
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the reversed proton relay mechanism: dual regulatory role of pendant carboxylates relevant to water oxidation
Taolue Liua,
Yu Weia,
Guojie Gaoa,
Mingxia Guoa,
Jinxuan Liu a,
Xin Ding*c,
Yingying Li*b and
Yan Gao*a
a,
Xin Ding*c,
Yingying Li*b and
Yan Gao*a
aDalian University of Technology, State Key Laboratory of Fine Chemicals, Dalian 116024, China. E-mail: dr.gaoyan@dlut.edu.cn
bZhengzhou Normal University, School of Chemistry and Chemical Engineering, Zhengzhou 450044, China. E-mail: liyingying@zznu.edu.cn
cQingdao University, College of Chemistry and Chemical Engineering, Qingdao 266071, China. E-mail: dingxin@qdu.edu.cn
First published on 30th October 2025
Abstract
To elucidate the “reversed proton relay” mechanism in mediating water oxidation under neutral conditions, we designed two Ru(II) complexes: [RuII(tda-κ-N3O)(isoq)2] (Ru1), featuring a coordination-oversaturated pendant carboxylate, and [RuII(tpc-κ-N3O)(isoq)2][PF6] (Ru1′), lacking this structural motif. In Ru1, coordination oversaturation renders the carboxylate dynamically accessible, enabling it to function as a proton switch that extracts protons from the metal center. Density functional theory (DFT) calculations, along with comparative experimental data, confirm that the carboxylate facilitates catalytic switching: in its deprotonated state, it stabilizes key intermediates and promotes O2 evolution via reversed proton transfer. Online high-resolution mass spectrometry (HRMS) further identifies key intermediates, providing direct experimental evidence for this unconventional proton relay pathway. Collectively, these findings uncover a previously unrecognized mode of proton transfer and offer valuable mechanistic insights for the rational design of efficient water oxidation catalysts under neutral conditions.
Introduction
The increasing global demand for sustainable energy has made the development of new energy storage forms a crucial research topic, with the conversion of solar energy into chemical energy being a central focus of sustainable energy projects.1–3 Solar fuel conversions produce a wide variety of products, from simple molecules like hydrogen (H2) and ammonia (NH3) to complex substances such as hydrocarbons, alcohols, and acids.4–7 The design of such conversion processes—particularly those for producing chemical fuels using solar energy—often draws inspiration from the principles of natural photosynthesis, with the core mechanism being the 4e−/4H+ water oxidation half-reaction driven by Photosystem II (PSII): this reaction efficiently facilitates energy transfer and electron transfer processes, providing a key energy foundation for subsequent carbon fixation. In nature, this reaction is catalyzed by the Mn4CaO5 cluster in PSII, with an overpotential of approximately 160 mV and a reaction rate of 100–400 s−1.8–11 However, its catalytic performance deteriorates significantly when removed from the natural environment, highlighting the urgent need for efficient artificial photosynthetic catalysts.The proton-electron coupled transfer mechanism observed in natural photosynthesis offers valuable guidance for the design of artificial catalysts. In the hydrogen evolution reaction (HER), the “proton relay” effect improves hydrogen production efficiency by regulating the local proton concentration near the metal center. Functional groups such as amino and carboxylic acids facilitate proton delivery, thereby promoting H–H bond formation.12–17 In the case of the oxygen evolution reaction (OER), pendant groups are often introduced to enhance catalytic performance. However, most studies have focused on empirical correlations between pendant group structures and catalytic activity, while overlooking the fact that these modifications represent a proton relay mechanism in reverse: a directional shift from proton donation (as in HER) to proton extraction, tailored to meet the kinetic requirements of water oxidation.18–27 This oversight highlights a fundamental design dilemma in early research: excessive reliance on saturated coordination environments to stabilize the metal center, while improving catalyst stability, can impede the approach of H2O to the metal site. On the other hand, it also affects the rate of directional transfer of protons (H+) from the metal center to the solvent, which is a key factor limiting the efficiency of many catalytic systems. The metal center's coordination environment regulates proton-transfer capability (by providing open sites or hindering migration), thereby determining catalytic efficiency (Scheme 1a). For example, pendant ligands create additional proton migration channels (LEqM + H2O → LEq(OH)–MH → LEq(OH)–M + H+; LEq(OH)–M → LEq(O)–MH → LEq(O)–M + H+; LEq = equatorial ligand), similar to their role in H2-splitting (LEqM + H2 → LEq(H)–MH), where both pathways optimize the transport efficiency of reaction intermediates by modulating the coordination environment.
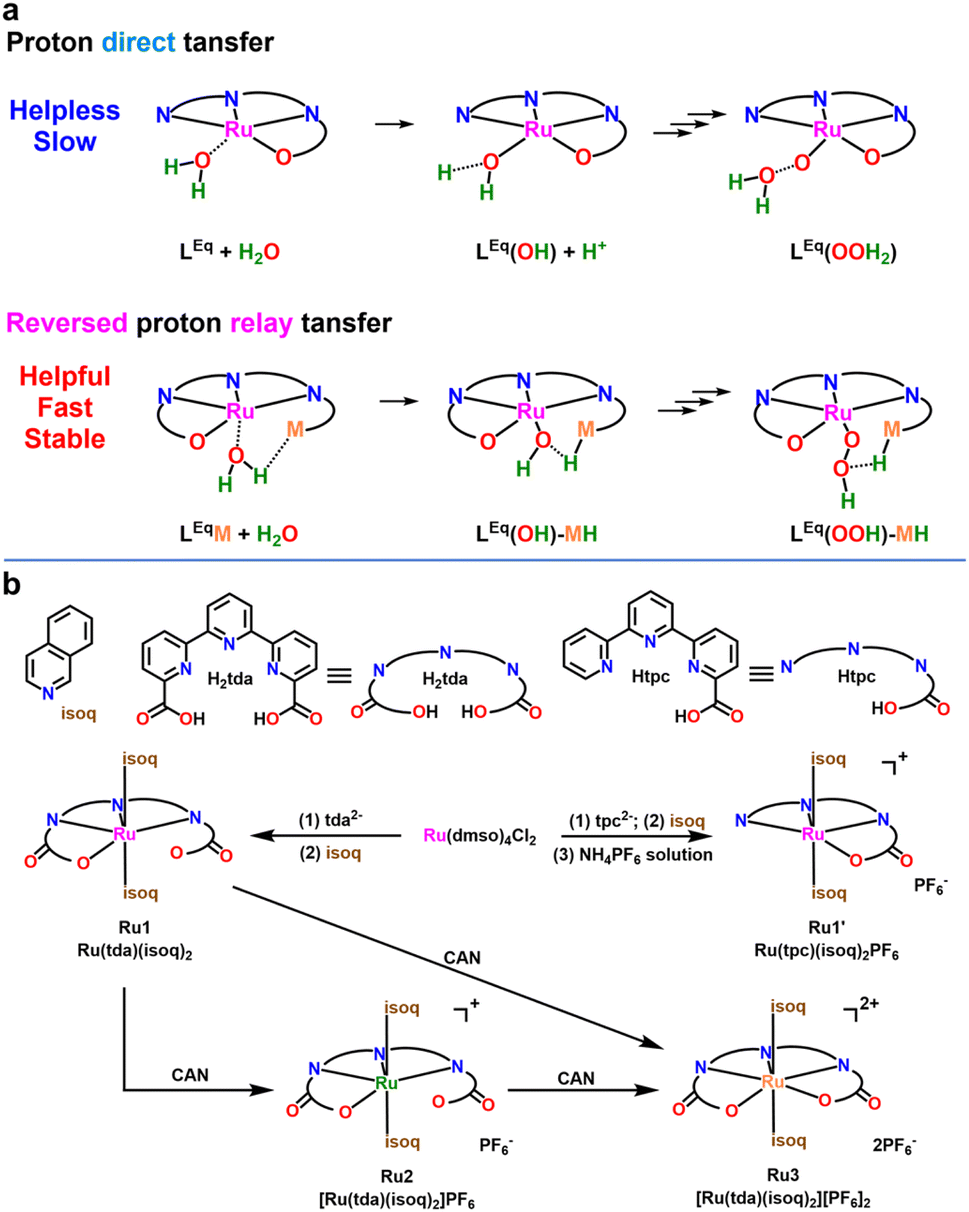 | ||
| Scheme 1 (a) Direct proton transfer in the absence of reversed proton relay and efficient proton transfer via reversed proton relay mechanism.a (b) Synthetic routes of complexes. aSchematic illustration. Actual conditions may vary. Axial ligands are omitted for clarity. | ||
Herein, we propose a “reversed proton relay” mechanism, wherein modulation of ligand field electron density enables efficient proton abstraction without disrupting a fully coordinated metal center. To verify this, two Ru(II) complexes were designed: Ru(tda)(isoq)2 (Ru1), with a pendant carboxylate group formed via coordination oversaturation, and Ru(tpc)(isoq)2PF6 (Ru1′), with saturated coordination (Scheme 1b). Experimental and computational results showed that the carboxylate in Ru1 functions as a bifunctional switch, facilitating proton transfer and modulating catalytic activity. Under neutral/alkaline conditions, this group mediates reversed proton transfer through hydrogen-bonded water networks, establishing an optimal reaction microenvironment to drive Ru oxidation state evolution. This process enables efficient water oxidation with significantly enhanced catalytic performance compared to the saturated-coordination Ru1′. More critically, online intermediate capture experiments (Fig. 4) visually elucidate the directional proton transfer pathway from the carboxylate group to the Ru center. This experimental visualization not only directly validates the theoretical predictions of the “reversed proton relay” mechanism but also provides a foundational basis for understanding the catalytic performance data. This study revises the traditional catalyst design that excessively pursues saturated coordination, and establishes a new design strategy for neutral water oxidation catalysts through the “reversed proton relay” mechanism of oversaturated coordination.
Experimental
Materials
All solvents were purified and dried by standard methods prior to use. Commercial reagents (Shanghai Aladdin) were used without further purification, except for trimethylsilyl cyanide (Anhui Energy Chemical). All organometallic reactions were performed using a Schlenk line under strictly oxygen-free conditions, with a continuous argon flow maintained throughout the process and a gas outlet sealed by a glycerol trap to prevent air ingress. Key reagents including Ru(dmso)4Cl2, 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylic acid (H2tda), and 2,2′:6′,2′′-terpyridine-6-carboxylic acid (Htpc), were synthesized and purified according to literature procedures.18,28–30 High-purity deionized water was prepared via Milli-Q nanopure filtration. 1H-NMR and 13C-NMR spectra were acquired on Bruker Avance II 400 or III 500 spectrometers. MS analysis was performed at Dalian University of Technology using a Micromass Q-TOF Micro and Bruker MALDI-TOF spectrometer. More details are available in the SI.Preparation of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 727), 326 (37455), 377 (15727) and 530 (5591).136), 311 (36182), 381 (15818).
727), 326 (37455), 377 (15727) and 530 (5591).136), 311 (36182), 381 (15818).Characterization and analysis methods
Mass spectrometry (MS) analyses were conducted using electrospray ionization (ESI) on three instruments: a Micromass UK Limited Q-TOF Micro, a Thermo Scientific LTQ Orbitrap XL, and a Bruker MALDI-TOF spectrometer. UV-vis absorption spectra were recorded on a Shimadzu UV-1900 spectrophotometer (Japan) with 1 cm quartz cuvettes. NMR spectra were acquired using Bruker Avance II 400 or Avance III 500 spectrometers. Solution pH values were measured by a calibrated Shanghai Leici PHS-3C pH meter, using standard solutions at pH 4.00, 6.86, and 9.18 for calibration prior to analysis. More details are available in the SI.Single crystals of Ru1 and Ru1′ were obtained by dissolving the complexes in ethanol-based solvent systems (with trifluoroethanol or water), placing the solutions in sealed flasks containing diethyl ether for vapor diffusion crystallization, and isolating dark red crystals after slow evaporation. Crystal structures were determined by mounting the crystals on a Bruker D8 Venture single-crystal diffractometer, collecting diffraction data with Cu or Mo target, solving the structures using Olex2, and refining with SHELXL.31
Electrochemical experiments were performed on a CHI 630E workstation (Shanghai Chenhua) using a three-electrode system. Glassy carbon disk electrodes (Φ = 2 mm) served as the working electrode, platinum wires (Φ = 1 mm) as the counter electrode, and pH-dependent reference electrodes were employed: Hg/Hg2SO4 (saturated K2SO4) for acidic, Ag/AgCl (saturated KCl) for neutral, Hg/HgO (0.1 M KOH) for basic conditions, and all referenced to NHE. Reference electrodes were calibrated with Ru(bpy)32+ (RuIII/RuII = 1.26 V vs. NHE). Cyclic voltammetry (CV), differential pulse voltammetry (DPV), and coulometry were applied to analyze redox behaviors across pH ranges.
The oxygen evolution was monitored by a NEOFOX-KIT-PATCH oxygen sensor kit (Ocean Optics, USA), and the catalytic water oxidation reaction was driven by ceric ammonium nitrate (CAN, CeIV) as the oxidant.
Mass spectrometry measurements to capture water oxidation intermediates were performed using Q-TOF, LTQ Orbitrap XL, and MALDI-TOF spectrometers. The reaction solution, prepared by dissolving the catalyst in water containing 27% CF3CH2OH (1.1 mM), was mixed with a certain amount of CeIV aqueous solution under stirring. After thorough mixing, the reaction solution was immediately transferred to a HRMS for analysis. The pH meter indicated that the pH of the reaction is 5.7 approximately after mixing with 4 eq CeIV.
DFT calculations
DFT calculations were performed with the Gaussian 16 program to elucidate the mechanism of the Ru-catalyzed water oxidation.32 All calculations were carried out using the B3LYP-D3 functional.33,34 The 6-31G(d,p) basis set was employed for non-metal atoms, and the Stuttgart-Dresden effective core potential (SDD) was used for the Ru atom.35 Analytical frequency calculations were performed at the same level as the geometry optimization to verify the local minima nature of various stationary points (no imaginary frequency) and saddle-point property of the transition state (only one imaginary frequency). The final energies were from single-point calculations employing the SMD implicit solvation model.36 All non-metal elements were depicted by 6–311 + G(2df,2p). The value of −6.3 kcal mol−1 of solvation energy of water in aqueous solution from the experiment was used.37 All species were corrected with a concentration-dependent energy term (1.9 kcal mol−1), with an additional 4.3 kcal mol−1 adjustment specifically for water. Protonation states were determined via pKa calculations for all plausible intermediates. The solvation energy of a proton was taken from the experiment (−264.0 kcal mol−1). Redox potentials were referenced to the standard hydrogen electrode (SHE, 4.281 V).Results and discussion
Catalyst synthesis and structural characterization
We designed two catalysts—[RuII(tda-κ-N3O)(isoq)2] (Ru1) and [RuII(tpc-κ-N3O)(isoq)2] (Ru1′)—to systematically investigate the dual role of the reversed relay group (Scheme 1b): (1) as a molecular switch regulating catalytic activity, and (2) as a dynamic reversed proton relay modulating the catalytic microenvironment.38,39 The ligands H2tda and Htpc were synthesized from 2,2′:6′,2′′-terpyridine (trpy) via three steps (Fig. S1–S6).28–30 Ru(dmso)4Cl2 first reacted with H2tda, followed by isoquinoline (isoq), to yield the red-wine complex Ru1 in 82.7% yield. Ru1' was synthesized similarly in 52.7% yield (Fig. S7–S14). X-ray crystallography confirmed both of complexes are low-spin d6 Ru(II) distorted octahedral structure (Fig. 1a, b, S15, S16, and Table S1).20,22,40 | ||
| Fig. 1 (a) X-ray ORTEP plots of [RuII(tda-κ-N3O)(isoq)2], Ru1. (b) X-ray ORTEP plots of [RuII(tpc-κ-N3O)(isoq)2]+, Ru1′. Ellipsoids are plotted at 50% probability. Color codes: Ru, purple; N, blue; O, red; C, gray. H are not shown. (c) Variable temperature 1H-NMR spectra of Ru(tda)(isoq)2 in CD3OD. | ||
This structural feature of Ru1 shows consistency with the previously reported Ru(tda)(py)2 catalyst in terms of crystal structure: the carbon skeletons of the tda2− ligands in the equatorial planes of both catalysts exhibit similar bond angles and bond lengths.18 However, notable differences are observed in the ligand – related carboxylate groups (COO−). The C–O bond length of the carboxylate group shortens from 1.284 Å in the pyridine-containing system to 1.132 Å in the isoquinoline-containing system, accompanied by an elongation of the Ru–O bond length at the metal center (from 2.198 Å (py) to 2.208 Å (isoq)). In the axial direction, the introduction of isoq leads to a significant increase in the Ru–N bond length (from 2.083 Å (py) to 2.093 Å (isoq)). This phenomenon suggests that when the axial ligand is switched from pyridine to isoquinoline, the resulting electronic effects may alter the state of the relay ligand of the catalyst in the solution microenvironment. Subsequent proton-coupled electron transfer (PCET) steps in the Pourbaix diagram and relative energies for Ru1 species in SI have repeatedly demonstrated that the relay ligand undergoes protonation in acidic solutions.
Furthermore, the intrinsic fast low-barrier dynamic exchange process of the pendant relay ligand in Ru1 enables it to stably function as a molecular switch. Specifically, the crystal structure of Ru1 reveals a significantly asymmetric feature: the O–Ru bond length is only 2.208 Å, while the adjacent non-coordinated O atom is 3.689 Å away from the Ru center (Fig. 1a and b). For an asymmetric complex like Ru1′, such structural disparities would typically manifest as asymmetric peak distributions in the 1H-NMR spectrum. However, Ru1 exhibits highly symmetric characteristic peaks in 1H-NMR. This proves that it is a highly stable, “resonance-like” structure. Variable temperature 1H-NMR experiments on Ru1 (198 K, CD3OD, Fig. 1c) further confirm its structural stability. Even at low temperatures, this complex maintains highly symmetric 1H-NMR features without any resonance peak splitting caused by slowed proton transfer kinetics in the side chains. For example, the overlapping hydrogen signals (7.5–8.0 ppm) at room temperature resolve into well-defined peaks upon cooling: the 7.95 ppm multiplet is assigned to hydrogens on the tda2− ligand (C1/C2) and the isoquinoline (C12), while the 7.70 ppm and 7.60 ppm multiplets dissociate into triplets and doublets at low temperatures, corresponding to hydrogens on the isoquinoline (C9/C7 and C8/C10), respectively. These findings conclusively establish that the two COO–Ru groups on the transverse ligands of Ru1 in solution maintain identical chemical states due to a rapid dynamic exchange process with an extremely low energy barrier, which is the fundamental mechanism underlying the observed symmetric 1H NMR spectrum.20,22 While this dynamic behavior inhibits stable water coordination in solution, it preserves robust metal-ligand integrity and ensures reversible switching essential for catalytic modulation.
In contrast, Ru1′ lacks this pendant group entirely. This key structural difference dictates their catalytic behavior: the free carboxylate in Ru1 forms hydrogen bonds with water, stabilizing protons and enhancing oxygen evolution. This structural difference underpins the distinct catalytic mechanisms of reversed proton relay: Ru1 can modulate Ru-Aqua species formation by regulating the carboxylate's electron density. Specifically, the deactivation of reversed proton relay (protonating COO−) not only disrupts hydrogen-bond network formation but also disables the mechanism entirely. This precise control over proton transfer pathways highlights the unique advantage when other pathways must compete with water oxidation. The ability to toggle the carboxylate's protonation state thus provides a powerful tool for optimizing catalytic efficiency in complex reaction environments.
Reversed proton relay effects control active species generation
A spectrophotometric redox titration experiment was designed to investigate the effect of protonation on the ligand's electronic density in the catalyst. Protonation of the carboxylate group was hypothesized to reduce Ru-Aqua formation (i.e., “switching off” the catalytic activity) and experimentally validated. During the redox titration under acidic conditions, incremental additions of the corresponding oxidant equivalents were performed, and UV-vis spectral changes were monitored (Fig. 2a and b). The stabilized solution spectra show strong alignment with the Ru2/Ru3 reference spectra independently prepared by introducing an equivalent CAN in Ru1 ethanol solution and isolating the products via saturated KPF6 precipitation (Scheme 1b and Fig. S17). Absorption peaks at 250–350 nm were attributed to π–π* transitions of the catalyst ligands, while the weaker visible-region absorption were identified as metal-to-ligand charge transfer (MLCT), Ru-d → bda-π*. Notably, under one catalytic cycle, no spectral evidence of oxidation states higher than RuIV (Ru3) was observed. At a Ce(IV) equivalent of 1.06, absorption peaks at 377 nm and 530 nm disappeared, with low absorbance remaining stable in subsequent titrations. The absorbance at 326 nm slowly decreased with modest change, while a sharp 270 nm peak appeared. Compared to UV-vis spectra of Ru3, when 2 eq. of CAN was added, the 326 nm oxidation absorption of Ru2 was not clearly distinguishable, likely due to insufficient oxidation time. This variation exhibits a significant difference compared to the previously reported Ru(tda)(py)2 system: after the RuII → RuIV oxidation occurs, RuIV(isoq) does not show a prominent peak-valley at 318 nm as observed in RuIV(py), but instead presents an observable concave feature. As the titration progressed, the 270 nm absorbance increased while absorbance beyond 300 nm decreased and stabilized. After adding oxidants for one catalytic cycle, the catalyst solution's UV-vis characteristics were monitored for 50 minutes and remained stable, showing no decay toward Ru2 and Ru3 trends. Under neutral conditions, Ru3 solution (RuIV) exhibited significant attenuation, which is attributed to the catalyst's redox potential exceeding the theoretical water oxidation potential leading to a disproportionation reaction in this chemical environment (Fig. 2c). However, its dissolution rate is slower than that of RuIV(tda)(py)2 catalyst, indicating that the introduction of the isoquinoline ligand modifies the aqueous microenvironment, thereby slowing the attack rate of water molecules on the relay-type catalyst and consequently delaying the formation of the Ru-Aqua active species during water oxidation.18 This characteristic effectively accounts for the absence of distinct new peaks attributable to the Ru-Aqua active species in subsequent repetitive cyclic voltammetry (RCV) scans (Fig. 2d), but concurrently enhancing the feasibility of online detection for reaction intermediates at various stages. | ||
| Fig. 2 (a) UV-vis spectra of complexes Ru1 (blue), Ru1′ (red). (b) UV-vis spectra of Ru1 (22 μM) (blue) in 0.1 M triflic acid, and successive additions of 0.152 equiv. of CeIV (1 mM) (grey) up to 2 equiv. Inset: to 4 equiv. (c) Stability test of Ru3, (37.5 μM) in pH 7. Inset: plot of absorbance vs. time for λ = 270 and 318 nm at pH 1.0 (black) and pH 7.0 (red). (d) RCV (v = 30 mV s−1) of 1.1 mM of Ru1 in pH 7 (27% CF3CH2OH). (e) Pourbaix diagram for the Ru species derived from Ru1. The detailed species states are simplified and represented solely by the oxidation state of Ru, the charge number, and the ligand status. (f) Plot of the icat – [cat] at 1.80 V in pH 7 (27% CF3CH2OH). (g) Comparison of long-term oxygen production capabilities for Ru1 and Ru1′. (h) Oxygen evolution curve at various concentrations of Ru1′. (i) Oxygen evolution curve of electrolysis (background-free). Inset point: theoretical O2 value calculated from the total charge quantity at this time. | ||
Reversed proton relay effects improve electrochemical performance
Redox titration indicates that Ru3 (oxidized from Ru1) undergoes neutral disproportionation in the absence of external oxidation, yet this does not imply a loss of catalytic activity. When the driving force is sufficient (RCV and electrolysis), Ru1 exhibits both greater stability and higher activity compared to Ru1′ (Fig. 2d and S20). Compared to Ru1 with an additional reverse proton transfer group, Ru1′ lacking this group exhibits catalytic center deactivation during cyclic water oxidation, accompanied by substantial electron loss. These findings confirm that the introduction of a pendant reversed proton relay enhances the stability of the catalyst during usage and extends its lifespan. Similarly, the stabilizing effect on the catalytic microenvironment through such characteristics has been reported in studies of analogous catalysts.20,22,41,42 CVs at different scan rates reveals that the surface redox processes of the Ru1 and Ru1' electrodes are diffusion-controlled according to the Randles–Sevcik equation (Fig. S18 and S19). The shift of Ru1 redox peak in CV from stable 0.51 V under neutral and alkaline conditions to 0.60 V under acidic conditions is because the carboxyl reversed proton transfer group on the equatorial ligand in the 0.1 M triflic acid solution undergoes protonation as previously discussed. The increase in current between 1.4 and 1.7 V in high pH is attributed to the catalytic oxidation of water by the active Ru-Aqua species produced by carboxylate reversed proton transport. Obviously, electrochemical tests confirm Ru1 exhibits higher OER performance in neutral conditions (Fig. S20) than Ru1′ (no regulation) or in acidic conditions (suppressed carboxylate). After repetitive cyclic voltammetry (RCV), Ru1 showed a significant increase in catalytic current under neutral Na2SO4 conditions, while the currents in acid solutions decreased (Fig. 2d). Combined with DPV and spectrophotometric redox titration results, these potential transitions are associated with the redox processes of RuII → RuIII + e− and RuIII → RuIV + e−. The Pourbaix diagram analysis also confirms that the redox behavior of Ru1 is pH-independent across most pH ranges (Fig. 2e). Notably, when the pH decreases, the single-electron transfer process (RuIII/RuII) transitions to a distinctly different PCET process (RuII–H → RuIII + H+ + e−). The pH-independent RuIII/RuIV step in Ru1 (Ru(tda)(isoq)2) exhibits a significantly lowered potential, decreasing from 1.10 V (Ru(tda)(py)2) to 1.04 V vs. NHE. This indicates that the Ru1 exhibits enhanced reducibility in its RuIV state. However, this characteristic renders the key reaction pathway, which relies on RuIV undergoing nucleophilic attack to form the RuIV–OH active species, thermodynamically unfavorable. This disadvantage consequently impedes the formation of active Ru-hydrate species. Specifically, under neutral conditions, the decay rate of the corresponding UV characteristic peak during the oxygen evolution process via high-valent disproportionation in Ru(tda)(py)2 is significantly faster than that in the corresponding process of Ru(tda)(isoq)2.18 Based on the DFT calculation data regarding the relative energies for Ru1 species from the SI, the protonation process of this catalyst exhibits a low energy barrier under acidic conditions (Fig. S31). Specifically, the pKa value of 6.14 for RuII–H effectively substantiates that isoquinoline (isoq) as an axial ligand induces a spatial effect distinctly different from that of pyridine. This spatial effect difference further modulates the microenvironment of the equatorial active site, thereby facilitating the formation of optimized PCET steps within the pH range of 1–4. Ru1 is highly stable in acidic aqueous solutions, making the formation of hydrated Ru-Aqua species difficult. On the other hand, Ru1′ without COO− shows greater stability in acidic environments, consistent with the preferences of the vast majority of catalysts. Consecutive detection of various oxidation states of RuII, such as RuII → RuIII and RuII–H2O → RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, was also observed in different pH (Fig. S21).43
O, was also observed in different pH (Fig. S21).43
Electrochemical analysis also confirms the oxygen evolution for two catalysts originated from a water nucleophilic attack (WNA): the icat of 1.8 V (NHE) in different pH linearly related to [cat] of two complexes and independent of scan rate (Fig. 2f, S22–S24 and Table S2).44 The corresponding experimental conditions and electrochemical methods are also suppled in SI. Ru1 exhibits higher TOF (0.0263 s−1) under acidic conditions than Ru1′ (0.0092 s−1) in electrochemical evaluations. Given the activation energy demand associated with the relay-type proton relay mechanism, this characteristic makes it essential to provide activation time for the catalysts and conduct synchronous online oxygen evolution monitoring. The macroscopic water oxidation performance evaluation results obtained in this way have more reliable reference value compared with the electrochemical activity data calculated from a single cyclic voltammetry test.
Reversed proton relay enables superior oxygen evolution
Electrochemical studies and spectroscopic evidence show that the reversed proton relay (the COO−) can enhance catalytic activity and stability under neutral conditions by optimizing the metal microenvironment during water oxidation. To validate this capacity of facilitating PCET through intermediate stabilization via hydrogen bonding, real time O2 concentration monitoring was performed in neutral via electrolysis tests. Ru1 achieved 18.16 μmol oxygen within the first 30 minutes, delivering an oxygen production 1.5 times that of Ru1′ (12.27 μmol), thereby confirming the critical role of the reversed proton relay mechanism in neutral water oxidation. On the other hand, as expected, the carboxyl group's protonation under acidic conditions disables its reversed proton transfer capacity, resulting in negligible oxygen evolution of Ru1 over 4 hours in triflic acid. In contrast, Ru1′, which exhibited favorable performance in acid solution tests, demonstrated a linear relationship between oxygen evolution rate and catalyst concentration, eventually reaching saturation over time. These macroscopic oxygen evolution experiments corroborate that the carboxylate molecular switch significantly enhances water oxidation reaction activity in low [H+] concentration.A four-hour study using cerium ammonium nitrate (Ce(IV)) as the oxidant in acidic homogeneous aqueous solutions and calculating the Turnover Number (TON, nO2/ncat) was conducted. Ru1′ achieved a TON of 934.3 after four hours, while Ru1 showed negligible activity (TON = 10.4, Fig. 2g). This stark contrast highlights the strong pH-dependence of reversed proton relay. Under strong acid conditions, despite Ce(IV)'s high oxidative capability, Ru1 fails to drive the water oxidation reaction effectively. The oxygen production level of Ru1 remains around the origin regardless of the catalyst content. In contrast, Ru1′ exhibits a strong response with oxygen production rates far outperforming Ru1. The linear relationship between oxygen production rate and catalyst concentration yields a high TOF = 0.39 s−1 for Ru1′, confirming its superior catalytic performance in acidic conditions (Fig. 2h, S25 and S26). The poor catalytic activity of reversed proton relay catalyat Ru1 in acidic solutions due to failure to form effective Ru-Aqua intermediates, while Ru1′ is highly active in acidic solutions. In contrast, the catalytic behavior reverses in neutral 0.1 M sodium sulfate solutions. When electrolyzing the catalysts on composite electrodes (CP) at 1.7 V vs. NHE for one hour, Ru1′ initially shows a higher current, but after subtracting the oxygen evolution from the blank carbon paper, Ru1 produces more oxygen overall in the initial stage (Fig. 2i, S27A and B). After 1.5 hours, Ru1 reaches 34.6 μmol oxygen, while Ru1′ peaks at 39.7 μmol. But Ru1 achieves a Faradaic efficiency of 90.53%, compared to 69.27% for Ru1′. This once again demonstrates the catalytic behavior of “reversed proton relay” varies with pH inseparably (Fig. 3a). Notably, Ru1′s stability under neutral conditions was further investigated by immersing a used composite electrode in CF3CH2OH for two weeks. UV-vis spectra confirmed the formation of divalent Ru1, indicating its stable state after catalysis termination is RuII state (Fig. S27C and D).
 | ||
| Fig. 3 (a) The water oxidation catalytic pathway of reversed proton relays. (b) Gibbs energy diagram of water oxidation catalyzed by 2. Axial ligands are omitted for clarity. | ||
Calculation of catalytic pathways and capture of intermediates
The properties and experimental results indicate that that the reversed proton relay operate through distinct mechanistic pathways depending on pH conditions. Further research from the perspective of energy using online experiments and computational modeling is needed to fully understand its mechanisms when reversed proton relay activated. DFT calculations reveal that Ru1 follows a WNA mechanism, where electron loss occurs on the peroxide's outer oxygen while Ru remains in the RuIII state (in Fig. 3b).32–37 It plays a crucial role uncovering the catalytic mechanism of Ru1 with free carboxylate group during water oxidation. Online high-resolution mass spectrometry (HRMS) validates mechanism by detecting key intermediates in different oxidation states.Although water coordination to Ru1 forming RuII–OH2 (1) is endergonic (ΔG = +8.7 kcal mol−1, Fig. S28), the active cycle initiates from the RuIII–OH2 (2) state, not low-valent RuII. The UV-vis spectra collected after the oxygen evolution electrolysis experiment confirmed that the RuII state merely represents the stabilized form of the catalyst in its inactive state. For RuIII–OH2 (2), a proton transfers to the carboxylate group with a single electron oxidation, with a calculated redox potential of 0.24 V. A PCET process (E° = 1.08 V) from 2 forms RuIV–OH (3, Ru–O1 = 1.85 Å) through a thermodynamically favorable process (ΔG = −14.8 kcal mol−1), which then forms RuV = O (4) through another PCET step (E° = 1.34 V; ΔG = −8.8 kcal mol−1) in Fig. S29. RuV = O triggers O–O bond formation via nucleophilic water attack at a transition state TS1 (Ru–O1 = 1.72 Å, O1–O2 distance = 2.28 Å, ΔG* = 15.7 kcal mol−1) that collapses into hydroperoxide Int1 (RuIII–OOH) and superoxide Int2 (RuIII–OO˙) intermediates before final O2 release (Fig. S30). Then closing the catalytic cycle and involving the combination of the second water with the Ru center to form RuIII–OH2 (2). Compared to Ru-Aqua, the direct oxidation of Ru1 to Ru2 has a potential of 0.16 V, the calculated redox potential from Ru2 to Ru3 is only 0.78 V (Fig. S31). Previous experiments indicate that Ru1 oxidation to Ru3 serves as a prerequisite step for generating the active Ru-Aqua species prior to catalytic cycle initiation. Thus, catalytic pathway of reversed proton relay Ru1 involves single-electron oxidation of Ru1 to Ru3, water attack to form 3, and entry into the Ru-Aqua O2 evolution cycle, solely cycling between RuIII/IV/V-Aqua states. Real-time mass spectrometry reveals sequential intermediates of Ru1 in water oxidation (Fig. 4), validating DFT predictions. Initially, Ru1 [RuII + Na]+ appears at m/z = 702.32 (Q-TOF, calcd. 702.0691). After the first Ce(IV) addition, Ru2 [RuIII]+ emerges (m/z = 679.33 [Q-TOF], 679.0796 [LTQ XL], calcd. 679.0803), confirmed as RuIIIPF6 (m/z = 824.0434, MALDI, calcd. 824.0417). A second Ce(IV) equivalent produces [RuIV]2+ (m/z = 339.5412, LTQ, calcd. 339.5931). Under reduced acidity, four Ce(IV) additions yield [RuIV–OH]+ (m/z = 696.0809, LTQ, calcd. 696.0821) via hydroxide attack. And PCET forms [RuV = O]+ (m/z = 695.0772, LTQ, calcd. 695.0743) a high-valence ruthenium intermediate with a seven-coordination geometry. Water attack on [RuV = O]+ generates Int1 [RuIII–OOH]+ (m/z = 713.0891, LTQ, calcd. 713.0843), which evolves into Int2 [RuIII–OO˙+ H]+ (m/z = 712.0853, LTQ, calcd. 712.0770) through proton loss. Final oxidation forms transient Int3, completing the cycle via OO bond cleavage.
 | ||
| Fig. 4 “Reversed Proton Relay” catalytic pathway and intermediate capture. The red potential comes from experiments and the blue potential comes from calculations. | ||
HRMS/MS data confirm all intermediates, elucidating the actively regulatory pathway of reversed proton relay mechanism (COO− as experimental example) in water oxidation. The experimental verification of key intermediates not only validates the DFT theoretical model, but also deepens the understanding of the proton transfer dynamic process during the activation of such molecular catalytic switches. Moreover, intermediates data and the theoretical calculation of pKa for each intermediate from DFT are presented (Fig. S32–S36 and Scheme S1). Furthermore, this study, referencing the research findings of the Ahlquist group, analyzed the feasibility of the “the carboxyl peroxide mechanism” for the catalyst with a microenvironment influenced by spatial effects when the isoquinoline substitutes the pyridine ligand, from an energetic perspective in SI.42,45 The alternative pathway TS1′ involving direct coupling of the oxyl radical with the carboxylate oxygen atom exhibits a low activation barrier of 2.1 kcal mol−1 (Fig. S37). This situation may arise when acid radical salts act as transport groups for reverse proton relay oxygen production. However, this pathway is thermodynamically unfavorable due to the subsequent challenges in C–O bond cleavage and O2 release, and is therefore rejected.
Conclusions
In summary, Ru1 and Ru1′, with carboxylate mediated group as the variable, were designed to validate the “reversed proton relay” mechanism, the core driver of neutral water oxidation. These complexes systematically elucidated the dual role of pendant carboxylate groups, formed via coordination oversaturation, as both a molecular switch and a reversed proton relay in water oxidation catalysis. The tda2− ligand framework forms a six-coordinate Ru(II) complex with a dynamic coordination site, enabling the carboxylate to toggle between protonated, catalytically inhibitory states under acidic conditions and deprotonated, proton-transfer-facilitating states under neutral to alkaline conditions. HRMS and DFT analyses confirmed that this dual regulation optimizes proton transfer and catalytic activity, effectively resolving the trade-off between coordination saturation and dynamic proton transport. This work establishes a new design paradigm that integrates molecular switching and proton relay functionalities for neutral water oxidation catalysts.Author contributions
Y. Gao conceived the idea for this study and designed the research. T. Liu conducted the research. Y. Li and X. Ding helped with the analysis and interpretation of the data. T. Liu wrote the manuscript and all authors assisted with editing, analysis, and interpretation.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures; compounds synthesis and characterization (NMR, HRMS, and crystal data); electrochemical data. See DOI: https://doi.org/10.1039/d5sc07030a.CCDC 2250881 and 2383557 contain the supplementary crystallographic data for this paper.46a,b
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21875030, 22278055, 22088102) and the Fundamental Research Funds for China Central Universities (DUT22LAB608). The authors thank Dr Qinqin Du(DUT) and Dr Rong Zhang (DUT) for supporting in MS measurement.Notes and references
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- P. Chen, Y. Xiao, S. Li, X. Jia, D. Luo, W. Zhang, H. J. Snaith, Q. Gong and R. Zhu, Chem. Rev., 2024, 124, 10623–10700 CrossRef CAS.
- R. M. Evans, B. Siritanaratkul, C. F. Megarity, K. Pandey, T. F. Esterle, S. Badiani and F. A. Armstrong, Chem. Soc. Rev., 2019, 48, 2039–2052 RSC.
- D. He, G. Huang, J. Hu, J. Ding, W. Liu, L. Chen, W. Yan, J. Zhu, S. Zhu, Q. Chen, X. Jiao and Y. Xie, Adv. Energy Mater., 2024, 15, 2402889 CrossRef.
- A. Agosti, Y. Nakibli, L. Amirav and G. Bergamini, Nano Energy, 2020, 70, 104510 CrossRef.
- S. Li, Y. Zhou, X. Fu, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. Enemark-Rasmussen, P. J. Kempen, C. D. Damsgaard, A. Xu, R. Sažinas, J. B. V. Mygind, N. H. Deissler, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nature, 2024, 629, 92–97 CrossRef CAS PubMed.
- N. Vereshchuk, M. Gil-Sepulcre, A. Ghaderian, J. Holub, C. Gimbert-Suriñach and A. Llobet, Chem. Soc. Rev., 2023, 52, 196–211 RSC.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nat., 2011, 473, 55–60 CrossRef CAS.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, ACS Catal., 2019, 9, 10115–10125 CrossRef CAS.
- S. N. Chowdhury, S. Biswas, P. Das, S. Paul and A. N. Biswas, Inorg. Chem., 2020, 59, 14012–14022 CrossRef CAS.
- S. Lin, S. Banerjee, M. T. Fortunato, C. Xue, J. Huang, A. Y. Sokolov and C. Turro, J. Am. Chem. Soc., 2022, 144, 20267–20277 CrossRef CAS PubMed.
- Y. Qiu, D. Ray, L. Yan, X. Li, M. Song, M. H. Engelhard, J. Sun, M.-S. Lee, X. Zhang, M.-T. Nguyen, V.-A. Glezakou, Y. Wang, R. Rousseau and Y. Shao, J. Am. Chem. Soc., 2023, 145, 26016–26027 CrossRef CAS PubMed.
- F. Droghetti, F. Begato, M. Raulin, G. Musiu, G. Licini, M. Natali and C. Zonta, Angew. Chem., Int. Ed., 2024, 63, e202408316 CrossRef CAS.
- M. Haake, B. Reuillard, M. Chavarot-Kerlidou, C. Costentin and V. Artero, Angew. Chem., Int. Ed., 2024, 63, e202413910 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Suriñach, M. Z. Ertem and A. Llobet, J. Am. Chem. Soc., 2020, 142, 5068–5077 CrossRef CAS.
- N. Vereshchuk, J. Holub, M. Gil-Sepulcre, J. Benet-Buchholz and A. Llobet, ACS Catal., 2021, 11, 5240–5247 CrossRef CAS.
- T. Liu, S. Zhan, N. Shen, L. Wang, Z. Szabó, H. Yang, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2023, 145, 11818–11828 CrossRef CAS.
- S. Bhunia, A. Rana, S. Hematian, K. D. Karlin and A. Dey, Inorg. Chem., 2021, 60, 13876–13887 CrossRef CAS.
- Q. Huo, J. Cao, L. Mi, J. Shao, M. Lv, X. Chen, H. Yang, X. Chai, Q. Hu and C. He, J. Mater. Chem. A, 2023, 11, 1335–1342 RSC.
- N. Wang, X.-P. Zhang, J. Han, H. Lei, Q. Zhang, H. Zhang, W. Zhang, U.-P. Apfel and R. Cao, Chin. J. Catal., 2023, 45, 88–94 CrossRef CAS.
- M. Li, T. Zhang, Y. Shi, C. He and C. Duan, Angew. Chem., Int. Ed., 2024, 63, e202406161 CrossRef CAS.
- D. Chen, W. Li, J. Liu and L. Sun, Energy Environ. Sci., 2025, 18, 3120–3128 RSC.
- V.-M. Mukkala, C. Sund, M. Kwiatkowski, P. Pasanen, M. Högberg, J. Kankare and H. Takalo, Helv. Chim. Acta, 1992, 75, 1621–1632 CrossRef CAS.
- C. Galaup, J.-M. Couchet, S. Bedel, P. Tisnès and C. Picard, J. Org. Chem., 2005, 70, 2274–2284 CrossRef CAS PubMed.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204–209 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03., Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- D. Andrae, U. HaiuBermann, M. Dolg, H. Stoll and H. PreuB, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- D. M. Camaioni and C. A. Schwerdtfeger, J. Phys. Chem. A, 2005, 109, 10795–10797 CrossRef CAS.
- T. Liu, CSD Commun., 2024, Deposited Date: 22 March 2023.
- T. Liu, CSD Commun., 2024, Deposited Date: 12 September 2024.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS.
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039–2048 CrossRef CAS.
- S. Zhan, J. A. De Gracia Triviño and M. S. G. Ahlquist, J. Am. Chem. Soc., 2019, 141, 10247–10252 CrossRef CAS.
- T. Fan, L. Duan, P. Huang, H. Chen, Q. Daniel, M. S. G. Ahlquist and L. Sun, ACS Catal., 2017, 7, 2956–2966 CrossRef CAS.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- A. Kundu, S. K. Barman and S. Mandal, Inorg. Chem., 2022, 61, 1426–1437 CrossRef CAS PubMed.
- (a) CCDC 2250881: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2fk72b; (b) CCDC 2383557: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l08yv.
| This journal is © The Royal Society of Chemistry 2025 |