
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelection of a DNA aptamer for aflatoxin B1 and the development of a lateral flow assay for the detection of aflatoxins in spiked peanut extract†
Fiona
Ebanks
,
Erin M.
McConnell
 ,
Emily
Mastronardi
,
Daniel
Goudreau
,
Hadi
Nasrallah
,
Velu
Ranganathan
and
Maria C.
DeRosa
*
,
Emily
Mastronardi
,
Daniel
Goudreau
,
Hadi
Nasrallah
,
Velu
Ranganathan
and
Maria C.
DeRosa
*
Laboratory for Aptamer Discovery and Development of Emerging Research (LADDER), Department of Chemistry, Carleton University, 1125 Colonel By Dr., Ottawa, ON K1S 5B6, Canada. E-mail: maria.derosa@carleton.ca
First published on 2nd July 2025
Abstract
Aflatoxins are a family of highly toxic compounds that contaminate food pre- and post-harvest, negatively impacting the agroeconomic system. Their severe acute and chronic health effects pose a threat to human health. Interest has been shown in developing rapid, reliable and cost-effective on-site detection tools for aflatoxins. A capture SELEX experiment to discover aptamers for total aflatoxin (B1, B2, G1 and G2) was conducted. The selection library was designed to be immobilized to a solid support matrix through complementary binding of a capture probe to the central capture region of the library template. From this selection the aptamer candidate DZA3 was selected because of its high affinity for AFB1, the most toxic of the aflatoxins. The dissociation constant (Kd) was elucidated using microscale thermophoresis which yielded results of Kd: 42.1 ± 23.8 nM. The aptamer was integrated into a colorimetric solution assay that achieved a detection limit as low as 2.28 nM in water. For ease and portability, a colorimetric lateral flow assay was developed that was tested in a non-complex sample and in spiked peanut extract. The limits of detection achieved were 14.67 nM in water and 1.90 μM (5.93 mg kg−1) in the spiked peanut extract. The limits of quantification achieved in water and the spiked peanut extract were 44.44 nM and 5.75 μM (17.80 mg kg−1), respectively.
Introduction
Foodborne diseases significantly threaten human health and the agroeconomic system. Certain of these can result from naturally occurring mycotoxins.1–3 Mycotoxins are small molecules produced by fungi that cause adverse or fatal effects in humans and animals when inhaled, absorbed through the skin, or ingested at low concentrations. Aflatoxins are a class of mycotoxin of great public, agricultural, and economic concern.4–6 There are four major naturally occurring aflatoxins (AFs), AFB1, AFB2, AFG1 and AFG2 and of these mycotoxins, AFB1, is the most potent due to its carcinogenicity.7 Changes in temperature and humidity pre- and post-harvest can promote the growth of Aspergilli, resulting in AFB1 contamination.6,8,9 AFB1 can be found in peanuts, maize, cottonseed, tree nuts, rice, and oilseeds.4,5,10 Overexposure to this toxin can lead to several acute and long-term adverse effects. AFM1 is a product of AFB1 metabolism and is excreted in the milk of mammals. This is especially problematic for children, where immune dysfunction due to AF exposure leads to increased infection risk and poor nutrient absorption.11–13 The extent of AFB1 toxicity varies according to dose, length of exposure, nutritional factors, and exposure to previous viruses.14–17The occurrence of AFB1 at low concentrations in food and feed requires detection methods with high sensitivity. Current detection methods for AFB1 include thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC), gas chromatography (GC), mass spectrometry (MS), and enzyme-linked immunosorbent assay (ELISA).18,19 These techniques have disadvantages, including dependence on expensive equipment, need for skilled technicians, lengthy analysis times, in some cases low sensitivity, limited accessibility, and susceptibility to false positive or negative results.20 Researchers have also proposed aptamer-based assays for aflatoxin detection.20,21 Xiong et al. also developed a solution-based colorimetric assay to detect AFB1 using AuNPs and NaCl, achieving a LOD of 7.32 ng mL−1.22 Lerdsri et al. developed a solution-based colour changing detection scheme using and alternative to NaCl, N,N/-bis(ethylene trimethylammonium)-3,4,9,10-perylene diimide, which achieved a LOD of 0.36 ng mL−1.23 Though sensitive due to the use of spectrophotometers, solution-based assays are not ideal for field application by non-experts. Researchers Mousivand et al.24 and Zhang et al.25 developed competitive lateral flow assay (LFA) aptasensing platforms for the detection of AFB1 in corn and municipal water with LODs of 0.10 ng mL−1 and 1.05 ng mL−1 respectively. Even with these advancements, there is a need to develop rapid, user-friendly, accurate and sensitive on-site detection methods to mitigate the growing problem of AFB1 contamination in food.7,26,27 The new paper-based technology proposed would be independent of additional instrumentation and produce results easily visible by the naked eye, while making use of limited yet robust components and reagents.
Aptamers, short single-stranded DNA or RNA molecules that bind their target with high-selectivity and specificity, have garnered increasing attention as a promising nanomaterial for therapeutics and detection.28–30 Aptamers present several advantages to other affinity tools such as antibodies; they are cost-effective to produce with low batch-to-batch variability, negate animal use, and are robust and stable over wide pH and temperature ranges.29,31,32 Aptamers can be chemically modified with relative ease to improve their affinity, specificity, immobilization, detection, and stability.29 Additionally, aptamers can be introduced to a wide range of targets, from small molecules to whole cells, with the ability to distinguish differences in functional groups and surface proteins.33–36
Aptamers are selected using an in vitro process called systematic evolution of ligands by exponential enrichment (SELEX).31,37,38 The reader is referred to DeRosa et al., for a comprehensive primer and review on SELEX.31,37,39 The aptamer sequence explored in this work was selected using capture SELEX (Fig. S1†).40,41 This type of SELEX yields the selection of high-affinity structure switching aptamer candidates while decreasing selection time.40,41
Following selection, aptamers can be integrated into a variety of detection methods that can include noble metal nanoparticles such as gold nanoparticles (AuNPs).42–44 AuNPs have been utilized in sensing, therapeutics, and catalysis.45–49 The ruby red colour of spherical AuNPs results from localized surface plasmon resonance (LSPR).49–51 The intensity and frequency of LSPR are dependent on the nanoparticle's shape, size, composition, and dielectric environment.52–55 The colours associated with the dispersed and aggregated states of noble metal nanoparticles (NMNPs) make them ideal for colorimetric assays.50
This research combines the sensing and detection properties of aptamers and AuNPs to create colorimetric in-solution and lateral flow assays. When sodium chloride (NaCl, salt) is added to AuNPs in-solution, it can induce aggregation of AuNPs by reducing the repulsion between particles, shifting the colour from bright red to blue/violet.55,56 DNA aptamers can be adsorbed onto the surface of commonly synthesized citrate-capped AuNPs non-specifically via coordination interaction between the DNA bases and the nanoparticles (NP).32,57,58 The adsorption of DNA can prevent the salt-induced aggregation of AuNPs. When the aptamer's target is present, the conformational changes that occur upon binding leave AuNPs open to salt-induced aggregation, resulting in a colour change from red to blue (Fig. 1a). This research exploits this mechanism to produce the in-solution colour-changing method for detecting AFB1.
 | ||
| Fig. 1 Illustration of a) the colorimetric in-solution assay and b) the colorimetric lateral flow assay for the detection of aflatoxin b1. a) The in-solution assay showing that in the presence of AFB1 the aptamer is preferentially bound to the toxin, due to the aptamer's change in conformation the AuNPs undergo NaCl-induced aggregation and the colour changes from red to blue. In the absence of AFB1 the aptamer passivates the surface of the AuNPs, and the colour remains red. b) The LFA showing that in the presence of target, preferential binding leads to a disappearance of colour at the test zone but in the absence of target, aptamer-AuNP stick at the test zone resulting in a red line. | ||
The colorimetric signalling and detection properties of aptamer-AuNPs can be further exploited to develop a lateral flow assay (LFA). In this work, a LFA was developed using AuNPs and the adsorption–desorption approach. In this approach, the lateral flow strip is made using a nitrocellulose membrane, an absorption pad, a streptavidin test line, and a PDDA control line. The aptamer is modified with biotin and adsorbed to the surface of AuNPs. In the absence of AFB1, the aptamer-AuNP complex will be trapped at the test line due to the biotin–streptavidin interaction, producing a visible red colour. In the presence of AFB1, the colour at the test line will disappear because the aptamer desorbs from the AuNPs to bind to AFB1. The control line will always appear as the cationic PDDA polymer captured the negatively charged AuNPs (Fig. 1b).
In the work described here, a new aptamer for AFB1 was discovered, characterized, and incorporated into an LFA. The LFA was challenged with both simple (buffer) and complex (peanut) matrices and shows promise for real-world screening of AFB1.
Materials and methods
A detailed list of chemicals and instruments are included in the ESI.†Capture-SELEX process
Aptamers were selected for total aflatoxin (B1, B2, G1 and G2) via capture SELEX using a central region capture domain (outlined in Fig. S1†). The selection template was 5′-AGCAGCACAGAGGTCAGATG-N15-TTTTGTGGGTAGGGCGGGTTGGTTTT-N15-CCTATGCGTGCTACCGTGAA-3′ and the capture probe sequence was 5′-TTTTTTTTTTTTAACCCGCCCTAC-3′. Regions of complementarity between the selection template and the capture probe are underlined. Primarily, the selection library was immobilized on agarose beads via biotin–avidin interaction where the capture probe was biotinylated, and the agarose beads were coated with avidin (Pierce™ Avidin Agarose. ThermoFisher Scienctific, Ottawa, Canada). The capture probe and library were hybridized in solution prior to immobilization by heating to 95 °C for 10 min, cooling to room temperature (RT), and then incubating at 4 °C overnight. Details of the selection round conditions are summarized in Table S1.† In general, negative selection was done in selection buffer (20 mM Tris–HCl (pH = 7.4), 100 mM NaCl, 5 mM KCl, 1 mM CaCl2), and the counter elution contained deoxynivalenol (DON), fumonisin B1 (FB1), ochratoxin A (OTA) and zearalenone at 8 pmol total (5 nM each or 1.48, 3.61, 2.02 and 1.59 ppb respectively) in selection buffer. Toxins were obtained from Millipore Sigma, (Oakville, ON, Canada). The positive elution decreased from 57.8 pmol to 1.6 pmol total aflatoxin over 5 selection rounds. Specific details of each selection round are summarized in Table S1.† Washing of the beads was achieved using cellulose acetate membrane, pore size 0.22 μm, sterile spin-X columns (Millipore Sigma, Canada). The final selection was subjected to 5% BSA and corn extract in selection buffer. To separate sequences that were bound to aflatoxin and either BSA or other proteins from the complex matrix a nitrocellulose filtration step was added during the 5th round of selection (see Fig. S2†). Following elution, the selection libraries were amplified via PCR (200 mM Tris–HCl, PH = 9.0, 100 mM KCl, 2% Triton X-100) using the following cycle: 95 °C for 5 min, 25 × 95 for 30 s, 56 °C for 30 s, 72 °C for 30 s, then 72 °C for 5 min. The sequences of the forward and reverse primers were 5′-6FAM-AGCAGCACAGAGGTCAGATG and 5′-AAAAAAAAAAAAAAAAAAAAAAAAA-HEG-TTCACGGTAGCACGCATAGG respectively. The amplicons were purified via 8% denaturing PAGE, where the fluorescently labeled enriched library was isolated. Selection enrichment was monitored by measuring fluorescence of the 6FAM modification (Ex: 495 nm and Em: 520 nm). Aptamer sequence candidates were identified by high-throughput sequencing, counting and clustering via the AptaSuite tools,59 secondary structure prediction,60 and common motif analysis of the top sequence candidates identified via AptaSuite. The workflow is summarized in Fig. S3.† Aptamer candidate sequences are included in Table S2.† Sequence identities are compared within the top five candidates (Table S3†) and to the top 27 remaining sequences (Fig. S4†).Aptamer characterization
For the gel shift assay, 20 μL of the hybridized DNA samples was mixed with 20 μL of 0.1 μM of total aflatoxin stock (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 AFB1, AFB2, AFG1, AFG2) and vortexed for two hours. After, an equal volume of 50% glycerol was added to each sample. Non-denaturing polyacrylamide gels were prepared by mixing 27 mL of acrylamide solution (bisacrylamiderylamide, 40% solution, 19:1), 18 mL of 5× TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA), and 44 mL of deionized water. The solution was stirred at 37 °C and filtered through a Whatman No. 1 filter paper. To the cooled solution, 900 μL of 10% ammonium persulfate and 90 μL of TEMED were added. After casting and pre-running, the power supply was set to a constant 200 V. To cool the system, cold water was circulated through the vertical heat exchanger. The gels were loaded with sample and ran three hours. Imaging was done on a 20 × 20 cm TLC plate (F254) using a multi-image light cabinet on the epi-UV setting (λex = 254 nm). The fluorescent DNA was visualized using a 302 nm excitation wavelength. For specificity analysis with FB1, OTA, DON and zearalenone (Z) were done. The results of this are presented in Fig. S5.†
:1:1:1 AFB1, AFB2, AFG1, AFG2) and vortexed for two hours. After, an equal volume of 50% glycerol was added to each sample. Non-denaturing polyacrylamide gels were prepared by mixing 27 mL of acrylamide solution (bisacrylamiderylamide, 40% solution, 19:1), 18 mL of 5× TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA), and 44 mL of deionized water. The solution was stirred at 37 °C and filtered through a Whatman No. 1 filter paper. To the cooled solution, 900 μL of 10% ammonium persulfate and 90 μL of TEMED were added. After casting and pre-running, the power supply was set to a constant 200 V. To cool the system, cold water was circulated through the vertical heat exchanger. The gels were loaded with sample and ran three hours. Imaging was done on a 20 × 20 cm TLC plate (F254) using a multi-image light cabinet on the epi-UV setting (λex = 254 nm). The fluorescent DNA was visualized using a 302 nm excitation wavelength. For specificity analysis with FB1, OTA, DON and zearalenone (Z) were done. The results of this are presented in Fig. S5.†
:1 serial dilution was prepared. Following this, 10 μL of each dilution was mixed with 10 μL of FAM-aptamer resulting in a final concentration of 5 nM FAM-aptamer. The concentrations of AFB1 ranged from 50 μM to 1.5 nM. The capillaries were filled with final reaction mixtures. Analysis was done using a Monolith NT.115 Pico at 25 °C with 7% LED and 80% laser power. The NanoTemeper MO. Affinity analysis software was used for curve fitting and Kd determination.
Control toxins were used to assess the specificity and selectivity of the assay. To do this, 10 μM of each toxin (AFB1, AFB2, AFG1, AFG2, DON, OTA, and FB1) was used and the colorimetric assay was conducted using the aforementioned approach. The results of the in-solution selectivity and specificity assay can be found in the supplemental in Fig. S10.†
AuNP concentration was further optimized by incubating 0.99, 1.05, 1.11, or 1.20 nM AuNP with 1.5 μM aptamer for 30 minutes. This was followed by the addition of 100 μL of toxin, resulting in AFB1 at concentrations ranging from 0.025 nM to 500 nM and an additional 30-minute incubation. The LFA was dipped in 30 μL of the mixture for 2 minutes and then photographed.
The assay was repeated with AFB1 and AuNP only to deduce the effects of the target and solvent on the test zone. Additionally, tests were carried out exactly as before but with a scrambled version of the aptamer (biotin-scram) to test the validity of aptamer. The strips were photographed and uploaded to ImageJ to analyze the intensity of the test zones. Graph Pad Prism 10 using ordinary one-way ANOVA with multiple comparison analysis was used to establish statistical significance.
Results and discussion
Selection of aptamers for aflatoxin via capture-SELEX
Multiple modifications and optimizations to the original in vitro selection protocols have been developed to decrease selection time and increase the probability of selecting high affinity aptamers.62 One such adaptation was to indirectly immobilize the nucleic acid library to a solid support matrix via a capture probe. Inspiration for the design of the SELEX protocol came from work done by Nutiu and Li (2005) who described the selection of “structure-switching” aptamers.63 Aptamers that bind to AFB1 (and AFM1) have been selected with affinities in the nM range (summarized in Table S5†). This work was motivated by the desire to specifically select high affinity structure switching aptamers to exploit this property for biosensing applications. In this protocol the nucleic acid selection pool was based on a template where a known central region was flanked by two randomized regions. This design, shown in Fig. S1,† allowed the pool to be immobilized on beads via hybridization of the complementary capture probe (purple) to the known region (blue) of the selection library template. A schematic diagram of the partitioning step of the selection procedure is shown in Fig. S2.† The actual sequences are shown in Table S2.†The aptamer template was 96 bases long with two 15-nucleotide (nt) random regions flanked by a primer region. Between the two random regions, the template also contained a known 18-nt biased sequence domain, PS2.M (5′-GTGGGTAGGGCGGGTTGG-3′), that was previously reported to form a parallel G-quadruplex.64 The PS2.M sequence was used to aid in structure switching so that high-affinity aptamers could be selected and to facilitate the immobilization of the sequence during selection to decrease selection time.62
Selection details and identification of aptamer candidates by high-throughput sequencing (HTPS)
Five selection rounds were completed, and the experimental details are summarized in Table S1.† The first four selection rounds were performed in non-complex selection buffer (20 mM Tris–HCl (pH = 7.4), 100 mM NaCl, 5 mM KCl, 1 mM CaCl2). To increase selective pressures the stringency was increased each round. The final round of selection was done in complex media (20 mM Tris–HCl (pH = 7.4), 100 mM NaCl, 5 mM KCl, 1 mM CaCl2, 5% BSA and corn extract). This allowed for the reduction of sequences having affinity for the components of the complex matrix from the library. To separate sequences that were bound to aflatoxin and either BSA or other proteins from the complex matrix an additional filtration step was added during the 5th round of selection. After the positive selection the eluate was passed through a nitrocellulose membrane (Fig. S2†). BSA was filtered from the solution by binding to the nitrocellulose membrane. DNA and aflatoxin were able to pass through the filter membrane and were collected and used for HTPS analysis to determine aptamer candidates.The sequences produced by HTPS were analyzed by AptaCluster.59 AptaCluster is a clustering algorithm that efficiently parses, clusters and analyzes sequencing data from aptamer selections. The program runs based on user-defined parameters and is fully automated, the workflow used is summarized in Fig. S3.† Enrichment scores, the count, the actual sequence, and the appearance of repetitive sequence domains were considered to narrow down the number of aptamer candidates to be experimentally characterized. Between sequences comparisons were facilitated by multiple alignment software. These analyses are shown in Tables S2 and S3 and Fig. S4.†
Screening of aptamer candidates using electrophoretic gel shift
The abilities of the top five aptamer candidates to undergo structural transition in the presence of AFB1 were screened by an electrophoretic mobility gel shift assay (EMSA) where capture probe displacement was monitored (Fig. 2a). | ||
| Fig. 2 a) The ability of the aptamer to undergo capture probe displacement in the presence of aflatoxin was screened by an electrophoretic gel shift assay where AFB1 binding was indicated by displacement of the fluorescent capture probe. b) Fluorescent images of the aptamer candidate screening by gel shift assay. Abbreviations: capture probe (CP), aptamer candidate (AC, ex: DZA_1), control hybridization reaction (C). c) SpotDenso analysis of fluorescence band intensity of aptamer candidates where the red X indicates no difference in intensity of the CP + AC band in the absence and presence of the target, and conversely the green arrows indicate a decrease in fluorescence due to displacement of the fluorescent capture probe in the presence of the target. | ||
Prior to the gel shift assay, aptamers were hybridized with the capture probe. For each aptamer candidate (AC), a control reaction (no aflatoxin) and aflatoxin reaction were prepared. The differences in intensity of the top (CP + AC) and bottom (CP) bands were evaluated and used to assess both the hybridization efficiency and target binding. The expected results are shown in Fig. 2a. Lane 1 shows the migration of the capture probe control. Lane 2 represents the control hybridization reaction. If the CP and AC were fully hybridized, then fluorescence would be mainly in the top band. In the presence of aflatoxin (lane 3) some of the CP is removed from the AC by the binding of aflatoxin to the aptamer (expansion at right). Therefore, upon target binding the intensity of the CP + AC band decreased and the intensity of the CP band increased.
The intensity of the CP + AC bands in the absence and presence of the target were compared using the SpotDenso analysis feature of an AlphaImager instrument (AlphaInnotech) to identify the top three aptamer candidates from this single trial for further characterization. The fluorescent gel images and the graphical analysis of fluorescent intensity for the aptamer candidates are shown in Fig. 2b. Upon examination of the fluorescent gel images, DZA_1 (lanes 2 and 3) and DZA_2 (lanes 4 and 5) did not hybridize well, most of the fluorescence was observed in the CP lane in both the control hybridization reaction (C1 and C2) and the DZA + AF reactions (lanes 3 and 5). Aptamer candidates DZA_3 (lane 6), DZA_5 (lane 8) and DZA_7 (lane 10) all hybridized with varying efficiencies. However, visual examination of the top (CP + AC) and bottom (CP) bands in lanes 7, 9 and 11 suggest that these sequences have some affinity to the target. Especially for DZA_3 and DZA_7 the intensity of the top band visually decreased in the presence of target.
To confirm these trends the intensity of each band was analyzed by the SpotDenso analysis tool. The Intensity of the top band (CP + AC), the intensity of an equivalent area of the background in the fluorescent image were measured. A graphical summary of the intensity difference is shown in Fig. 2c. Based on the gel shift assay and the graphical analysis of the fluorescent data, aptamer candidates DZA_3, DZA_5 and DZA_7 were chosen for further analysis as the decrease in fluorescence, corresponding to release of the capture probe from the hybridized CP + AC complex upon target binding, was observed. Preliminary specificity screening was also achieved using this method (results are shown in Fig. S5†). Of the three candidates, DZA-3 showed the least non-specific displacement of the capture probe.
Microscale thermophoresis
Further characterization of DZA3, DZA5 and DZA7 was done through in-solution MST experiments (Fig. S6†). MST is an advantageous characterization method because of its sensitivity, low sample consumption and rapid measurements.65 The aptamer candidates were challenged with the four major aflatoxins (AFB1, AFB2, AFG1, and AFG2). Fluorescently labelled aptamer was introduced to concentrations of AFB1. From this experiment, DZA3 showed high affinity to AFB1 and AFG2 with dissociation constants of 42.1 ± 23.8 nM and 15.3 ± 4.8 nM, respectively. DZA7 displayed affinity towards AFG2 with a Kd of 3.3 ± 0.2 μM. DZA5 did not display measurable affinity to either of the four aflatoxins tested. The normalized MST traces for DZA3 and DZA7 are shown in Fig. S6.† DZA3 was selected for further characterization due to its strong apparent affinity for AFB1 and AFG2.It is sometimes challenging to determine the binding affinity of an aptamer to a small molecule, as target-binding is not accompanied by a large mass increase.66 MST data analysis involves fitting thermophoresis curves to mathematical models to extract binding parameters. The complexity of these models and the assumptions made during data analysis can introduce errors, especially in cases where binding kinetics or multiple binding sites are involved.67 When coupled with the low solubility of aflatoxins, it becomes difficult to ascertain a Kd value.
Structural prediction
RNAstructure was used to predict the secondary structure of DZA3 (Fig. S7†). The most plausible secondary structure can be forecasted by comparing the free energy prediction and the maximum expected structure. The folding free energy structure prediction is achieved using the Watson–Crick interaction of DNA bases.68 The maximum expected accuracy structure uses the sum of pairing probabilities and has been deemed more accurate.68,69 The MaxExpect accuracy structures predicted are, on average, more precise than those predicted using the minimum folding free energy.69 For DZA3, software predicts two similar stem-loop structures with a minimum folding free energy (ΔG, kcal mol−1) of −11.9 (Fig. S7a†), and a MaxExpect score of 7.4 (Fig. S7b†).DZA3 is a guanine-rich (G-rich) sequence, with structure switching potential. G-rich sequences can form a highly stable G-quadruplex.70 To help determine the likelihood of the DZA3 sequence forming a G-quadruplex, the quadruplex forming G-rich sequences (QGRS) Mapper software was used. The software reports a G-score based on the prevalence of shorter loops to longer loops, that the loops are equal in size and that the stability of the structure increases with an increasing number of G-quartets.71 QGRS forecasts the likelihood of a G-quadruplex forming at position 42 within the sequence. This location corresponds to the known G-quadruplex forming region PS2.M.
Another tool for predicting the formation of intramolecular G-quadruplexes is the intramolecular G-quadruplex (imGQ) finder.72 This software allows the user to define the required number of G-quartets, loop length, and the number of substitutions or defects allowed.73 The analysis for imperfect G-quadruplexes was done for tetrads of three and a smaller loop size. Similarly, a G-quadruplex was predicted at position 42.
Structural prediction and binding using circular dichroism
Circular dichroism was used to confirm whether DZA3 would form a G-quadruplex. The sequence was resuspended in a buffer containing K+ to stabilize potential guanine tetrads.74 The spectral data obtained by monitoring ellipticity at between 240–245 nm and 260–280 nm is shown in Fig. 3. | ||
| Fig. 3 CD spectrum of DZA3 in 20 mM Tris buffer (100 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2), showing the 245 nm minimum and 280 nm maximum corresponding to B-form DNA. | ||
DNA sequences known to fold into a parallel G-quadruplex contain a maximum at 260 nm and a minimum at 240 nm. The experimental result did not correspond to a G-quadruplex, instead, the results more closely match that of commonly found B-form DNA. The PS2.M is a G-quadruplex forming sequence, the spectrum for which is displayed in Fig. S8.† The DZA3 sequence showed a minimum at 245 nm and a maximum at 280 nm. In the presence of 50 nM AFB1, the spectrum looked almost identical. The presence of AFB1 resulted in a slight increase at 280 nm. The peak similarities in both spectra confirm the absence of a significant conformational change of DZA3 in the presence of AFB1. Based on the characterization method and the length of the aptamer conformational change may not be observed as not all regions of the aptamer may display affinity. CD relies on substantial structural changes upon aptamer binding. CD spectroscopy primarily detects changes in global conformation of biomolecules. It may not be sufficiently sensitive to detect subtle conformational changes or binding events involving small molecules, especially when these changes do not significantly alter the overall secondary structure of the aptamer.74
Colorimetric detection of AFB1
The development of an LFA using aptamers as the molecular recognition element and AuNPs as the signal reporter requires preliminary screening of the aptamer in an in-solution assay. To begin, the stability of the AuNPs in the presence of AFB1 alone (with and without salt) was assessed. It was found that AFB1 was able to induce the aggregation of AuNPs at concentrations above 10 μM (Fig. S9A†), and that AFB1 did not exhibit protective effects against salt induced aggregation (Fig. S9B†). This may be due to the ability of the toxin to coordinate with the citrate groups on the surface of the AuNPs. Additionally, the sensor demonstrated good selectivity to AFB1 over other toxins (Fig. S10†).To determine the sensitivity of the assay, varying concentrations of AFB1 (0.02 nM–10.00 μM) were introduced to the DZA3 aptamer, and the spectral changes were analysed using UV-vis spectroscopy. Well-dispersed 10–15 nm AuNPs exhibit a plasmon resonance at 520 nm, while aggregated AuNPs result in a red shift in absorbance and the appearance of a peak at 600–700 nm that is highly dependent on the extent of aggregation that is induced (Fig. 4).
 | ||
| Fig. 4 In-solution colorimetric assay showing the increase in aggregation as more AFB1 is added. Concentration increases from right (0 nM, control) to left (10 μM). With increasing target, the assay changes from red to blue. | ||
As the concentration of AFB1 increased, there was a decrease in the 520 nm plasmon resonance and an increase in the new 640 nm peak. A calibration curve was created by plotting the ratio of the absorbance at 640 nm to the absorbance at 520 nm (Fig. 5a and b respectively). The aptamer showed good linearity at two points between 0.2 nM and 20 nM AFB1 and 200 nM to 10 μM (Fig. 5c and d), resulting in LODs of 2.28 nM and 1.81 μM.
 | ||
| Fig. 5 A) Spectral changes to AuNP visible absorbance spectrum due to the aptamer's interaction with varying concentrations of AFB1 target ([0, 0.02, 0.2, 2, 20, 200 nM], [2, 4, 6, 8, 10 μM]); B) ratio of the absorbance at 640 nm and 520 nm; C) and D) points of linearity used to determine the LOD, which were 2.28 nM and 1.81 μM. Error bars represent standard error. | ||
TEM analysis
Transmission electron microscopy (TEM) was used to support the proposed mechanism. When 10 μM of AFB1 was added in the presence of DZA3 and NaCl, the preferential binding of the aptamers to the target left the AuNPs susceptible to salt-induced aggregation. This could be seen by the clustering of the spherical AuNPs in Fig. S11a.† Fig. S11b† shows that in the absence of AFB1, the aptamer-coated AuNPs remained well dispersed in the presence of salt. The high-resolution TEM studies support the colorimetric assay that utilizes the modified adsorption–desorption approach, confirming the DZA3 aptamer's affinity to the AFB1 target.AFB1 aptamer-based biosensor
As previously mentioned, current detection methods for AFB1 have several disadvantages, including dependence on expensive equipment, need for skilled technicians, lengthy analysis times, accessibility and susceptibility to false positive or negative results.75,76 We have shown that utility of an aptamer-based assay for the detection of AFB1 in solution. Though promising, this format is not ideal for portability of the detection tool and requires additional wait time. As a result, there is an urgent need to develop rapid, user-friendly, portable, accurate and sensitive detection methods to mitigate the growing problem of AFB1 contamination.29,77,78 Here, we developed a low-cost aptamer-based lateral flow assay (LFA) to detect aflatoxins, specifically AFB1, in spiked peanut extract.Optimization of DNA concentration
The final construct of the lateral flow assay included a streptavidin concentration of 1.5 mg mL−1 mixed with 0.5% pullulan. Streptavidin was the capture bioreceptor of choice because of its availability, fast association kinetics and strong affinity towards the biotin-labelled aptamer.79 The concentration of streptavidin was increased to facilitate a higher density of biotin-labelled aptamer at the test zone and, thus, a higher maximum signal.79 With the increase in streptavidin, the amount of biotin labelled aptamer needed was estimated based on an assumed amount of available binding pockets after adhesion to the surface of the strip.Concentrations of 1, 1.5 and 2 μM were chosen for testing with a small range of AFB1. It was found that 1 μM of biotin-labelled DNA resulted in insufficient fading of the test zone in the presence of AFB1 (Fig. 6a). This could be due to an insufficient amount of biotin-labelled DNA, leaving binding pockets of streptavidin open to interaction with the AuNPs. Two (2) μM of biotin-labelled aptamer resulted in consistent fading as the concentration of AFB1 was increased. When the concentration of DNA seemed to be more than what was needed at the test zone there was increased smearing at the control zone (Fig. 6c). Hence, 1.5 μM biotin labelled aptamer was chosen as it had a reproducible signal change in the presence of increasing concentrations of AFB1 with minimal smearing of the control zone (Fig. 6b). Fig. 6c shows the trend in the test zones as calculated by ImageJ.
 | ||
| Fig. 6 LFA strips tested with a small range of AFB1 using a) 1 μM DZA3 aptamer, b) 1.5 μM DZA3 aptamer and c) 2 μM DZA3 aptamer. d) Quantification of the signal change at the test zones using ImageJ. | ||
Optimization of AuNP to improve sensitivity
The mechanism proposed for the LFA requires the release of DNA from the surface of AuNPs upon the addition of target. This is made possible due to the conformational change that occurs during high affinity binding events. Though DZA3 presents a low dissociation constant at high concentrations of AFB1 the test zone does not completely fade. It is possible that an aptamer-target-AuNP complex was formed.74 DZA3 is a lengthy aptamer, that is 96 bases. After AFB1 binds to its epitope zone, regions of the sequence where conformational change was unlikely could adhere non-covalently to AuNPs resulting in an DZA3-AFB1-AuNP complex. A similar scenario was explored by Mirón-Mérida et al.,74 the researchers proposed the formation of an aptamer-target-complex involving a 96-base FB1 aptamer.The concentration of AuNPs was reoptimized to improve the sensitivity of the assay (Fig. 7a–d) and mitigate the formation of the potential DZA3-AFB1-AuNP complex. The idea behind this was that reducing the amount of AuNP available for non-specific interaction with the aptamer would increase the sensitivity of the assay.74 To do this, 0.99, 1.05, 1.11 and 1.20 nM AuNP were tested with 1.5 μM DZA3 and varying amounts of AFB1.
 | ||
| Fig. 7 Optimization of the concentration of AuNP to reduce the formation of DZA3-AFB1-AuNP complex to improve sensitivity. Image shows 1.5 μM DZA3 with varying concentration of AFB1 an a) 0.99 nM AuNP, b) 1.05 nM AuNP, c) 1.11 nM AuNP and d) 1.20 nM AuNP. | ||
In Fig. 7a, the assay performed with 0.99 nM AuNP resulted in a consistent and drastic fading as low as 0.025 nM AFB1 and produced a LOD and LOQ of 11.43 nM and 34.63 nM, respectively. At this concentration, there was almost complete fading at the test zone as AFB1 concentration was increased proving that finetuning the amount of AuNP added to the assay can reduce for formation of DZA3-AFB1-AuNP complex. This concentration of AuNP was not suitable due to how faint the test zone was in the absence of AFB1. Concentrations of 1.05 nM and 1.11 nM AuNP delivered comparable LODs and LOQs (Table 1).
| AuNP, nM | LOD, nM | LOQ, nM |
|---|---|---|
| 0.99 | 11.4 | 34.6 |
| 1.05 | 26.2 | 79.5 |
| 1.11 | 37.6 | 113 |
| 1.20 | 4250 (4.25 μM) | 12880 (1.29 μM) |
1.20 nM AuNP was not ideal as the test zone did not completely fade possibly explained by two theories: either with increased particles there is decreased steric hinderance and an aptamer-target-AuNP complex is formed that is captured at the test line or the presence of the line is due to the difference in actual number of particles. Where the number of aptamer particles per reaction stays the same, there is an increase in the number of gold nanoparticles per reaction from 3.3 × 1019 (1.11 nM) to 3.6 × 1019 (1.20 nM). At 1.20 nM AuNP, the number of particles passes the threshold where there is a favourable number of aptamers to AuNPs to see the line disappear (enough aptamer is being desorbed in the presence of target to lose the line). In this case, the number of aptamers per AuNP is decreased, but the number of particles that have aptamers on the surface and can interact with the test line is increased, so enough are captured on the test line to mask the aptamer target binding. This results in a high LOD of 4.25 μM and a LOQ of 12.88 μM. At this concentration of AuNP the assay would not be useful in detecting AFB1. As a result of these data, 1.11 nM AuNP was chosen for further analysis (Fig. 7c).
Implementation of optimized parameters for assessment of the assay with control toxins and real sample
To ensure that the presence of AFB1 is not responsible for the fading occurring at the test zone, it was tested on the LFA strip alone. As mentioned, without DNA, streptavidin can recruit AuNP at the test zone resulting in a red line (Fig. 8a and b 0 nM). Optimization components of the LFA resulted in an assay with higher sensitivity and limits of detection. This was assessed using concentrations of AFB1 ranging from 0.25 pM to 50 nM that were prepared in water and tested on the strip in the presence of 1.5 μM DZA3 and 1.11 nM AuNP (Fig. 8a). The resulting LOD and LOQ were 14.67 nM and 44.44 nM, respectively. The LFA performed most reliably at concentrations above 0.25 nM. A scrambled version of DZA3 was created and tested with AFB1. The test zone faded slightly in comparison to the 0 nM AFB1 sample.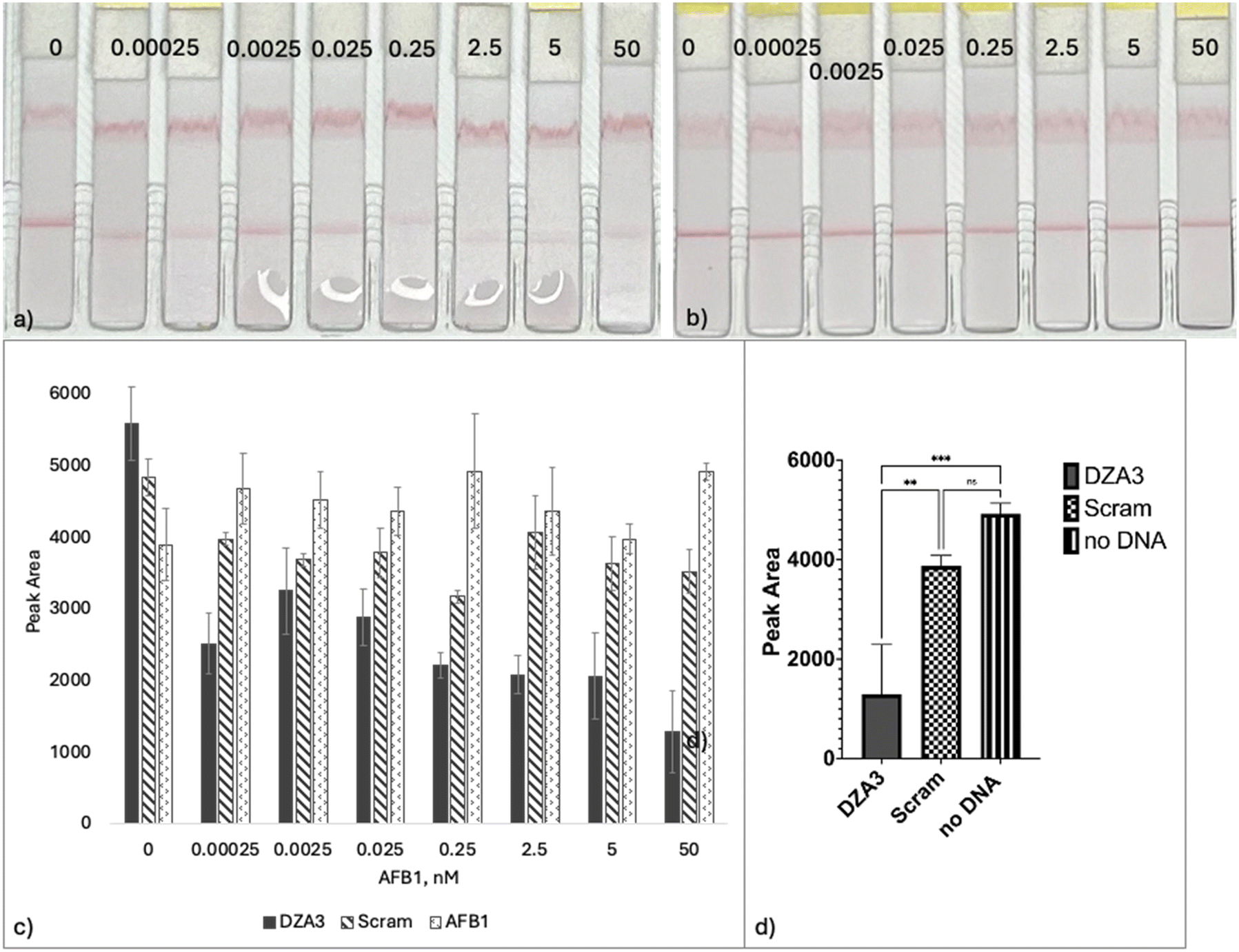 | ||
| Fig. 8 a) Increasing concentrations of AFB1 (0–50 nM) with 1.5 μM biotin-DZA3 and 1.11 nM AuNP were introduced to the lateral flow assay to assess the performance of the LFA, from this the resulting LOD and LOQ were 14.67 nM and 44.44 nM, respectively. b) A scrambled version of DZA3 was used as a control to test the specificity of the sequence in comparison to the 0 nM AFB1 sample. c) Comparison of the test zone signals for LFA conducted with biotin-DZA3, biotin-scram and no DNA (AFB1 only). d) A visual summary of the ordinary one-way ANOVA at 50 nM AFB1. F(2, 6) = 28.93, ** = 0.0046 and *** = 0.0008. | ||
To compare the performance of the aptamer (Fig. 8a) with the scrambled sequence (Fig. 8b) and assay without DNA sequences present, ordinary one-way ANOVA was performed. A visual summary of the ordinary one-way ANOVA is shown in Fig. 8c. After evaluating the signal at the various AFB1 concentrations, the performance of the DZA3 aptamer in comparison to the scrambled sequence was deemed significant at 50 nM, p = 0.0046. In comparison to the scenario without DNA (AFB1 only) the performance of the DZA3 aptamer was significant at concentrations of 0.25 nM to 50 nM (Fig. 8d).
Selectivity tests with control toxins
To test the selectivity of the assay AFB1 and a mixture of AFB1, AFB2, AFG1 and AFG2 were tested against ochratoxin A (OTA), deoxynivalenol (DON), fumonisins B1 (FB1) and atrazine (ATR) (Fig. 9a). The mixture of AFB1:AFB2:AFG1:AFG2 was set at 7:1:1:1, to approximate the abundance of AFB1 compared to other aflatoxins in peanut products.80,81 Ordinary one-way ANOVA was used to perform a Šídák's multi-comparison analysis of the test zone signals. It was found that the changes in the test zone intensity of the sample containing 50 nM AFB1 was comparable to the total AFs mixture (35 nM AFB1, 5 nM AFB2, AFG1, and AFG2) with no significant difference between the two (p > 0.999) (Fig. 9b). The disappearance at the test zone in the total AFs sample AFs was significant in comparison to the other toxins tested, highlighting the selectivity of the aptamer (p < 0.05).
 | ||
| Fig. 9 a) The AuNP based LFA selectivity experiments for the DZA3 aptamer with AFB1 and total AFs in comparison to OTA, DON, FB1, ATR. b) ImageJ analysis of the test zones and a summary of the ordinary one-way ANOVA results establishing significance between the signal of AFs and OTA, DON, FB1, ATR. F(6, 14) = 8.165. | ||
Analysis in spiked peanut extract as a practical application of the AuNP-based LFA, the assay was tested spiked peanut extract (Fig. 10a). The matrix of ground peanuts was extracted using 70% methanol in water. It is important to mention that aflatoxins are commonly extracted from foodstuff in organic solvents, which can pose specific challenges for DNA-AuNP systems, namely the presence of organic solvents can cause non-specific desorption of DNA from AuNPs. The work of Lu et al., resultant aggregation state was affected by both organic solvent concentration and DNA length (shorter was less stable).82 To mitigate these challenges, a low % methanol was used, and the aptamer was very long. Additionally, the aptamers were selected in the presence of trace amounts of methanol (<1%). Aliquots of the extract were spiked with 5, 50, 500 and 1000 nM AFB1. ImageJ was used to analyze the intensity of the test zones, and the results plotted in Fig. 10b. The LOD and LOQ were calculated to be 1.90 μM (5.93 mg kg−1) and 5.75 μM (17.80 mg kg−1), respectively. In Canada, the regulatory limits for aflatoxin in food and animal feed are 15 μg kg−1 and 20 μg kg−1 respectively.83 These values are consistent with those set globally either for AFB1 independently (lowest limit set by European Union at 2.0 μg kg−1), or total aflatoxins (lowest limit set by European Union at 4.0 μg kg−1).84 These values are above the regulatory limits however, inclusion of a complex matrix resulted in a decrease in sensitivity of the assay. The construct serves as a proof of concept that a rapid and user-friendly detection tool for AFB1, and total aflatoxins can be developed using aptamers. To improve the detection limits of the assay the addition of a short and inexpensive clean-up step will be explored. With further refinement the sensitivity of the assay can be improved to result in lower LODs and LOQs.
 | ||
| Fig. 10 Analysis of spiked peanut samples for the presence of AFB1. A) The AuNP-based LFA showing the decrease in test zone intensity as AFB1 concentration is increased. B) Histogram of peak area intensities calculated using ImageJ. | ||
Conclusions
Here, a novel aptamer for AFB1 with sensitivity towards total aflatoxins was selected and characterized. The DZA3 aptamer showed high affinity to AFB1. Using MST the dissociation constant was (Kd) of 42.1 ± 23.8 nM. The modified in-solution assay adsorption–desorption approach was successful in the detection of AFB1 and gave an LOD as low as 2.28 nM (0.71 ppb). The LFA format is more suitable for mass distribution, and ease of use. The aptamer was incorporated into the LFA format using the adsorption–desorption approach. The LFA was challenged with other potent mycotoxins (OTA, DON, FB1) and a common herbicide (atrazine). The construct responded significantly (p < 0.05) in the presence of AFB1 and total aflatoxins, with minimal fading in the presence of OTA, DON, FB1 and atrazine. In an aqueous solution the construct had a LOD and LOQ of 14.67 nM (4.60 ppb) and 44.44 nM (13.88 ppb), respectively. The LFA was tested using an extract from peanuts. The extract was spiked with AFB1 and tested on the strip. The resulting LOD and LOQ were 1.90 μM (0.59 ppm) and 5.75 μM (1.80 ppm), respectively. Though the detection limits of the LFA were not sufficient for monitoring at or below the regulatory limits, this is an important proof of concept demonstration that a paper-based colorimetric approach for the detection of AFB1 is viable. Future work will focus on extract concentration, LFA optimization, and aptamer truncation. Though differences in individual aptamer-AuNP interactions can affect the crispness of the test lines in the LFA assay, batch-to-batch variations in printing of the strips can lead to variations in the sharpness of the lines observed on the LFA. This parameter will need to be optimized for reproducibility quality. Though aptamer truncation can be challenging for small molecule targets, truncation is a practical strategy to increase affinity of the aptamer-target interaction, and decrease costs of final LFA test.85Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Aptamer selection, bioinformatics and screening EMSA were conceptualized by Erin M. McConnell and Maria C. DeRosa and performed by Erin M. McConnell and Emily Mastronardi. Characterization experiments with the aptamers were conducted by Erin M. McConnell, Daniel Goudreau, Velu Ranganathan, Hadi Nasrallah and Fiona Ebanks. Lateral flow assays were conceptualized by Fiona Ebanks and Maria C. DeRosa and performed by Fiona Ebanks. Manuscript was prepared by Fiona Ebanks and Erin M. McConnell. The manuscript was edited by Fiona Ebanks, Erin McConnell, Hadi Nasrallah and Maria C. DeRosa. Revisions were completed by Erin M. McConnell, Fiona Ebanks, and Maria C. DeRosa.Conflicts of interest
The authors state that there are no conflicts to declare.Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Council, the Saskatchewan Ministry of Agriculture Development Fund, The Alberta Wheat and Barley Commission, Western Grains Research Foundation, Saskatchewan Wheat Development Commission, Alberta Wheat Commission, and Manitoba Crop Alliance, the Ontario Agrifood Research Initiative, and 3M for support.Notes and references
- World Health Organization (WHO), Food Safety, 2022, https://www.who.int/news-room/fact-sheets/detail/food-safety.
- H. S. Hussein and J. M. Brasel, Toxicity, Metabolism, and Impact of Mycotoxins on Humans and Animals, Toxicology, 2001, 167(2), 101–134, DOI:10.1016/S0300-483X(01)00471-1.
- A. G. Marroquín-Cardona, N. M. Johnson, T. D. Phillips and A. W. Hayes, Mycotoxins in a changing global environment – A review, Food Chem. Toxicol., 2014, 69, 220–230, DOI:10.1016/j.fct.2014.04.025.
- J. I. Pitt, Mycotoxins, Elsevier Inc., 2013, DOI:10.1016/B978-0-12-416041-5.00030-5.
- J. D. Miller, Fungi and mycotoxins in grain: Implications for stored product research, J. Stored Prod. Res., 1995, 31(1), 1–16, DOI:10.1016/0022-474X(94)00039-V.
- M. E. Zain, Impact of mycotoxins on humans and animals, J. Saudi Chem. Soc., 2011, 15(2), 129–144, DOI:10.1016/j.jscs.2010.06.006.
- D. Lu, X. Tian and Y. Yang, et al., Rapid and sensitive aptamer-linked immunosorbent assay with aptamer-templated silver nanoparticles for detection of aflatoxin B1 in medicinal and edible food, Food Biosci., 2024, 60, 104387, DOI:10.1016/j.fbio.2024.104387.
- J. C. Netto-Ferreira, B. Heyne and J. C. Scaiano, Photophysics and photochemistry of aflatoxins B1 and B2, Photochem. Photobiol. Sci., 2011, 10(10), 1701–1708, 10.1039/c1pp05103b.
- J. B. Renaud, J. P. Walsh and M. W. Sumarah, Optimization of Aflatoxin B1-Lysine Analysis for Public Health Exposure Studies, Toxins, 2022, 14(10), 672, DOI:10.3390/toxins14100672.
- M. H. Taniwaki, J. I. Pitt, M. V. Copetti, A. A. Teixeira and B. T. Iamanaka, Understanding Mycotoxin Contamination Across the Food Chain in Brazil: Challenges and Opportunities, Toxins, 2019, 11(7), 411, DOI:10.3390/TOXINS11070411.
- Eighty-third report of the Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Contaminants in Food, 2017.
- J. B. Renaud, J. P. Walsh and M. W. Sumarah, Simplified Synthesis and Stability Assessment of Aflatoxin B1-Lysine and Aflatoxin G1-Lysine, Toxins, 2022, 14(1), 56, DOI:10.3390/toxins14010056.
- B. Srinivasan, S. Ghosh and P. Webb, et al., Assessing an aflatoxin exposure biomarker: Exploring the interchangeability and correlation between venous and capillary blood samples, Environ. Res., 2022, 215, 114396, DOI:10.1016/j.envres.2022.114396.
- A. E. Rogers, Nutritional modulation of aflatoxin carcinogenesis, in The toxicology of aflatoxins: human health, veterinary and agricultural significance, ed. D. L. Eaton and J. D. Groopman, Academic Press, San Diego, USA, 1994, pp. 207–230 Search PubMed.
- A. Dhakal, M. F. Hashmi and E. Sbar, Aflatoxin Toxicity, StatPearls, 2023, https://www.ncbi.nlm.nih.gov/books/NBK557781/ Search PubMed.
- M. Adhikari, G. Ramjee and P. Berjak, Aflatoxin, kwashiorkor, and morbidity, Nat. Toxins, 1994, 2(1), 1–3, DOI:10.1002/NT.2620020102.
- D. L. Eaton, S. Ramsdell and G. E. Neal, Biotransformation of aflatoxins, The toxicology of aflatoxins: human health, veterinary and agricultural significance, 1993, pp. 45–72 Search PubMed.
- S. A. Tittlemier, J. Brunkhorst and B. Cramer, et al., Developments in mycotoxin analysis: an update for 2019-2020, World Mycotoxin J., 2021, 14(1), 3–26, DOI:10.3920/WMJ2020.2664.
- X. Shkembi, M. Svobodova, V. Skouridou, A. S. Bashammakh, A. O. Alyoubi and C. K. O'Sullivan, Aptasensors for mycotoxin detection: A review, Anal. Biochem., 2022, 644, 114156, DOI:10.1016/J.AB.2021.114156.
- N. M. Danesh, H. B. Bostan and K. Abnous, et al., Ultrasensitive detection of aflatoxin B1 and its major metabolite aflatoxin M1 using aptasensors: A review, TrAC, Trends Anal. Chem., 2018, 99, 117–128, DOI:10.1016/J.TRAC.2017.12.009.
- Q. Wang, Q. Yang and W. Wu, Progress on Structured Biosensors for Monitoring Aflatoxin B1 From Biofilms: A Review, Front. Microbiol., 2020, 11, 408, DOI:10.3389/fmicb.2020.00408.
- J. Xiong, S. He, L. Qin, S. Zhang, W. Shan and H. Jiang, Aptasensor-based assay for dual-readout determination of aflatoxin B1 in corn and wheat via an electrostatic force–mediated FRET strategy, Microchim. Acta, 2023, 190(2), 80, DOI:10.1007/s00604-023-05641-1.
- J. Lerdsri, W. Chananchana, J. Upan, T. Sridara and J. Jakmunee, Label-free colorimetric aptasensor for rapid detection of aflatoxin B1 by utilizing cationic perylene probe and localized surface plasmon resonance of gold nanoparticles, Sens. Actuators, B, 2020, 320, 128356, DOI:10.1016/j.snb.2020.128356.
- M. Mousivand, M. Javan-Nikkhah, L. Anfossi, F. Di Nardo, M. Salina and K. Bagherzadeh, High performance aptasensing platform development through in silico aptamer engineering for aflatoxin B1 monitoring, Food Control, 2023, 145, 109418, DOI:10.1016/j.foodcont.2022.109418.
- S. Zhang, S. Zhao, S. Wang, J. Liu and Y. Dong, Development of Lateral Flow Immunochromatographic Strips for Micropollutant Screening Using Colorants of Aptamer-Functionalized Nanogold Particles, Part II: Experimental Verification with Aflatoxin B1 and Chloramphenicol, J. AOAC Int., 2018, 101(5), 1408–1414, DOI:10.5740/jaoacint.18-0056.
- H. B. Patel and T. M. Jefferies, Eluotropic strength of solvents: Prediction and use in reversed-phase high-performance liquid chromatography, J. Chromatogr. A, 1987, 389, 21–32, DOI:10.1016/S0021-9673(01)94407-3.
- J. Griffiths, A Brief History of Mass Spectrometry, Anal. Chem., 2008, 80, 5678–5683 CrossRef CAS PubMed , http://masspec.scripps.edu/mshistory/perspectives/sborman.php.
- M. Liu, L. Wang, Y. Lo, S. C. Shiu, A. B. Kinghorn and J. A. Tanner, Aptamer-Enabled Nanomaterials for Therapeutics, Drug Targeting and Imaging, Cells, 2022, 11(1), 159, DOI:10.3390/cells11010159.
- G. A. De, M. McKeague, J. D. Miller, M. C. DeRosa and A. Visconti, Determination of ochratoxin A in wheat after clean-up through a DNA aptamer-based solid phase extraction column, Food Chem., 2011, 127(3), 1378–1384, DOI:10.1016/j.foodchem.2011.01.107.
- S. Reverdatto, D. S. Burz and A. Shekhtman, Peptide Aptamers: Development and Applications, Curr. Top. Med. Chem., 2015, 15(12), 1082–1101 CrossRef CAS PubMed.
- M. McKeague and M. C. DeRosa, Challenges and opportunities for small molecule aptamer development, J. Nucleic Acids, 2012, 2012, 748913, DOI:10.1155/2012/748913.
- J. Liu, Adsorption of DNA onto gold nanoparticles and graphene oxide: surface science and applications, Phys. Chem. Chem. Phys., 2012, 14(30), 10485–10496, 10.1039/C2CP41186E.
- J. Zhou and J. Rossi, Aptamers as targeted therapeutics: current potential and challenges, Nat. Rev. Drug Discovery, 2017, 16(3), 181–202, DOI:10.1038/nrd.2016.199.
- J. Qu, S. Yu, Y. Zheng, Y. Zheng, H. Yang and J. Zhang, Aptamer and its applications in neurodegenerative diseases, Cell. Mol. Life Sci., 2017, 74(4), 683–695, DOI:10.1007/s00018-016-2345-4.
- J. G. Bruno, M. P. Carrillo, A. M. Richarte, T. Phillips, C. Andrews and J. S. Lee, Development, screening, and analysis of DNA aptamer libraries potentially useful for diagnosis and passive immunity of arboviruses, BMC Res. Notes, 2012, 5, 633, DOI:10.1186/1756-0500-5-633.
- D. Shangguan, T. Bing and N. Zhang, in Cell-SELEX: Aptamer Selection Against Whole Cells BT - Aptamers Selected by Cell-SELEX for Theranostics, ed. W. Tan and X. Fang, Springer, Berlin Heidelberg, 2015, pp. 13–33, DOI:10.1007/978-3-662-46226-3_2.
- H. Gu, N. Duan and S. Wu, et al., Graphene oxide-assisted non-immobilized SELEX of okdaic acid aptamer and the analytical application of aptasensor, Sci. Rep., 2016, 6(1), 21665, DOI:10.1038/srep21665.
- D. S. Wilson and J. W. Szostak, In Vitro Selection of Functional Nucleic Acids, Annu. Rev. Biochem., 1999, 68, 611–647, DOI:10.1146/ANNUREV.BIOCHEM.68.1.611.
- M. C. DeRosa, A. Lin and P. Mallikaratchy, et al., In vitro selection of aptamers and their applications, Nat. Rev. Methods Primers, 2023, 3(1), 54, DOI:10.1038/s43586-023-00238-7.
- A. Ruscito and M. C. DeRosa, Small-molecule binding aptamers: Selection strategies, characterization, and applications, Front. Chem., 2016, 4, 1–14, DOI:10.3389/fchem.2016.00014.
- S. Y. Lam, H. L. Lau and C. K. Kwok, Capture-SELEX: Selection Strategy, Aptamer Identification, and Biosensing Application, Biosensors, 2022, 12(12), 1142, DOI:10.3390/bios12121142.
- K. Rahimizadeh, Q. u. a. Zahra, S. Chen, B. T. Le, I. Ullah and R. N. Veedu, Nanoparticles-assisted aptamer biosensing for the detection of environmental pathogens, Environ. Res., 2023, 238, 117123, DOI:10.1016/j.envres.2023.117123.
- Y. He, C. Y. Wen, Z. J. Guo and Y. F. Huang, Noble metal nanomaterial-based aptasensors for microbial toxin detection, J. Food Drug Anal., 2020, 28(4), 508–520, DOI:10.38212/2224-6614.1155.
- T. C. Chiu and C. C. Huang, Aptamer-functionalized nano-biosensors, Sensors, 2009, 9(12), 10356–10388, DOI:10.3390/s91210356.
- G. Vinci and M. Rapa, Noble Metal Nanoparticles Applications: Recent Trends in Food Control, Bioengineering, 2019, 6(1), 10, DOI:10.3390/bioengineering6010010.
- T. S. Rodrigues, A. G. M. Da Silva and P. H. C. Camargo, Nanocatalysis by noble metal nanoparticles: controlled synthesis for the optimization and understanding of activities, J. Mater. Chem. A, 2019, 7(11), 5857–5874, 10.1039/C9TA00074G.
- N. Farkhari, S. Abbasian, A. Moshaii and M. Nikkhah, Mechanism of adsorption of single and double stranded DNA on gold and silver nanoparticles: Investigating some important parameters in bio-sensing applications, Colloids Surf., B, 2016, 148, 657–664, DOI:10.1016/J.COLSURFB.2016.09.022.
- P. Talarska, M. Boruczkowski and J. Żurawski, Current Knowledge of Silver and Gold Nanoparticles in Laboratory Research—Application, Toxicity, Cellular Uptake, Nanomaterials, 2021, 11(9), 2454, DOI:10.3390/nano11092454.
- G. Alberti, C. Zanoni, L. R. Magnaghi and R. Biesuz, Gold and Silver Nanoparticle-Based Colorimetric Sensors: New Trends and Applications, Chemosensors, 2021, 9(11), 305, DOI:10.3390/CHEMOSENSORS9110305.
- H. Wang, H. Rao, M. Luo, X. Xue, Z. Xue and X. Lu, Noble metal nanoparticles growth-based colorimetric strategies: From monocolorimetric to multicolorimetric sensors, Coord. Chem. Rev., 2019, 398, 113003, DOI:10.1016/j.ccr.2019.06.020.
- C. Wu, X. Zhou and J. Wei, Localized Surface Plasmon Resonance of Silver Nanotriangles Synthesized by a Versatile Solution Reaction, Nanoscale Res. Lett., 2015, 10(1), 1–6, DOI:10.1186/S11671-015-1058-1/TABLES/2.
- K. A. Willets and R. P. Van Duyne, Localized Surface Plasmon Resonance Spectroscopy and Sensing, Annu. Rev. Phys. Chem., 2007, 58, 267–297, DOI:10.1146/ANNUREV.PHYSCHEM.58.032806.104607.
- X. Ma, S. He, B. Qiu, F. Luo, L. Guo and Z. Lin, Noble Metal Nanoparticle-Based Multicolor Immunoassays: An Approach toward Visual Quantification of the Analytes with the Naked Eye, ACS Sens., 2019, 4(4), 782–791, DOI:10.1021/acssensors.9b00438.
- X. Ma, S. He, B. Qiu, F. Luo, L. Guo and Z. Lin, Noble Metal Nanoparticle-Based Multicolor Immunoassays: An Approach toward Visual Quantification of the Analytes with the Naked Eye, ACS Sens., 2019, 4(4), 782–791, DOI:10.1021/ACSSENSORS.9B00438/ASSET/IMAGES/LARGE/SE-2019-004384_0008.JPEG.
- V. A. Ogarev, V. M. Rudoi and O. V. Dement'eva, Gold Nanoparticles: Synthesis, Optical Properties, and Application, Inorg. Mater., 2018, 9(1), 134–140, DOI:10.1134/S2075113318010197.
- F. L. Heredia, P. J. Resto and E. I. Parés-Matos, Fast Adhesion of Gold Nanoparticles (AuNPs) to a Surface Using Starch Hydrogels for Characterization of Biomolecules in Biosensor Applications, Biosensors, 2020, 10(8), 99, DOI:10.3390/bios10080099.
- B. Liu and J. Liu, Methods for preparing DNA-functionalized gold nanoparticles, a key reagent of bioanalytical chemistry, Anal. Methods, 2017, 9(18), 2633–2643, 10.1039/C7AY00368D.
- J. M. Carnerero, A. Jimenez-Ruiz, P. M. Castillo and R. Prado-Gotor, Covalent and Non-Covalent DNA–Gold-Nanoparticle Interactions: New Avenues of Research, ChemPhysChem, 2017, 18(1), 17–33, DOI:10.1002/CPHC.201601077.
- J. Hoinka, R. Backofen and T. M. Przytycka, AptaSUITE: A Full-Featured Bioinformatics Framework for the Comprehensive Analysis of Aptamers from HT-SELEX Experiments, Mol. Ther.--Nucleic Acids, 2018, 11, 515–517, DOI:10.1016/J.OMTN.2018.04.006.
- S. E. Ali, A. Mittal and D. H. Mathews, RNA Secondary Structure Analysis Using RNAstructure, Curr. Protoc., 2023, 3(7), e846, DOI:10.1002/CPZ1.846.
- D. H. Mathews, RNA secondary structure analysis using RNAstructure, Curr. Protoc. Bioinf., 2006, 13, 12.6, DOI:10.1002/0471250953.bi1206s13.
- M. McKeague, E. M. McConnell and J. Cruz-Toledo, et al., Analysis of In Vitro Aptamer Selection Parameters, J. Mol. Evol., 2015, 81(5–6), 150–161, DOI:10.1007/S00239-015-9708-6/FIGURES/8.
- R. Nutiu and Y. Li, In vitro selection of structure-switching signaling aptamers, Angew. Chem., Int. Ed., 2005, 44(7), 1061–1065, DOI:10.1002/ANIE.200461848.
- W. Liu, H. Zhu and B. Zheng, et al., Kinetics and mechanism of G-quadruplex formation and conformational switch in a G-quadruplex of PS2.M induced by Pb2+, Nucleic Acids Res., 2012, 40(9), 4229, DOI:10.1093/NAR/GKR1310.
- M. McKeague, R. Velu and A. De Girolamo, et al., Comparison of In-Solution Biorecognition Properties of Aptamers against Ochratoxin A, Toxins, 2016, 8(11), 336, DOI:10.3390/TOXINS8110336.
- M. McKeague and M. C. DeRosa, Challenges and Opportunities for Small Molecule Aptamer Development, J. Nucleic Acids, 2012, 2012(1), 748913, DOI:10.1155/2012/748913.
- A. M. Mueller, D. Breitsprecher, S. Duhr, P. Baaske, T. Schubert and G. Längst, MicroScale Thermophoresis: A Rapid and Precise Method to Quantify Protein–Nucleic Acid Interactions in Solution, Methods Mol. Biol., 2017, 1654, 151–164, DOI:10.1007/978-1-4939-7231-9_10.
- J. S. Reuter and D. H. Mathews, RNAstructure: software for RNA secondary structure prediction and analysis, BMC Bioinf., 2010, 11, 129, DOI:10.1186/1471-2105-11-129.
- Z. J. Lu, J. W. Gloor and D. H. Mathews, Improved RNA secondary structure prediction by maximizing expected pair accuracy, RNA, 2009, 15(10), 1805–1813, DOI:10.1261/RNA.1643609.
- C. Platella, C. Riccardi, D. Montesarchio, G. N. Roviello and D. Musumeci, G-quadruplex-based aptamers against protein targets in therapy and diagnostics, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861(5), 1429–1447, DOI:10.1016/J.BBAGEN.2016.11.027.
- O. Kikin, L. D'Antonio and P. S. Bagga, QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences, Nucleic Acids Res., 2006, 34(suppl_2), W676–W682, DOI:10.1093/nar/gkl253.
- L. Donato, C. Scimone and S. Alibrandi, et al., Investigating G-quadruplex structures in RPGR gene: Implications for understanding X-linked retinal degeneration, Heliyon, 2024, 10(8), e29828, DOI:10.1016/J.HELIYON.2024.E29828.
- A. Varizhuk, D. Ischenko and V. Tsvetkov, et al., The expanding repertoire of G4 DNA structures, Biochimie, 2017, 135, 54–62, DOI:10.1016/J.BIOCHI.2017.01.003.
- V. A. Mirón-Mérida, Y. González-Espinosa, M. Collado-González, Y. Y. Gong, Y. Guo and F. M. Goycoolea, Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1, Biosensors, 2021, 11(1), 18, DOI:10.3390/bios11010018.
- S. Hu, P. J. J. Huang, J. Wang and J. Liu, Dissecting the Effect of Salt for More Sensitive Label-Free Colorimetric Detection of DNA Using Gold Nanoparticles, Anal. Chem., 2020, 92(19), 13354–13360, DOI:10.1021/ACS.ANALCHEM.0C02688/ASSET/IMAGES/LARGE/AC0C02688_0007.JPEG.
- J. Xiong, S. He and S. Zhang, et al., A label-free aptasensor for dual-mode detection of aflatoxin B1 based on inner filter effect using silver nanoparticles and arginine-modified gold nanoclusters, Food Control, 2023, 144, 109397, DOI:10.1016/j.foodcont.2022.109397.
- W. A. Mula-Abed, 25- Hydroxyvitamin D: Explosion in Clinical Interest and Laboratory Requests, Oman Med. J., 2009, 24(4), 239–241, DOI:10.5001/omj.2009.49.
- D. Lu, X. Tian and Y. Yang, et al., Rapid and sensitive aptamer-linked immunosorbent assay with aptamer-templated silver nanoparticles for detection of aflatoxin B1 in medicinal and edible food, Food Biosci., 2024, 60, 104387, DOI:10.1016/J.FBIO.2024.104387.
- C. Parolo, A. Sena-Torralba and J. F. Bergua, et al., Tutorial: design and fabrication of nanoparticle-based lateral-flow immunoassays, Nat. Protoc., 2020, 15(12), 3788–3816, DOI:10.1038/s41596-020-0357-x.
- L. de Almeida, R. Williams, D. M. Soares, H. Nesbitt, G. Wright and W. Erskine, Aflatoxin levels in maize and peanut and blood in women and children: The case of Timor-Leste, Sci. Rep., 2019, 9(1), 13158, DOI:10.1038/s41598-019-49584-1.
- K. W. Lien, X. Wang, M. H. Pan and M. P. Ling, Assessing Aflatoxin Exposure Risk from Peanuts and Peanut Products Imported to Taiwan, Toxins, 2019, 11(2), 80, DOI:10.3390/toxins11020080.
- W. Lu, L. Wang and J. Li, et al., Quantitative investigation of the poly-adenine DNA dissociation from the surface of gold nanoparticles, Sci. Rep., 2015, 5(1), 10158, DOI:10.1038/srep10158.
- RG-8 Regulatory Guidance: Contaminants in Feed (formerly RG-1, Chapter 7) - inspection.canada.ca. Accessed December 3, 2024, https://inspection.canada.ca/en/animal-health/livestock-feeds/regulatory-guidance/rg-8.
- American Peanut Council. Aflatoxin Limits in Peanuts and Peanut Products (as of April 15th, 2020), 2020.
- K. Wei, Z. Ye and W. Dong, et al., Generating robust aptamers for food analysis by sequence-based configuration optimization, Talanta, 2024, 275, 126044, DOI:10.1016/j.talanta.2024.126044.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00271g |
| This journal is © The Royal Society of Chemistry 2025 |