
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of CpG methylation based on the change in amplification efficiency of strand-displacement DNA polymerase by CpG methylation†
Mizuki
Tomizawa
,
Kiwako
Watanabe
,
Kaori
Tsukakoshi
and
Kazunori
Ikebukuro
 *
*
Department of Biotechnology and Life Science, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184-8588, Japan. E-mail: ikebu@cc.tuat.ac.jp
First published on 19th March 2025
Abstract
A method for detecting CpG methylation is required in clinical settings because CpG methylation is associated with various diseases. CpG methylation leads to structural changes in single-stranded DNA and also changes the stability of double-stranded DNA. We hypothesized that the amplification efficiency of DNA polymerase, with its strand displacement ability, might be altered by CpG methylation. We chose loop-mediated isothermal amplification (LAMP), which uses strand displacement DNA synthesis, for its validation. The LAMP products from the synthetic DNA of the upstream region of the dopamine receptor D2 (DRD2) and the androgen receptor (AR) promoter region were detected by turbidity and fluorescence intensity measurements. The methylated synthetic DNA was amplified more slowly than the unmethylated synthetic DNA. The LAMP products from the human genomic DNA were detected by fluorescence intensity measurement and electrophoresis. The highly methylated genomic DNA was amplified slower than the less methylated genomic DNA in the AR promoter region. CpG methylation detection through differences in the amplification efficiency of LAMP reaction may be used for a rapid and easy detection method of CpG methylation.
1. Introduction
DNA methylation is the most common eukaryotic DNA modification and is one of many epigenetic phenomena.1 It alters gene expression without changing the nucleotide sequences and plays an important role in maintaining genome stability.2–5 DNA methylation in eukaryotes involves the addition of a methyl group to the carbon 5 position of the cytosine ring, followed by a guanine, and is called CpG methylation. This reaction is catalyzed by DNA methyltransferases at CpG dinucleotides.6 Aberrant CpG methylation is associated with various conditions, such as diseases, inborn disorders, cancers, and mental disorders.7–9 Therefore, the detection of DNA methylation is important for disease diagnosis.Assays based on sodium bisulfite conversions, such as bisulfite sequencing, methylation-specific PCR, combined bisulfite restriction analysis (COBRA), and methylation-sensitive high-resolution melting (MS-HRM), have been widely used for DNA methylation analysis.10–13 Such procedures are based on the bisulfite-induced oxidative deamination of genomic DNA under conditions in which cytosine is converted to uracil, but methylated cytosine remains unchanged.14,15 The DNA fragment bearing the target sequence is amplified using PCR and sequenced. By comparing the bisulfite-treated and untreated genomic DNA sequences, the locations of the methylated cytosines can be identified. Although such assays are widely used for DNA methylation analysis, they require several hours to convert all unmethylated cytosine residues in the genomic DNA to uracil and to analyze the sequences. An early or definitive diagnosis of some diseases, such as cancer and mental disorders, requires a rapid and easy detection of DNA methylation levels in target genomic regions. To rapidly detect DNA methylation levels, an assay without bisulfite treatment is required. Some assays can detect DNA methylation without bisulfite treatment, such as methylation-sensitive restriction enzyme-based assays or affinity enrichment assays.16–18 However, these assays cannot target all genomic regions owing to limitations in recognizing the sequences.
In a previous study, we reported that CpG methylation affects non-canonical steric structures, the guanine quadruplex (G4) and i-motif, formed by guanine- and cytosine-rich oligonucleotides, respectively.19–21 G4s are formed by G-tetrad stacking and Hoogsteen bonding in G-rich sequences.22,23 The i-motif is formed by cytosine–cytosine hydrogen bonding in C-rich sequences and complementary strands of G4.24,25 On the basis of these findings, we developed a method for detecting CpG methylation.20,21,26 In G4, methylated cytosines can be detected using G4 ligands that recognize changes in G4 topology due to CpG methylation. In i-motifs, CpG methylation increases the transitional pH of several i-motif-forming DNAs and their thermal stability. Furthermore, we showed that the initial elongation efficiency of PCR decreases with increasing CpG methylation levels in G-quadruplex and i-motif-forming sequences. Thus, DNA methylation changes the structure of single-stranded DNA (ssDNA), as well as the properties of double-stranded DNA (dsDNA). In particular, CpG methylation is known to reduce the flexibility of dsDNA and make dsDNA more difficult to dissociate.27–31 These changes in DNA properties due to CpG methylation are considered to be related to the CH/π interaction between the methyl group of the methylated cytosine and the neighboring base.32
Loop-mediated isothermal amplification (LAMP), a type of strand displacement DNA synthesis method, has been widely used in infection testing and can detect specific genomic DNA sequences.33 The expected reactions for methylated DNA amplification by LAMP are shown in Fig. 1. Four primers are used in the initial steps of the LAMP reaction: two inner primers, a forward inner primer (FIP) and a backward inner primer (BIP), and two outer primers, F3 and B3. DNA is amplified with high efficiency from a dumbbell-like DNA structure, which is finally created by the four primers (Fig. 1D). At the beginning of the LAMP reaction, the target strand and the newly synthesized strand from the LAMP reaction form a complementary strand, as shown in Fig. 1A. As shown in Fig. 1B, the new strand is displaced by DNA polymerase with strand displacement ability. When the target is methylated DNA, the ease of dsDNA dissociation affects the elongation reaction of the strand-displacement DNA polymerase because the strand is displaced from the methylated DNA (Fig. 1E).
 | ||
| Fig. 1 Schematic representation of DNA amplification using LAMP (A–D). DNA amplification using LAMP may be affected by DNA methylation because DNA methylation leads to stability changes in dsDNA (E). The black line indicates the target DNA and the orange line indicates the newly synthesized DNA from LAMP. LAMP; loop-mediated isothermal amplification. | ||
In this study, we hypothesized that changes in the structural stability of dsDNA caused by CpG methylation would appear as differences in the amplification efficiency of LAMP using strand-displacement DNA polymerase, which is different from the DNA polymerase used in conventional PCR (Fig. 1E). This is also likely to be significantly affected early in the LAMP reaction, which uses the target methylated DNA as a template to synthesize the DNA strand. To prove this hypothesis, we compared the differences in the amplification efficiency of LAMP between unmethylated and methylated DNA using synthetic and genomic DNA and turbidity measurement, fluorescence intensity measurement, and electrophoresis.
2. Materials and methods
2.1. Preparation of full-length synthetic DNA
The synthetic DNA was purchased from Eurofins Genomics (Tokyo, Japan), Hokkaido System Science Co., Ltd. (Hokkaido, Japan), and Tsukuba Oligo Service Co., Ltd. (Ibaraki, Japan). We chose the upstream region of the dopamine receptor D2 (DRD2) and the androgen receptor (AR) promoter region as the target gene for LAMP. To perform LAMP, target DNA over 200 bp was required; however, it is difficult to chemically synthesize such long DNA oligonucleotides. Therefore, we prepared the target DNA in two ways.In the first method, the target DNA for DRD2 was constructed by ligation after the hybridization of complementary short synthetic oligonucleotides. To prepare full-length template DNA, we mixed 8.4 μL of 0.8 M NaCl in TE buffer, and 2.8 μL of two 10 μM DNA strands. The sequences and pairs of hybridized DNA strands used for ligation are shown in Tables S1 and S2.† Schematic representations of the preparation of the full-length template DNA are shown in Fig. S1A.† The synthetic DNA with a phosphate-modified 5′-end was purchased, except for DRD2_target 1 and DRD2_complement 1. This mixture was heated at 95 °C for 10 min and then slowly cooled to 25 °C for 60 min. Then, 5 μL of ligation buffer and 2.5 μL of T4 DNA ligase (Promega) were added and the mixture was incubated at 16 °C for 30 min. The mixture was purified by phenol–chloroform extraction. Then, 11.2 μL of 3 M sodium acetate, 1.0 μL of ethachinmate (Nippon Gene Co., Ltd., Tokyo, Japan), and 336.6 μL of 100% ethanol were added to 100 μL of this solution, and was then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g and 4 °C for 5 min. Finally, the supernatant was discarded, and the precipitate was dissolved in TE buffer. DNA concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). This solution was adjusted to 100 nM and used as a template for the LAMP reaction. Methylated target 2, and methylated complement 2 each have five methylated CpG sites, so the full-length dsDNA has a total of ten methylated cytosines. These full-length unmethylated and methylated DNA samples were confirmed by 1% agarose gel electrophoresis (Fig. S1B†).
000 × g and 4 °C for 5 min. Finally, the supernatant was discarded, and the precipitate was dissolved in TE buffer. DNA concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). This solution was adjusted to 100 nM and used as a template for the LAMP reaction. Methylated target 2, and methylated complement 2 each have five methylated CpG sites, so the full-length dsDNA has a total of ten methylated cytosines. These full-length unmethylated and methylated DNA samples were confirmed by 1% agarose gel electrophoresis (Fig. S1B†).
In the second method, PCR products were enzymatically methylated for AR. Schematic representations of the preparation of enzymatically methylated template DNA are shown in Fig. S1C.† We amplified the AR inserted in plasmid DNA (pEX-A2J2) by PCR using TaKaRa Ex Taq® Hot Start Version (Takara). The sequence of AR and primers for PCR is shown in Table S3.† For PCR, 50 μL of the reaction mix contained 5 μL of 10 × Ex Taq buffer, 4.0 μL of 2.5 mM of each dNTP, 1.25 units of TaKaRa Ex Taq HS, 1.0 μM primers, and 2 μL of plasmid DNA. The PCR conditions were 30 cycles of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 30 s. The PCR products were purified using the FastGene® Gel/PCR Extraction Kit (NIPPON Genetics Co., Ltd., Tokyo, Japan) and enzymatically methylated with CpG methyltransferase (New England BioLabs, Ipswich, MA, USA). For enzymatic methylation, the enzyme was added to both unmethylated and methylated samples, and unmethylated and methylated DNA samples were prepared with or without the substrate S-adenosylmethionine to equalize the effect of the enzyme. For unmethylated DNA, the reaction mix contained 2.0 μL of ultrapure water, 2.0 μL of NE buffer, 15.0 μL of PCR product, and 1.0 μL of CpG methyltransferase, and for methylated DNA, 2.0 μL of NE buffer, 2.0 μL of S-adenosylmethionine, 15.0 μL of PCR product, and 1.0 μL of CpG methyltransferase were used as the reaction mixture, and incubated at 37 °C for 1 h and 65 °C for 20 min. These samples were purified by phenol–chloroform extraction and ethanol precipitation. Finally, the supernatant was discarded, and the precipitate was dissolved in TE buffer. DNA concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop 2000 (Thermo Fisher Scientific). This solution was adjusted to 100 nM and used as a template for the LAMP reaction.
2.2. Preparation of genomic DNA
Prostate cancer cell lines, LNCaP and Du145, were obtained from the RIKEN Bio-resource Research Center (Ibaraki, Japan). These cells were cultured in 15 mL of RPMI 1640 medium (FUJIFILM Wako Chemicals, Osaka, Japan) containing 10% fetal bovine serum (GIBCO, Uxbridge, UK), 100 μg mL−1 streptomycin, and 100 U mL−1 penicillin (GIBCO) at 37 °C in 5% CO2. Genomic DNA was extracted from each cultured cell line using NucleoSpin® Tissue (Macherey-Nagel, Düren, Germany). The genomic DNA was treated with restriction enzyme NheI-HF (New England BioLabs) and the treated genomic DNA was purified by phenol–chloroform extraction and ethanol precipitation. The genomic DNA was dissolved in TE buffer, and DNA concentrations were determined by measuring the absorbance at 260 nm using a Nanodrop 2000 (Thermo Fisher Scientific). This solution was adjusted to 1 pM and used as a template for the LAMP reaction.2.3. LAMP primer design and LAMP reaction
Primers were designed using Primer Explorer V5 software (Fujitsu, Tokyo, Japan) (Table S4†). The FIP primer contained sequences of F1c (complementary to F1) and F2. The BIP primer contained sequences of B1c (complementary to B1) and B2. First, FIP hybridizes to F2c at the 3′-end of the target DNA, and DNA elongation by polymerase begins (Fig. 1A). Next, F3, which is a few bases shorter and lower in concentration than FIP, slowly hybridizes to F3c on the target DNA, and strand displacement and DNA elongation begin (Fig. 1B). The elongation of the strand displacement using F3 releases a FIP-linked complementary strand that forms a looped structure at one end (Fig. 1C). The same elongation occurs with BIP and B3 at the 5′-end of the elongated strand. The FIP- and BIP-linked complementary strands resulted in the formation of dumbbell-like DNA structures (Fig. 1D). DNA was amplified with high efficiency from this dumbbell-like DNA structure.LAMP reactions were performed using the Loopamp DNA Amplification Kit (Eiken Chemical Co., Ltd., Tokyo, Japan). The final LAMP reaction solution contained 2 μL template DNA (synthetic DNA: 100 nM, genomic DNA: 1 pM), 1 μL of Bst DNA polymerase, 1.6 μM of FIP and BIP primers, and 0.2 μM each of outer primers F3 and B3, in a 1× reaction mix. The final volume was adjusted to 25 μL. The mixture was incubated at 58 °C for DRD2 and 65 °C for AR. Reaction times were 0, 10, 20, and 30 min for synthetic DNA and 0, 30, 40, and 50 min for genomic DNA. Incubation was started at the same time for all samples, and the LAMP reaction solution was incubated at 80 °C for 5 min to terminate the reaction after each incubation time. The sequences that were expected to be amplified by LAMP in AR and DRD2 are shown in Table 1.
| Gene name | LAMP-amplified sequences (5′ to 3′) | Size (bp) | Number of methylated CpG sites |
|---|---|---|---|
| Bold and underlined text indicates methylated cytosines. For the AR gene, the potential position of methylation in the genomic DNA is described. LAMP; loop-mediated isothermal amplification. | |||
| DRD2 | GCCTGGAACGGGTAGGAGGGGTTGGGGGATTCCGCCATCCCTTGTTTTGAGG![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) GGGAAGCAACCCTGACGCCCACTGGCTCCCACCCACACCCAGAGTAATAAGCTGTGATTGCAGGCTGGGTCCTCACCGTCTGCTCGCCAGTCTTCTCCTTTGAGGACTCAGAAGCCAAGGGTTG GGGAAGCAACCCTGACGCCCACTGGCTCCCACCCACACCCAGAGTAATAAGCTGTGATTGCAGGCTGGGTCCTCACCGTCTGCTCGCCAGTCTTCTCCTTTGAGGACTCAGAAGCCAAGGGTTG |
181 | 5 |
| AR | GGGAGGGGAGAAAAGGAAAGGGGAGGGGAGGGAAAAGGAGGTGGGAAGGCAAGGAGGCGGCCGGTGGGGGGGGACCGACTGCAAACTGTTGCATTTGCTCTCCACCTCCCAGGCCCCCTCGAGATCCGGGGAGCCA |
144 | 8 |
2.4. Turbidity measurement
The absorbance at 650 nm was measured as turbidity to detect precipitates of magnesium pyrophosphate. The mixture of 25 μL LAMP products and 45 μL distilled water was measured using GeneQuant1300 (Biochrom, Cambridge, UK). The turbidity was determined by taking the difference between the absorbance of the sample solution and that of distilled water as a reference.2.5. Circular dichroism (CD) spectroscopy
CD spectral measurements were performed using a J-820 (JASCO, Tokyo, Japan). The prepared full-length synthetic DNA of DRD2 was adjusted to 1 μM for measurement. Each spectrum of unmethylated or methylated dsDNA was subtracted from that of the TE buffer at 25 °C as a blank. Thermal stability was measured from 5 to 95 °C by increasing the temperature in steps of 0.1 °C min−1. The Tm value of each dsDNA sample was calculated using the VWTP-959 melting analysis program (JASCO) with a double-strand cleavage model.2.6. Fluorescence intensity measurement
The LAMP products (2 μL) were mixed with 300 μL of 1/10000 diluted SYBR Green I (Takara, Shiga, Japan) in 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA, and incubated at 25 °C for 30 min. Fluorescence intensity was measured using Varioskan Flash (Thermo Fisher Scientific) (excitation at 494 nm, detection at 521 nm). The fluorescence intensity of the LAMP reaction at 0 min was subtracted from that of the background. To generate a calibration curve, synthetic unmethylated and methylated DNA samples were mixed in different ratios to generate samples with different levels of methylation (ranging from 0% to 100%).
2.7. Bisulfite sequencing
Genomic DNA was treated with bisulfite using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). The AR promoter region was amplified by PCR using non-bisulfite-treated DNA with PrimeSTAR® GXL (Takara) and bisulfite-treated DNA with EpiTaqTM HS (Takara). The two primer sets are listed in Table S5.† For PCR using non-bisulfite-treated DNA, 50 μL of the reaction mix contained 10 μL of 5× PrimeSTAR® GXL buffer, 200 μM of each dNTP, 1.25 units of PrimeSTAR® GXL DNA polymerase, 0.2 μM primers, and 2 μL of non-bisulfite-treated DNA. For PCR using bisulfite-treated DNA, 50 μL of the reaction mix contained 5 μL of 10× EpiTaq PCR buffer, 300 μM of each dNTP, 1.25 units of TaKaRa EpiTaq HS, 2.5 mM MgCl2, 0.4 μM primers, and 2 μL of bisulfite-treated DNA. The PCR conditions for PrimeSTAR® GXL were 35 cycles of 95 °C for 10 s, 58 °C for 30 s, and 72 °C for 40 s, and those for EpiTaq™ HS were 40 cycles of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 30 s. The PCR product was cloned using a pGEM-T Easy vector system (Promega) and transformed into Escherichia coli DH5α strain. White colonies were collected, and plasmids were extracted using the FastGene™ Plasmid Mini Kit (NIPPON Genetics Co., Ltd., Tokyo, Japan). The sequence of the inserted PCR product was verified by DNA sequencing from the Eurofins sequencing service (Eurofins Genomics).2.8. Electrophoresis analysis
LAMP products of genomic DNA from LNCaP and Du145 cells reacted for 40 min were electrophoresed on 2% agarose gel. A 100 bp ladder was used as a size marker (FUJIFILM Wako Chemicals, Osaka, Japan). We used plasmid DNA containing the AR gene as a positive control for the LAMP reaction because it was difficult to synthesize oligo DNA of more than 200 bases and distilled water as a negative control. The band intensities between 200 and 1000 bp were analyzed using ImageJ software (National Institutes of Health, MD, USA).3. Results and discussion
3.1. Turbidity measurement of LAMP products of synthetic DNA in DRD2
To investigate whether changes in DNA amplification efficiency in LAMP are due to CpG methylation or not, DNA samples with known cytosine methylation were required. We first used synthetic dsDNA from the upstream region of the DRD2 gene, which could potentially be used for the early diagnosis of schizophrenia. DRD2 is associated with schizophrenia, and CpG methylation rates were significantly lower in the blood of patients with schizophrenia than in controls.34–36The target DNA from the upstream region of DRD2 (260 bp) was required to perform LAMP; however, it is difficult to chemically synthesize such long DNA oligonucleotides. Therefore, in this work, we first split the full-length dsDNA of 260 bp into six short oligonucleotides with lengths of 71, 69, and 120 bases as the target strands and of 97, 75, and 88 bases as the complementary strands (Table S1†). Subsequently, each of the six short oligonucleotides was chemically synthesized, followed by hybridization and ligation, to obtain the 260 bp full-length dsDNA (Fig. S1A†). dsDNA with 10 methylated cytosines was used as the methylated DNA, and dsDNA with no methylated cytosines was used as the unmethylated DNA. Full-length dsDNA was obtained by ligation and analyzed by agarose gel electrophoresis (Fig. S1B†). The LAMP reaction was performed using 2 μL of 100 nM dsDNA.
One of the most popular detection methods for LAMP products is turbidity measurement.37 Turbidity due to precipitates such as magnesium pyrophosphate was produced as the LAMP reaction progressed. We performed the LAMP reaction for 0, 10, 20, and 30 min and measured turbidity as absorbance at 650 nm. The absorbances of unmethylated and methylated DNA samples increased at 30 min, but that of methylated DNA was lower than that of unmethylated DNA (Fig. 2). Statistical analysis was performed using One-way ANOVA with Bonferroni correction, and the p-value for unmethylated and methylated DNAs at 30 min of reaction was less than 0.01.
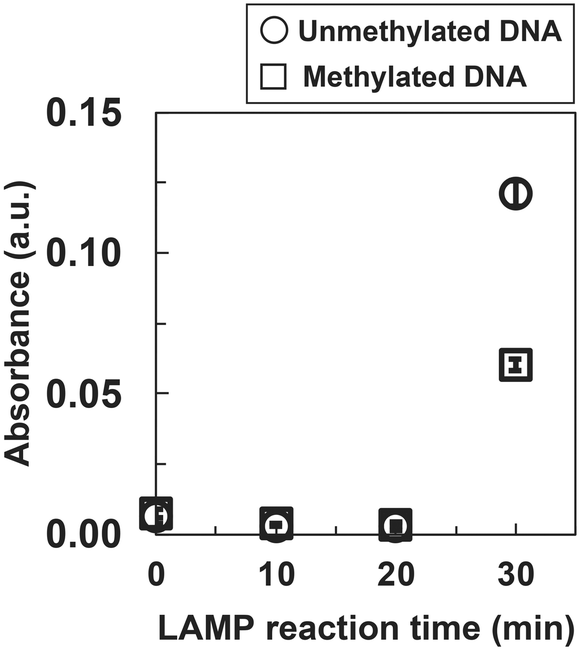 | ||
| Fig. 2 Absorbance measurement of the LAMP product of synthetic DRD2 DNA. Absorbance data are shown as the mean ± SD on the graph (N = 3). LAMP; loop-mediated isothermal amplification, a.u.; arbitrary unit. | ||
There is a difference in absorbance despite using the same DNA sequence and primer set. This may reflect differences in methylation.
To confirm dsDNA stability by CpG methylation in the DNA sample used for the LAMP reaction, we performed temperature-dependent CD spectroscopy on a 260 bp dsDNA sample used in the LAMP reaction of DRD2 (Fig. S2A and B†). The Tm values were 72.2 ± 0.5 °C for unmethylated dsDNA and 71.4 ± 0.6 °C for methylated dsDNA, which were similar. Furthermore, since the LAMP reaction of DRD2 was performed at 58 °C, we compared the spectral data of unmethylated and methylated DNAs at 60 °C, and found that all obtained spectra were similar; thus, there was almost no difference here neither (Fig. S2C†). We also found no difference in spectral data between unmethylated and methylated DNAs in this measurement because it is difficult to find differences due to methylation in the measurement using long DNA sequences (>200 bp).
For short DNA sequences (10–20 bp), methylation results in a more tightly bound base pairing and an increased thermal melting temperature.30,31 The partial stabilization of dsDNA by methylation may have prevented the strand displacement reaction of strand-displacement DNA polymerase, resulting in a lower LAMP reaction efficiency in methylated DNA.
3.2. Fluorescence intensity measurement of LAMP products of synthetic DNA in DRD2 and AR
We measured the fluorescence intensity of SYBR Green I for a more detailed analysis of the DNA amplification process concerning the amplification time. SYBR Green I can bind to dsDNA via intercalation and dsDNA can be detected with higher sensitivity than by the turbidity test. The fluorescence intensity was calculated by subtracting the background intensity at 0 min (Δfluorescence intensity).For DRD2, the Δfluorescence intensity for both the unmethylated and methylated DNA samples increased with amplification time and reached saturation at 30 min (Fig. 3A). In addition, the Δfluorescence intensity of the methylated DNA at 20 min was lower than that of the unmethylated DNA at 20 min. Statistical analysis was performed using One-way ANOVA with Bonferroni correction, and the p-value for unmethylated and methylated DNAs at 20 min of reaction was less than 0.01. The results of the fluorescence intensity measurements were in good agreement with those of the turbidity measurement, which showed that methylated DNA was amplified more slowly than unmethylated DNA.
 | ||
| Fig. 3 Fluorescence intensity measurement results of the LAMP product of DRD2 (A) and AR (B) synthetic DNA. Fluorescence intensity data are shown as the mean ± SD on the graph (N = 3). LAMP; loop-mediated isothermal amplification, a.u.; arbitrary unit. | ||
This indicates that the LAMP reaction of methylated DNA saturates slowly. There appears to be a large difference in initial amplification rate because the template DNA is methylated first, whereas the elongated strand does not contain methylated cytosine. The time points at which the signal increased in the turbidity and fluorescence intensity measurements may differ because the sensitivity of the fluorescence measurement is higher than that of the turbidity measurement and the response is faster.
In addition, we next detected synthetic dsDNA from the AR promoter region, which is correlated with hormone-independent prostate cancer.38,39 We used the AR promoter region as a model because prostate cancer cell lines with different CpG methylation levels are readily available. To obtain the target DNA from the AR promoter region (246 bp) to perform LAMP, we first performed PCR from the AR-promoter-region-inserted plasmid DNA. We then obtained methylated and unmethylated DNA samples by methylating the PCR products or not with CpG methyltransferase. The LAMP reaction was performed with 2 μL of 100 nM dsDNA similar to DRD2.
In AR, Δfluorescence intensity increased for both the unmethylated and methylated DNA samples, depending on the amplification time (Fig. 3B). In addition, the Δfluorescence intensity of methylated DNA at 20 min was lower than that of unmethylated DNA at 20 min. Statistical analysis was performed using One-way ANOVA with Bonferroni correction, and the p-value for unmethylated and methylated DNAs at 20 min of reaction was less than 0.01. The results of the fluorescence intensity measurements were in good agreement with those of DRD2, which showed that methylated DNA was amplified more slowly than unmethylated DNA.
In DRD2 and AR, it was confirmed that methylated DNA was amplified more slowly than unmethylated DNA. We assumed that the strand displacement and elongation reaction of strand-displacement DNA polymerase were rather inhibited because CpG methylation increases the thermal stability of dsDNA and makes it more difficult to dissociate.
To investigate whether the fluorescence intensity measurement for LAMP products is dependent on the amount of methylated DNA, we prepared samples with different methylation levels of synthetic DNA from AR. The LAMP products that stopped the reaction at 20 min were used because they showed the greatest difference between the unmethylated and methylated DNA samples, as seen in the results in Fig. 3B. The results show that the fluorescence intensity decreased depending on the methylation level, which ranged from 0% to 100% (Fig. 4). Thus, we obtained the linear calibration curves (R2 = 0.975), and it may be possible to calculate the methylation level of the target DNA by drawing a calibration curve using DNA at the same concentration as the sample concentration.
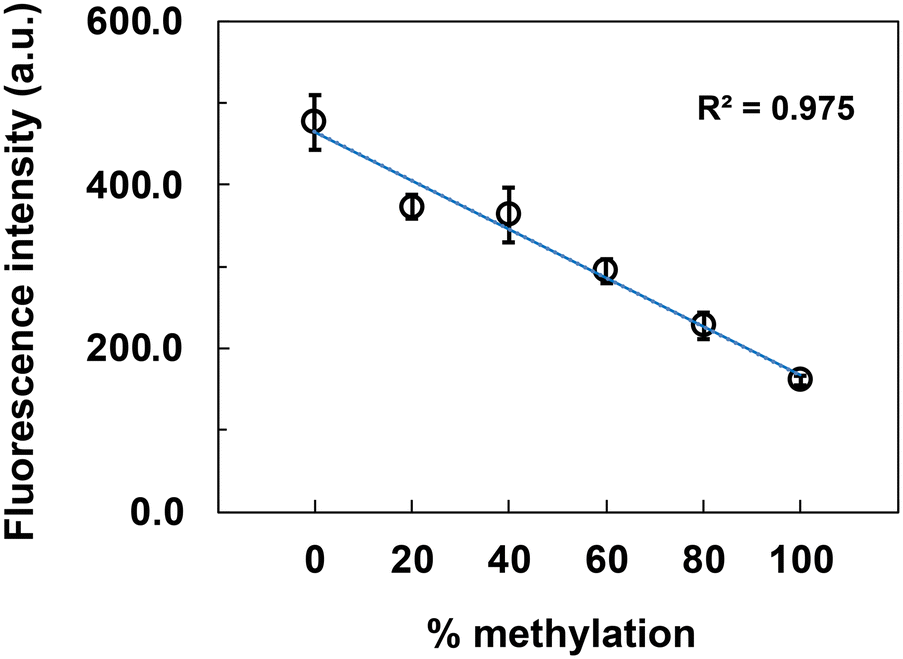 | ||
| Fig. 4 Calibration curve for LAMP products from AR synthetic DNA. Unmethylated and methylated DNA samples were mixed to prepare samples with different methylation levels. Fluorescence intensity data are shown as the mean ± SD on the graph (N = 3). LAMP; loop-mediated isothermal amplification, a.u.; arbitrary unit. | ||
3.3. Amplification of genomic DNA from LNCaP and Du145 cells of AR gene by LAMP
We performed the LAMP reaction with genomic DNA to confirm that CpG methylation also affects the efficiency of the LAMP reaction in the genomic DNA samples. Genomic DNA was extracted from LNCaP and Du145 cells. These cell lines are well-known models of prostate cancer; LNCaP shows low methylation, and Du145 shows high methylation in the AR promoter region.38,39First, we determined the CpG methylation levels in the LAMP amplification region of the AR gene from the genomic DNA of LNCaP and Du145 cells via bisulfite sequencing. The CpG methylation levels were as follows: LNCaP 0% and Du145 78.3% (N = 5) (Fig. 5A). In addition, the methylation levels were confirmed by outsourced testing at Funakoshi Co., Ltd. (Fig. S3†). The CpG methylation levels were as follows: LNCaP 4.2% and Du145 71.9% (N = 8). We used these genomic DNAs because the outsourced results were almost identical to ours. The LAMP reaction was performed with 2 μL of 1 pM genomic DNA. The LAMP products were detected by fluorescence intensity measurement and electrophoresis analysis.
 | ||
| Fig. 5 Analysis of the LAMP-amplified AR promoter region in genomic DNA. (A) Methylation status was analyzed by bisulfite sequencing. Percentages in the parentheses indicate methylation levels. Each circle corresponds to one CpG site. The closed circles indicate methylated CpG sites and the open ones indicate unmethylated CpG sites. The set of horizontally aligned circles represents an individual clone and the vertical number represents an individual CpG site in these tables. “N” refers to the number of sequenced clones containing genomic DNA from each cell. (B) Fluorescence intensity measurement. Fluorescence intensity data are shown as the mean ± SD on the graph (N = 3). (C) Electrophoresis analysis of the LAMP-amplified genomic DNA. LAMP products were electrophoresed in 2% agarose gel. Lane M, 100 bp ladder used as the size marker; lanes 1–4, all LAMP products from genomic DNA of LNCaP cells, Du145 cells, plasmid DNA inserted in the AR gene as a positive control of the LAMP reaction, and distilled water as a negative control of LAMP reaction, respectively. LAMP; loop-mediated isothermal amplification, a.u.; arbitrary unit. | ||
In contrast to the synthetic DNA (100 nM), 1 pM genomic DNA was used for LAMP because the genomic DNA extracted from the cells was not soluble in buffer at concentrations above 1 pM. We measured the turbidity of the LAMP product from LNCaP and Du145 cell genomic DNA (data not shown). There were no significant differences between the LNCaP and Du145 cell genomic DNA samples. The reason why the turbidity measurement showed no differences may be the much lower concentration of genomic DNA (1 pM) than of synthetic DNA (100 nM) and the lower sensitivity to detect small differences, such as methylation in high background due to unrelated DNA sequences in the genomic DNA. First, the LAMP products were detected by measuring the fluorescence intensity of SYBR Green I, which intercalates into the dsDNAs produced by LAMP. The fluorescence intensity was calculated by subtracting the background intensity at 0 min (Δfluorescence intensity). The Δfluorescence intensity increased for both the LNCaP and Du145 cell genomic DNA samples depending on the amplification time, and that of Du145 cell genomic DNA was lower than that of LNCaP cell genomic DNA at 40 min (Fig. 5B). Statistical analysis was performed using One-way ANOVA with Bonferroni correction, and the p-value for LNCaP and Du145 cell genomic DNAs at 40 min of reaction was less than 0.01. This indicated that the LAMP reaction of highly methylated genomic DNA was slow to saturate. This result was similar to that of synthetic DNA, in that methylated DNA slowly saturated (Fig. 3B). The time points at which the signal increased in the synthetic and genomic DNA samples differed possibly because the amounts of DNA in the samples were different (synthetic DNA: 100 nM, genomic DNA: 1 pM). Fig. 4 shows the results of the calibration curve of the LAMP product from 100 nM synthetic DNA, so it cannot be compared to 1 pM genomic DNA. However, it might be possible to determine the methylation rate of samples with unknown methylation rates by drawing a calibration curve of the LAMP product from 1 pM genomic DNA using genomic DNA from cells with different methylation rates. Next, we performed the electrophoresis analysis of the LAMP products to confirm that the LAMP reaction of Du145 cell genomic DNA saturates slowly in another method. The LAMP products from the 40 min reaction were used because they showed the greatest difference between the LNCaP and Du145 cell genomic DNA samples. When the LAMP reaction is successful, DNA bands of different sizes are produced. Plasmid DNA containing the AR gene was used as a positive control for the LAMP reaction because it was difficult to synthesize oligo DNA of more than 200 bases. The LAMP reaction was performed without any problems with the positive control, and there was no nonspecific amplification from the negative control with distilled water (Fig. 5C). There appeared to be a difference between the band intensities of the LAMP products of the LNCaP and Du145 cell genomic DNA samples; therefore, the intensities of the bands with dimensions ranging between 200 and 1000 bp were analyzed using ImageJ. The band intensity was higher in the genomic DNA of LNCaP cells (band intensity: 29.7 arbitrary unit [a.u.]) than in Du145 cells (band intensity: 23.4 a.u.). The band intensity of the LAMP products from the Du145 cell genomic DNA was lower despite the same LAMP reaction time, indicating that the high methylation of the genomic DNA interfered with the amplification reaction.
The amount of genomic DNA used to detect genomic DNA in this study was 2 amol. When CpG methylation testing is performed clinically, leukocyte genomic DNA in blood is often used as the sample. It was reported that the number of leukocytes in Japanese blood is approximately 6000 cells per μL, and it is calculated that 1 mL of such blood is sufficient to obtain the required amount of genomic DNA by this method.40 Therefore, this method can potentially detect CpG methylation with high sensitivity using blood as the sample.
4. Conclusions
We demonstrated that the amplification efficiency of LAMP was reduced by CpG methylation. This was confirmed not only with synthetic DNA but also with genomic DNA extracted from cells. This may be because CpG methylation increases the structural stability of dsDNA, preventing strand displacement by DNA polymerase in the LAMP reaction.There are several limitations of this method for the detection of CpG methylation. The first is that the concentrations of DNA samples used for detection must be pre-adjusted. The time point of signal change was different between the synthetic and genomic DNA samples, possibly depending on the concentrations of DNA. Therefore, to detect CpG methylation by this method, a procedure to adjust the DNA concentration of the target DNA is required. The second is the primer design. The LAMP reaction is a method of amplifying DNA using multiple primers in a complex structure. Particularly when using genomic DNA, which contains a large amount of DNA other than the target sequence, primers that bind nonspecifically to DNA other than the target are expected to adversely affect the efficiency of amplification by CpG methylation. Therefore, primers for the LAMP reaction should be designed carefully. The third is the density of CpG methylation in the target sequence. The target sequence must be a CpG-rich sequence, such as a CpG island with a high density of CpGs, or the difference in the DNA amplification efficiency of the LAMP reaction is not detectable. Therefore, similarly to the primer design, the target sequence should be carefully considered. If these conditions are met, this method can detect CpG methylation within 1 h by performing a LAMP reaction using commercially available reagents. It may be possible to apply it as a rapid and easy method for detecting CpG methylation.
Data availability
All key data supporting the conclusions made in this paper have been included either in the main text or in the ESI.†Author contributions
M. T. and K. I. conceived and designed the study. M. T. and K. W. performed the experiments. M. T., K. T., and K. I. analyzed the data. M. T., K. T., and K. I. wrote the manuscript. All the authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr. Taiji Oyama, JASCO Corporation, for calculating the Tm value. This work was supported by the Support for Pioneering Research Initiated by the Next Generation (FL-SPRING), Tokyo University of Agriculture and Technology, Japan.References
- R. Holliday, Epigenetics: A historical overview, Epigenetics, 2006, 1, 76–80 Search PubMed.
- S. C. Barton, M. A. H. Surani and M. L. Norris, Role of paternal and maternal genomes in mouse development, Nature, 1984, 311, 374–376 CAS.
- R. S. Hansen and S. M. Gartler, 5-Azacytidine-induced reactivation of the human X chromosome-linked PGK1 gene is associated with a large region of cytosine demethylation in the 5′ CpG island, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4174–4178 CAS.
- R. Z. Chen, U. Pettersson, C. Beard, L. Jackson-Grusby and R. Jaenisch, DNA hypomethylation leads to elevated mutation rates, Nature, 1998, 395, 89–93 CAS.
- A. P. Wolffe and M. A. Matzke, Epigenetics: Regulation through repression, Science, 1999, 286, 481–486 CAS.
- T. H. Bestor, The DNA methyltransferases of mammals, Hum. Mol. Genet., 2000, 9, 2395–2402 CAS.
- D. J. Driscoll, M. F. Waters, C. A. Williams, R. T. Zori, C. C. Glenn, K. M. Avidano and R. D. Nicholls, A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes, Genomics, 1992, 13, 917–924 CAS.
- J. F. Costello, M. C. Frühwald, D. J. Smiraglia, L. J. Rush, G. P. Robertson, X. Gao, F. A. Wright, J. D. Feramisco, P. Peltomäki, J. C. Lang, D. E. Schuller, L. Yu, C. D. Bloomfield, M. A. Caligiuri, A. Yates, R. Nishikawa, H.-J. Su Huang, N. J. Petrelli, X. Zhang, M. S. O'Dorisio, W. A. Held, W. K. Cavenee and C. Plass, Aberrant CpG-island methylation has non-random and tumour-type–specific patterns, Nat. Genet., 2000, 24, 132–138 CrossRef CAS PubMed.
- S. Numata, K. Ishii, A. Tajima, J. Iga, M. Kinoshita, S. Watanabe, H. Umehara, M. Fuchikami, S. Okada, S. Boku, A. Hishimoto, S. Shimodera, I. Imoto, S. Morinobu and T. Ohmori, Blood diagnostic biomarkers for major depressive disorder using multiplex DNA methylation profiles: Discovery and validation, Epigenetics, 2015, 10, 135–141 CrossRef PubMed.
- M. Frommer, L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Paul, A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1827–1831 CrossRef CAS PubMed.
- J. G. Herman, J. R. Graff, S. Myöhänen, B. D. Nelkin and S. B. Baylin, Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS PubMed.
- Z. Xiong and P. W. Laird, COBRA: A sensitive and quantitative DNA methylation assay, Nucleic Acids Res., 1997, 25, 2532–2534 CrossRef CAS PubMed.
- T. K. Wojdacz and A. Dobrovic, Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation, Nucleic Acids Res., 2007, 35, e41 CrossRef PubMed.
- R. Shapiro, R. E. Servis and M. Welcher, Reactions of uracil and cytosine derivatives with sodium bisulfite, J. Am. Chem. Soc., 1970, 92, 422–424 CrossRef CAS.
- H. Hayatsu, Y. Wataya and K. Kai, Addition of sodium bisulfite to uracil and to cytosine, J. Am. Chem. Soc., 1970, 92, 724–726 CrossRef CAS PubMed.
- A. O. H. Nygren, Methylation-specific MLPA (MS-MLPA): Simultaneous detection of CpG methylation and copy number changes of up to 40 sequences, Nucleic Acids Res., 2005, 33, e128 CrossRef PubMed.
- M. Weber, J. J. Davies, D. Wittig, E. J. Oakeley, M. Haase, W. L. Lam and D. Schübeler, Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells, Nat. Genet., 2005, 37, 853–862 CrossRef CAS PubMed.
- D. Hiraoka, W. Yoshida, K. Abe, H. Wakeda, K. Hata and K. Ikebukuro, Development of a method to measure DNA methylation levels by using methyl CpG-binding protein and luciferase-fused zinc finger protein, Anal. Chem., 2012, 84, 8259–8264 CrossRef CAS PubMed.
- K. Tsukakoshi, S. Saito, W. Yoshida, S. Goto and K. Ikebukuro, CpG methylation changes G-quadruplex structures derived from gene promoters and interaction with VEGF and SP1, Molecules, 2018, 23, 944 CrossRef PubMed.
- D. Oshikawa, S. Inaba, Y. Kitagawa, K. Tsukakoshi and K. Ikebukuro, CpG methylation altered the stability and structure of the i-motifs located in the CpG islands, Int. J. Mol. Sci., 2022, 23, 6467 CAS.
- H. Hasegawa, I. Sasaki, K. Tsukakoshi, Y. Ma, K. Nagasawa, S. Numata, Y. Inoue, Y. Kim and K. Ikebukuro, Detection of CpG methylation in G-quadruplex forming sequences using G-quadruplex ligands, Int. J. Mol. Sci., 2021, 22, 13159 CAS.
- M. Gellert, M. N. Lipsett and D. R. Davies, Helix formation by guanylic acid, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 CAS.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Quadruplex DNA: Sequence, topology and structure, Nucleic Acids Res., 2006, 34, 5402–5415 CAS.
- G. Manzini, N. Yathindra and L. E. Xodo, Evidence for intramolecularly folded i-DNA structures in biologically relevant CCC-repeat sequences, Nucleic Acids Res., 1994, 22, 4634–4640 CAS.
- H. A. Day, P. Pavlou and Z. A. E. Waller, i-motif DNA: Structure, stability and targeting with ligands, Bioorg. Med. Chem., 2014, 22, 4407–4418 CAS.
- W. Yoshida, H. Yoshioka, D. H. Bay, K. Iida, K. Ikebukuro, K. Nagasawa and I. Karube, Detection of DNA methylation of G-quadruplex and i-motif-forming sequences by measuring the initial elongation efficiency of polymerase chain reaction, Anal. Chem., 2016, 88, 7101–7107 CAS.
- D. Nathan and D. M. Crothers, Bending and flexibility of methylated and unmethylated EcoRI DNA, J. Mol. Biol., 2002, 316, 7–17 CAS.
- P. M. D. Severin, X. Zou, H. E. Gaub and K. Schulten, Cytosine methylation alters DNA mechanical properties, Nucleic Acids Res., 2011, 39, 8740–8751 CAS.
- C. M. R. López, A. J. Lloyd, K. Leonard and M. J. Wilkinson, Differential effect of three base modifications on DNA thermostability revealed by high resolution melting, Anal. Chem., 2012, 84, 7336–7342 Search PubMed.
- P. J. Sanstead, B. Ashwood, Q. Dai, C. He and A. Tokmakoff, Oxidized derivatives of 5-methylcytosine alter the stability and dehybridization dynamics of duplex DNA, J. Phys. Chem. B, 2020, 124, 1160–1174 CAS.
- C. Rausch, P. Zhang, C. S. Casas-Delucchi, J. L. Daiß, C. Engel, G. Coster, F. D. Hastert, P. Weber and M. C. Cardoso, Cytosine base modifications regulate DNA duplex stability and metabolism, Nucleic Acids Res., 2021, 49, 12870–12894 CAS.
- Y. Umezawa and M. Nishio, Thymine-methyl/π interaction implicated in the sequence-dependent deformability of DNA, Nucleic Acids Res., 2002, 30, 2183–2192 CAS.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Loop-mediated isothermal amplification of DNA, Nucleic Acids Res., 2000, 28, e63 CAS.
- A. Zhang, J. Yu, J. Liu, H. Zhang, Y. Du, J. Zhu, G. He, X. Li, N. Gu and G. Feng, The DNA methylation profile within the 5′-regulatory region of DRD2 in discordant sib pairs with schizophrenia, Schizophr. Res., 2007, 90, 97–103 Search PubMed.
- S. Numata, T. Ye, M. Herman and B. K. Lipska, DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia, Front. Genet., 2014, 5, 280 Search PubMed.
- Y. Funahashi, Y. Yoshino, K. Yamazaki, Y. Ozaki, Y. Mori, T. Mori, S. Ochi, J. Iga and S. Ueno, Analysis of methylation and -141C Ins/Del polymorphisms of the dopamine receptor D2 gene in patients with schizophrenia, Psychiatry Res., 2019, 278, 135–140 CAS.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CAS.
- D. F. Jarrard, H. Kinoshita, Y. Shi, C. Sandefur, D. Hoff, L. F. Meisner, C. Chang, J. G. Herman, W. B. Isaacs and N. Nassif, Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells1, Cancer Res., 1998, 58, 5310–5314 CAS.
- H. Kinoshita, Y. Shi, C. Sandefur, L. F. Meisner, C. Chang, A. Choon, C. R. Reznikoff, G. S. Bova, A. Friedl and D. F. Jarrard, Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer1, Cancer Res., 2000, 60, 3623–3630 CAS.
- S. Sakuragi, J. Moriguchi, F. Ohashi and M. Ikeda, Reference value and annual trend of white blood cell counts among adult Japanese population, Environ. Health Prev. Med., 2012, 18, 143 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sd00012b |
| This journal is © The Royal Society of Chemistry 2025 |