
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAvoiding commercial kit-based DNA isolation and purification steps: a rapid method for Cryptosporidium oocyst detection†
Rabbee G.
Mahmudunnabi
 ab,
Amandeep Singh
Pannu
c,
Nam-Trung
Nguyen
ab,
Helen M.
Stratton
*ab and
Muhammad J. A.
Shiddiky
*c
ab,
Amandeep Singh
Pannu
c,
Nam-Trung
Nguyen
ab,
Helen M.
Stratton
*ab and
Muhammad J. A.
Shiddiky
*c
aSchool of Environment and Science, Griffith University, Nathan, QLD 4111, Australia. E-mail: h.stratton@griffith.edu.au
bQueensland Micro- and Nanotechnology Centre, Griffith University, Nathan, QLD 4111, Australia
cRural Health Research Institute (RHRI), Charles Sturt University, Orange, NSW 2800, Australia. E-mail: mshiddiky@csu.edu.au
First published on 16th January 2025
Abstract
Current routine diagnostic tests for Cryptosporidium oocysts in water are performed in centralised laboratories using the National Association of Testing Authorities (NATA) approved USEPA Method 1623.1. This method uses fluorescent microscopy, which suffers from artefacts and false positive responses from contaminating oocyst-size particles. Additionally, existing molecular detection methods based on real-time PCR (qPCR) require purified nucleic acid, primarily relying on laborious, time-consuming, and expensive centralised laboratory-based DNA isolation procedures using commercial kits. Both the microscopy and PCR-based molecular techniques are not suitable for rapid detection due to the nature of the experiment and instrumentation. This study reports a rapid and simple method that eliminates the need for multi-step DNA isolation and purification procedures. The method involves the direct heat lysis of magnetically isolated Cryptosporidium oocysts from water samples, followed by a loop-mediated isothermal amplification (LAMP)-based detection. The analytical performance of this assay reveals a LOD of 0.17 copies per μL of genomic DNA (gDNA) with a dynamic range from 1.05 × 104 copies per μL to 1.05 copies per μL. We simulated the matrix effect by putting mud into tap water and spiked oocysts to demonstrate the practical applicability of the assay. The designed LAMP detected as low as 5 and 10 oocysts per 10 mL of tap water without and with simulated matrices, respectively. The ultrasensitive nature of this assay can be attributed to its acceleration due to targeting an intron-less gene. We propose that this simple and rapid method can be extended to detect various types of pathogens.
1. Introduction
Cryptosporidium species are obligate intestinal pathogenic protozoa that infect a broad range of hosts, including humans.1 This pathogen is transmitted through the faecal-oral route via contaminated food and water, causing diarrhoea and diarrhoea-associated disease, commonly referred to as cryptosporidiosis.2 In developing countries, this pathogen is endemic due to the lack of sanitation and scarcity of safe drinking water. It has gained public attention because of its frequent appearance in waterborne outbreaks in different parts of the world.3,4 Generally, infections are self-limiting. However, they can be life-threatening for infants and immunocompromised individuals. According to the Global Burden of Diseases (GBD) 2019, cryptosporidiosis was one of the leading causes of diarrhea-related mortality in children under five, with 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 422 deaths and 8.2 million disability-adjusted life-years (DALYs) worldwide.5,6 Both symptomatic and asymptomatic cryptosporidiosis in early childhood lead to malnutrition, stunted growth, and poor cognitive development.7,8
422 deaths and 8.2 million disability-adjusted life-years (DALYs) worldwide.5,6 Both symptomatic and asymptomatic cryptosporidiosis in early childhood lead to malnutrition, stunted growth, and poor cognitive development.7,8
To mitigate the burden of Cryptosporidium-borne disease, testing and monitoring public water supplies and environmental samples is imperative. Currently, the USEPA 1623.1 method stands as the approved method for detecting Cryptosporidium spp. in water. This method consists of three major steps: (i) filtration of water sample for concentrating Cryptosporidium oocysts, (ii) immunomagnetic separation (IMS) for selectively capturing oocysts from the filtrate, and (iii) fluorescent-monoclonal antibody tagging of oocysts for detection through microscopy.9,10 However, microscopic detection in the USEPA method is plagued by autofluorescence and false positive responses from similar-sized debris or algal cells.11 While well-trained personnel may overcome these issues by observing the DAPI-stained nuclei, the limited sensitivity and laborious, time-consuming sample labelling process restrict the number of samples that can be processed per day.2,12 Although enzyme-linked immunosorbent assay (ELISA) is rapid, it offers limited specificity and sensitivity.12 To improve the sensitivity of whole oocyst detection, various biosensor-based techniques, such as electrochemical sensors,13–15 CRISPR-based sandwich ELISA,16 gold nanoparticle-based immunoblot assay,17 have been introduced. While most of these methods provide adequate sensitivity, the electrochemical sensor and dot blot still struggle with false positive signals from nonspecific adsorption. The CRISPR-based system is affected by the GC content of the target sequence, and both activity and specificity depend on the guide RNA.18
Over the years, molecular methods have gained popularity over whole oocyst detection using microscopy.9,12 Polymerase chain reaction (PCR),19 nested PCR,20 quantitative PCR (qPCR),21 and TaqMan probe-based qPCR,22,23 have been used widely for the detection of Cryptosporidium spp. While PCR-based approaches are powerful techniques to study the epidemiology and transmission of the parasite, their sensitivity and specificity are significantly affected by the quantity and quality of DNA preparations. Moreover, they are reported to be influenced by various ionic inhibitors present in environmental and water samples.24,25 TaqMan probe increased the selectivity, whereas droplet digital PCR (ddPCR)26,27 offers high sensitivity without being affected by PCR inhibitors. However, both techniques increase the cost per reaction and are unsuitable for on-site application.
Besides the ionic inhibitors, molecular detection of pathogens in environmental samples, especially from water, is heavily limited by preconcentration/filtration steps. Therefore, methods with innate sensitivity that are less affected by inhibitory molecules must be considered. In this regard, loop-mediated isothermal amplification (LAMP) is suitable for rapid and sensitive molecular detection of viruses,28 bacteria,29 parasites,30 and other pathogens. The strand displacement activity of Bst polymerase is key for LAMP reactions that amplify DNA/RNA within 30–60 minutes at a constant temperature, typically at 65 °C.31 As this enzyme is resistant to most ionic inhibitors, LAMP has been successfully applied to environmental sample analysis.27 Moreover, LAMP does not require temperature cycling, making it highly suited for resource-limited settings. Results can be obtained through colorimetric detection, allowing for simple and cost-effective diagnostics without the need for sophisticated equipment. When applied to Cryptosporidium spp., LAMP offers significant advantages in both sensitivity and ease of use, especially when targeting oocysts in complex environmental samples. Karanis et al.32 first reported the preliminary evaluation of a LAMP assay for Cryptosporidium spp. targeting the 60 kDa glycoprotein (gp60) gene. Following this, Bakheit et al.30 designed a highly sensitive LAMP assay targeting the S-adenosyl-L-methionine synthetase (SAM) gene and demonstrated that LAMP successfully detected Cryptosporidium spp. that had previously tested negative by PCR. Later, a separate group reported reverse transcription LAMP for 18S rRNA transcript, which is more sensitive than previously mentioned LAMP assays.33
Until now, all these LAMP assays for detecting Cryptosporidium spp. have relied on commercial extraction kits or complicated nucleic acid extraction procedures to obtain purified or semi-purified nucleic acids. This limitation hinders the potential use of LAMP in point-of-care or field settings. This study presents an approach for Cryptosporidium detection, wherein the nucleic acid is extracted through heat lysis in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5). A portion of prepared lysate, without nucleic acid purification, is then used for subsequent LAMP amplification of specific target sequences. This proof-of-concept study successfully detected Cryptosporidium spp. in tap water. It can easily be adapted to detect Cryptosporidium spp. in other types of water samples (e.g., wastewater, drinking water, environmental waters etc.).
2. Experimental
2.1 Materials and instrument
All oligonucleotide sequences (Table S1†) were ordered from Integrated DNA Technologies (USA). Dynabeads® MyOne™ Streptavidin C1, PBS buffer tablet were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, United States). The anti-Cryptosporidium monoclonal antibody (catalog number ab54066) and the Biotin Conjugation Kit (Fast, Type B) – Lightning-Link® (catalog number ab201796) were purchased from Abcam (Cambridge, MA, USA). Viable Cryptosporidium oocysts (107 oocysts per mL) were purchased from Biopoint Pty. LTD (Sydney, NSW, Australia) and Cryptosporidium purified genomic DNA (1.05 × 105 copies per μL) was purchased from ATCC (Manassas, Virginia, USA). SensiFAST™ SYBR® No-ROX Kit (Meridian Bioscience, Cincinnati, Ohio, USA) and Luna® Universal One-Step RT-qPCR Kit (NEB, Ipswich, MA, USA) were used for qPCR and RT-qPCR, respectively. The WarmStart® Colorimetric LAMP 2× Master Mix (DNA & RNA) and WarmStart® Fluorescent LAMP Kit (DNA & RNA) were ordered from NEB for LAMP. DNA and RNA isolation from oocysts was performed using the DNeasy Blood & Tissue Kit (Qiagen, USA), FastDNA™ SPIN Kit for Soil (MP Biomedicals, USA), and Monarch® Total RNA Miniprep Kit (NEB, USA). The Invitrogen™ DNA-free™ DNA Removal Kit was obtained from Thermo Fisher Scientific. All other reagents were of analytical grade and purchased from Sigma-Aldrich (USA).Temperature cycling and LAMP reactions were performed in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Gel electrophoresis and imaging were conducted on the Horizontal Electrophoresis System (Bio-Rad) and the Gel Doc XR+ Imaging System (Bio-Rad). Bead beating for DNA and RNA extraction was performed using the FastPrep-24 5G (MP biomaterials, USA).
2.2 Nucleic acid extraction and Cryptosporidium lysate preparations
To extract DNA and RNA from Cryptosporidium oocysts, initially, oocysts were subjected to two rounds of bead beating (6 m s−1 for 40 s each) using 1.0 mm glass beads in FastPrep-24 5G. After this step, DNA and RNA were isolated according to the protocol provided with the DNeasy Blood & Tissue Kit, FastDNA™ SPIN Kit for Soil, and Monarch® Total RNA Miniprep Kit, respectively. Subsequently, DNA and RNA preparations underwent additional treatment with RNase and DNase to eliminate co-purified RNA and DNA.For the lysis of Cryptosporidium oocyst without using a commercial kit, an appropriate number of oocysts was suspended in 1× TE buffer (10 mM Tris, 1 mM EDTA, pH 7.5), and the samples were heated to 95 °C for 3, 5, 10 and 15 minutes to optimise the appropriate incubation time.
2.3 LAMP, qPCR, and RT-qPCR of nucleic acid
LAMP primers used in this study were reported by Bakheit et al.30 According to that report, the LAMP primer set (Table S1†) can amplify the SAM gene of C. parvum (accession no. AB119646.1 and AY161084.1) C. hominis (accession no. XM_662396) and C. meleagridis (accession no. AB119648.1), enabling the simultaneous detection of these species. The LAMP forward (F3) and backward (B3) primers were used as qPCR and RT-qPCR primers, generating an amplicon of 216 bp. However, multiple sequence alignment of four sequences of the mentioned accession numbers was performed in MEGA software (versions MEGA 11), and the complementary relationships of each primer were tested, as shown in Fig. S1.†All six LAMP primer stocks were prepared at a concentration of 100 μM (Table S2†). To minimize variability in the LAMP reaction, a 10× concentrated primer stock was created based on calculations provided in Table S1.† The stock was then aliquoted into small volumes and stored at −20 °C for extended use. The original protocols and calculations for LAMP, qPCR, and RT-qPCR were slightly modified and adjusted to suit the objective of the current study. For all amplification reactions (qPCR, RT-qPCR, and LAMP), 2.5 μL of Cryptosporidium genomic DNA target (ranging from 1.05 × 104 copies per μL to 1.05 × 10−1 copies per μL) or lysate were used, and the final volume was adjusted with nuclease-free water (Table S3†). The temperature cycling conditions for each type of molecular amplification are detailed in Table S4.† The colourimetric and fluorescent LAMP reactions were set for approximately 45 minutes in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Fluorescent readings were programmed in the instrument every 53 seconds (× 50 cycles ≈ 44.15 or 45 minutes), and the color change in the cLAMP was captured using a mobile camera (Samsung S21).
2.4 Gel electrophoresis
A 1.5% agarose gel was made in 1× TAE buffer to do the gel electrophoresis. 3 μL SYBR Safe (Invitrogen, Australia) was added to every 100 μL of gel during the gel preparation. Subsequently, 4 μL of the sample was loaded into each gel well, and electrophoresis was conducted at 90 volts for 1 hour.2.5 Modification of magnetic beads with anti-Cryptosporidium monoclonal antibody
First, the anti-Cryptosporidium monoclonal antibody (Abcam) was biotinylated according to the protocol of Biotin Conjugation Kit (Fast, Type B) – Lightning-Link® kit (Abcam). Briefly, after gently mixing 10 μL of antibody (1.0 mg mL−1) with 1.0 μL of modifier reagent, the whole solution was added to a vial of lyophilised biotin. Following the incubation of 15 minutes at dark, 1.0 μL of quencher reagent was added to the biotinylated antibody. Subsequently, Dynabeads MyOne™ streptavidin (magnetic beads) was modified with biotinylated anti-Cryptosporidium monoclonal antibody following the protocol provided with Dynabeads. In brief, 5.0 mg (500 μL) of Dynabeads were washed three times. Then, 20 μg of biotinylated anti-Cryptosporidium monoclonal antibody was added, followed by incubation at room temperature for 30 minutes with gentle rotation. Subsequently, Dynabeads were washed three times with PBS/BSA (PBS, pH 7.4 containing 0.01% [w/v] BSA) to remove biotinylated antibodies and, finally, resuspended in 500 μL of PBS (10 mM, pH 7.4).2.6 Determination of optimal bead concentration for immuno-magnetic isolation
To determine the optimal number of antibody-modified magnetic beads for the immune-magnetic isolation (IMS) of oocysts, 50 μL (4.25 × 108 beads) of monoclonal antibody-modified magnetic beads were diluted to 1080 μL of PBS. Then, four replicates of 60 μL, 50 μL, 40 μL, 30 μL, 20 μL and 10 μL of antibody-modified beads (equivalent to 2.36 × 107 beads, 1.97 × 107 beads, 1.57 ×107 beads, 1.17 × 107 beads, 7.86 × 107 beads, 3.93 × 107 beads respectively) were incubated with 104Cryptosporidium oocysts in 5 mL PBS (pH = 7.4) buffer at room temperature for 30 minutes. After that, oocysts-bound magnetic beads were resuspended in 15 μL of TE buffer and heated at 95 °C for 10 minutes, followed by magnetic removal of magnetic beads. 2.5 μL of this lysate was used for fLAMP reactions, and the optimal bead concentration was determined based on the lowest Cq.2.7 Analysis of spiked tap water
For the analysis of Cryptosporidium spiked tap water, different numbers of Cryptosporidium oocysts (1000-5) were spiked into 10 mL of tap water. Following this, samples were centrifuged at 1500g for 15 min. After discarding the supernatant, volumes were adjusted to 5 mL with PBS (pH = 7.4), and samples were transferred to a Leighton tube. This was followed by incubation with an optimized amount of anti-Cryptosporidium-modified magnetic beads (40 μL ≈ 1.17 × 107 beads). Subsequently, the beads were recovered and heated to 95 °C for 10 minutes, followed by LAMP of the lysate as described in section 2.5. For DNA isolation using commercial kits, the captured oocysts bound to beads were directly processed through bead-beating, as outlined in section 2.2. To evaluate the effect of environmental matrices on the assay's performance, we generated a simulated matrix environment. This involved collecting 0.1 mL of packed mud pellet by centrifuging (2000g) forest runoff, which was then spiked into 10 mL of tap water with varying concentrations of oocysts (20, 15, 10, 5, and 3 oocysts). The procedures for IMS, lysate preparation, and the subsequent cLAMP assays were conducted following the previously described protocols.3. Results and discussion
3.1 Assay principle
The USEPA 1623.1 method or International Standard Method (ISO) 15553 (ISO, 15553) is designed to detect Cryptosporidium spp. in water samples. It consists of three main steps: filtering the water sample to concentrate Cryptosporidium oocysts, using immunomagnetic isolation to capture the oocysts selectively, and tagging them with fluorescent-monoclonal antibodies for detection under a microscope. In this study, we introduce a faster approach to detecting Cryptosporidium spp. We propose using LAMP detection following the IMS step in the USEPA method1623.1 or ISO, 15553, eliminating the need for microscopy. Our proof-of-concept demonstrates (Fig. 1) that the LAMP reaction can be accelerated and effectively utilised for highly sensitive detection of Cryptosporidium spp. in water samples without requiring nucleic acid isolation using a commercial kit.
 | ||
| Fig. 1 Schematic representation of the accelerated LAMP assay. During IMS, oocysts were captured by magnetic beads modified with monoclonal antibodies. The beads attached to oocysts were then resuspended in a small volume and heat lysed. Following this step, detection was performed using LAMP, where the lysate was directly added to the reaction without nucleic acid purification. | ||
The sensitivity of the reported approach is realized due to several factors. Direct lysis of oocysts after IMS allows for the extraction and concentration of all genomic DNA (gDNA) and RNA copies of captured oocysts in the desired buffer. Additionally, the designed LAMP primers target the SAM gene, which is intronless. Thus, the designed primers also amplify the corresponding SAM transcript (RNA). The whole lysate containing both gDNA and RNA facilitates the acceleration of the LAMP process (further explained in section 3.3).
3.2 LAMP primer's specificity
Two types of tests have been conducted to evaluate the selectivity of LAMP primers. First, the cLAMP reaction was set with the positive control (C. parvum gDNA, 1.05 × 103 copies per μL), C. parvum lysate, E. coli gDNA, and nuclease-free water as the target. The WarmStart® Colorimetric LAMP 2X Master typically exhibits a bright pink color due to the presence of phenol red, which maintains an overall pH slightly above 8. However, during amplification, the overall pH of the reaction drops to below 7 as excess protons are released by polymerase activity, causing a change in color from bright pink to orange. As shown in Fig. 2(A), the orange color was observed when the positive control and lysate were used as targets, while the color remained unchanged in the presence of E. coli gDNA or no target control. This suggests that the primers are selective for C. parvum. All the positive and negative reactions were run on a 1.5% agarose gel (Fig. 2(B)). Positive samples exhibited concatemers with some prominent bands (lanes 1 and 2 for the positive control; lanes 3 and 4 for C. parvum lysate) ranging between 100–700 bp, whereas negative samples showed faint bands below 100 bp of unamplified LAMP primers. An additional level of selectivity testing was conducted after immunomagnetic separation of oocysts and E. coli lysate (Fig. 2(C)). As expected, a positive color change in the Cryptosporidium lysate indicated the selectivity of the primers. Similar agarose gel band patterns (Fig. 2(D)) were observed for both positive and negative samples, as in Fig. 2(B). | ||
| Fig. 2 LAMP primer selectivity test in colorimetric reaction. (A) Testing the selectivity of primers against the positive control (1.05 × 103 gDNA copies per μL), C. parvum lysate, negative control (E. coli genomic DNA), and no target control (NTC). (C) IMS-LAMP with C. parvum and E. coli for testing selectivity. In both cases (A and B), reactions were analysed by gel electrophoresis, as represented in the lower panel of the fig. (B and D). DNase and RNase-free water were used as targets in NTC. Every colorimetric reaction was performed in duplicate. | ||
3.3 Optimisation of analytical parameters
After testing selectivity, sample incubation time at 95 °C was optimized for the maximum lysis of oocysts. 104 oocysts were resuspended in 50 μL of TE buffer and heated for 3, 5, 10, and 15 minutes. Following this, 2.5 μL of lysate was added to the fLAMP reaction. According to Fig. S2(A),† heating at 95 °C for 10 minutes was the best condition, as indicated by the lowest Cq values. Extending the incubation time beyond 10 minutes could result in the degradation of nucleic acids, which is reflected in the slight increase of the Cq value compared to the Cq value of the 10 minute heating period.Next, the amount of magnetic beads for each IMS was optimized based on the fLAMP response. According to the response of fLAMP (Fig. S2(B)†), four replicates of 60 μL, 50 μL, 40 μL, 30 μL, 20 μL, and 10 μL of antibody-modified magnetic beads were used to capture 104 oocysts. It is important to note that even the lowest amount, 10 μL of antibody-modified magnetic beads, contains 3.93 × 107 beads, which is sufficient to capture 104 oocysts from 1.0 mL of PBS (10 mM, pH 7.4). Fig. S2(B)† shows that Cq values were approximately the same from 60 μL to 30 μL of modified magnetic beads. Recorded Cq values for 20 μL and 10 μL beads were ≥13. However, the exact spots of magnetic accumulation for these two amounts were hard to observe, and resuspending in 15 μL was erroneous, which was reflected in the fLAMP data. For better visibility of the accumulated magnetic beads, the optimal amount for the IMS seemed to be 40 μL (≈ 1.57 × 107 beads) of modified beads.
3.4 Effect of lysate on sensitivity
To understand the cause of the high sensitivity of the LAMP assay using the whole lysate as a target, it is important to confirm the presence of the SAM transcript in the lysate, or in other words, ensure that the SAM gene remains under transcription in the oocyst. To do so, qPCR and RT-qPCR were conducted using gDNA and total RNA from 104 oocysts as targets. During qPCR (Fig. 3(A)), gDNA showed a Cq value of 23.15, whereas no Cq value was observed for RNA. This indicates that the primers were amplifying the gene appropriately, and RNA preparation was free from any carryover DNA contamination. Following this, 2.5 μL of total RNA was used for RT-qPCR, and as shown in Fig. 3(B), the SAM transcripts were amplified with an average Cq value of 26.97. All the products of qPCR and RT-qPCR were analysed by agarose gel electrophoresis (Fig. 3(C and D)), where bands appeared to have a size of 216 bp in all cases. Finally, Cq values of fLAMP with extracted genomic DNA, RNA, and whole lysate of oocyst were compared. WarmStart® LAMP 2X Master Mix contains Bst polymerase, which amplifies both the DNA and RNA. Oocyst lysate, DNA, and RNA were amplified in fLAMP with Cq values of 14.19, 14.89, and 21.07, respectively (Fig. 3(E)). It is evident that the LAMP of lysate is more sensitive than that of DNA or RNA due to the amplification of both the SAM gene and RNA transcript present in the lysate. | ||
| Fig. 3 (A) qPCR and (B) RT-qPCR of genomic DNA and total RNA isolated from 104 oocysts. Agarose gel electrophoresis of (C) qPCR and (D) RT-qPCR products. The product length is 216 bp. (E) LAMP of isolated genomic DNA, RNA, and lysate of 104 oocysts. All the reactions were performed in duplicate. | ||
3.5 LAMP assay sensitivity and analysis of oocysts from tap water
The analytical sensitivity of the LAMP assay was evaluated and compared with qPCR. Under optimized conditions, both cLAMP, fLAMP, and qPCR were conducted for 10-fold dilutions of C. parvum gDNA, ranging from 1.05 × 104 to 1.05 × 10−1 copies per μL (Fig. 4(A) and (B)), with four replicates for each data point. As indicated in Table 1, the reaction efficiency was 100% for all DNA inputs except for 1.05 × 10−1 copies per μL, where the efficiency dropped to 25% (positive reaction:total reaction = 1:4) for both cLAMP and fLAMP (Fig. 4(C)). In the case of qPCR, no amplification was observed at this concentration (Fig. S4†). The Cq values of fLAMP ranging from 1.05 × 104 copies per μL to 1.05 × 100 copies per μL were considered when calculating the LOD (lower limit of detection) due to successful amplification of all replicates. Both fLAMP assay and qPCR exhibited excellent analytical performance (%RSD ≤ 5% for n = 4) with a correlation coefficient, R2 = 0.99, and a detection limit (LOD) of 0.17 copies per μL and 0.3 copies per μL for fLAMP and qPCR, respectively. Here, LOD is defined as 3 × σ/S, where S is the slope of the curve, and σ is the standard deviation of the y-intercept. Although both fLAMP and qPCR amplified the same lowest concentration in this experiment, LAMP demonstrated higher sensitivity than qPCR, as reflected in their LOD. All concentrations were verified using cLAMP (Fig. 4(C)) and confirmed with agarose gel electrophoresis, which displayed concatemers for a positive reaction and exhibited similar prominent band positions between 100 bp and 700 bp (Fig. S3†). A total of 0.263 copies (= (1.05 × 10−1) copies per μL × 2.5 μL) of gDNA were utilized for the lowest concentration. Considering that the genome copy number cannot be a fraction, it is logical to conclude that the positive reaction detected a single copy of the genome.
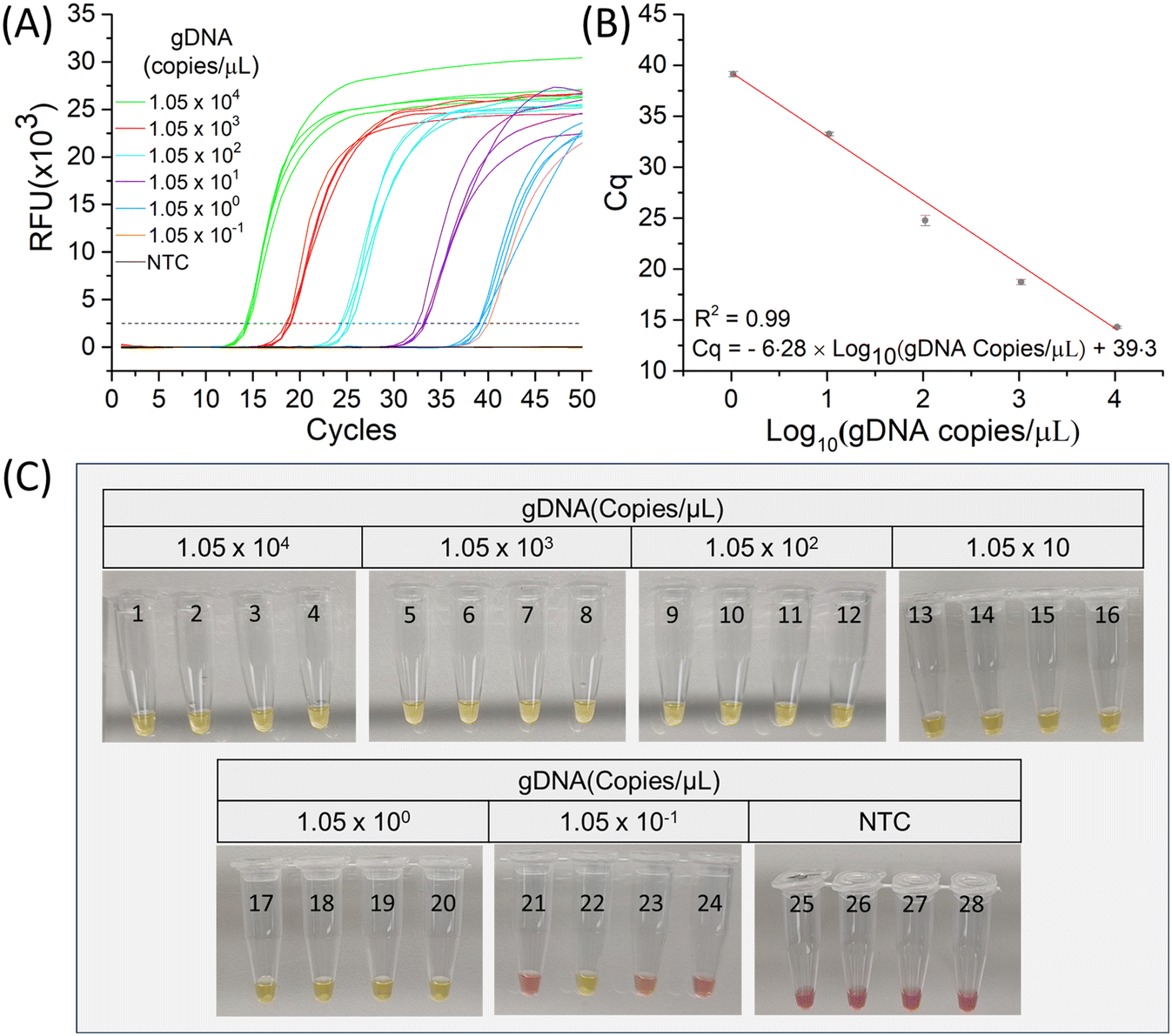 | ||
| Fig. 4 Testing of sensitivity. (A) Fluorescent LAMP (fLAMP) amplification curve for a 10-fold serial dilution of C. parvum genomic DNA, ranging from from 1.05 × 104 to 1.05 × 10−1 copies per μL. (B) Corresponding calibration curve. Each data point with an error bar in the calibration curve represents the mean Cq value and the standard deviation of four measurements. (C) Colorimetric LAMP (cLAMP) results for the same DNA concentration used in fLAMP. Similarly, four cLAMP reactions were performed for each concentration. | ||
| DNA (copies per μL) | 1.05 × 104 | 1.05 × 103 | 1.05 × 102 | 1.05 × 101 | 1.05 × 100 | 1.05 × 10−1 |
|---|---|---|---|---|---|---|
| Positive reaction | 100% (4/4) | 100% (4/4) | 100% (4/4) | 100% (4/4) | 100% (4/4) | 25% (1/4) |
To demonstrate the applicability of this, designated numbers of oocysts (ranging from 1000 to 2) were spiked in 10 mL of tap water (Fig. 5), and samples were processed as described in section 2.7. After the IMS, oocysts bound to magnetic beads were directly lysed by heating at 95 °C for 10 minutes, followed by performing fLAMP and cLAMP with 2.5 μL of lysate. All the fLAMP and cLAMP reactions appeared positive except for 2 oocysts. fLAMP achieved a calculated LOD of 0.47 oocysts (%RSD ≤ 5%, n = 4), or in other words, 1 oocyst per IMS from 10 ml of tap water; however, practically 5 oocysts were detected under the tested conditions. Depending on the age, storage, and environmental conditions of the oocysts, their viability may be compromised, halting their metabolism and impacting the analytical sensitivity of the assay.33,34 Further, we evaluated the effect of matrices on the LAMP assay. A 0.1 mL mud pellet collected from forest runoff was centrifuged at 2000g and used for spiking, with varying numbers of oocysts (20, 15, 10, 5, 3) added to 10 mL of tap water. As shown in Fig. S5,† the cLAMP reaction maintained 100% efficiency at a concentration of 10 oocysts. However, the efficiency dropped to 50% with five oocysts and was undetectable with three oocysts, highlighting a significant impact of the matrix during IMS. Reduced IMS recovery is common in matrix-laden samples, particularly those with high dissolved iron content, which can inhibit assay performance. This effect aligns with previous findings showing lower IMS recoveries in matrix-rich environments,34,35 especially when iron concentrations are elevated, interfering with antibodies binding to the target's epitope.36 In this case, aptamer can be used as a target recognition element instead of an antibody.37 Aptamers can be tailored for specific applications and have proven useful in environmental samples.37,38 Additionally, variability due to the low oocyst counts may contribute to the absence of detectable results at three oocysts in tap water with spiked matrices.
 | ||
| Fig. 5 Detection of oocysts from tap water. (A) Heat lysis combined with fLAMP amplification curve for 5 to 1000 oocysts. (B) Corresponding calibration curve. Each data point with an error bar in the calibration curve represents the mean Cq value and the standard deviation of four measurements. (C) cLAMP for the same number of oocysts used in fLAMP. | ||
We compared the cLAMP response from DNA preparations of two commercially available kits and lysate. To do this, different numbers of oocysts (ranging from 5 to 1000 oocysts) were spiked in 10 mL of tap water. Lysate or DNA was prepared after IMS according to the process described in section 2.7 or in section 2.2, respectively. Following this cLAMP reactions were set with lysate and DNA preparations. As shown in Table S5,† cLAMP with lysate outperformed cLAMP with purified DNA preparation (using DNeasy Blood and Tissue Kits and FastDNA™ SPIN Kit for Soil). While both extraction processes showed consistent cLAMP responses for as low as 30 oocysts, DNeasy exhibited slightly better efficiency (75%) than FastDNA™ (25%), as evident from cLAMP responses of 20 oocysts.
Previously, Inomata et al.33 reported a highly sensitive RT-LAMP (18S rRNA) for Cryptosporidium oocysts detection in serial dilutions of isolated RNA, assumed to be equivalent to a single oocyst. This sensitivity was attributed to an extremely high number of 18S rRNA copies (3.5 × 105) per oocyst.39 However, this sensitivity may be compromised when isolating RNA from an extremely low number of oocysts. Unlike the reported assay, which requires a complex RNA extraction process, our current method does not require any special nucleic acid extraction to achieve similar sensitivity. Sun et al.40 compared three direct DNA isolation processes (boiling, boiling in 1% Triton X-100, and treating with 0.02 M NaOH) against a commercial kit (DNAzol method) and found that boiling the adenovirus sample for 10 minutes provided superior LAMP detection over qPCR. Sowmya et al.41 found that boiling in 1% Triton X-100 was effective for DNA isolation from Gram-positive bacteria and subsequently effective for LAMP detection. Here, we utilized simple heat lysis in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5) for effective lysis of Cryptosporidium oocysts.
Regarding sensitivity and assay preparation time, the accelerated LAMP assay is comparable to the recently reported CRISPR/Cas12a-powered sensor (see Table S6† for a detailed comparison of methods).16,42 CRISPR/Cas12a-powered lateral flow strip (LFS)42 sensor consistently detected 10 oocysts per gram of fecal sample; however, a semi-purified DNA preparation was used, and it took a relatively longer time (1.5 hours) and needed two separate reactions followed by LFS for visual detection.42 Meanwhile, CRISPR/Cas12a-powered immunosensor16 showed a single oocysts detection limit per sample in saline solution. When complex matrices like mud were introduced, LOD compromised to 10 oocysts, depicting the interferences of matrices. Usually, it takes 1.5 hours to detect oocysts, except for a single oocyst detection, which needs to run the reaction with CRISPR/Cas12a enzymatic system enzymes for an extended period of time of nearly 2.5 hours.16 Unlike these sensors, our assay offers an almost similar sensitivity, as mentioned earlier, takes only approximately 40 minutes to detect, and has a comparatively simple detection procedure.
4. Conclusion
In conclusion, our study has successfully detected Cryptosporidium oocysts without the use of commercial kit-based DNA isolation and purification steps. The proposed method, which involves direct heat lysis after immunomagnetic separation for the LAMP reaction, holds the potential to detect 5 to 10 oocysts, depending on water quality. The enhanced sensitivity is attributed to the accelerated LAMP reaction from both DNA and RNA transcripts of an intron-less gene. Importantly, this method emerges as a viable alternative to microscopy or qPCR, as employed in the USEPA method 1623.1 for detecting Cryptosporidium oocysts. This method could be readily adapted to target various intron-lacking genes of Cryptosporidium, such as the heat shock protein 70 gene (accession number XM_001388291.1) and the oocyst wall protein 4 gene (accession number XP_627230.1), both of which are highly conserved and functionally significant in the organism's biology and pathogenesis. By selecting such genetic markers, the method ensures robust detection and enables precise, reliable diagnostics tailored to specific applications. Moreover, the modular design of the method allows seamless adaptation for other pathogens. This can be achieved by substituting the functional elements—such as pathogen-specific monoclonal antibodies and LAMP primers targeting organism-specific genetic markers—without requiring substantial modifications to the core workflow. This adaptability of this platform underscores its potential for broad application in detecting diverse pathogens across various diagnostic contexts.Data availability
The data supporting this article have been included as part of the main text and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the GUIPRS and GUPRS higher degree research scholarships awarded to R. G. M. by Griffith University.References
- A. Zahedi, A. Paparini, F. Jian, I. Robertson and U. Ryan, Public health significance of zoonotic Cryptosporidium species in wildlife: critical insights into better drinking water management, Int. J. Parasitol. Parasites Wildl., 2016, 5, 88–109 CrossRef PubMed.
- E. M. Hassan, B. Örmeci, M. C. DeRosa, B. R. Dixon, S. A. Sattar and A. Iqbal, A review of Cryptosporidium spp. and their detection in water, Water Sci. Technol., 2021, 83, 1–25 CrossRef PubMed.
- A. Efstratiou, J. E. Ongerth and P. Karanis, Waterborne transmission of protozoan parasites: review of worldwide outbreaks-an update 2011–2016, Water Res., 2017, 114, 14–22 CrossRef.
- U. Ryan, N. Hijjawi and L. Xiao, Foodborne Cryptosporidiosis, Int. J. Parasitol., 2018, 48, 1–12 CrossRef.
- IHME, Global burden of disease study, 2019, https://vizhub.healthdata.org/gbd-results/, (accessed 06 November 2024) Search PubMed.
- I. H. Gilbert, S. Vinayak, B. Striepen, U. H. Manjunatha, I. A. Khalil and W. C. Van Voorhis, Safe and effective treatments are needed for cryptosporidiosis, a truly neglected tropical disease, BMJ Glob. Health, 2023, 8, 12540 Search PubMed.
- W. Checkley, R. H. Gilman, L. D. Epstein, M. Suarez, J. Fernando Diaz, L. Cabrera, R. E. Black and C. R. Sterling, Asymptomatic and symptomatic cryptosporidiosis: their acute effect on weight gain in Peruvian children, Am. J. Epidemiol., 1997, 145, 156–163 CrossRef.
- D. I. Guerrant, S. R. Moore, A. A. M. Lima, P. D. Patrick, J. B. Schorling and R. L. Guerrant, Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil, Am. J. Trop. Med. Hyg., 1999, 61, 707–713 Search PubMed.
- USEPA Method 1623.1: Cryptosporidium and Giardia in Water by Filtration/IMS/FA, https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100J7G4.TXT (Accessed: 06 November 2024) Search PubMed.
- C. Delling, I. Holzhausen, A. Daugschies and M. Lendner, Inactivation of Cryptosporidium parvum under laboratory conditions, Parasitol. Res., 2016, 115, 863–866 CrossRef.
- M. W. LeChevallier, G. D. Di Giovanni, J. L. Clancy, Z. Bukhari, S. Bukhari, J. S. Rosen, J. Sobrinho and M. M. Frey, Comparison of method 1623 and cell culture-PCR for detection of Cryptosporidium spp. in source waters, Appl. Environ. Microbiol., 2003, 69, 971 CrossRef.
- U. Ryan, A. Paparini and C. Oskam, New technologies for detection of enteric parasites, Trends Parasitol., 2017, 33, 532–546 CrossRef.
- A. Iqbal, M. Labib, D. Muharemagic, S. Sattar, B. R. Dixon and M. V. Berezovski, Detection of Cryptosporidium parvum oocysts on fresh produce using DNA aptamer, PLoS One, 2015, 10, e0137455 CrossRef PubMed.
- G. S. Luka, H. Najjaran and M. Hoorfar, On-chip-based electrochemical biosensor for the sensitive and label-free detection of Cryptosporidium, Sci. Rep., 2022, 12, 1–11 CrossRef.
- C. Thiruppathiraja, V. Saroja, S. Kamatchiammal, P. Adaikkappan and M. Alagar, Development of electrochemical based sandwich enzyme linked immunosensor for Cryptosporidium parvum detection in drinking water, J. Environ. Monit., 2011, 13, 2782–2787 RSC.
- Y. Li, F. Deng, T. Hall, G. Vesey and E. M. Goldys, CRISPR/Cas12a-powered immunosensor suitable for ultra-sensitive whole Cryptosporidium oocyst detection from water samples using a plate reader, Water Res., 2021, 203, 117553 CrossRef CAS PubMed.
- C. Thiruppathiraja, S. Kamatchiammal, P. Adaikkappan and M. Alagar, An advanced dual labeled gold nanoparticles probe to detect Cryptosporidium parvum using rapid immuno-dot blot assay, Biosens. Bioelectron., 2011, 26, 4624–4627 CrossRef CAS.
- W. J. Jung, S. J. Park, S. Cha and K. Kim, Factors affecting the cleavage efficiency of the CRISPR-Cas9 system, Anim. Cells Syst., 2024, 28, 75–83 CrossRef CAS PubMed.
- U. M. Morgan, L. Pallant, B. W. Dwyer, D. A. Forbes, G. Rich and R. C. A. Thompson, Comparison of PCR and microscopy for detection of Cryptosporidium parvum in human fecal specimens: clinical trial, J. Clin. Microbiol., 1998, 36, 995 CrossRef CAS.
- S. Coupe, C. Sarfati, S. Hamane and F. Derouin, Detection of Cryptosporidium and identification to the species level by nested PCR and restriction fragment length polymorphism, J. Clin. Microbiol., 2005, 43, 1017–1023 CrossRef CAS.
- C. Mary, E. Chapey, E. Dutoit, K. Guyot, L. Hasseine, F. Jeddi, J. Menotti, C. Paraud, C. Pomares, M. Rabodonirina, A. Rieux and F. Derouin, Multicentric evaluation of a new real-time PCR assay for quantification of Cryptosporidium spp. and identification of Cryptosporidium parvum and Cryptosporidium hominis, J. Clin. Microbiol., 2013, 51, 2556–2563 CrossRef CAS PubMed.
- J. B. Burnet, L. Ogorzaly, A. Tissier, C. Penny and H. M. Cauchie, Novel quantitative TaqMan real-time PCR assays for detection of Cryptosporidium at the genus level and genotyping of major human and cattle-infecting species, J. Appl. Microbiol., 2013, 114, 1211–1222 CrossRef CAS.
- S. J. Hadfield, G. Robinson, K. Elwin and R. M. Chalmers, Detection and differentiation of Cryptosporidium spp. in human clinical samples by use of real-time PCR, J. Clin. Microbiol., 2011, 49, 918–924 CrossRef CAS PubMed.
- Z. Koloren, I. Sotiriadou and P. Karanis, Investigations and comparative detection of Cryptosporidium species by microscopy, nested PCR and LAMP in water supplies of Ordu, Middle Black Sea, Turkey, Ann. Trop. Med. Parasitol., 2011, 105, 607–615 CrossRef CAS PubMed.
- C. Gallas-Lindemann, I. Sotiriadou, J. Plutzer, M. J. Noack, M. R. Mahmoudi and P. Karanis, Giardia and Cryptosporidium spp. dissemination during wastewater treatment and comparative detection via immunofluorescence assay (IFA), nested polymerase chain reaction (nested PCR) and loop mediated isothermal amplification (LAMP), Acta Trop., 2016, 158, 43–51 CrossRef CAS PubMed.
- R. Yang, A. Paparini, P. Monis and U. Ryan, Comparison of Next-Generation Droplet Digital PCR (DdPCR) with Quantitative PCR (QPCR) for Enumeration of Cryptosporidium Oocysts in Faecal Samples, Int. J. Parasitol, 2014, 44, 1105–1113 CrossRef CAS PubMed.
- N. P. Mthethwa, I. D. Amoah, P. Reddy, F. Bux and S. Kumari, Fluorescence and colorimetric LAMP-based real-time detection of human pathogenic Cryptosporidium spp. from environmental samples, Acta Trop., 2022, 235, 106606 CrossRef CAS.
- M. P. Hang, C. Nakajima, K. Ohashi and M. Onuma, Loop-mediated isothermal amplification for rapid detection of Newcastle disease virus, J. Clin. Microbiol., 2005, 43, 1646–1650 CrossRef.
- S. Lee, V. S. L. Khoo, C. A. D. Medriano, T. Lee, S. Y. Park and S. Bae, Rapid and in-situ detection of fecal indicator bacteria in water using simple DNA extraction and portable loop-mediated isothermal amplification (LAMP) PCR methods, Water Res., 2019, 160, 371–379 CrossRef PubMed.
- M. A. Bakheit, D. Torra, L. A. Palomino, O. M. M. Thekisoe, P. A. Mbati, J. Ongerth and P. Karanis, Sensitive and specific detection of Cryptosporidium species in PCR-negative samples by loop-mediated isothermal DNA amplification and confirmation of generated LAMP products by sequencing, Vet. Parasitol., 2008, 158, 11–22 CrossRef PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Loop-mediated isothermal amplification of DNA, Nucleic Acids Res., 2000, 28, e63–e63 CrossRef PubMed.
- P. Karanis, O. Thekisoe, K. Kiouptsi, J. Ongerth, I. Igarashi and N. Inoue, Development and preliminary evaluation of a loop-mediated isothermal amplification procedure for sensitive detection of Cryptosporidium oocysts in fecal and water samples, Appl. Environ. Microbiol., 2007, 73, 5660–5662 CrossRef PubMed.
- A. Inomata, N. Kishida, T. Momoda, M. Akiba, S. Izumiyama, K. Yagita and T. Endo, Development and evaluation of a reverse transcription-loop-mediated isothermal amplification assay for rapid and high-sensitive detection of Cryptosporidium in water samples, Water Sci. Technol., 2009, 60, 2167–2172 CrossRef PubMed.
- P. A. Rochelle, R. De Leon, A. Johnson, M. H. Stewart and R. L. Wolfe, Evaluation of immunomagnetic separation for recovery of infectious Cryptosporidium parvum oocysts from environmental samples, Appl. Environ. Microbiol., 1999, 65, 841 CrossRef PubMed.
- J. C. Arona, T. J. Hall, F. Mckinnirey and F. Deng, Comparison of four commercial immunomagnetic separation kits for the detection of Cryptosporidium, J. Water Health, 2023, 21, 1580–1590 CrossRef PubMed.
- G. P. Yakub and K. L. Stadterman-Knauer, Evaluation of immunomagnetic separation for recovery of Cryptosporidium parvum and Giardia duodenalis from high-iron matrices, Appl. Environ. Microbiol., 2000, 66, 3628 CrossRef PubMed.
- S. Arshavsky-Graham, C. Heuer, X. Jiang and E. Segal, Aptasensors versus immunosensors—Which will prevail?, Eng. Life Sci., 2022, 22, 319–333 CrossRef PubMed.
- E. M. Hassan, B. R. Dixon, S. A. Sattar, A. Stalker, B. Örmeci and M. C. DeRosa, Highly sensitive magnetic-microparticle-based aptasensor for Cryptosporidium parvum oocyst detection in river water and wastewater: Effect of truncation on aptamer affinity, Talanta, 2021, 222, 121618 Search PubMed.
- D. Deere, G. Vesey, M. Milner, K. Williams, N. Ashbolt and D. Veal, Rapid method for fluorescent in situ ribosomal RNA labelling of Cryptosporidium parvum, J. Appl. Microbiol., 1998, 85, 807–818 CrossRef PubMed.
- Y. Sun, L. Zhao, M. Zhao, R. Zhu, J. Deng, F. Wang, F. Li, Y. Ding, R. Tian and Y. Qian, Four DNA extraction methods used in loop-mediated isothermal amplification for rapid adenovirus detection, J. Virol. Methods, 2014, 204, 49–52 CrossRef PubMed.
- N. Sowmya, M. S. Thakur and H. K. Manonmani, Rapid and simple DNA extraction method for the detection of enterotoxigenic Staphylococcus aureus directly from food samples: comparison of PCR and LAMP methods, J. Appl. Microbiol., 2012, 113, 106–113 CrossRef CAS PubMed.
- F. Yu, K. Zhang, Y. Wang, D. Li, Z. Cui, J. Huang, S. Zhang, X. Li and L. Zhang, CRISPR/Cas12a-based on-site diagnostics of Cryptosporidium parvum IId-subtype-family from human and cattle fecal samples, Parasites Vectors, 2021, 14, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00344f |
| This journal is © The Royal Society of Chemistry 2025 |