
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChelation-assisted multiple and relay C–H functionalization of unactivated aliphatic E-alkenes
Yini
Wang
a,
Xiaoli
Li
a,
Chengxing
Peng
a,
Yu
Chen
c,
Xi
Lu
d,
Yuhang
Zhu
a,
Peiyuan
Yu
 *c,
Guofu
Zhong
*ab and
Jian
Zhang
*a
*c,
Guofu
Zhong
*ab and
Jian
Zhang
*a
aCollege of Materials, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, China. E-mail: zhangjian@hznu.edu.cn
bDepartment of Chemistry, Eastern Institute of Technology, Ningbo, 315200, China. E-mail: gzhong@eitech.edu.cn
cDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: yupy@sustech.edu.cn
dPetroleum Exploration and Production Research Institute, Sinopec, Beijing 102206, China
First published on 6th November 2025
Abstract
Olefinic C–H modification has been a highly sought-after objective to upgrade commodity molecules into densely functionalized compounds. Unactivated aliphatic E-alkenes of electronically and sterically unbiased properties are ubiquitous compounds, but they are challenging targets for alkenyl C–H functionalization due to low reactivity and selectivity issues. We report the first sequential and relay C–H functionalization of unactivated aliphatic E-alkenes using alkenes and alkynes to afford complex polyenes and polyeneynes. The dienes, obtained from olefinic C–H functionalization by alkyne hydroalkenylation, underwent upgrading and controllable C–H functionalization by a six- or seven-membered palladacycle. The method allows for a range of functionalizations such as oxidative alkenylation, hydroalkenylation and alkynylation, via five- to eight-membered cyclopalladation. The robustness of the protocols was demonstrated by the preparative scale, chemical derivation and intra-molecular reactions to produce 12- to 16-membered macrocycles. Mechanistic experiments combined with DFT calculations explained the reactivities and selectivities of various alkenyl C–H bonds.
Introduction
Configurated multi-substituted alkenes are one of the most widely used organic compounds,1 and their stereo-selective synthesis remains a long-standing and challenging research topic. Alkenyl C–H functionalization affords atom- and step efficient synthesis of densely functionalized alkene compounds. One powerful method is non-directed alkenyl C–H functionalization such as the Heck type reaction2 and radical reaction,3 but they lead to extremely low reactivity and poor E/Z selectivity when applied to the reaction of internal alkenes. While chelation-assisted (directed) aromatic and aliphatic C–H functionalization have been well defined,4 directed alkenyl C–H functionalization has been demonstrated to be a powerful method toward the synthesis of multi-substituted alkene derivatives in a regio- and stereo-selective manner,4e,5 proceeding via a C–H cyclometallation event. However, previous reports generally employed disubstituted terminal alkenes6 and disubstituted Z-alkenes7 as substrates, reacted by the formation of endo- and exo-metallocycles respectively to give rise to tri-substituted alkenes, in which the competitive α- or β C–H positions were substituted (Scheme 1A(a and b)),5–8 and the essential problem in achieving regio-selectivity remains unsolved. There have been examples of β-C–H functionalization of electronically biased (activated) alkenes such as trans-acrylic acids/amides by five-membered endo-metallocycles;5a,6 however, α-C–H functionalization in these protocols is prevented due to difficulty in four-membered cyclometallation. Moreover, although there is a recent example of α-/β C–H functionalization of trans-styrenes (Scheme 1 A(d)),8 C–H functionalization of more general unactivated aliphatic (E)-alkenes still remains unexplored (Scheme 1A(c)). Tetra-substituted alkenes are valuable targets and their E/Z preparation by olefinic C–H functionalization is quite challenging. There are only sporadic reports on this topic. Previously, sequential olefinic C–H arylation of simple alkenes by the Pd-catalyzed Heck reaction was disclosed by Itami and Yoshida2d and Studer.2e Recently, the Su group demonstrated a synthesis of triarylated all-carbon tetrasubstituted olefins by a tandem remote-carbonyl-directed Heck arylation/isomerization/alkenyl C–H arylation of 1,1-disubstituted olefins.6u Directed sequential olefinic C–H functionalization of aliphatic E-alkenes without E/Z isomerization could also provide tetra-substituted alkenes which is unexplored. | ||
| Scheme 1 (A) Challenges for C–H functionalization of unactivated aliphatic E-alkenes. (B) Group-directed olefinic C–H functionalization. (C) Multiple and relay C–H functionalization of unactivated aliphatic E-alkenes. (D) Bioactive molecules containing homoallyl and bishomoallyl amines. | ||
Great efforts have been devoted to C–H functionalization of conjugated alkenes such as acrylic acids and their derivatives,6a–j enol6k–n or enamine derivatives6o–q and aryl alkenes.6r–u,7j,k,8 In contrast, C–H functionalization of nonconjugated aliphatic alkenes is much less explored due to their steric and electronic unbias, inert properties of aliphatic C–C double bonds, and the high degree of conformational flexibility for more challenging C–H cyclometallation.7b,8 There have been limited examples of conversion of aliphatic alkenes.6,7 Loh and Xu reported alkenyl C–H alkenylation of allyl and homoallyl alcohols.6r Engle7a and our group7b reported bidentate- and monodentate-chelation-assisted olefinic C–H alkenylation of Z-alkenes respectively. Carreira developed alkenyl C–H iodination7c and alkynylation7d of Z-alkenes. Also, there have been reports on C–H arylation7e of mono-substituted alkenes by exo-palladation. Unfortunately, these methods are restricted to aliphatic Z-alkenes, terminal alkenes and cyclic alkenes (Scheme 1A(a–d)). Furthermore, group-directed hydrofunctionalization and difunctionalization of aliphatic E-alkenes were well defined to afford complex alkanes (Scheme 1B(a)),9,10 and there was also oxidative Heck arylation using aliphatic E-alkenes (Scheme 1B(b)).11 However, chelation-assisted direct alkenyl C–H functionalization of aliphatic E-alkenes remains unexplored and elusive (Scheme 1B(c)).12
Polyenes such as dienes are occurring in countless natural products and drugs, and development of their selective alkenyl C–H functionalization could greatly expand the utility of the current strategy which also affords rapid construction of molecular libraries for new drug investigations. Unfortunately, selective C–H functionalization of polyenes such as 1,3-dienes bearing competitive olefinic C–H bonds has remained elusive and unexplored2–8 presumably due to difficulties in selectivity control, reaction complexity (cyclization, decomposition, isomerization, etc.), conformational flexibility (s-trans vs. s-cis) and undesirable metal-coordination that may prevent the catalytic reaction (Scheme 1A(e)).
Multiple and sequential C–H functionalization enabled by one directing group greatly promotes the synthetic efficiency and practical usage by avoiding the tedious installation and removal process.4,5 However, this research topic is really challenging due to problems rooted in both reactivity and selectivity. With our long-term interest in alkenyl C–H functionalization,6g,7b,7j,8 we wondered whether we could develop selective and multiple alkenyl C–H functionalization of aliphatic E-alkenes of low reactivity. However, there are several challenges for such a transformation. The existence of competitive olefinic and allyl C–H bonds in aliphatic (E)-alkenes as well as possible alkene isomerization and migration may lead to poor selectivity and more side reactions under transition-metal-catalysis.1b,6u,12 Additionally, group directed olefinic C–H activation of E-alkenes by exo-cyclometallation is really challenging due to disfavored steric/electronic effects, and there is only one related example of α- and β-C–H activation of trans-styrenes (Scheme 1B).8,12 Thirdly, selective C–H functionalization of conjugated polyenes is unexplored and elusive. Herein, we report the first selective and multiple alkenyl C–H functionalization of unactivated aliphatic E-alkenes, affording efficient synthesis of multi-substituted polyenes and polyeneynes with excellent regio- and E/Z ratio selectivity (Scheme 1C). Selective olefinic C–H functionalization of complex conjugated dienes is investigated for the first time.
Results and discussion
Alkenyl amines widely occur in countless natural products and drugs (Scheme 1D). At the beginning of our study, (E)-type allyl, homoallyl and bishomoallyl amine derivatives bearing two competitive olefinic C–H bonds were chosen as the substrates for C–H alkenylation using acrylate 2a. The reported Pd, Ir, Ru, Rh and Co based catalytic conditions showed no catalytic activity for E-alkenyl amine derivatives bearing monodentate directing groups such as amide, sulfonamide, urea, etc.4,5 We attributed the failure to the weak monodentate coordination and energetically unfavorable monocyclic C–H cyclometallation, also exhibiting the challenge of the C–H conversion of E-alkenes. In this regard, various bidentate directing groups were chosen in hope of promoting alkenyl C–H activation through the formation of more energetically favorable bicyclic metallocycles. We noticed that the reaction of bishomoallyl amide 1 bearing Daugulis's 8-aminoquinoline (DG1) with acrylate 2a led to desired product 3 in 26% yield with 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 Z/E ratio selectivity, using 10 mol% Pd(OAc)2, 10 mol% p-benzoquinone (p-BQ), 1.5 equivalents of PivOH, and 3.0 equivalents of MnO2 in DMSO at 80 °C (Cond. A) (Table 1, entry 1). Next, we turned to examine other N,N-bidentate-chelation-assisted directing groups such as pyridine-2-carboxamide (DG2), 2-pyrazine carboxamide (DG3), 4-pyrimidine carboxamide (DG4) and pyrimidine-2-carboxamide (DG5). While alkene bearing DG2 afforded 73% yield, incorporation of DG3 gave 81% yield with 93:7 Z/E ratio selectivity (entries 2–3). Notably, bishomoallyl amine bearing DG4 and DG5 also led to good results (85% and 73% yields respectively) (entries 4–5). Monodentate directing groups such as carboxylic acid failed to promote the reaction (entry 6). Decreasing Pd(OAc)2 to 5 mol% still led to 64% yield (entry 3). Changing the chain length between DG3 and the olefinic moiety remarkably influenced the reactivity of the alkene substrates. For example, while the homoallyl amine substrate led to 56% yield with 80:20 Z/E ratio selectivity, the non-5-ene-1-amine derivative afforded an alkenylation product with only 23% yield, demonstrating the more favorable formation of five-membered exo-palladacycles over seven-membered exo-palladacycles under Cond. A (entries 7 and 8). No detection of other regio-isomers also shows the much more favorable formation of five- to seven-membered exo-palladacycles over six- to eight-membered endo-palladacycles.
:15 Z/E ratio selectivity, using 10 mol% Pd(OAc)2, 10 mol% p-benzoquinone (p-BQ), 1.5 equivalents of PivOH, and 3.0 equivalents of MnO2 in DMSO at 80 °C (Cond. A) (Table 1, entry 1). Next, we turned to examine other N,N-bidentate-chelation-assisted directing groups such as pyridine-2-carboxamide (DG2), 2-pyrazine carboxamide (DG3), 4-pyrimidine carboxamide (DG4) and pyrimidine-2-carboxamide (DG5). While alkene bearing DG2 afforded 73% yield, incorporation of DG3 gave 81% yield with 93:7 Z/E ratio selectivity (entries 2–3). Notably, bishomoallyl amine bearing DG4 and DG5 also led to good results (85% and 73% yields respectively) (entries 4–5). Monodentate directing groups such as carboxylic acid failed to promote the reaction (entry 6). Decreasing Pd(OAc)2 to 5 mol% still led to 64% yield (entry 3). Changing the chain length between DG3 and the olefinic moiety remarkably influenced the reactivity of the alkene substrates. For example, while the homoallyl amine substrate led to 56% yield with 80:20 Z/E ratio selectivity, the non-5-ene-1-amine derivative afforded an alkenylation product with only 23% yield, demonstrating the more favorable formation of five-membered exo-palladacycles over seven-membered exo-palladacycles under Cond. A (entries 7 and 8). No detection of other regio-isomers also shows the much more favorable formation of five- to seven-membered exo-palladacycles over six- to eight-membered endo-palladacycles.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Step | DG | n | Conditionsa | Yieldb (%) | E/Zc | Metallacycle |
| a Conditions A: 1 (0.15 mmol, 1.0 equiv.), 2 (2.5 equiv.), Pd(OAc)2 (10 mol%), PivOH (1.5 equiv.), MnO2 (3.0 equiv.), BQ (10 mol%) in DMSO (0.15 M) at 80 °C for 36 h, under Ar. Conditions B: 3 (0.1 mmol, 1.0 equiv.), 2 (2.5 equiv.), Pd(OAc)2 (10 mol%), AcOH (2.0 equiv.), MnO2 (3.0 equiv.), BQ (10 mol%), in MeCN at 100 °C for 36 h, under Ar (1 atm). b Yields are isolated yields. c E/Z ratio was determined by 1H NMR. d 5 mol% Pd(OAc)2 was used. e 2b was used instead of 2a. f 2a was used instead of 2b for the second C–H alkenylation of diene 3 obtained in entry 10. | |||||||
| 1 | 1 | DG1 | 1 | A | 26 | 15:85 |
6-Exo |
| 2 | 1 | DG2 | 2 | A | 73 | 7:93 |
6-Exo |
| 3 | 1 | DG3 | 2 | A | 81(64)d | 7:93 |
6-Exo |
| 4 | 1 | DG4 | 2 | A | 85 | 7:93 |
6-Exo |
| 5 | 1 | DG5 | 2 | A | 73 | 6:94 |
6-Exo |
| 6 | 1 | CO2H | 1, 0 | A | 0 | — | 6, 5-Exo |
| 7 | 1 | DG3 | 1 | A | 56 | 20:80 |
5-Exo |
| 8 | 1 | DG3 | 3 | A | 23 | <1:99 |
7-Exo |
| 9 | 1 | DG3 | 0 | A | 0 | — | 4-Exo |
| 10e | 1 | DG3 | 2 | A | 80 | 5:95 |
6-Exo |
| 11 | 2 | DG2 | 2 | B | 35 | >99:1 |
7-Endo |
| 12 | 2 | DG3 | 2 | B | 60 | >99:1 |
7-Endo |
| 13 | 2 | DG4 | 2 | B | 48 | >99:1 |
7-Endo |
| 14 | 2 | DG5 | 2 | B | 52 | >99:1 |
7-Endo |
| 15 | 2 | DG3 | 1 | B | 55 | >99:1 |
6-Endo |
| 16f | 2 | DG3 | 2 | B | 49 | >99:1 |
7-Endo |

However, the allyl amine derived substrate showed no reactivity (entry 9), presumably due to the difficulty in forming the strained metallocycle intermediate. The reaction also proceeded well by using acrylate 2b instead (entry 10). Further C–H functionalization of the obtained diene 3 bearing three competitive olefinic C–H bonds was investigated. When diene 3 bearing DG2 was subjected to 10 mol% Pd(OAc)2, 10 mol% p-benzoquinone (p-BQ), 1.5 equivalents of PivOH, and 3.0 equivalents of MnO2 in MeCN at 100 °C (Cond. B), C–H alkenylation occurred at the β-C–H bond and triene product 4 was obtained in 35% yield with > 99:1 E/Z ratio selectivity (entry 11). To our delight, the reaction of diene 3 bearing DG3 led to the triene product in 60% yield with >99:1 E/Z ratio selectivity by the formation of a seven-membered endo-palladacycle, and no detection of other regio isomers exhibited the excellent site-selectivity of the protocol (entry 12). Other DGs such as DG4 and DG5 also performed well to provide triene products in 48% and 52% yields respectively with >99:1 E/Z ratio selectivity (entries 13 and 14). It is worth noting that the diene generated from homoallyl amine also underwent the second C–H alkenylation only at the β-position by a six-membered endo-palladacycle, and the triene 4 was successfully obtained with 55% yield and >99:1 E/Z ratio selectivity under Cond. B (entry 15). The second C–H alkenylation of diene 3 obtained in entry 10 also gave 49% yield using acrylate 2a instead (entry 16).
Once we obtained the optimized conditions, we turned to examine the conversion of various E-configurated alkenyl amides 1 bearing DG3 (Scheme 2). Oct-4-ene-amine derived amide reacted well with various acrylates to give dienes 3a–3f in 40–81% yields with excellent Z/E ratio selectivity. Gram-scale preparation of 3a (1.67 g) is successful (93% yield, 94:6 Z/E ratio) to demonstrate its robustness. Other electron-deficient alkenes such as vinyl ketone, acrylamide, vinyl phosphonate, vinyl sulphone and styrenes were all reacted well to give dienes 3g–3o in 27–85% yields. Branched aliphatic E-alkenes bearing alkyl or aryl were suitable substrates to afford C–H alkenylation products 3p–3u in 43–99% yields with up to 99:1 Z/E ratio selectivity. E-Alkenes bearing various groups such as methyl, pentyl, hexyl, isobutyl, isopropyl, arylethyl and cyclohexyl were examined, and all of them successfully led to desired products 3v–3ag in 60–98% yields with up to >99/1 Z/E selectivity. Compound 3ah was determined to be in (Z,E)-configuration and s-trans conformation (which is of lower energy) by X-ray crystallographic analysis (CCDC 2419583). Homoallyl amine derived substrates bearing i-Pr and n-Pr and n-hexyl were smoothly converted to give rise to 3ai–3al in moderate to good yields. However, branched homoallyl amide led to 3am in 33% yield with 90:10 Z/E ratio selectivity. While non-5-ene-1-amide afforded product 3an in 23% yield, allyl amide showed no reactivity.
 | ||
| Scheme 2 Substrate scope of olefinic C–H alkenylation of aliphatic E-alkenes. (a) Reaction conditions: 1 (0.1 mmol), 2 (0.25 mmol), Pd(OAc)2 (10 mol%), PivOH (1.5 equiv.), MnO2 (3.0 equiv.), BQ (10 mol%) in DMSO (0.15 M) at 80 °C for 36 h. (b) 48 h. (c) 100 °C. The yields are isolated yields based on 1. Z/E ratios in parentheses were determined by 1H NMR. | ||
With the dienes 3 in hand, we then turned to examine their further C–H alkenylation by seven- and six-membered endo-cyclopalladation to prepare trienes 4 (Scheme 3A). Dienes 3 generated from alkenes 1 and acrylate 2a reacted well with different acrylates and acrylamides, and C–H functionalization all occurred at the β-C–H bond far away from the ester to give corresponding trienes 4a–4e in 33–60% yields with >99:1 E/Z ratio selectivity by the formation of seven-membered palladacycles. Other dienes bearing alkyl groups such as methyl, pentyl and hexyl were all converted smoothly to give 4f–4h in moderate yields with 30–40% substrate recovery. Notably, even diene bearing a bulky iso-butyl was still able to give triene 4i in 30% yield with >99:1 E/Z ratio selectivity. Incorporated phenyl ethyl bearing ortho, meta- or para-Me, Cl and CF3 into alkene substrates reacted well with acrylate to provide trienes 4j–4n in 37–54% yields with > 99:1 E/Z ratio selectivity. Dienes containing branched aliphatic chains also afforded alkenylation products 4o–4q in moderate yields with > 99:1 E/Z ratio selectivity. (E,E,E)-Configuration and s-trans conformation of conjugated triene 4j were determined by X-ray crystallographic analysis (CCDC 2419584), and all of the configurations and conformations of triene products 4 were assigned analogously to this compound. Diene 3e also underwent C–H alkenylation with tert-butyl acrylate 2a to produce 4r in 49% yield. To our delight, diene 3aj derived from homoallyl amine successfully led to triene 4s in 55% yield with >99:1 E/Z ratio selectivity, by the formation of a six-membered palladacycle. Large scale preparation of 4a (0.58 g) and 4s (0.46 g) is also successful to show the robustness of the protocol.
 | ||
| Scheme 3 Substrate scope of olefinic C–H alkenylation to prepare trienes. aConditions B: 1 (0.1 mmol), 2 (0.25 mmol), Pd(OAc)2 (10 mol%), AcOH (2.0 equiv.), MnO2 (3.0 equiv.), BQ (10 mol%) in CH3CN (0.15 M) at 100 °C for 36 h. The yields are isolated yields based on 1 or 3. E/Z ratio in parentheses was determined by 1H NMR. | ||
Multiple C–H alkenylation of E-alkenes in “one-pot” fashion was also successful under Cond. B at 100 °C, using 5.0 equivalents of electron-deficient alkenes (Scheme 3B). This protocol allowed an efficient approach toward the preparation of conjugated trienes 5 in 33–61% yields from simple E-monoenes, with the generation of diene 3 in 12–67% yields. Notably, all of these reactions afforded no regioisomeric products from β′- and γ′-C–H functionalization, exhibiting their excellent regio-selectivity.14
Other coupling partners were also investigated (Scheme 4). Internal alkynes 6 reacted with E-alkenes 1a smoothly to produce mixed complex dienes 7a–7c in 45–63% yield with the formation of trienes 8a–8c in 24–32% yields with excellent E/Z ratio selectivity (up to >99:1), using 10 mol% Pd(OAc)2 and 1.5 equivalents of MesCO2H in mixed dioxane/MeCN at 100 °C (Cond. C). (Z,E,Z)-Configuration of triene 8c was determined by X-ray crystallographic analysis (CCDC 2419699). If 7b was subjected to Cond. C, 8b was obtained in 29% yield with 42% recovery. These results indicated a sequential α- and β C–H activation/cis-hydroalkenylation with alkynes 6. Next, we turned to investigate other types of C–H functionalizations of dienes 7a–7c bearing two competitive olefinic C–H bonds Ha and Hb. Diene 7a from bishomoallyl amine exhibited no reactivity under either oxidative alkenylation (Cond. B) or alkynylation conditions (Cond. D). Interestingly, C–H alkenylation and alkynylation of dienes 7b and 7c underwent smoothly at the olefinic C–H bond Ha farther away from the directing group to afford trienes (9b and 9c) and eneynes (11a and 11b), with the alkenyl C–H bond Hb closer to the directing group intact (Scheme 4B). Herein, functionalization of C–H bond Ha proceeding by a seven-membered C–H cyclopalladation was previously regarded to be energetically more disfavored than the activation of the C–H bond Hb by a six-membered palladacycle.5–8 These results are different from the reactions of dienes 3 in Scheme 3 and dienes 7 in Scheme 4A.15
 | ||
| Scheme 4 Olefinic C–H functionalization to prepare polyenes and polyeneynes. | ||
If aliphatic alkene 1 was subjected to alkynylation Cond. D, a bis-alkynylation reaction occurred to afford 12a and 12b in 59% and 35% yields, with the formation of E/Z isomers 12a′ and 12b′ in 25% and 20% yields respectively (Scheme 4C). (E)-Configuration of polyeneyne 12b was confirmed by X-ray crystallographic analysis (CCDC 2419743). Interestingly, α-alkynylation products 12c and 12d were smoothly obtained in 44% and 20% yields respectively by using MeCN as a solvent instead. Notably, further alkenyl C–H alkenylation of 12c was also successful under Cond. B, giving rise to 12d in moderate yield with >99:1 E/Z ratio selectivity.
Competition experiments were performed to obtain additional mechanistic insight, using various E- and Z-configurated alkenes under Cond. A (Scheme 5A). While both alkene 1a-Z and 1a were converted by six-membered C–H exo-cyclometallation, Z-type alkene 1a-Z reacted much faster than the E-alkene 1a (3a-Z 53%, 3a 33%) due to decreased distortion energy which is consistent with our previous observations using aryl alkenes.8 Competition experiments with alkene 1ai-Z and 1ai also demonstrated the Z-type alkene 1ai-Z to be more reactive (3aj-Z 24% and 3aj 16%) which were converted by five-membered C–H exo-cyclopalladation. If Z-type alkenes 1a-Z and 1ai-Z were subjected to Cond. A, they showed comparable reactivity toward acrylate (3a-Z 49% and 3aj-Z 46%). Competition reactions using E-type alkenes 1a and 1ai were investigated. Product yield of 3aj is higher than that of 3a (3a 27% and 3aj 34%), showing that the formation of the five-membered exo-palladacycle is more kinetically favored than that of the six-membered exo-palladacycle, which is different from the previous observations.7b Competition experiments using aliphatic alkene 1a and styrene 13 showed that styrene 13 reacted much more quickly to outcompete the alkene 1a (3a 2% and 14 21%), also supporting the challenge of C–H cyclopalladation of aliphatic alkene substrates. Deuterium incorporation experiments using 1a showed the six-membered C–H exo-cyclometallations to be reversible (13% D) with 65% recovery, and no E/Z isomerization was observed (Scheme 5B).16 Kinetic isotope effect (KIE) experiments confirmed the olefinic C–H activation of 1a by C–H exo-cyclopalladation to be the rate-determining step (Scheme 5C). These results exhibited the directed reversible C–H bond activation to originate the regioselectivity and exclude other possible pathways such as nucleometallation elimination and π-allyl Pd(II) mechanism.13
 | ||
| Scheme 5 Mechanistic experiments and DFT calculations. The relative free energies in DFT calculations are given in kcal mol−1. | ||
To further investigate the origin of the observed high/exclusive regioselectivity of the reaction, DFT calculations were performed on the key carboxylate-assisted Pd-catalyzed alkenyl C–H activation steps (Scheme 5D–H). We selected three substrates with various distances between the directing group and the alkene to calculate the relative free energies of key transition states leading to different regioisomeric products. It was found that for transition states TS(n) for exo-cyclopalladation, when n = 2, the reactivity is highest with an energy barrier of 16.4 kcal mol−1. This can be attributed to the formation of a six-membered metallocycle with lower ring strain, and this speculation is further supported by comparing the activation energy barriers in the TS′(n) for endo-cyclopalladation, where the lowest energy barrier (20.8 kcal mol−1) is observed when n is 1, in which a five-membered palladacycle is formed in the transition state TS1′. The computational results also indicate that for the olefin substrates 1 (n = 1, 2 and 3), the C(sp2)–H bond closer to the directing group is more readily activated, which is consistent with the experimental results. Given that the two C(sp2)–H bonds in the alkene of selected substrates have similar chemical environments, we speculate that the differential ring strain in these key transition states also dominates the regioselectivity. Interestingly, for diene substrates 7b and 7c, the transition state TS4′ for the activation of the olefinic C(sp2)–H bond by a six-membered palladacycle was 3.5 kcal mol−1 higher than the transition state TS4 for the activation of the olefinic C(sp2)–H bond far away from the directing group by a seven-membered palladacycle, which is in accordance with the experimental results in Scheme 4B.17 The unique selectivity may be attributed to the significant difference in the chemical environment of the two C–H bonds, which are dominated by the C–H bond activation or the following insertion step (Scheme 5H).15,17
We also examined the compatibility of the protocols using natural products (Scheme 6A). Notably, acrylates bearing natural geraniol, menthol, borneol and β-sitosterol were all smoothly converted to afford corresponding dienes 3ao–3ar in 40–80% yields with excellent E/Z ratio selectivity, showing the robustness of the protocol. Next, we turned to examine the chemical derivation of the obtained polyenes and eneynes. If diene 3a was treated with m-CPBA (3-chloroperoxybenzoic acid), epoxidation occurred successfully to produce oxirane 15 in 80% yield, with the other olefinic moiety adjacent to the ester intact. Interestingly, diene 3a reacted well with nitromethane by Michael addition to give olefin 16 in 57% yield. Hydrogenation of diene 3a went well to produce alkane 17 in 51% yield. The Diels–Alder reaction using 3l was successful in constructing bicyclic products 18. Also, directing group PC was easily removed by subjecting 3l to EtOH/NaOH to afford free amine 19 (Scheme 6B). The directing group in triene 4a was also readily removed by Boc protection and reduction by LiAlH4 to give Boc-NH amine 20 in quantitative yield (Scheme 6C). If eneyne 12 was treated with TBAF, desiliconization occurred smoothly to afford terminal alkyne 21 in 83% yield, which coupled with PhI to give 22 in 63% yield under palladium catalysis (Scheme 6D). Intramolecular cross-coupling reactions were also investigated. If alkenes 23a–c were subjected to Cond. A in a low concentration (0.01 M in DMSO), 12-, 14- and 16-membered macrocycles 24a–c were obtained in up to 41% yield with >99:1 E/Z ratio selectivity (Scheme 6E). We also examined the photophysical properties of products 8 and 11 with large conjugating structures. The fluorescence emission of compounds 8a–8c, 11a and 11b was also investigated and the emission maxima altered from 387 to 468 nm, exhibiting good luminescent properties which may be potentially applied in optical materials (see the SI for details).
 | ||
| Scheme 6 Synthetic demonstrations and chemical derivations. | ||
Conclusions
In summary, we report palladium-catalyzed multiple olefinic C–H functionalization of aliphatic monoenes and dienes to afford complex polyenes and eneynes. C–H functionalizations such as oxidative alkenylation, hydoalkenylation and alkynylation were all enabled by the N,N-bidentate directing group of 2-pyrazine carboxamide through the formation of five- to eight-membered endo- and exo-palladacycles. This protocol allowed olefinic C–H functionalization of a wide range of substrates including bishomoallyl amine, homoallyl amine and 5-ene-1-amine derived alkenyl amides which showed wide functionality tolerance. The synthetic applicability was demonstrated by the preparative scale, chemical derivation of the polyenes and eneynes, and primary success in intra-molecular reactions to produce 12–16 membered macrocycles. Selective C–H functionalization of dienes by a six- or seven-membered palladacycle is investigated for the first time which is controlled by both steric and electronic effects. Mechanistic experiments and DFT calculations were performed to explain the relative reactivities and selectivities of various E- and Z-alkenes.Author contributions
Y. W., X. L., C. P., X. L. and Y. Z. performed all of the experiments. J. Z. conceived and supervised the project. J. Z. prepared this manuscript with the assistance of G. Z., Y. W. and X. L. P. Y. and Y. C. conducted DFT calculations. All of the authors participated in data analysis and discussions. Y. W. and X. L. contributed equally.Conflicts of interest
The authors declare that they have no conflict of interest.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary information (SI). Supplementary information: materials and methods, experimental procedures, characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d5sc07408h.CCDC 2419583 (3ah), 2419584 (4j), 2419699 (8c) and 2419743 (12b) contain the supplementary crystallographic data for this paper.18a–d
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 22278103, 21502037, 22171130 and 21672048), Natural Science Foundation of Zhejiang Province (ZJNSF) (LY19B020006, LY15B020008), Major Project of Hangzhou Health Science and Technology Plan (Z20200046), Key Subject of Stomatology in Hangzhou, and Hangzhou Normal University.Notes and references
- (a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS PubMed; (b) J. Wang, in Stereoselective Alkene Synthesis, New York, Springer-Verlag, 2012 CrossRef; (c) F. Buttard, J. Sharmaa and P. A. Champagne, Chem. Commun., 2021, 57, 4071–4088 RSC; (d) N. Mukherjee, S. Planera and K. Grela, Org. Chem. Front., 2018, 5, 494–516 RSC.
- (a) M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874–884 CrossRef CAS PubMed; (b) A. Deb and D. Maiti, Eur. J. Org Chem., 2016, 2017, 1239–1252 CrossRef; (c) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253–3260 RSC; (d) K. Itami, M. Mineno, N. Muraoka and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 11778 CrossRef CAS PubMed; (e) Z. He, S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 3699 CrossRef CAS PubMed.
- (a) S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070–1082 RSC; (b) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (b) B. Liu, A. W. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (c) C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS PubMed; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, B. U. W. Maes and M. Schnürch, et al. , Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (f) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS PubMed; (g) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed.
- (a) J. Zhang, X. Lu, C. Shen, L. Xu, L. Ding and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263–3314 RSC; (b) M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J. S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed; (c) Y. Zhu, Y. Liao, S. Jin, L. Ding, G. Zhong and J. Zhang, Chem. Rec., 2023, 23, e202300012 CrossRef CAS PubMed.
- (a) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem. –Eur. J., 2011, 17, 7167–7171 CrossRef CAS PubMed; (b) X. Zhang, X. Liu, L. Li, J. Li, L. Zhao, R. He, C. Shen, G. Zhong and J. Zhang, Chin. J. Chem., 2025, 43, 3569–3574 Search PubMed; (c) K. Meng, Y. Sun, J. Zhang, K. Zhang, X. Ji, L. Ding and G. Zhong, Org. Lett., 2019, 21, 8219–8224 CrossRef CAS PubMed; (d) T. Li, C. Shen, Y. Sun, J. Zhang, P. Xiang, X. Lu and G. Zhong, Org. Lett., 2019, 21, 7772–7777 CrossRef CAS PubMed; (e) X.-H. Hu, J. Zhang, X.-F. Yang, Y.-H. Xu and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 3169–3172 CrossRef CAS PubMed; (f) B. Jiang, M. Zhao, S.-S. Li, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2018, 57, 555–559 CrossRef CAS PubMed; (g) K. Meng, J. Zhang, F. Li, Z. Lin, K. Zhang and G. Zhong, Org. Lett., 2017, 19, 2498–2501 CrossRef CAS PubMed; (h) J.-P. Krieger, D. Lesuisse, G. Ricci, M.-A. Perrin, C. Meyer and J. Cossy, Org. Lett., 2017, 19, 2706–2709 CrossRef CAS PubMed; (i) F. Li, C. Yu, J. Zhang and G. Zhong, Org. Lett., 2016, 18, 4582–4585 CrossRef CAS PubMed; (j) J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, ACS Catal., 2020, 10, 3790–3796 CrossRef CAS; (k) M. Boultadakis-Arapinis, M. N. Hopkinson and F. Glorius, Org. Lett., 2014, 16, 1630–1633 CrossRef CAS PubMed; (l) X.-H. Hu, X.-F. Yang and T.-P. Loh, Angew. Chem., Int. Ed., 2015, 54, 15535–15539 CrossRef CAS PubMed; (m) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430–11433 CrossRef CAS PubMed; (n) A. Seoane, C. Comanescu, N. Casanova, R. García-Fandiño, X. Diz, J. L. Mascareñas and M. Gulías, Angew. Chem., Int. Ed., 2019, 58, 1700–1704 CrossRef CAS PubMed; (o) M. Font, B. Cendón, A. Seoane, J. L. Mascareñas and M. Gulías, Angew. Chem., Int. Ed., 2018, 57, 8255–8259 CrossRef CAS PubMed; (p) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 834–837 CrossRef CAS PubMed; (q) Y.-C. Luo, C. Yang, S.-Q. Qiu, Q.-J. Liang, Y.-H. Xu and T.-P. Loh, ACS Catal., 2019, 9, 4271–4276 CrossRef CAS; (r) Q.-J. Liang, C. Yang, F.-F. Meng, B. Jiang, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2017, 56, 5091–5095 CrossRef CAS PubMed; (s) R. Luan, P. Lin, K. Li, Y. Du and W. Su, Nat. Commun., 2024, 15, 1723 CrossRef CAS PubMed; (t) X. Zhang, X. Liu, X. Li, Y. Wang, R. He, G. Zhong and J. Zhang, Org. Lett., 2025, 27, 4074–4078 CrossRef CAS PubMed; (u) R. Luan, P. Lin, K. Li, Y. Du and W. Su, Nat. Commun., 2024, 15, 1723 CrossRef CAS PubMed.
- (a) M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805–5813 CrossRef CAS PubMed; (b) K. Meng, T. Li, C. Yu, C. Shen, J. Zhang and G. Zhong, Nat. Commun., 2019, 10, 5109–5119 CrossRef PubMed; (c) B. S. Schreib and E. M. Carreira, J. Am. Chem. Soc., 2019, 141, 8758–8763 CrossRef CAS PubMed; (d) B. S. Schreib, M. Fadel and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 7818–7822 CrossRef CAS PubMed; (e) B. Han, B. Li, L. Qi, P. Yang, G. He and G. Chen, Org. Lett., 2020, 22, 6879–6883 CrossRef CAS PubMed; (f) Z. Wu, N. Fatuzzo and G. Dong, J. Am. Chem. Soc., 2020, 142, 2715–2720 CrossRef CAS PubMed; (g) C. Shen, X. Lu, J. Zhang, L. Ding, Y. Sun and G. Zhong, Chem. Commun., 2019, 55, 13582–13585 RSC; (h) Y. Zhu, F. Chen, D. Cheng, Y. Chen, X. Zhao, W. Wei, Y. Lu and J. Zhao, Org. Lett., 2020, 22, 8786–8790 CrossRef CAS PubMed; (i) Y.-C. Wang, Y.-H. Huang, H.-C. Tsai, R. S. Basha and C.-M. Chou, Org. Lett., 2022, 22, 6765–6770 CrossRef PubMed; (j) C. Shen, Y. Zhu, W. Shen, S. Jin, L. Zhong, S. Luo, L. Xu, G. Zhong and J. Zhang, Org. Chem. Front., 2022, 9, 4834–4839 RSC; (k) M. Liu, J. Sun, T. G. Erbay, H.-Q. Ni, R. Martín-Montero, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2022, 61, e202203624 CrossRef CAS PubMed.
- While trans aryl alkenes reacted well, aliphatic (E)-alkenes showed very low reactivity and poor E/Z selectivity, see: Y. Zhu, Y. Wang, W. Shen, X. Chen, Q. Liu, L. Yang and J. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202315273 CrossRef CAS PubMed.
- Hydrofunctionalization of alkenes, see: (a) C. Lee, H.-J. Kang, H. Seo and S. Hong, J. Am. Chem. Soc., 2022, 144, 9091–9100 CrossRef CAS PubMed; (b) J. Jeon, H. Ryu, C. Lee, D. Cho, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 10048–10059 CrossRef CAS PubMed; (c) J. A. Gurak, J. K. S. Yang, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 5805–5808 CrossRef CAS PubMed.
- Difunctionalization of alkenes, see: (a) Y. Li, Y. Liang, J. Dong, Y. Deng, C. Zhao, Z. Su, X. Bi, Q. Liu and J. Fu, J. Am. Chem. Soc., 2019, 141, 18475–18485 CrossRef CAS PubMed; (b) T. Kang, N. Kim, P. T. Cheng, H. Zhang, K. Foo and K. M. Engle, J. Am. Chem. Soc., 2021, 143, 13962–13970 CrossRef CAS PubMed; (c) T. Kang, J. M. González, Z.-Q. Li, K. F. Peter, T. W. Cheng and K. M. Engle, ACS Catal., 2022, 12, 3890–3896 CrossRef CAS.
- (a) A. M. Romine, K. S. Yang, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2019, 9, 7626–7640 CrossRef CAS PubMed; (b) H. Lv, H. Kang, B. Zhou, X. Xue, K. M. Engle and D. Zhao, Nat. Commun., 2019, 10, 5025 CrossRef PubMed.
- There is one example of alkenyl C–H functionalization of aliphatic E-alkene, a tandem reaction proceeded by E/Z isomerization and C–H alkenylation using (E)-hex-4-enamide, which is actually a conversion of Z-alkene (ref. 7a) (eqn (1)). Aliphatic E-alkene is also demonstrated to be far less reactive for direct C–H functionalization in this work
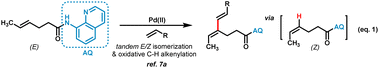 .
. - R. Matsuura, M. K. Karunananda, M. Liu, N. Nguyen, D. G. Blackmond and K. M. Engle, ACS Catal., 2021, 11, 4239–4246 CrossRef CAS PubMed.
- It is proposed that electron-withdrawing groups such as ester on dienes 3 remarkably decrease the reactivity of the adjacent olefinic C–H bond, and the olefinic C–H bond farther away is electronically richer and easier to be C–H functionalized (Scheme 3). These results are in accordance with previous reports on conversion of aryl alkenes, also see ref. 8.
- Herein, transition state TS4′ of the olefinic C–H activation via a six-membered palladacycle was 3.5 kcal mol−1 higher than the transition state TS4 of the olefinic C–H activation by a seven-membered palladacycle (Scheme 5H), and the C–H activation step may be rate-determining which dominates the selectivity in oxidative alkenylation and alkynylation reactions in Scheme 4B. In Scheme 4a, however, the C–H activation step should not be rate-determining and alkyne insertion of the palladacycle intermediate dominates the reaction selectivity to give 8b and 8c (alkyne insertion of a six-membered palladacycle is energetically more favorable than that of a seven-membered palladacycle).
- For substrate 3a, 13% deuterium incorporation was observed under Cond. B with E/Z isomerization (Z/E = 64:36) (98% recovery). For substrate 7b (Z/E = 82:18), 32% deuterium incorporation occurred at the olefinic C–Ha bond and 9% deuterium incorporation occurred at the olefinic C–Hb bond with E/Z isomerization (Z/E = 79:21) (67% recovery) (see the SI for details).
- These primary results exhibited electronic effects such as conjugation of the aromatic ring that may promote the olefinic C–H activation via larger-sized cyclopalladation (also see competition experiments in Scheme 5A, 1avs.13), and the detailed mechanistic studies will be discussed in a later report.
- (a) CCDC:2419583: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m6s2q; (b) CCDC:2419584: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m6s3r; (c) CCDC:2419699: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m6wtk; (d) CCDC: 2419743: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m6y71.

| This journal is © The Royal Society of Chemistry 2025 |