371 CrossRef CAS.
- W. Liao, S. Zhou, Z. Wang, J. Long, M. Chen, Q. Zhou and Q. Wang, J. Mater. Chem. A, 2022, 10, 25–40 Search PubMed.
- W. Liao, S. Zhou, Z. Wang, F. Liu, H. Pan, T. Xie and Q. Wang, ChemCatChem, 2021, 13, 4925–4930 CrossRef CAS.
- Y. Zeng, J. Liang, B. Li, H. Yu, B. Zhang, K. S. Reeves, D. A. Cullen, X. Li, D. Su, G. Wang, S. Zhong, H. Xu, N. Macauley and G. Wu, ACS Catal., 2023, 13, 11871–11882 CrossRef CAS.
- J. Li, Q. Zhou, M. Yue, S. Chen, J. Deng, X. Ping, Y. Li, J. Li, Q. Liao, M. Shao and Z. Wei, Appl. Catal., B, 2021, 284, 119728 CrossRef CAS.
- L. Mao, K. Fu, J. Jin, S. Yang and G. Li, Int. J. Hydrogen Energy, 2019, 44, 18083–18092 CrossRef CAS.
- S. Hanif, X. Shi, N. Iqbal, T. Noor, R. Anwar and A. M. Kannan, Appl. Catal., B, 2019, 258, 117947 CrossRef CAS.
- X. Shi, N. Iqbal, S. S. Kunwar, G. Wahab, H. A. Kasat and A. M. Kannan, Int. J. Hydrogen Energy, 2018, 43, 3520–3526 CrossRef CAS.
- M. Zhang, T. Zhou, D. Bukhvalov, F. Han, C. Wang and X. Yang, Appl. Catal., B, 2023, 337, 122976 CrossRef CAS.
- Y. Zhou, J. Chen, Z. Huang, Y. Peng, L. Xing, C. Tang, N. Wang, L. Meng, M. Wu, L. Du and S. Ye, Nanoscale, 2024, 16, 5215–5221 RSC.
- N. Iqbal, Catalysts, 2020, 10, 25–31 Search PubMed.
- S. Zaman, X. Tian, Y. Q. Su, W. Cai, Y. Yan, R. Qi, A. I. Douka, S. Chen, B. You, H. Liu, S. Ding, X. Guo and B. Y. Xia, Sci. Bull., 2021, 66, 2207–2216 CrossRef CAS.
- Y. Xiong, Y. Yang, F. DiSalvo and H. Abruña, ACS Nano, 2020, 14, 15–41 Search PubMed.
- L.-L. Ling, W.-J. Liu, S.-Q. Chen, X. Hu and H. Jiang, ACS Appl. Nano Mater., 2018, 1, 3331–3338 CrossRef CAS.
- J. Wang, G. Wu, W. Wang, W. Xuan, J. Jiang, J. Wang, L. Li, W.-F. Lin, W. Ding and Z. Wei, J. Mater. Chem. A, 2019, 7, 25–41 Search PubMed.
- W. Guo, X. Gao, M. Zhu, C. Xu, X. Zhu, Z. Xuyan, R. Sun, Z. Xue, J. Song, L. Tian, J. Xu, W. Chen, Y. Lin, Y. Li, H. Zhou and Y. Wu, Energy Environ. Sci., 2022, 16, 335–341 Search PubMed.
- L. Huang, Y.-Q. Su, R. Qi, D. Dang, Y. Qin, S. Xi, S. Zaman, B. You, S. Ding and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 25530–25537 CrossRef CAS PubMed.
- S. Zaman, Y.-Q. Su, C.-L. Dong, R. Qi, L. Huang, Y. Qin, Y.-C. Huang, F.-M. Li, B. You, W. Guo, Q. Li, S. Ding and B. Yu Xia, Angew. Chem., Int. Ed., 2022, 61, e202115835 CrossRef CAS.
- Z. Chen, C. Hao, B. Yan, Q. Chen, H. Feng, X. Mao, J. Cen, Z. Q. Tian, P. Tsiakaras and P. K. Shen, Adv. Energy Mater., 2022, 12, 2201600 CrossRef CAS.
- M. Chen, S. Hwang, J. Li, S. Karakalos, K. Chen, Y. He, S. Mukherje, D. Su and G. Wu, Nanoscale, 2018, 10, 17318–17326 RSC.
- S. Yin, Y.-N. Yan, L. Chen, N. Cheng, X. Cheng, R. Huang, H. Huang, B. Zhang, Y.-X. Jiang and S.-G. Sun, ACS Nano, 2024, 18, 551–559 CrossRef CAS.
- L. Chong, H. Zhou, J. Kubal, Q. Tang, J. Wen, Z. Yang, I. D. Bloom, D. Abraham, H. Zhu, J. Zou and W. Ding, Chem Catal., 2023, 3, 100541 CrossRef CAS.
- N. Zhou, R. Zhang, R. Wang and Y. Li, Chem. Eng. J., 2023, 474, 146010 CrossRef CAS.
- Y. Pan, J. Gao, Y. Li, E. Lv, U. Khan, X. Yang, J. Yao, A. Nairan and Q. Zhang, Small, 2024, 20, 2304594 CrossRef CAS.
- J. Lai, S. Chen, X. Liu, X. Yan, Z. Qin, L. Xie, Z. Lin, Z. Cai, Y. Zhao, H.-L. Wang, Y. Huang and Q. Li, ACS Catal., 2023, 13, 11996–12006 CrossRef CAS.
- Y. Zhou, Y. Zhang, Z. Li, C. Hao, Y. Wang, Y. Li, Y. Dang, X. Sun, G. Han and Y. Fu, Chemosphere, 2020, 259, 127463 CrossRef CAS PubMed.
- X. Feng, H.-J. Zhang, Z. Zhou, C. Zhu, L. Jia, Z. Ma and Y. Xue, J. Electrochem. Soc., 2024, 171, 125–141 Search PubMed.
- J. Gao, X. Zhou, Y. Wang, Y. Chen, Z. Xu, Y. Qiu, Q. Yuan, X. Lin and H.-J. Qiu, Small, 2022, 18, 2202071 CrossRef CAS.
- T. Yoo, J. M. Yoo, A. Sinha, M. Bootharaju, E. Jung, H. Lee, B.-H. Lee, J. Kim, W. Antink, Y. Kim, J. Lee, E. Lee, D. Lee, S.-P. Cho, S. Yoo, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2020, 5, 6–15 Search PubMed.
- L. Huang, M. Wei, R. Qi, C.-L. Dong, D. Dang, C.-C. Yang, C. Xia, C. Chen, S. Zaman, F.-M. Li, B. You and B. Y. Xia, Nat. Commun., 2022, 13, 6703 CrossRef CAS.
- W. Xiao, D. Yan, Q. Zhao, D. Bukhvalov and X. Yang, Appl. Catal., B, 2024, 346, 123740 CrossRef CAS.
- Y. Zeng, J. Liang, C. Li, Z. Qiao, B. Li, S. Hwang, N. Kariuki, C.-W. Chang, M. Wang, M. Lyons, S. Lee, Z. Feng, G. Wang, J. Xie, D. Cullen, D. Myers and G. Wu, J. Am. Chem. Soc., 2023, 145, 25–36 CrossRef.
- W. Xu, Z. Zhu, Y. Wang, P. Cui, L. Tong, K. Zhao, J. Yuan, Z.-Y. Zhou, H.-W. Liang, N. Tian and S.-G. Sun, J. Mater. Chem. A, 2023, 11, 36–46 Search PubMed.
- Y. Yan, G. Li, X. Cheng, S. Yin, H. Zeng, R. Huang, C. Wang, Y. Jiang and S. Sun, Int. J. Hydrogen Energy, 2023, 48, 19522–19531 CrossRef CAS.
- F. Xiao, Q. Wang, G.-L. Xu, X. Qin, I. Hwang, C.-J. Sun, M. Liu, W. Hua, H.-w. Wu, S. Zhu, J.-C. Li, J.-G. Wang, Y. Zhu, D. Wu, Z. Wei, M. Gu, K. Amine and M. Shao, Nat. Catal., 2022, 5, 503–512 CrossRef CAS.
- Z. Qiao, C. Wang, C. Li, Y. Zeng, S. Hwang, B. Li, S. Karakalos, J. Park, A. Kropf, E. Wegener, Q. Gong, H. Xu, G. Wang, D. Myers, J. Xie, J. Spendelow and G. Wu, Energy Environ. Sci., 2021, 14, 36–49 RSC.
- K. Wan, H. Chen, J. Wang, B. Li, M. Chai, P. Ming and C. Zhang, J. Catal., 2023, 427, 115124 CrossRef CAS.
- R. Mo, X. Zhang, Z. Chen, S. Huang, Y. Li, L. Liang, Z. Q. Tian and P. K. Shen, Int. J. Hydrogen Energy, 2021, 46, 15991–16002 CrossRef CAS.
- Z. Cui, G. Fu, Y. Li and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 9901–9905 CrossRef CAS PubMed.
- F. Zhou, Y. Ruan, M. Zhu, X. Gao, W. Guo, X. Liu, W. Wang, M. Chen, G. Wu, T. Yao, H. Zhou and Y. Wu, Small, 2023, 19, 2302328 CrossRef CAS.
- X. Ao, W. Zhang, B. Zhao, y. ding, G. Nam, L. Soule, A. Abdelhafiz, C. Wang and M. Liu, Energy Environ. Sci., 2020, 13, 12–17 RSC.
- W. Jung, W. Lee, H.-S. Oh and B. Popov, J. Mater. Chem. A, 2020, 8, 4624–4633 Search PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- X. Han, Q. Wang, Z. Zheng, Z. Nan, X. Zhang, Z. Song, M. Ma, J. Zheng, Q. Kuang and L. Zheng, ACS Sustain. Chem. Eng., 2021, 9, 3–12 CrossRef.
- X. X. Wang, S. Hwang, Y.-T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 CrossRef CAS.
- P. Guo, X. Yunfei, B. Liu, M. Ma, L. Shen, Y. Dai, Z. Zhang, Z. Zhao, Y. Zhang, L. Zhao and Z. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 503–512 Search PubMed.
- Y.-F. Xia, B. Liu, P. Guo, F.-D. Tu, L.-X. Shen, M. Ma, Y.-K. Dai, J. Liu, B. Xu, Y.-L. Zhang, L. Zhao, Y. Wang and Z.-B. Wang, J. Catal., 2024, 429, 115296 CrossRef CAS.
- Y. Xue, H. Li, X. Ye, S. Yang, Z. Zheng, X. Han, X. Zhang, L. Chen, Z. Xie and Q. Kuang, Nano Res., 2019, 12, 8–16 Search PubMed.
- J. Chen, J. Dong, J. Huo, C. Li, L. Du, Z. Cui and S. Liao, Small, 2023, 19, 2301337 CrossRef CAS.
- X. Li, Y. He, S. Cheng, B. Li, Y. Zeng, Z. Xie, Q. Meng, L. Ma, K. Kisslinger, X. Tong, S. Hwang, S. Yao, C. Li, Z. Qiao, C. Shan, Y. Zhu, J. Xie, G. Wang, G. Wu and D. Su, Adv. Mater., 2021, 33, 2106371 CrossRef CAS.
- Y. Zhou, J. Li, Q. Wu, N. Wang, L. Xing, L. Wang, L. Du and S. Ye, Small, 2024, 20, 2312011 CrossRef CAS PubMed.
- Y. Luo, K. Li, Y. Chen, J. Feng, L. Wang, Y. Jiang, L. Li, G. Yu and J. Feng, Adv. Mater., 2023, 35, 2300624 CrossRef CAS.
- Y. Hu, X. Guo, T. Shen, Y. Zhu and D. Wang, ACS Catal., 2022, 12, 5380–5387 CrossRef CAS.
- J. Xu, K. Feng, C. Lu, X. Wang, J. Chen, Z. Wang, J. Zhong, Y. Huang and T. K. Sham, J. Phys. Chem. Lett., 2023, 14, 8296–8305 CrossRef CAS.
- B. Sun, Y.-C. Jiang, Q.-L. Hong, X. Liu, F.-M. Li, D.-S. Li, Y. Yang and Y. Chen, J. Energy Chem., 2023, 85, 3077–3087 CrossRef.
- S. Y. Lim, S. Martin, G. Gao, Y. Dou, S. B. Simonsen, J. O. Jensen, Q. Li, K. Norrman, S. Jing and W. Zhang, Adv. Funct. Mater., 2021, 31, 2006771 CrossRef CAS.
- H. Niu, L. Huang, Y. Qin, R. Qi, B. Mei, D. Wu, F.-M. Li, B. You, Q. Li, Y. Yao, Z. Wang, T. Yao, S. Ding, W. Guo, Y. Chen, Y. Su, F. Song and B. Y. Xia, J. Am. Chem. Soc., 2024, 146, 22650–22660 CrossRef CAS PubMed.
- P. Guo, B. Liu, F. Tu, Y. Dai, Z. Zhang, X. Yunfei, M. Ma, Y. Zhang, L. Zhao and Z. Wang, Energy Environ. Sci., 2024, 17, 3077–3087 RSC.
| This journal is © The Royal Society of Chemistry 2025 |