
Advanced diesel from ethanol: a pathway to produce sustainable and high-quality drop-in biofuels†
Juan-Manuel
Restrepo-Flórez‡
 abc,
Javier E.
Chavarrio‡
a,
Emmanuel
Canales‡
a,
Dustin
Witkowski
b,
Srinath
Subramanian
b,
Paolo
Cuello-Peñaloza
a,
David A.
Rothamer
*b,
Christos T.
Maravelias
*de and
George W.
Huber
*a
abc,
Javier E.
Chavarrio‡
a,
Emmanuel
Canales‡
a,
Dustin
Witkowski
b,
Srinath
Subramanian
b,
Paolo
Cuello-Peñaloza
a,
David A.
Rothamer
*b,
Christos T.
Maravelias
*de and
George W.
Huber
*a
aDepartment of Chemical and Biological Engineering, University of Wisconsin–Madison, USA. E-mail: gwhuber@wisc.edu
bDepartment of Mechanical Engineering, University of Wisconsin–Madison, USA. E-mail: rothamer@wisc.edu
cDepartment of Chemical Engineering, University of Florida, USA
dDepartment of Chemical and Biological Engineering, Princeton University, USA. E-mail: maravelias@princeton.edu
eAndlinger Center for Energy and the Environment, Princeton University, USA
First published on 7th October 2024
Abstract
In this work, we develop a novel technology for the transformation of ethanol into diesel via Guerbet coupling and etherification. Our strategy overcomes the limitations of previous studies, namely, the low yield of diesel #2, and the complex separation network required. To overcome these limitations, we rely on the use of hydrogenolysis for the removal of esters, and the implementation of butanol recycling. Herein, we present a thorough analysis of this strategy integrating the experimental evaluation of catalysts for the involved reactions, process synthesis, technoeconomic analysis, lifecycle analysis, fuel property modelling, and characterization of the fuels produced in a diesel engine. In contrast to other catalytic strategies, in this work diesel #2 constitutes the main product (92% of the produced fuels). The diesel produced has excellent cold flow properties (cloud point ∼ −28 °C) and a very high cetane number (∼94) while satisfying flash point requirements. A technoeconomic analysis leads to a minimum fuel selling price (MFSP) between $4.6–8.4 per GDE for ethanol prices between $1.5 per gal and $3.4 per gal (in 2021 dollars). Depending on the carbon intensity of the ethanol used as feedstock, and the energy consumption of the process, we found that reductions >70% in GHG emissions are feasible in comparison with fossil diesel. The diesel fuel can become carbon negative if an ethanol feedstock with a sufficiently low carbon footprint is used and the ethanol upgrading biorefinery uses renewable hydrogen and produces steam using renewable natural gas.
1. Introduction
The consumption of diesel fuel is projected to remain constant in the U.S. at ∼4 million barrels per day for the next 30 years.1 With CO2 emissions of 10.18 kg gal−1,2 this consumption would result in the release of ∼1.7 million tons of CO2 into the environment every day (∼10% of the total US emissions). Diesel fuel primarily powers ships, trucks, and heavy-duty vehicles, which pose challenges for electrification, unlike lighter vehicles. This makes it a complex sector to decarbonize.3 Biofuels will play a significant role in these applications during the energy transition and are part of the long-term vision of the U.S. DOE.4 A significant amount of research has been devoted to analyzing different renewable diesel production pathways5–9 (Table 1). These efforts have been focused on identifying biofuel alternatives satisfying three requirements: (1) economic feasibility, (2) low greenhouse gas emissions (<60% of fossil diesel), and (3) operability, which has been assessed based on the fuel properties.5 Ideal production pathways should lead to the production of sustainable diesel with properties equal to or superior to those of fossil diesel. For reference, at a commercial scale in the U.S., the most widely produced renewable alternatives to diesel are hydroprocessed esters and fatty acids (HEFA) and biodiesel, both of which are derived from vegetable oils, a feedstock with limited availability and high prices.11| CN [—] | Flash point [°C] | Cloud point [°C] | Energy density [MJ L−1] | Viscosity [mm2 s−1] | Density [kg m−3] | MFSPa [$ per GDE] | ΔGHG [%] | |
|---|---|---|---|---|---|---|---|---|
| a MFSP is in 2021 dollars. b Diesel spot price. | ||||||||
| Diesel | >40 | >52 | ∼35 | 1.9–4.1 | >820 | 1.8–3.5b | [—] | |
| Biodiesel | >47 | >93 | −5 to 15 | 33 | 1.9–6 | 880 | 5.3 | 39–90 |
| Renewable diesel HEFA | >70 | >61 | −39 | 34.4 | 2–4 | 770–790 | 4.7–7.8 | 60–80 |
| Fischer Tropsch diesel | >70 | >61 | −34 to 10 | 34 | 2–4 | 770–790 | 5.5 | 89 |
| HTL fuel | 30–68 | >55 | −60 to 20 | 34.5–36.9 | 2.3–2.7 | 800–879 | 4.3–6.7 | 62–73 |
| Farnesane | 58.6 | 110 | −73 | 33.5 | 14 | 773 | 7.8–9.4 | 61 |
| Isoalkanes from VFA | 48 | 74 | −80 | 34.6 | 1.49 | 780 | 12.5 | <60% |
| Ethanol-to-distillate | 55–68 | >54 | −60.1 | 35 | 2.0–4.8 | 786 | 4.7–7.2 | >90% |
| Fatty acid fuel esters | 50–60 | >130 | −10 | >30 | 2.9–3.7 | 817–861 | 3.8–5.3 | 53% |
| Short chain esters from oilseed crops | 52 | 111 | −18 | 29.6 | 1.7 | 871 | 25.1 | 20–53 |
| Polyoxymethylene ethers | 73–75 | 62–63 | −27–19 | 20–32 | 1.9 | 1.0662 | 6.4–7 | 81 |
| 4-Butoxyheptane | 80 | 64 | −80 | 30.8 | 0.795 | 791 | 11.0 | 27 |
| Alkoxyalkanoates from lactate esters | 44–62 | 65–117 | <−50 | 23–33 | 1.2–2.3 | 900–930 | 7.8 | 65 |
| Fatty alkyl ethers | 74–104 | >150 | −5 to −16 | 34–36 | 830–850 | 6.3 | 57–75 | |
| Ethanol Guerbet coupling and etherification | 73.2 | >50 | −37 | 27.5 | 1.92 | 815 | 4–7.7 | >50 |
| This work | 94 | ∼52 | −28.9 | 29.36 | 1.7 | 794.7 | 4.6–8.4 | 0–144 |
Among the pathways listed in Table 1, we are interested in ethanol upgrading by Guerbet coupling and etherification.10 This pathway has several advantages in terms of fuel properties, GHG emission reduction, and feedstock availability, while simultaneously showing technoeconomic feasibility.10 From a fuel property perspective, it has been demonstrated that it is possible to produce a renewable diesel #2 that satisfies most ASTM requirements with a high cetane number (>70) and good cold flow properties (cloud point < −37 °C).10 These outstanding fuel properties are the result of using an ether rich blend as diesel fuel. Ethers are known for their high cetane number and have been identified as a potential diesel replacement in several studies.12–14 In terms of GHG mitigation potential, the possibility of obtaining more than 60% reduction in comparison with fossil diesel has been shown.10 Furthermore, depending on the carbon intensity of the ethanol used, it is possible to produce carbon neutral or carbon negative diesel fuel. We note that a pathway based on ethanol upgrading is particularly well positioned in terms of feedstock availability. Not only is there already an ethanol infrastructure in the U.S., with a production capacity close to 17 billion gallons per year,15,16 but also, there are emerging technologies (e.g., fermentation of lignocellulosic residues17–19 and syngas20) that may disrupt the ethanol market by increasing the supply while reducing the environmental impacts and costs of ethanol production.
Despite the advantages of ethanol upgrading via Guerbet coupling and etherification, we have identified three limitations hindering the deployment of this technology. First, the Guerbet coupling reaction of ethanol produces 1-butanol as the main product.21–24 The excess of 1-butanol in the etherification reaction leads to the production of a significant fraction of dibutyl ether, with a flash point (25 °C) well below the diesel #2 requirement (>52 °C).25 Consequently, the yield of diesel #2 gets is reduced; for reference, in our previous work, we found a maximum diesel #2 yield of ∼50%.10 Second, the most effective Guerbet coupling catalysts for diesel production (i.e., those that produce the highest amount of C6+ alcohols) also produce significant amounts of esters, aldehydes, and ketones.21,26 Esters significantly affect the ether selectivity in etherification reactions, while the aldehydes and ketones produced are not suitable for use in diesel due to their physicochemical properties (low molecular weight aldehydes and ketones have a low flash point). Third, while it is expected that the diesel #2 produced by Guerbet coupling and etherification would improve the emission profile in comparison with diesel fuel based on results obtained with di-butyl-ether,27 the blends developed in this work have not been characterized in terms of engine performance.
In this work, we address the previous limitations and present a new strategy for the catalytic upgrading of ethanol into diesel #2. We present laboratory experimental results that are incorporated into process design, technoeconomic analysis (TEA), lifecycle assessment (LCA), and characterization of the fuels produced in terms of their physicochemical properties and their experimental evaluation in a diesel engine. The developed process uses four catalytic steps: Guerbet coupling, hydrogenolysis, etherification, and oligomerization. Importantly, we implement a butanol recycling strategy that significantly improves the production of C6+ alcohols. While this strategy has been suggested in the literature,23 this is the first time that experimental evidence supporting the effect of butanol recycling on alcohol distribution is presented. Furthermore, the hydrogenolysis step used, also employed for the first time in this type of application, allows us to efficiently remove esters, aldehydes, and ketones, transforming them into alcohols. This work is the result of a collaborative effort among catalyst experts, fuel property modelers, process and systems engineers, and engine researchers. This broad and convergent synergy has enabled us to couple all the biofuel production stages, presenting a comprehensive view of this novel and promising diesel production strategy.
2. Catalytic pathway development and technology overview
A schematic of the developed technology is shown in Fig. 1. Anhydrous ethanol is used as the feedstock of a Guerbet reaction, in which higher alcohols (C4+) are obtained as a main product along with esters, aldehydes, and ketones as byproducts. Wastewater is a byproduct of this process. The outlet from the Guerbet coupling reaction is used as the feedstock for hydrogenolysis in which esters, aldehydes, and ketones are transformed into their parent alcohols. The hydrogenolysis reaction product consists mainly of higher alcohols, and unconverted ethanol. In this work, we implement a recycling strategy in which unconverted ethanol and a significant fraction of the butanol produced are returned to the Guerbet reactor. This recycling leads to an operation in which ethanol and butanol are cofed to the Guerbet reactor. The partial recycling of butanol reduces the butanol content in the etherification reaction. Alternatively, one can use catalysts with very high selectivity toward C6+ alcohols such as the one developed by Gu and co-workers.28 The fraction of unrecycled butanol, along with the C6+ alcohols produced are split into two fractions: (1) the C4–C8 alcohols, which are fed into an etherification reaction in which ethers with 8–16 carbons are produced and (2) the larger alcohols (C10+), which are used directly in the diesel blend. Olefins (C4–C8) are produced in the etherification process and are oligomerized to increase their average molecular weight so that they can be blended into diesel.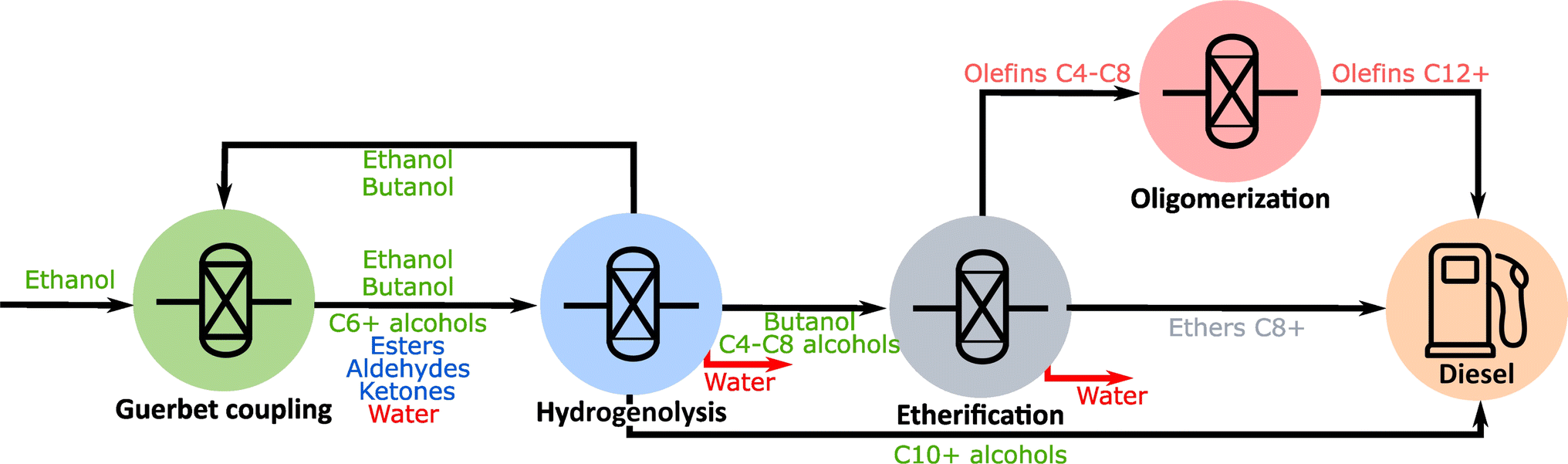 | ||
| Fig. 1 Schematic of the technology developed in this work. | ||
2.1 Producing higher alcohols from ethanol
 | ||
Fig. 2 (a) Conversion of ethanol and butanol into C6 alcohols and esters. Ratio of the molar flow of alcohols in the cofeed strategy/alcohols in control experiments, normalized by unit time and mass of the catalyst: (b) hexanol and 2-ethyl-butanol, and (c) octanol and 2-ethyl-hexanol. T = 325 °C, Ptot = 300 psig, (ethanol + butanol)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 4:1, 100–300 mg 0.1% Cu/Mg3AlO. :H2 = 4:1, 100–300 mg 0.1% Cu/Mg3AlO. | ||
The motivation for recycling butanol into the Guerbet coupling reactor is to increase the production of hexanol by facilitating the overall reaction ethanol + butanol → hexanol + H2O. However, there are many side reactions, for example, 2-ethyl-butanol is also synthesized from these two reactants when ethanol acts as the electrophile.29 Esterification reactions produce butyl acetate and ethyl butyrate as shown in Fig. 2(a). The product pool becomes even more complex since alcohols can react with themselves. For example, two ethanol molecules can produce butanol or ethyl acetate and two butanol molecules can produce 2-ethyl-hexanol or butyl butyrate.
Previous etherification studies using acid catalysts have demonstrated that linear alcohols preferentially undergo bimolecular dehydration to produce ethers, whereas branched alcohols form olefins through monomolecular dehydration.10,29 Therefore, tracking the ratio of linear to branched alcohols is important. Fig. S1(a) and (b)† show this ratio for C6 and C8 alcohols, respectively. Our results indicate that the catalyst is selective for forming linear alcohols (values > 1). For C6 alcohols, the ratio of hexanol to 2-ethyl-butanol is slightly higher in the cofeed experiments than in the control, indicating that cofeeding butanol facilitates the production of hexanol more than its branched counterpart. On average, the outlet hexanol molar flowrate is 3.4 and 3.2 higher than the molar flow of 2-ethyl-butanol for the cofeed and pure ethanol feed experiments, respectively (see Table S1† for carbon flowrates in each run). For C8 alcohols, a more noticeable difference is observed. Fig. S1(b)† shows that cofeeding ethanol and butanol produces more 2-ethyl-hexanol in comparison with the control experiment. For reference, in the co-feed case, the octanol/2-ethyl-hexanol ratio is close to one; in contrast, in the control experiments, it ranges between ∼1.5–3 depending on the WHSV. This increase in the relative concentration of 2-ethyl-hexanol in the cofeed experiments can be explained by considering that octanol is formed only from the coupling of ethanol and hexanol, with the former acting as the nucleophile and the latter acting as the electrophile. In contrast, 2-ethyl-hexanol is produced either when hexanol is the nucleophile and ethanol is the electrophile or by coupling of two butanol molecules. Thus, cofeeding butanol enhances butanol self-condensation to form 2-ethyl-hexanol. Fig. S2(a)† shows that overall linear alcohols are formed preferentially, demonstrating that a cofeed strategy is selective for producing the most relevant diesel fuel precursors. A comparison of molar production of linear and branched alcohols between cofeed and control experiments is presented in Fig. 2(b) and (c). The data indicate that cofeeding alcohols boosts the formation of hexanol and 2-ethyl-butanol by a factor of 2.2 (Fig. 2(b)). This suggests that the presence of butanol in the feed does not change the nucleophilic/electrophilic role of ethanol in the reaction. Similarly, Fig. 2(c) shows that the production of C8 alcohols is enhanced by cofeeding butanol to the reactor, with the caveat that the formation of the branched alcohol is more accelerated than the formation of octanol.
Fig. S3(a)† shows the production of esters from ethanol/butanol coupling. Production of ethyl acetate remained unchanged in cofeed and control experiments. In contrast, the production of other esters like ethyl butyrate, butyl acetate, and butyl butyrate is enhanced. The data show that butyl acetate is produced preferentially over ethyl butyrate, which is due to the large excess of ethanol (70% mol in feed) that induces the formation of acetaldehyde and subsequently dehydrogenative coupling between acetaldehyde and butanol. The presence of butanol in cofeed experiments leads to a higher production of butyl butyrate compared to the control experiments. The overall selectivity of C6+ alcohols with respect to esters is presented in Fig. S2(b).† This figure suggests that the Guerbet coupling stage will benefit from a cofeed strategy since the ratio of C6+ alcohols to esters improves. We note that the production of long chain alcohols competes strongly with ester formation, given that the values in Fig. S2(b)† are around 1.
The carbon yield for diesel fuel precursors (alcohols, aldehydes, ester, ethers, and ketones) is presented in Table 2. The values reported have been calculated by using the information contained in Table S1† and eqn (1). For simplicity, the information has been condensed into categories based on functional groups. In eqn (1), nC,in refers to the total carbon flow rate entering the Guerbet reactor, while nC,i,out refers to the carbon flowrate at the outlet of the reactor specifically for compound i.
 | (1) |
:P(H2) = 4
| Experiment | Cofeed 1 | Control 1 | Cofeed 2 | Control 2 | Cofeed 3 | Control 3 |
|---|---|---|---|---|---|---|
| WHSV (h−1) | 26.27 | 26.27 | 6.53 | 6.53 | 1.33 | 1.33 |
| C balance (%) | 99.7 | 100.0 | 94.5 | 96.5 | 95.8 | 91.1 |
| EtOH conversion (%) | 26.57 | 33.16 | 41.75 | 41.87 | 66.04 | 73.88 |
| Diesel fuel precursor yield (%) | 7.76 | 6.18 | 13.41 | 10.68 | 28.90 | 23.54 |
| C6+ alcohol (%) | 4.04 | 3.08 | 6.67 | 5.77 | 7.36 | 5.05 |
| C6+ aldehyde (%) | 0.90 | 0.83 | 1.26 | 1.26 | 3.17 | 2.80 |
| C6+ ester (%) | 2.28 | 1.18 | 3.67 | 1.90 | 11.43 | 10.43 |
| C4+ ketones (%) | 0.47 | 0.65 | 1.46 | 0.75 | 6.86 | 4.92 |
As presented in Table 2, the yield of diesel fuel precursors increases in our cofeed strategy in comparison to the control experiments. Percentage increments were between 1.58 and 5.36. The main contributors for this indicator are alcohols and esters, which show an average increment of ∼1 percentage point each.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, with the ability to perform C–O scissions while exhibiting negligible activity for C–C bond cleavage.32 We initially conducted catalytic studies with a model feed of 5% hexyl-acetate and 95% butanol at WHSV = 0.05, 0.16, 0.40, and 0.81 gester gcat−1 h−1. Experimental results for the catalytic reduction of esters through hydrogenolysis are shown in Fig. 3 for the hexyl acetate–butanol model feed. We note that in these experiments the poisoning effect of water has not been studied; this would be the subject of future work. The concentrations presented in panels (a) and (b) represent the liquid phase after condensation of the effluents leaving the hydrogenolysis reactor.
O bonds, with the ability to perform C–O scissions while exhibiting negligible activity for C–C bond cleavage.32 We initially conducted catalytic studies with a model feed of 5% hexyl-acetate and 95% butanol at WHSV = 0.05, 0.16, 0.40, and 0.81 gester gcat−1 h−1. Experimental results for the catalytic reduction of esters through hydrogenolysis are shown in Fig. 3 for the hexyl acetate–butanol model feed. We note that in these experiments the poisoning effect of water has not been studied; this would be the subject of future work. The concentrations presented in panels (a) and (b) represent the liquid phase after condensation of the effluents leaving the hydrogenolysis reactor.
 | ||
| Fig. 3 (a) and (b) mole concentration (M) of the reactants and most abundant products in the liquid product at the outlet of the hydrogenolysis reactor and (c) total ester conversion as a function of the inverse of the WHSV for a blend of 5% hexyl acetate and 95% butanol (mol mol−1), T = 200 °C, Ptot = 420 psig, hexyl acetate:H2 = 1:480 ratio defined on a molar basis, 100–300 mg of 10% wt Cu/ZrO2. Data at 1/WHSV = ∞ correspond to the equilibrium concentrations from Aspen Plus. | ||
As observed in Fig. 3(a), butanol concentration presents a rapid decrease from 9.97 M to 9.6 M (1/WHSV = 1.23). Interestingly, butyl acetate exhibits the opposite trend (see Fig. 3(b)) by increasing from 0 to 0.3 M in the same contact time frame. In addition, hexanol concentration is observed to increase rapidly reaching values close to those of hexyl acetate at contact time zero, while ethanol increases more modestly as contact time increases, reaching similar concentrations to hexanol at high contact times (∼20 h). The observed trend is an indication of a two-stage process, where initially hexyl acetate undergoes a transesterification reaction governed by hexyl acetate + butanol → butyl acetate + hexanol, and then butyl acetate undergoes hydrogenolysis, releasing the ethanol and butanol moiety, explaining the increase in concentration of butanol and ethanol at 1/WHSV > 1.23 h. We believe that butyl acetate is the main species that undergoes the hydrogenolysis reaction; however, we do not preclude that a fraction of the hexyl acetate fed to the system can also undergo catalytic reduction. Interestingly, butyl acetate emerges as the predominant ester between 1/WHSV = 1.33 and 6.25 h, while butyl butyrate concentration becomes more important at contact times higher than 6.25 h. A complete description of the molar concentrations of all the species identified in the outlet of the hydrogenolysis reactor is presented in Table S3.†
Based on the species identified experimentally and considering the poor ability of copper to cleave C–C bonds, we postulate that the system can be accurately described by reactions (R1) and (R2). (R1) is the hydrogenolysis of the ester. (R2) involves hydrogenation of the aldehyde.
 | (R1) |
 | (R2) |
We performed thermodynamic equilibrium calculations under the reaction conditions in Aspen Plus V12.1 by implementing a linearly independent set of equilibrium reactions derived from the systematic combination of ethyl, butyl, and hexyl chains in reactions (R1) and (R2) (see Table S4†). Thermodynamic equilibrium calculations represent the limiting case at which WHSV = 0 h−1. For comparison purposes, the results of the thermodynamic calculations are depicted in Fig. 3(a) and (b) at 1/WHSV = ∞, and complete data for all the products are presented in Table S3.†Fig. 3(a) and (b) show that the extrapolated experimental molar concentrations of all the species identified in the outlet of the hydrogenation reactor agree with the thermodynamic equilibrium calculations performed in Aspen.
In reactions (R1) and (R2), it is observed that bond transformations result from C–O cleavage and hydrogenation of CO, which implies no change in the identity of alkyl chains Ri and Rj. In other words, the moles of each alkyl chain are conserved (alkyl chain balance is usually >95% for all our experiments (see Table S3†)). This fact allows us to easily identify the fate of each alkyl chain and represent our results in terms of alkyl chain selectivity as defined by eqn (2).
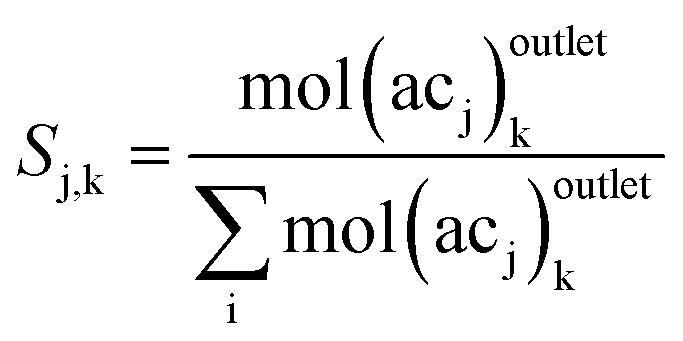 | (2) |
Other species containing ethyl chains like ethyl acetate, ethyl butyrate and acetaldehyde were detected as products with low preference for ethyl chains. Fig. S4(b),† shows the selectivity for butyl chains. The results point out that reacted butanol molecules preferentially formed butyl acetate, butyl butyrate and butyraldehyde. Finally, Fig. S4(c)† shows the selectivity for hexyl chains. The data indicate that hexyl chains were hydrogenated to form hexanol, while butyl hexanoate was identified with a hexyl selectivity lower than 2%. Once again, our experimental data were compared with thermodynamic equilibrium calculations, demonstrating that at low WHSV values the alkyl chains tend to follow the equilibrium distribution.
Overall, the data presented in Fig. 3 show that at high contact times (1/WHSV) the system converts most of the hexyl and acetate moieties into hexanol and ethanol, respectively. Fig. S5† shows that butanol conversion remains low (<4%), which indicates that the goal of performing the catalytic reduction of esters, while maintaining the butanol unreacted was satisfactorily achieved. Fig. S5† also depicts the stability of the Cu/ZrO2 catalysts by tracking the conversion of the reactants as a function of time on stream (TOS). For both hexyl acetate and butanol the conversion remains unchanged up to 60 h. Deactivation of the catalyst in this timeframe and conversion regime is then assumed to be negligible, since carbon and mass balances were typically between 93 and 100%.
While it is known that esters have a negative impact on the etherification reaction,10 the effect of each ester is still unclear. Thus, to track the effectiveness of the hydrogenolysis reaction, we define total ester conversion (eqn (3)). This function tracks the fraction of ester functionality removed without tracking particular esters. Experimental results are shown in Fig. 3(c) along with the equilibrium value obtained through simulations (1/WHSV = ∞). The fraction of esters removed increases monotonically as the contact time increases, approaching the thermodynamic limit (89.1%) when 1/WHSV is 20 h (ester removal ∼87.7%).
 | (3) |
After the initial characterization, we tested the catalyst with a realistic Guerbet coupling stream, consisting of a complex mixture of alcohols and esters with varying alkyl chain lengths. The composition of this stream is based on previous work, which demonstrated a 66% single-pass conversion of ethanol.10 Herein, the complexity of the stream is reduced by neglecting compounds containing alkyl chains higher than eight carbons (see Table S2† for composition). Equilibrium calculations were also carried out for this system using Aspen Plus®. Given the presence of secondary alcohols in the Guerbet coupling stream, new chemical reactions to define the equilibrium between secondary alcohols and their respective ketones were added (eqn (R3) and Table S5†).
 | (R3) |
Fig. 4 shows the results of a catalytic ester reduction test using the simulated Guerbet coupling stream. The molar concentration distribution of the feed used in the experiment, the experimental liquid phase molar concentrations obtained in this study, and the equilibrium concentrations achieved in our simulations are shown in the figure. The results were calculated for the liquid phase after condensation of the products. The molar concentration of linear alcohols increased due to the hydrogenolysis of esters. Negligible dehydrogenation of branched alcohols was observed as predicted by thermodynamics. In contrast, the dehydrogenation of secondary alcohols to their respective ketones was observed. Overall, branched and secondary alcohols remained almost unchanged, while the main catalytic activity was due to hydrogenolysis of esters and hydrogenation of aldehydes. Our results suggest that butyl butyrate, hexyl butyrate, and butyl hexanoate became the predominant esters in the outlet stream of the reactor, owing to butanol and hexanol being the most abundant species in the feed. Overall, the ester mol fraction was reduced from 7.6% to 0.3%, the linear alcohols mol fraction increased from 80.81 to 89.1%, and the total alcohols mol composition increased from 92.3 to 99.3%. The total ester conversion calculated using eqn (3) is 96.0 ± 0.2%, while the thermodynamic limit predicted in Aspen is 96.35%. This result is shown in Fig. 4, where the molar concentration of esters from our experiment and the thermodynamic equilibrium concentration are virtually the same. A complete description of the molar concentration of each species identified in the outlet of the reactor can be found in Table S6.† These results highlight the feasibility of using hydrogenolysis to remove the esters produced in the Guerbet coupling reactor.
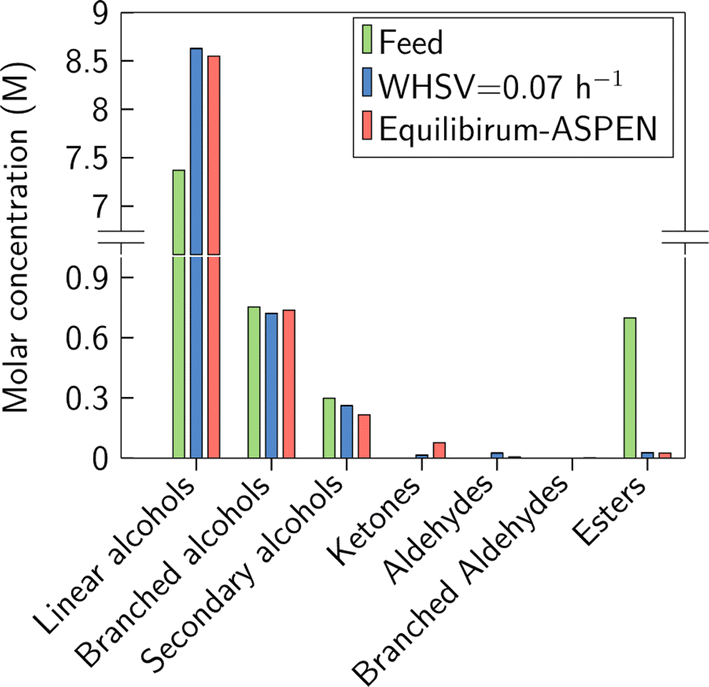 | ||
| Fig. 4 Molar concentration in the liquid phase for the hydrogenation of a simulated Guerbet coupling stream. The experiments were conducted at WHSV = 0.07 h−1, T = 200 °C, Ptot = 420 psig, ester:H2 = 1:480, 500 mg 10% wt. Cu/ZrO2. The detailed feed composition is provided in Table S2.† | ||
:3, using the results for a 30% single-pass ethanol conversion in the Guerbet step (see Table S10†). Additionally, we implement a hydrogenolysis step to remove the esters. We select operational conditions (temperature, pressure, and the hydrogen to alcohol ratio) to ensure full conversion of ketones and aldehydes and 95% conversion of esters. The Guerbet coupling reactor operates at 325 °C and 25 bar with hydrogen as a carrier gas in a 1:4 molar ratio of hydrogen:(ethanol + butanol). The hydrogenolysis reactor operates at 200 °C and 25 bar, with a hydrogen:ester ratio of 400:1. Both reactors operate at a slightly higher pressure than that reported experimentally to facilitate subsequent separations. The products of the hydrogenolysis reactor are partially condensed in a flash tank, enabling hydrogen recycling. A sequence of distillation columns and a molecular sieving unit are used to recover and recycle the reactants (ethanol and butanol) (streams 34 and 36) as well as obtain a higher alcohol rich stream (stream 37), and wastewater (streams 16 and 21). We note that column 3 splits stream 37, rich in higher alcohols, into a heavy product at the bottom containing alcohols with more than 10 carbons, and a light product, at the top, rich in C4–C9 alcohols which is the feedstock for the etherification area. The heavy stream is blended directly into the diesel product. The composition of the etherification feedstock stream is shown in Table 3. The most abundant alcohol is hexanol, in contrast to the results of Restrepo-Flórez et al., where butanol was the main alcohol.10 The ester fraction is 0.32% mol; thus, the etherification feed consists almost entirely of alcohols.
 | ||
| Fig. 5 Layout of the Guerbet and hydrogenolysis area. Chemical species labeling: A: alcohols, ES: esters, K: ketones, AL: aldehydes, OP: olefins/paraffins, H: hydrogen, and W: water. The numerical characters indicate the carbon length. | ||
| Chemical species | Symbol | Mol percentage |
|---|---|---|
| N-Butanol | A4 | 34.622 |
| 2-Butanol | A4-2N | 1.324 |
| 2-Methyl-1-butanol | A5-2 | 3.612 |
| 1-Hexanol | A6 | 40.221 |
| 2-Ethyl-1-butanol | A6-2 | 8.329 |
| 2-Heptanol | A7-2N | 3.406 |
| 2-Ethyl-1-hexanol | A8-2 | 3.592 |
| 1-Octanol | A8 | 4.245 |
| 2-Octanol | A8-2N | 0.109 |
| 2-Nonanol | A9-2N | 0.219 |
| N-Butyl-N-butyrate | ES8 | 0.210 |
| Ethyl-caproate | ES8-2 | 0.080 |
| N-Hexyl-acetate | ES8-3 | 0.032 |
| L/B | 5 | |
| L/S | 16 |
2.2 Producing ethers and higher olefins from higher alcohols
The addition of n-butanol to the ethanol oligomerization reactor leads to increased C6+ alcohol content, compared to when only ethanol is used. Here, the C6+ alcohol mol fraction is at ∼60%, compared to our previously reported work, where we used a dehydration feed stream containing 30 mol% of C6+ alcohols.10 We also note that the addition of n-butanol in the oligomerization reactors leads to an increase in branched alcohols. This is expected, as alcohols larger than ethanol react as nucleophiles to produce larger branched products over alcohol coupling catalysts.23 Furthermore, the size of the secondary alcohols increases with the introduction of the n-butanol recycling stream, leading to an increase in the average size of the final olefin fuel precursors obtained from the dehydration reactor.
A detailed breakdown of the selectivity obtained in etherification reaction (defined as the percentage of total moles of carbon in a product to the total moles of carbon converted) is shown in Table 4. Compared to our previous results,10,33 we observe a reduction in the selectivity toward light ethers (e.g., n-butyl ether), showing a shift toward the production of larger distillate-range molecules. We note that while the C10+ ether selectivity slightly changes between previous results and the findings of this paper (∼50% vs. ∼56%),33 there is also a noticeable change in the yield of C10+ ethers. We previously reported that a relevant economic factor for our ethanol to diesel technology is fuel yield.10 In the current work, we increase the yield of distillate range molecules by reducing the butyl ether yield from 14% to 4%.33 This can be attributed to an increase in the fraction of C6+ alcohols in the feed stream. These results are an indication that the butanol recycling strategy implemented succeeded in reducing the amount of low molecular weight ethers formed. This demonstrates the possibility of using the butanol recycle fraction as a control variable to tune the molecular weight distribution of the products. In addition to the ethers obtained, we also observed a fraction of olefins (4 to 9 carbons). These olefins result from the dehydration of β-branched alcohols and secondary alcohols in the reaction blend.10 Finally, we note that a large fraction of coke is produced (∼18%).33 This indicates that further research is needed to develop tailored catalysts to reduce the coke yield. The catalyst used was tested by running a flow reactor for 21 h continuously. The results are shown in Fig. 6, where we show conversion, carbon balance, and product selectivity as a function of time on stream. These values remain relatively stable throughout the test. More detailed information on the dehydration chemistry, reactor yields, and the pure alcohol feedstock used can be found in Canales et al.33
| Compound | Carbon # | Selectivity |
|---|---|---|
| Ethers | ||
| Butyl ether | 8 | 5.28 |
| Butyl ethyl-butane ether | 10 | 1.56 |
| C10 linear ethers | 10 | 20.98 |
| Butyl ethyl-hexane ether | 12 | 0.85 |
| Hexyl ethyl-butane ether | 12 | 3.18 |
| C12 linear ethers | 12 | 22.20 |
| Hexyl ethyl-hexane ether | 14 | 1.72 |
| Octyl ethyl-butane ether | 14 | 0.47 |
| C14 linear ethers | 14 | 4.81 |
| C16 linear ethers | 16 | 0.30 |
|
||
| Olefins | ||
| Butenes | 4 | 1.70 |
| C5 olefins | 5 | 0.62 |
| C6 olefins | 6 | 5.05 |
| Heptenes | 7 | 4.41 |
| C8 olefins | 8 | 2.76 |
| Nonenes | 9 | 0.26 |
| Unknown products | — | 5.19 |
| Coke | — | 18.66 |
 | ||
| Fig. 6 TOS data for EtOH/oligomerization dehydration products. Reaction conditions: T = 170 °C, P = 110 psig, feedstock flowrate = 0.040 mL min−1, Ar flowrate = 10 mL min−1, WHSV = 1.085 h−1. Data reproduced from ref. 33 with the permission from the Royal Society of Chemistry. | ||
Table S7† shows the conversion rates for each of the reactions that are happening in the dehydration step. Each alcohol in the feedstock can undergo different reactions leading to the formation of ethers or olefins. The observed ether and olefin carbon selectivity is shown in Fig. 7. The carbon selectivity (Si,j = nouti,j/nConvi) is defined as the ratio of the moles of carbon in product j derived from alcohol i (nouti,j), to the moles of alcohol i converted (nConvi). It is important to note that this selectivity is a function of the feed composition, since the presence of other alcohols determines the type of products that can be formed. We highlight that in the case of 1-butanol, around 50% of the carbon is converted into E10+ ethers (linear and branched).
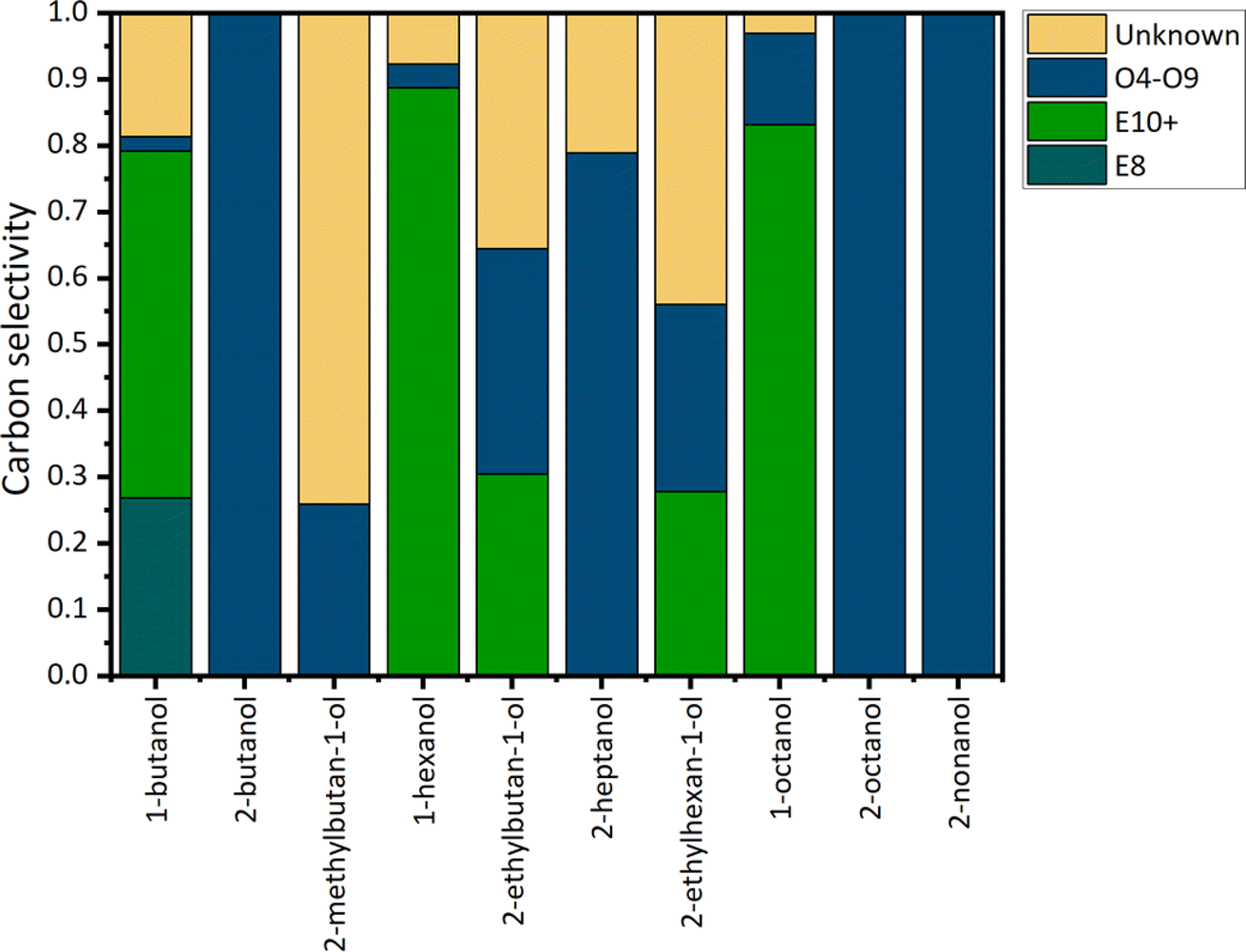 | ||
| Fig. 7 Carbon selectivity for the etherification reaction when HY zeolite is used as a catalyst. Reaction conditions: T = 170.1 °C, P = 110 psig, feedstock flowrate = 0.040 mL min−1, Ar flowrate = 10 mL min−1, WHSV = 1.085 h−1. Chemical species labeling in the legend: the letter indicates the species type, and the number denotes the carbon length. O: olefins and E: ethers. Data taken from ref. 33. | ||
 | ||
| Fig. 8 Layout of the etherification (green), oligomerization (red), and final production fractionation (blue) areas. Chemical species labeling: the letter indicates the species type, and the number denotes the carbon length. A: alcohols, ES: esters, K: ketones, AL: aldehydes, OP: olefins/paraffins, H: hydrogen, and W: water. | ||
3. Fuel characterization
The physicochemical properties of the fuels produced are estimated based on literature reported blending rules and in-house developed methods, as we have previously described.8,10,34 The results obtained are shown in Table 5. The diesel #2 fraction produced (our primary product) is characterized by a very high cetane number (∼94) and outstanding cold flow properties (cloud point of −28.9 °C). While its density is slightly lower than that of typical fossil diesel (795 kg m−3vs. 815–840 kg m−3 for diesel #2) we note that this value is not regulated by an ASTM standard.25 In the case of viscosity and flash point, we observe marginally lower values than those in the specification25 and the diesel fuel minimum values are within the uncertainty of the estimation method. The only property that is substantially below the ASTM standard requirement is T90 (246.4 °C vs. a minimum of 282 °C for diesel #2 (ref. 25)). The fact that this value is lower implies that to meet the ASTM standard, the produced fuel needs to be used in a blend. However, we note that a lower T90 value can be beneficial from an operating perspective if the flash point requirement is satisfied, as it is in this case.37 Thus, there are no operational constraints for using the produced fuel as drop-in diesel. If the diesel product in this work is used as a blend, an improvement in the cetane number of the base fuel may be achieved. For reference, the expected cetane number obtained for a 20% (vol/vol) blend between our diesel and base diesel with a cetane number of 40 is ∼50.8 (based on a linear blending rule by volume). Although the diesel #1 and gasoline fractions produced are a minority, accounting for less than 10% of the total product, their physicochemical characteristics are more suitable for use in blends rather than as drop-in fuels. In the case of diesel #1, the main limitation is the flash point which is significantly lower than the requirement (22.2 °C vs. 38 °C). In the case of gasoline, the octane number (estimated based on the calculated cetane38) is too low (∼59 vs. 89).| Fuel | Source | CN | ρ [kg m−3] | μ [mm2 s−1] | FP [°C] | CP [°C] | T90 [°C] |
|---|---|---|---|---|---|---|---|
| a This property is not part of the ASTM standard, but we present typical values. | |||||||
| Diesel #2 | Fossil | 40 (ref. 25) | 815–840 | 1.9–4.1 (ref. 25) | 52 (ref. 25) | — | 282–338 (ref. 25) |
| Previous work | 73.2 | 789.7 | 1.92 | 49.9 | −37.3 | 241.9 | |
| This work | 94.4 | 794.7 | 1.71 | 51.4 | −28.9 | 246.4 | |
| Diesel #1 | Fossil | 40 (ref. 25) | 815–840 | 1.3–2.4 (ref. 25) | 38 (ref. 25) | — | <288 (ref. 25) |
| Previous work | 69.6 | 815.3 | 0.7 | 20.7 | −102.7 | 139.8 | |
| This work | 56.2 | 781.3 | 0.87 | 22.2 | −108.1 | 134.3 | |
| Gasoline | Fossil | <10 | 715–770 | <2a | — | — | <190 (ref. 35) |
| Previous work | 11 | 827.9 | 0.64 | −19.75 | −98.25 | 87.9 | |
| This work | 33.1 | 707.4 | 0.50 | −13.67 | −99.4 | 110.2 | |
4. Economics
To study the economics of the process, a detailed discounted cash flow analysis is performed. The main assumptions are listed in Table S8;† these assumptions are consistent with those used in NREL reports for bioethanol production; likewise the ethanol capacity is selected such that the proposed refinery can process an output of the same scale as the NREL lignocellulosic ethanol refinery.17 Equipment costs are estimated using an Aspen process economic analyzer. The installation factors are taken from the work of Humbird and co-workers.17 Operating costs such as feedstocks, waste treatment, and utilities are calculated based on simulation results. The lignocellulosic ethanol price used in the base case ($2.85/Gal) corresponds to the value estimated by NREL.17 For the catalyst cost estimation, we use either the recently developed tool CatCost39 (Guerbet coupling and hydrogenolysis catalysts), or commercially available data for zeolites40,41 (etherification and oligomerization catalysts).Utility consumption is reduced by more than 40% in heating and cooling duties by implementing heat integration based on pinch analysis.42 The associated heat exchanger network (HEN) consists of 83 heat exchangers, and it is designed using the Aspen Energy Analyzer®. Furthermore, energy rich purge streams are used to produce steam (∼138 GJ h−1 assuming 80% efficiency with respect to the low heating value), partially offsetting the energy needs of the refinery.
In Fig. 9, we show the minimum selling price (Fig. 9(a)), annualized capital and operating costs (Fig. 9(b)), and a breakdown of the installed costs per area (Fig. 9(c)). The minimum selling price is $6.52 per gal ($6.94 per GDE, where GDE stands for gallon of diesel equivalent). These results are obtained for an ethanol price of $2.85 per gal; if a cheaper feedstock is used (e.g., corn ethanol), then a significantly lower MFSP is obtained. For reference, the current price of corn ethanol is ∼$2.41 per gal, and in the last 20 years it has reached a low price of ∼$1.5 per gal.43 If these prices are used, the MFSP is 5.78 $ per gal (6.15 $ per GDE) and 4.39 $ per gal ($4.66 per GDE), respectively. To put these results in perspective, the spot price of diesel in the last 5 years has ranged from $2.03 to 4.90 per gal.44 The main competing technologies available at a commercial scale are biodiesel produced by transesterification of vegetable oils and hydrotreated vegetable oils (HVO) produced by hydrogenating vegetable oils. Vegetable oils are a more expensive feedstock than ethanol with limited availability. Based on the average transesterification yield, methanol cost, and assuming product credits from glycerol amounting to $0.4 per kg, the feedstock cost of biodiesel has ranged between $2.0 and 7.1 per gal in the last 5 years.45 For reference, the cost of feedstock represents ∼81% of the total production cost.45 Biodiesel also receives US federal subsidies of a $1.00 per gal blenders credit and a D4 RIN that ranged from $1.46 to 1.81 per gal in 2022.46 In contrast, the process that we propose leads to feedstock costs ranging from $2.3 to 4.6 per gal based on corn ethanol price values. This highlights the advantages of using ethanol to produce diesel fuel as opposed to vegetable oils. At a low-to-medium Technology Readiness Level (TRL), several candidate technologies (see Table 1) have emerged for sustainable diesel fuel production. Among these technologies, gasification followed by Fischer–Tropsch synthesis received significant research attention. 47–51 In comparison to this technology, our system has similar economic potential, with two added advantages (1) it does not need to deal with tar contamination, an important bottleneck in biomass gasification systems,52 and (2) it offers a higher flexibility in tailoring fuel properties, as the recycling strategy developed in this work ensures that one can tune the ether distribution to achieve higher cetane numbers. For comparison of the MSP obtained in this work with other renewable technologies for the production of diesel fuel see Table 1.
 | ||
| Fig. 9 (a) Minimum fuel selling price (MFSP), (b) annualized capital and operating costs and (c) total installed cost per area. GC: Guerbet coupling, E: etherification, OLIG: oligomerization, FRAC: final fractionation, and HEN: heat exchanger network. An annualization factor of 0.1061 is used. | ||
The total operational costs, shown in Fig. 9(b), are dominated by feedstock costs, ∼92% of which are due to ethanol and the remaining 8% are due to hydrogen. The total operational cost is ∼208.2 $MM per year (∼$5.7 per gal) and the annualized capital cost is ∼28.34 $MM. The total capital investment (TCI) is ∼267 $MM. The breakdown of capital costs per area in Fig. 9(c) shows that the Guerbet coupling area is the most capital intensive with the cost of reactors and pressure changing equipment (mainly compressors required in the gas recycle streams) being the most significant. This result points toward the need to operate at higher conversion, while still maintaining high product selectivity. In addition, the operation of the hydrogenolysis reactor at a lower hydrogen to ester ratio would be beneficial because it would result in reduced compressor size.
To further understand the impact of the different parameters and assumptions made in the estimation of the MFSP, we performed a sensitivity analysis as shown in Fig. 10(a). Reference values are shown on the y-axis legend. The parameters are varied within a range (shown at the sides of each bar) that represents reasonably optimistic and pessimistic scenarios with respect to the base case value. The corresponding percentage change in the MFSP is shown on the x-axis. The parameters are grouped into three categories: (1) operating costs, (2) capital costs, and (3) financial assumptions. Among the parameters in the first group, the costs of ethanol, hydrogenolysis catalyst, and hydrogen are the more influential, in that order. Changes in the other parameters result in marginal changes in the MFSP. In terms of catalysis, the largest consumption is for the hydrogenolysis reaction – 613 ton per year, in comparison to 3.3 ton for Guerbet, 19 ton for etherification, and 32 ton for oligomerization. This is because the hydrogenolysis catalyst has a very low WHSV (0.07 h−1) and processes a large stream. Among the parameters in the second group, the total capital cost of the Guerbet area is the most significant. Reductions in its capital cost can be achieved, for example, by operating the Guerbet reactor at higher conversion. Finally, among the financial parameters the discount rate appears to be the most important one. In the base case scenario, we assume a value of 10%, consistent with other reference studies on biofuel production.17 Based on Fig. 10(a), we conclude that the two most influential parameters are the price of ethanol and the cost of the hydrogenolysis catalyst. Accordingly, the impact of these two parameters is explored in more detail in Fig. 10(b) where we use the WHSV as a proxy of catalyst cost (if the WHSV increases, the amount of catalyst decreases and the size of the hydrogenolysis reactor diminishes). The figure is based on the range in which the price of corn ethanol has oscillated in the last 10 years43 and our reference case, based on lignocellulosic ethanol cost.
 | ||
| Fig. 10 (a) Tornado plot showing the sensitivity of the MSFP to different parameters. The reference value is shown on the y-axis. Optimistic and pessimistic values for each parameter are used (shown at the sides of each bar). (b) Heat map showing the minimum selling price as a function of the ethanol price and the WHSV of the hydrogenolysis catalyst. The range of corn ethanol prices in the last 10 years is shown. | ||
5. Life cycle analysis
A well-to-wheels attributional lifecycle analysis (LCA) of the proposed technology is performed using the GREET model (Fig. 11).53 The goal of the study is to establish the GHG emissions of the proposed technology in comparison to fossil diesel. A functional unit of 1 MJ of fuel (before combustion) is used. The boundaries of the system, as well as the foreground and background processes are shown in Fig. S6.† The coproduction of gasoline is accounted for using a system expansion approach as suggested by the ISO14040 standard.54 For the end of life, we model the vehicle using the default parameters for a long-haul truck in the GREET model.53 We evaluate five different ethanol feedstocks (corn, corn stover, switchgrass, miscanthus, and poplar). Pathways for ethanol production from these feedstocks are available in GREET and are used without modifications. Additionally, we explore the case where ethanol is produced using corn in a biorefinery in which carbon capture is implemented (labeled “Corn w/CC”); the data for this pathway are also obtained from the GREET model. Eight different scenarios are evaluated for each feedstock. These scenarios are defined considering the sources of natural gas (shale gas or food waste anaerobic digestion), electricity (current US mix or wind), and hydrogen (methane or water electrolysis using renewable electricity) used in the ethanol upgrading process. Natural gas is used in the upgrading refinery to produce steam. The life cycle inventory of the upgrading process is constructed using data for feedstocks and energy consumption obtained from Aspen Plus simulations (Table S11†). The impact assessment is performed using the GREET model for GHG emissions.53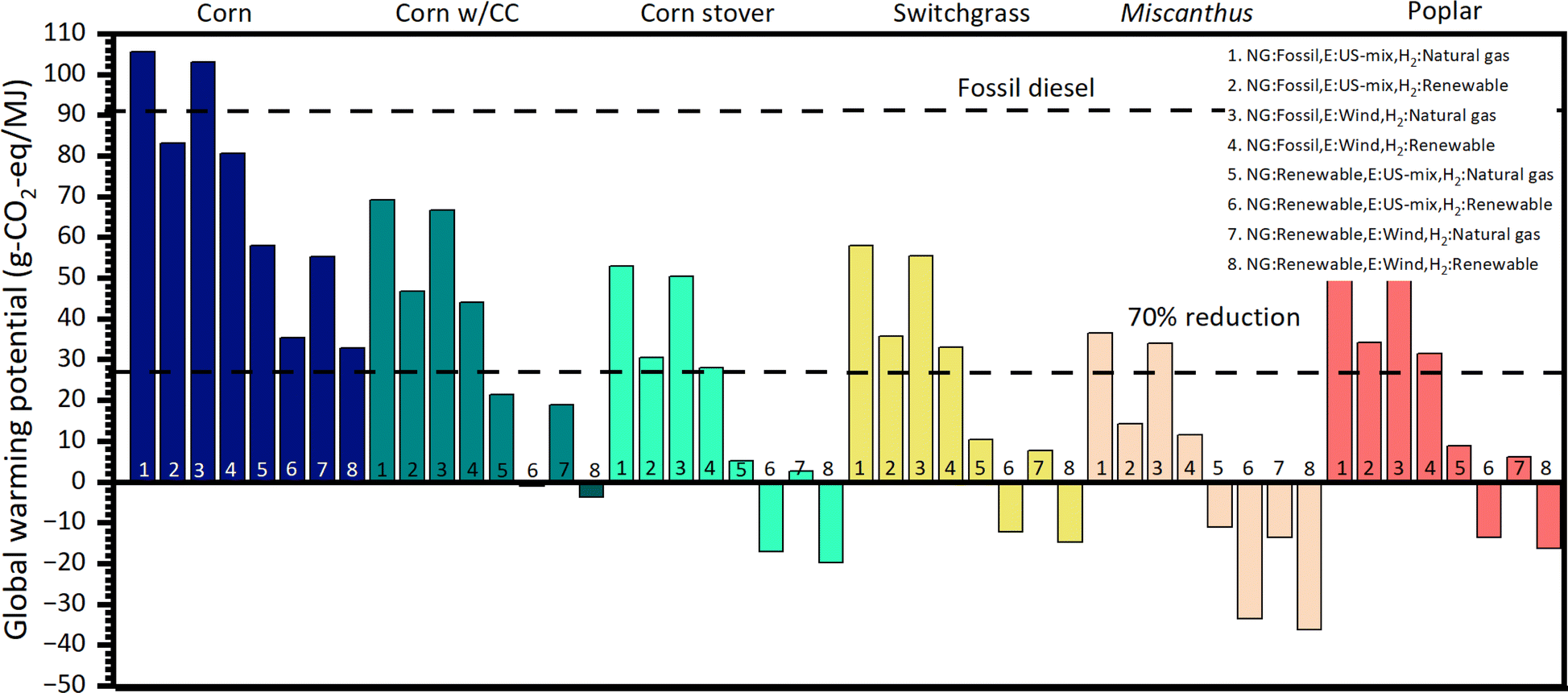 | ||
| Fig. 11 Well-to-wheels GHG emissions of the diesel fuel produced in this work. Six different ethanol sources are evaluated. The numbers in the bars correspond to different scenarios (see the legend for the key). | ||
The interpretation of the results obtained after the impact assessment leads to the conclusion that in the most conservative scenario (#1, which assumes that natural gas and hydrogen are produced from nonrenewable resources while electricity is obtained from the grid) only ethanol from Miscanthus is close to the target of reducing GHG emissions by more than 70% in comparison to fossil diesel. On the other hand, in the most optimistic scenario (#8, which assumes that natural gas, electricity, and hydrogen used in the upgrading process are produced from renewable sources), all feedstocks, except for corn, can attain carbon negative emissions. We note that in the optimistic scenario, we can significantly benefit from developments in renewable hydrogen production which is a priority area in the U.S. research agenda.55 Importantly, corn ethanol has significant environmental benefits only if renewable natural gas is used in the upgrading process (scenarios 5–8) or if it is coupled with a carbon capture strategy. A comparison of the GHG emission potential of the proposed technology with respect to other biofuels is presented in Table 1.
While the process that we have developed has been optimized to reduce cost and environmental impacts, potential future technological developments may lead to even lower environmental impacts. To explore the impact of these potential improvements on GHG emissions, we perform a parametric analysis shown in Fig. S7.† One advantage of the fuels produced in this work is that they may lead to a moderate increase in energy efficiency (see the Engine performance section). In Fig. S8,† we explore how changes in vehicle energy efficiency lead to moderate improvements in the well-to-wheels GHG emissions.
6. Engine performance
The performance of the mono-ether bioblendstock was evaluated, using a simple three component surrogate (composed of 65 vol% dibutyl ether, 33 vol% dihexyl ether, and 2 vol% diisopentyl ether), in a single-cylinder compression-ignition direct-injection research engine.56–58 The surrogate was blended with #2 diesel fuel (Haltermann Solutions, 2007 emissions certification fuel) at 20 vol% and 37 vol%. These volume fractions of bioblendstock, were selected to yield a final fuel with cetane numbers of 50 and 60, respectively. Additional details on the fuel properties of the surrogate-diesel blends can be found in ref. 57. Under different engine load and speed conditions, the blends with the surrogate bioblendstock were found to reduce engine-out soot emissions in comparison to the baseline #2 diesel fuel (Haltermann Solutions, 2007 emissions certification fuel). The decrease in engine-out soot emissions was observed to increase as the blend fraction of the surrogate ether bioblendstock increased. Fig. 12(a) shows the soot-nitrogen oxides (NOx) trade-off for the fuels for a C50 operating mode (10 bar gross indicated mean effective pressure and 2200 rpm engine load and speed, respectively), which is one of the four operating modes evaluated in this study. At this operating point, the cetane number 60 fuel blend exhibited 48% lower soot emissions than #2 diesel at the most advanced injection timing evaluated, shifting the trade-off in a favorable direction despite a small increase in NOx emissions. This reduction in soot emissions for the bioblendstock surrogate was expected as the mono-ether fuel blends had a lower chemical sooting propensity, as was quantified by the yield sooting index (YSI).59 The cetane number 60 fuel, a blend with 37% (by volume) bioblendstock surrogate, had a YSI value of 165, significantly lower than the measured value for #2 diesel (246).57 The engine results at all four warmed up operating modes resulted in similar or slightly increased engine efficiency relative to the baseline #2 diesel fuel. The ether bioblendstock diesel blends also had a broader operating range where emission constraints could be met. These results indicate overall equal or improved performance relative to the baseline #2 diesel fuel for warmed up operation. Detailed information about the operating modes can be found in Subramanian 2024.58 | ||
| Fig. 12 (a) Indicated specific particulate matter (soot) and NOx for #2 diesel and ether–diesel blends with cetane numbers 50 and 60 for C50 (10 bar gross indicated mean effective pressure and 2200 rpm). The data point corresponding to the most advanced injection timing is highlighted. (b) Combustion efficiency, (c) indicated specific NOx emissions, and (d) exhaust enthalpy for #2 diesel and di-butyl ether–diesel blend as a function of targeted NOx emissions. A combustion efficiency of 94.5%, along with three NOx emissions of 0.5, 0.8 and 1 g kW−1 h−1, was targeted to compare the fuels. | ||
The performance of the bioblendstock was also evaluated during operation designed to increase the exhaust enthalpy to bring the aftertreatment components to their operating temperatures. The critical trade-off during this operation is the increase in fuel consumption and engine-out emissions of partial products of combustion.56,60 A large proportion of the total emissions of regulated pollutants during the federal certification test cycle occurs during this phase of engine operation.61 A blend of di-butyl ether (Sigma-Aldrich, RegentPlus, >99%), a mono-ether expected to be present in the bioblendstock, was compared with #2 diesel with a derived cetane number (DCN)62 of 60 at matched engine-out emissions (see Fig. 12(b) and (c)). More details about the engine operating conditions and fuel composition can be found in ref. 57 and 58. This single component mono-ether surrogate was expected to replicate the combustion performance of the actual mono-ether bioblendstock at the matched cetane number, as the volatility level of the fuel was found to have limited sensitivity on engine operation under these conditions. Additionally, comparing fuels with different chemical compositions (alkane vs. diesel blended with oxygenated components) at matched reactivity demonstrated similar engine and emission performance.1 The higher-reactivity fuel was observed to have ∼13% higher exhaust enthalpy than #2 diesel at matched combustion efficiency and NOx emissions,57,58 as shown in Fig. 12(d). This increase was driven by operating the higher reactivity fuel at a more retarded injection timing and a lower exhaust gas recirculation (EGR) rate in comparison to the baseline fuel, leading to an increase in exhaust temperature and exhaust mass flow. The higher exhaust enthalpy at matched engine-out emissions for the mono-ether fuel blend was expected to reduce the overall fuel and emissions penalty associated with this operation, thereby, improving aftertreatment thermal management operation.57,58
7. Conclusions
In this work, we have developed a new catalytic pathway for the production of diesel from ethanol and comprehensively studied its economic and GHG mitigation potential. Our results show that the proposed process is economically promising and leads to the production of fuels with 70% lower greenhouse gas emissions than their fossil counterparts, while in the most optimistic scenario carbon neutrality can be achieved. The process primarily leads to the production of diesel #2 (∼92%), which can be used as a drop-in biofuel. The properties of this fuel are outstanding, with a cetane number of ∼94 and a cloud point ∼−28.9 °C. We showed that a limitation of previous approaches can be overcome by a hydrogenolysis step which removes esters produced in the Guerbet reaction with significant impacts on the performance of etherification reactions. The strategy presented herein can play a key role in the energy transition by enabling the decarbonization of the heavy-duty transportation sector.8. Experimental methods
8.1 Guerbet coupling
A 0.3% wt. Cu/Mg2.9AlO catalyst was prepared through co-precipitation of Cu(NO3)2·3H2O (Sigma-Aldrich 61194), Al(NO3)3·9H2O (Sigma-Aldrich 237973) and Mg(NO3)3·6H2O (Sigma-Aldrich 237175) precursors at pH 10. The resulting cake was filtered, washed with deionized water, dried overnight at 110 °C (Lab-line, 3511) and calcined for 2 h at 600 °C with a heating ramp of 4 °C min−1. A thorough description of the synthesis procedure accompanied by reagent proportions and catalyst characterization can be found in our previous publications.21,268.2 Hydrogenolysis
Zirconia (ZrO2) support was prepared through the oxidative treatment of zirconium(IV) hydroxide (Zr(OH)4, Aldrich 46417–1, 97%) as described elsewhere.63 Briefly, Zr(OH)4 was calcined at 500 °C for 5 h with a 4 °C min−1 heating ramp in a static air atmosphere in a muffle furnace (ThermoFisher Scientific, Thermolyne) to obtain a white powder, which is assumed to be zirconia (ZrO2). Textural characterization by nitrogen physisorption was carried out using an ASAP 2020 (Micromeritics), yielding a BET surface area of 146 m2 g−1 and a BJH pore volume of 0.14 cm3 g−1. The Cu/ZrO2 catalyst with a theoretical metal load of 10% wt. was synthesized through incipient wetness impregnation by dissolving Cu(NO3)2·3H2O (Sigma-Aldrich 61194, 99%) in Milli-Q water, heating the solution up to 60 °C for complete dissolution of the salt29 and then adding it dropwise to the zirconia. After impregnation of the support, it was dried overnight (at least 12 h) in an oven (Lab-line, 3511) at 110 °C and subsequently crushed and calcined (4 °C min−1) at 500 °C for 5 h in a static air atmosphere.8.3 Reaction conditions
Guerbet coupling and hydrogenolysis reactions were performed independently in the same reaction setup, meaning that the description of the reaction procedure varies only in the reaction conditions. For simplicity, reaction conditions for hydrogenolysis will be described in the main text, while those for Guerbet coupling will be given inside brackets. The calcined catalyst was sieved to 177–354 μm (mesh 80–45) and packed into a stainless-steel fix bed reactor (16 in long, 3/8 in outer diameter) by using 100–500 mg of the catalyst. This powder was fixed at the center of the reactor by placing layers of glass wool (Acros organics, 393611000) and silica chips (Sigma-Aldrich, 342831) at each end, and then reduced in situ at 300 °C (1 °C min−1) for 2 hours under 20 mL min−1 of pure hydrogen flow (101 kPa)63 [325 °C (4 °C min−1) for 12 hours under 50 mL min−1 of pure hydrogen flow (101 kPa)21 for Guerbet coupling]. An isothermal profile along the catalytic bed was achieved by using aluminum blocks between the reactor and the electric tube furnace (Thermo Fisher, Lindberg blue M Mini-Mite); the temperature was measured with a K-type thermocouple, which was embedded in the aluminum heating block. After reduction, the reactor was cooled down to 200 °C and pressurized to 420 psig [325 °C and 300 psig] with a back-pressure regulator (Equilibar, ZF0SVN8).64 A blend of butanol–hexyl acetate of composition 95–5% mol [ethanol–butanol 70–30% mol] was fed with a syringe pump (Teledyne ISCO) at 6–18 μL min−1 [23–93 μL min−1], with pure H2 cofed at 36–109 mL min−1 [2.5–10.3 mL min−1] to a preheated section maintained at ∼190 °C [>200 °C] to ensure that the feed was in the gaseous phase upon contacting the catalyst. The molar ratio of esters to hydrogen of the gaseous phase entering the reactor was kept constant at 1:480 for hydrogenolysis experiments, while for Guerbet coupling reactions the molar ratio of alcohols to hydrogen was maintained at 4:1. After reaction, products were collected in a removable 110 mL glass condenser (Ace glass) immersed in a dry ice bath. With the aim of reducing the sampling error due to low product mass collection, 15 mL of 1-propanol (Sigma-Aldrich, 96566) was loaded to the condenser before collecting products with a typical sample collection time of 1–2 h. Gases that did not condense were sent to a three-valve system, which allowed the gaseous flow to vent or towards an online gas GC (Shimadzu 2010) equipped with a flame ionization detector (FID) and thermal conductivity detector (TCD) for gas phase sampling. Liquid samples were prepared for analysis through gas chromatography by diluting them with 1-propanol and adding a known amount of 1-pentanol as the internal standard. Such liquid products were analyzed via GC-FID (Shimadzu 2014), and quantification was performed by using external standards. Product's identity was further supported by gas chromatography–mass spectrometry (Shimadzu GCMS-QP2010). For further description of analysis methods for liquid and gas phases, please refer to our previous publications.21,23 The selection of temperature and pressure for operating our Guerbet coupling reactor was based on the previous work of Guo et al.,64 who studied the conversion of ethanol to 1-butanol over CuMgAlOx catalysts. After their publication, we have consistently performed coupling experiments at 325 °C and 300 psig. The selection of the P(EtOH + ButOH)/P(H2) ratio is based on our previous publication,26 where we found that at a ratio of 4, the alcohol propagation probability, meaning the probability of forming long chain alcohols, is maximized. The reaction conditions for our hydrogenolysis reactor were guided by the work presented in one of our publications,65 where we studied the model mixture hexyl acetate–butanol in more detail.
8.4 Etherification
The etherification of the ethanol/butanol Guerbet coupling products was carried out in an upward configuration continuous flow reactor made of stainless steel. The bed was packed with 1.8 g of powder HY catalyst (Si/Al = 30). The composition of the catalyst bed, reactor dimensions, and collection procedure can be found elsewhere.10 Ar gas was flowed at 10 mL min−1, and the liquid flow rate was varied to obtain WHSV = 1 h−1. For determining cross-etherification species, the analytical techniques for product identification were also taken from elsewhere.108.5 Engine testing
The experiments were performed in a single-cylinder research version of the General Motors 1.9L (Z19DTH) engine. The test cell has a customized fuel system to deliver high-pressure fuel to the engine and it includes a HORIBA 5-gas emissions bench and a hydrocarbon analyzer to measure common engine-out pollutants. The test cell has precise control over intake (air temperature and pressure) and boundary conditions (oil and coolant temperature). More details about the test cell setup and engine geometry can be found in Subramanian 2024.58The fuels used in the study were obtained from manufacturers with high purity levels.
Data availability
The data supporting this article has been included as part of the ESI.† The methods and conditions required for the experiments are described in the Methods section. Aspen Plus models used in the TEA will be made available upon request. (S1) shows data for cofeeding experiments, (S2) shows data for hydrogenolysis experiments, (S3) shows data for etherification experiments, (S4) shows data for technoeconomic analysis, and (S5) shows data for lifecycle analysis.Author contributions
Juan-Manuel Restrepo-Flórez, Javier E. Chavarrio, and Emmanuel Canales: conceptualization, data curation, formal analysis, investigation, methodology, software, visualization, writing – original draft. Dustin Witkowski and Srinath Subramanian: conceptualization, formal analysis, investigation, methodology, software, writing – original draft. Paolo Cuello-Peñaloza: conceptualization, investigation, methodology. David A. Rothamer: conceptualization, resources, funding acquisition, supervision, project administration, writing – review & editing. Christos T. Maravelias: conceptualization, methodology, resources, funding acquisition, supervision, project administration, writing – review & editing. George W. Huber: conceptualization, methodology, resources, funding acquisition, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Co-Optima program, award number E-EE0008480. The views expressed herein do not necessarily represent the views of the U.S. Department of Energy or the United States Government.References
- EIA, Annual Energy Outlook 2021, Washington DC, 2021, vol. 2021 Search PubMed.
- EPA, Greenhouse Gases Equivalencies Calculator – Calculations and References, https://www.epa.gov/energy/greenhouse-gases-equivalencies-calculator-calculations-and-references, accessed 19 September 2022.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Science, 2018, 360, 1419 CrossRef CAS PubMed.
- US DOE, US Department of Transport, US EPA and US Department of Housing, The U.S. National Blueprint for Transportation Decarbonization, 2050.
- D. J. Gaspar, Top 13 Blendstocks Derived from Biomass for Mixing-Controlled Compression-Ignition (Diesel) Engines: Bioblendstocks with Potential for Lower Emissions and Increased Operability, Richland, WA, 2021 Search PubMed.
- J. M. Restrepo-Flórez and C. T. Maravelias, Computer Aided Chemical Engineering, Elsevier Masson SAS, 2022, vol. 49 Search PubMed.
- J. M. Restrepo-Flórez and C. T. Maravelias, Energy Environ. Sci., 2021, 14, 493–506 RSC.
- J.-M. Restrepo-Flórez, J. Ryu, D. Witkowski, D. A. Rothamer and C. T. Maravelias, Energy Environ. Sci., 2022, 15, 4376–4388 RSC.
- N. M. Eagan, M. D. Kumbhalkar, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Nat. Rev. Chem, 2019, 3, 223–249 CrossRef CAS.
- J.-M. Restrepo-Flórez, P. A. Cuello-penaloza, E. Canales, D. Witkowski, D. A. Rothamer, G. W. Huber and C. T. Maravelias, Sustainable Energy Fuels, 2023, 7, 693–707 RSC.
- U. Energy Information Administration, Monthly Energy Review – January 2024, 2024 Search PubMed.
- N. A. Huq, X. Huo, G. R. Hafenstine, S. M. Tifft, J. Stunkel, E. D. Christensen, G. M. Fioroni, L. Fouts, R. L. McCormick, P. A. Cherry, C. S. McEnally, L. D. Pfefferle, M. R. Wiatrowski, P. T. Benavides, M. J. Biddy, R. M. Connatser, M. D. Kass, T. L. Alleman, P. C. St John, S. Kim and D. R. Vardon, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26421–26430 CrossRef CAS.
- J. E. Rorrer, A. T. Bell and F. D. Toste, ChemSusChem, 2019, 12, 2835–2858 CrossRef CAS.
- M. Dahmen and W. Marquardt, Energy Fuels, 2017, 31, 4096–4121 CrossRef CAS.
- R. A. Dagle, A. D. Winkelman, K. K. Ramasamy, V. Lebarbier Dagle and R. S. Weber, Ind. Eng. Chem. Res., 2020, 59, 4843–4853 CrossRef CAS.
- EIA, Monthly Energy Review-June 2021, Washington DC, 2021, vol. 159 Search PubMed.
- D. Humbird, R. Davis, L. Tao, C. Kinchin, D. Hsu, A. Aden, P. Schoen, J. Lukas, B. Olthof, M. Worley, D. Sexton and D. Dudgeon, Process Design and Economics for Biochemical Conversion of Lignocellulosic Biomass to Ethanol: Dilute-Acid Pretreatment and Enzymatic Hydrolysis of Corn Stover, Golden, Colorado, 2011 Search PubMed.
- D. R. Keshwani and J. J. Cheng, Bioresour. Technol., 2009, 100, 1515–1523 CrossRef CAS.
- N. Sarkar, S. K. Ghosh, S. Bannerjee and K. Aikat, Renewable Energy, 2012, 37, 19–27 CrossRef CAS.
- X. Ou, X. Zhang, Q. Zhang and X. Zhang, Front. Energy, 2013, 7, 263–270 CrossRef.
- P. A. Cuello-Penaloza, R. G. Dastidar, S. C. Wang, Y. Du, M. P. Lanci, B. Wooler, C. E. Kliewer, I. Hermans, J. A. Dumesic and G. W. Huber, Appl. Catal., B, 2022, 304, 120984 CrossRef CAS.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008, 259, 183–189 CrossRef CAS.
- N. M. Eagan, B. J. Moore, D. J. McClelland, A. M. Wittrig, E. Canales, M. P. Lanci and G. W. Huber, Green Chem., 2019, 21, 3300–3318 RSC.
- O. V. Larina, K. V. Valihura, P. I. Kyriienko, N. V. Vlasenko, D. Y. Balakin, I. Khalakhan, T. Čendak, S. O. Soloviev and S. M. Orlyk, Appl. Catal., A, 2019, 588, 1–11 CrossRef.
- ASTM D975-20c, Standard specification for diesel fuel, 2014.
- P. A. Cuello-penaloza, J. Chavarrio-Canas, Y. Du, M. P. Lanci, D. A. Maedke, J. A. Dumesic and G. W. Huber, Appl. Catal., B, 2022, 318, 121821 CrossRef CAS.
- S. Subramanian and D. Rothamer, in SAE Technical Paper Series, 2023 Search PubMed.
- J. Gu, W. Gong, Q. Zhang, R. Long, J. Ma, X. Wang, J. Li, J. Li, Y. Fan, X. Zheng, S. Qiu, T. Wang and Y. Xiong, Nat. Commun., 2023, 14, 7935 CrossRef CAS PubMed.
- N. S. Kim, S. Y. Hwang, E. Y. Kim and K. N. Han, Jpn. J. Appl. Phys., 2010, 49, 05EA04 CrossRef.
- J. Schittkowski, K. Tölle, S. Anke, S. Stürmer and M. Muhler, J. Catal., 2017, 352, 120–129 CrossRef CAS.
- W. Zhang, Y. Yao, S. Xie, K. Gubsch, Y. Yang, X. Lan and H. Lin, Catal. Today, 2021, 374, 53–60 CrossRef CAS.
- T. K. Phung, Int. J. Hydrogen Energy, 2022, 47, 42234–42249 CrossRef CAS.
- E. Canales, S. C. Hower, D. P. Li, A. Tambe, D. Rothamer and G. W. Huber, Sustainable Energy Fuels, 2024, 8, 3036–3047 RSC.
- D. Witkowski, M. Groendyk and D. A. Rothamer, Combust. Flame, 2023, 255, 112883 CrossRef CAS.
- ASTM D4814-21c, Standard Specification for Automotive Spark-Ignition Engine Fuel, 2022, DOI:10.1520/D4814-21C.
- E. Mark, D. Schwietert, M. J. R and S. Nagatsuka, World-Wide Fuel Charter Gasoline and Diesel Fuel, Truck and Engine Manufacturers Association, 6th edn, 2019 Search PubMed.
- M. Groendyk and D. Rothamer, Fuel, 2017, 194, 195–210 CrossRef CAS.
- J. Yanowitz, M. A. Ratcliff, R. L. McCormick, J. D. Taylor and M. J. Murphy, Compendium of Experimental Cetane Numbers, NREL/TP-5400-67585, 2017 Search PubMed.
- F. G. Baddour, L. Snowden-Swan, J. D. Super and K. M. Van Allsburg, Org. Process Res. Dev., 2018, 22, 1599–1605 CrossRef CAS.
- HZSM5 Zeolite cost, https://www.alibaba.com/product-detail/Zeolite-Zsm5-Zsm-5-Price-Zeolite_62579342984.html?spm=a2700.galleryofferlist.topad_creative.d_title.2d7761c2HmAN7FHighlight, accessed 1 May 2023.
- HY zeolite cost, https://www.alibaba.com/product-detail/high-purity-hy-zeolite-catalyst-micro_1600062317816.html?spm=a2700.galleryofferlist.normal_offer.d_title.2f391e6b3Ao4kp, accessed 1 May 2023.
- B. Linnhoff and E. Hindmarsh, Chem. Eng. Sci., 1983, 38, 745–763 CrossRef CAS.
- Trading economics, https://tradingeconomics.com/commodity/ethanol, accessed 30 April 2023.
- EIA, Diesel spot price, https://www.eia.gov/petroleum/gasdiesel/, accessed 1 May 2023.
- Don Hofstrand, Tracking biodiesel profitability, https://www.extension.iastate.edu/agdm/energy/html/d1-15.html, accessed 24 September 2023.
- US EPA, RIN Trades and Price Information, RIN Trades and Price Information, accessed 9 June 2023.
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89(1), S29–S35 CrossRef CAS.
- R. M. Swanson, A. Platon, J. A. Satrio and R. C. Brown, Fuel, 2010, 89(1), S11–S19 CrossRef CAS.
- R. M. Swanson, A. Platon, J. A. Satrio and R. C. Brown, ACS National Meeting, Book of Abstracts, 2009 Search PubMed.
- S. Safarian, R. Unnpórsson and C. Richter, Renewable Sustainable Energy Rev., 2019, 110, 378–391 CrossRef CAS.
- M. Rafati, L. Wang, D. C. Dayton, K. Schimmel, V. Kabadi and A. Shahbazi, Energy Convers. Manage., 2017, 133, 153–166 CrossRef CAS.
- D. C. de Oliveira, E. E. S. Lora, O. J. Venturini, D. M. Y. Maya and M. Garcia-Pérez, Renewable Sustainable Energy Rev., 2023, 172, 113047 CrossRef CAS.
- M. Q. Wang, GREET 1.5-Transportation Fuel-Cycle Model Volume 1: Methodology, Development, Use, and Results, ANL/ESD-39, 1999, vol. 1 Search PubMed.
- ISO 14040, Environmental management-Life cycle assessment-Principles and framework.
- A. Pareek, R. Dom, J. Gupta, J. Chandran, V. Adepu and P. H. Borse, Mater. Sci. Energy Technol., 2020, 3, 319–327 CAS.
- S. Subramanian and D. Rothamer, SAE Technical Papers, 2023, pp. 1–18 Search PubMed.
- S. Subramanian, Investigation of the Use of High-Cetane Mono-Ether Bioblendstocks to Improve Catalyst Heating and Warmed Up Operation of MCCI Engines, The University of Wisconsin, Madison, PP-United States-Wisconsin, 2024 Search PubMed.
- S. Subramanian and D. A. Rothamer, Fuel, 2024, 373(10), 132139 CrossRef CAS.
- C. S. McEnally and L. D. Pfefferle, Combust. Flame, 2007, 148, 210–222 CrossRef CAS.
- S. Busch, A. Wu and S. Cho, SAE Technical Papers, DOI:10.4271/2021-01-1182.
- C. Sharp, C. C. Webb, G. Neely, M. Carter, S. Yoon and C. Henry, SAE Int. J. Engines, 2017, 10, 1697–1712 CrossRef.
- P. Products and T. Methods, ASTM D7170-16, Standard Test Method for Determination of Derived Cetane Number ( DCN ) of Diesel Fuel Oils – Fixed Range Injection Period, Constant Volume Combustion Chamber Method 1, ASTM International, 2007, pp. 1–12 Search PubMed.
- I. C. Freitas, S. Damyanova, D. C. Oliveira, C. M. P. Marques and J. M. C. Bueno, J. Mol. Catal. A: Chem., 2014, 381, 26–37 CrossRef CAS.
- M. F. Guo, M. J. Gray, H. Job, C. Alvarez-Vasco, S. Subramaniam, X. Zhang, L. Kovarik, V. Murugesan, S. Phillips and K. K. Ramasamy, Green Chem., 2021, 23, 8030–8039 RSC.
- J. E. Chavarrio, K. Kirkendall-Jones, R. G. Dastidar and G. W. Huber, ACS Catal., 2024, 11411–11424 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00943f |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |