
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of vicinal amino alcohols promoted by fluorine-containing counteranions
Yuchen
Yang
a,
Jia-Long
Wu
a,
Sheng-Ye
Zhang
b,
Xu
Liu
a,
Teng
Sun
a,
Yanan
Zhao
 *b and
Lijia
Wang
*a
*b and
Lijia
Wang
*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Frontiers Science Center of Molecule Intelligent Syntheses, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: ljwang@chem.ecnu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China
First published on 27th November 2025
Abstract
Chiral vicinal amino alcohols are pivotal building blocks in organic synthesis and pharmaceutical research. Herein, we report a chiral copper-catalyzed two-step one-pot strategy for the efficient synthesis of diaryl chiral vicinal amino alcohols, featuring excellent diastereoselectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and enantioselectivity (up to 99.5% ee). A key discovery is the critical role of fluorinated counteranions, which significantly enhance both reactivity and stereocontrol—an underappreciated effect in copper-catalyzed asymmetric reactions. DFT calculations reveal that secondary Cu–F interactions, combined with the spatial confinement of hexafluorophosphate, fine-tune the catalytic chiral environment, enabling precise stereocontrol via modulation of π–π stacking and weak non-covalent interactions. This strategy exhibits broad substrate scope, accommodating diverse aryl, heteroaryl, and functionalized substituents, and allows gram-scale synthesis with facile deprotection to free amino alcohols. The mechanistic insights into counterion effects highlight counterion engineering as a powerful tool for optimizing asymmetric catalysis.
:1 dr) and enantioselectivity (up to 99.5% ee). A key discovery is the critical role of fluorinated counteranions, which significantly enhance both reactivity and stereocontrol—an underappreciated effect in copper-catalyzed asymmetric reactions. DFT calculations reveal that secondary Cu–F interactions, combined with the spatial confinement of hexafluorophosphate, fine-tune the catalytic chiral environment, enabling precise stereocontrol via modulation of π–π stacking and weak non-covalent interactions. This strategy exhibits broad substrate scope, accommodating diverse aryl, heteroaryl, and functionalized substituents, and allows gram-scale synthesis with facile deprotection to free amino alcohols. The mechanistic insights into counterion effects highlight counterion engineering as a powerful tool for optimizing asymmetric catalysis.
Introduction
Chiral vicinal amino alcohols, endowed with unique structural and chiral features, occupy an irreplaceable pivotal position in organic synthesis, pharmaceutical research, chirotechnology, and materials science as key functional molecules bridging chemistry, biology, and materials science.1 Since Kumada and co-workers first reported the asymmetric hydrogenation of amino ketones in 1979,2 researchers have successively developed a variety of elaborate asymmetric catalytic strategies for the synthesis of chiral vicinal amino alcohols.3 Among these, Sharpless asymmetric aminohydroxylation stands out as a landmark contribution, as it enables the direct, highly enantioselective construction of vicinal amino alcohol moieties from simple alkenes, revolutionizing the efficient and stereocontrolled synthesis of this important class of compounds.4 However, the synthesis of chiral vicinal amino alcohols is constrained by several factors, including the scope of applicable substrates, functional group compatibility, and the simplicity of reaction conditions. Meanwhile, there is a growing demand for diverse chiral vicinal amino alcohols. As a result, new synthetic methods continue to emerge.5–8 This highlights the critical need to develop novel asymmetric catalytic strategies that should be able to expand the structural diversity of chiral vicinal amino alcohols while enhancing their synthetic efficiency and accessibility.The strategy of synthesizing chiral vicinal amino alcohols via nucleophilic asymmetric ring opening of epoxides or aziridines with nitrogen or oxygen nucleophiles represents one of the most straightforward and atom-economical approaches. Among these strategies, a powerful one is the desymmetrization of meso-substrates, as depicted in Fig. 1a. Pioneering work in this area was reported by Jacobsen and coworkers in 1998, who described the first chromium-catalyzed enantioselective nitrogen-nucleophilic ring opening of meso-epoxides.9 Subsequently, Schneider et al.10 and Feng et al.11 independently developed asymmetric nitrogen-nucleophilic ring opening of meso-epoxides using chiral scandium(III) and indium(III) complexes, respectively. Alternatively, Toste et al.12 and List et al.13 utilized chiral phosphoric acid (CPA) catalysts to achieve the synthesis of chiral vicinal amino alcohols via alcohol-nucleophilic ring opening of meso-aziridiniums and meso-aziridines, respectively. More recently, Feng et al.14 and Wang et al.15 disclosed the oxygen-nucleophilic ring opening of meso-aziridines catalyzed by chiral magnesium complexes, further expanding the scope of this desymmetrization strategy. A complementary approach involves the kinetic resolution of racemic unsymmetric epoxides or aziridines (Fig. 1b). For example, Yamamoto and co-workers developed a chiral tungsten-catalyzed kinetic resolution of racemic 2,3-epoxy alcohols with aromatic amines for the construction of chiral vicinal amino alcohols.16 In a related study, Cao et al. reported an efficient chiral phosphoric acid-catalyzed water-nucleophilic ring opening of racemic aziridines, which also proceeds via a kinetic resolution pathway.17 Although the aforementioned strategies for synthesizing chiral vicinal amino alcohols are direct and efficient, they suffer from limitations: being restricted to desymmetrization of meso-type substrates and, for unsymmetric substrates, requiring more than 2 equivalents of racemic starting materials to proceed via kinetic resolution.
 | ||
| Fig. 1 Synthesis of chiral vicinal amino alcohols via asymmetric ring-opening of epoxides/aziridines. | ||
In 1991, Evans and co-workers first discovered the Cu(I)-catalyzed amination of trimethylsilyl enol ethers to afford N-Ts amino ketones, postulating that the reaction proceeds via an aziridine ring-opening rearrangement pathway analogous to Rubottom oxidation.18 This pioneering work initiated research into the asymmetric catalysis of this reaction.19 In 2018, Read de Alaniz et al. developed a highly efficient Cu(I)-catalyzed asymmetric electrophilic α-amination of silyl enol ether derivatives via the nitrosocarbonyl hetero-ene reaction (Fig. 1c)20 More recently, a landmark study by Matsunaga and colleagues demonstrated the chiral ruthenium-catalyzed amination of alkyl silyl enol ethers (Fig. 1c).19f However, no successful examples have been reported for diaryl-substituted silyl enol ether substrates. We conceived that developing a strategy involving asymmetric aziridination of silyl enol ethers with metal nitrenes,21 combined with one-pot stereoselective reduction, could provide an alternative and efficient pathway for the synthesis of chiral diaryl vicinal amino alcohols (Fig. 1d). Although the hexafluorophosphate anion (PF6−) is widely employed as a counterion in copper-catalyzed asymmetric reactions, its role in asymmetric catalytic processes has long been overlooked.22 This is likely due to the fact that PF6− is typically classified as a weakly coordinating anion (WCA) and was specifically designed to minimize interference with catalytic processes. Herein, we found that fluoro-containing counteranions can promote both the reactivity and enantioselectivity of the reaction. DFT calculations revealed that the interaction between a fluorine atom and the central copper metal can fine-tune the chiral environment of the catalytic center, thereby enabling the chiral copper-catalyzed one-pot synthesis of a series of diaryl chiral vicinal amino alcohols with excellent diastereo- and enantioselectivity.
Results and discussion
Initially, we investigated the reaction of the sterically hindered polysubstituted silyl enol ether 1a with nitrene precursor 2 under catalysis by various chiral copper salts, followed by a one-pot reduction with zinc borohydride to directly afford the N-Ts protected chiral amino alcohol (Table 1, entries 1–9). When copper sulfate and copper phosphate were employed, no desired product was detected (entries 1–2). In the case of cuprous cyanide, copper diphenylphosphinate (CuDPP), copper 2-thiophenecarboxylate (CuTC) and CuBr·SMe2, only trace amounts (<5% yield isolated by column chromatography) of the desired products were observed (entries 3–6). Cu(ClO4)2·6H2O could lead to 32% yield with 75% ee and a 96:4 dr (entry 7). Notably, during this process, we observed that the introduction of fluorine atoms into the counteranion significantly enhanced both the yield and enantioselectivity of the reaction. Using copper trifluoromethanesulfonate, the desired product 3a was afforded in 26% yield with 77% ee and a 90:10 dr (entry 8). In contrast, cuprous trifluoromethanesulfonate improved the yield of 3a to 44% with enhanced stereoselectivity (86% ee and 96:4 dr, entry 9). We further explored other fluorine-containing counteranions. The use of Cu(CH3CN)4BF4 led to a slight improvement in enantioselectivity to 88% ee (entry 10), while Cu(CH3CN)4PF6 resulted in further enhancements in both the yield and ee value, affording 3a in 87% yield with 96% ee and a 97:3 dr (entry 11). To further verify the influence of fluorine atoms on the catalytic center, we tested the counteranion BArF−, which might be spatially remote from the reaction center, and observed a significant decrease in both the yield and enantioselectivity of 3a (59% yield and 76% ee, entry 12). Subsequently, we examined the effect of the chiral backbone of the ligands on the reaction and found that, among the chiral bisoxazoline (BOX) ligands tested, the one with a tert-butyl backbone (L1) outperformed those with other chiral backbones, such as isopropyl, benzyl, phenyl, and indenyl (L2–L5, entries 13–16). Thus, we identified Cu(CH3CN)4PF6 as the optimal copper salt and L1 as the optimal chiral ligand. Notably, we also investigated the effects of different reducing agents on the diastereoselectivity of the product. Although a 97:3 dr can also be achieved when LiAlH4 is used as the reducing agent (entry 17), subsequent studies revealed that the diastereoselectivity of the reaction proved to be unsatisfactory when this reducing agent is applied to substrates with other substituents. Optimization of other reaction conditions can be found in the SI.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | L | Yieldb (%) | eec (%) | drd |
|
a Unless otherwise noted, all reactions were carried out with 1a (0.2 mmol), 2 (0.3 mmol), 4 Å MS (100 mg), and Cu salt/L (1:1.2, 10 mol%) in PhCl (2.0 mL) under a N2 atmosphere at 0 °C for 24 h, and then with Zn(BH4)2 (1.0 mmol, 1.0 M in THF), TBAF (0.6 mmol, 1.0 M in THF) and EtOH (1.0 mL) at 0 °C to rt for 4 h.
b Isolated yield.
c Determined by chiral HPLC.
d Determined by crude 1H NMR.
e LiAlH4 (1.0 mmol) was used instead of Zn(BH4)2.
|
|||||
| 1 | CuSO4 | L1 | N.D. | — | — |
| 2 | Cu3(PO4)2 | L1 | N.D. | — | — |
| 3 | CuCN | L1 | Trace | 59 | — |
| 4 | CuDPP | L1 | Trace | 68 | — |
| 5 | CuTC | L1 | Trace | 76 | — |
| 6 | CuBr·SMe2 | L1 | Trace | 49 | — |
| 7 | Cu(ClO4)2·6H2O | L1 | 32 | 75 | 96:4 |
| 8 | Cu(OTf)2 | L1 | 26 | 77 | 90:10 |
| 9 | CuOTf | L1 | 44 | 86 | 96:4 |
| 10 | Cu(CH3CN)4BF4 | L1 | 34 | 88 | 94:6 |
| 11 | Cu(CH3CN)4PF6 | L1 | 87 | 96 | 97:3 |
| 12 | Cu(BArF) | L1 | 59 | 76 | 97:3 |
| 13 | Cu(CH3CN)4PF6 | L2 | 34 | 71 | 93:7 |
| 14 | Cu(CH3CN)4PF6 | L3 | 55 | 78 | 96:4 |
| 15 | Cu(CH3CN)4PF6 | L4 | 51 | 78 | 94:6 |
| 16 | Cu(CH3CN)4PF6 | L5 | 32 | 70 | 964 |
| 17e | Cu(CH3CN)4PF6 | L1 | 80 | 97 | 97:3 |
|
|||||
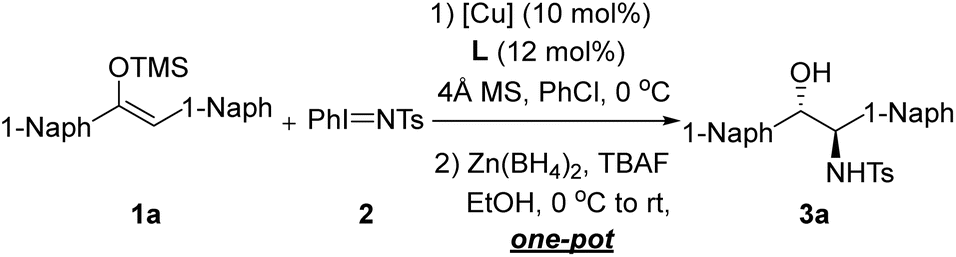
This one-pot strategy for the synthesis of chiral amino alcohols exhibits good substrate adaptability. Under the optimized reaction conditions (Table 1, entry 8), we explored structurally diverse diaryl-substituted enol silyl ethers (Table 2). When R1 is 1-naphthyl and R2 is 2-naphthyl, the corresponding amino alcohol 3b was obtained in 93% yield with a 96:4 dr and 97% ee. Replacing the 2-naphthyl group with 5-benzofuranyl, 5-benzothienyl, or 5-benzodihydrofuranyl resulted in negligible changes in enantioselectivity, and the corresponding amino alcohols 3c–3e were still afforded with 97–98% ee. Substrates 1f and 1g bearing more complex 3-fluorenyl and 2-dibenzofuranyl substituents were also compatible with this reaction, achieving 97% ee and 96% ee, respectively. When R2 is a simple phenyl group, 3h was obtained in 96% yield with a 98:2 dr and 98% ee. For substrate 1h, we investigated the effect of different reducing agents on diastereoselectivity. NaBH4 only resulted in diminished diastereoselectivity (74:26 dr), which is presumably due to the fact that zinc metal might be more ready to form chelating interactions with the substrate, which facilitates the control of diastereoselectivity during the reduction process. Interestingly, an inversion of diastereoselectivity (20:80 dr) can be achieved when DIBAL-H is used instead of Zn(BH4)2. Detailed results are available in the SI. Introduction of halogen substituents at different positions of the benzene ring (including fluorine, chlorine, and bromine) enabled the synthesis of the corresponding chiral amino alcohols 3i–3n with 94–99% yields, 96:4 to >99:1 dr, and 93–96% ee. Incorporation of electron-withdrawing trifluoromethyl groups at the para- and meta-positions of the benzene ring led to a slight decrease in yield and enantioselectivity, giving 3o and 3p in 72% yield with 92% ee and 85% yield with 90% ee, respectively. Introduction of methyl groups at the ortho-, para-, and meta-positions of the benzene ring had almost no impact on the enantioselectivity of the reaction, and 3q–3s were obtained with 97–98% ee. Although the introduction of an electron-rich methoxy group at the ortho-position of the benzene ring caused a decrease in enantioselectivity (3t, 90% ee), the corresponding product was obtained with exclusive enantioselectivity when the methoxy group was introduced at the meta-position (3u, 99.5% ee). For the para-methoxy-substituted substrate (3v), the reaction activity decreased slightly (84% yield), while the diastereoselectivity remained exclusive (>99:1 dr) and the enantioselectivity showed no significant change (97% ee). Other substituents on the benzene ring were also well-tolerated. For example, the 3,4-dimethoxy-substituted product 3w was obtained in 73% yield with a 93:7 dr and 95% ee; the para-trifluoromethoxy-substituted substrate afforded the corresponding amino alcohol 3x in 67% yield with a 98:2 dr and 92% ee. Reactive functional groups such as carboxyl, protected alcohol and amine derivatives were tolerated, affording 3y–3aa with 90–95% ee. Bpin- and TMS-substituted substrates were also explored to give the corresponding 3bb and 3cc with 97–99% ee. Introduction of phenyl groups at the ortho-, meta-, and para- positions of the benzene ring led to the corresponding biphenyl amino alcohols 3dd–3ff with 93–95% ee. When the phenyl group was replaced with a heteroaromatic ring, the reaction system remained compatible, and the thiophene-substituted amino alcohol 3gg was synthesized in 96% yield with a 97:3 dr and 97% ee. The tert-butyl-aryl silyl enol ether was tried as substrate, and the desired product 3hh could be obtained in 91% ee and >99:1 dr with 31% yield.
|
a Unless otherwise noted, all reactions were carried out with 1 (0.2 mmol), 2 (0.3 mmol), 4 Å MS (100 mg), and Cu(CH3CN)4PF6/L1 (1:1.2, 10 mol%) in PhCl (2.0 mL) under a N2 atmosphere at 0 °C for 24 h, and then Zn(BH4)2 (1.0 mmol, 1.0 M in THF), TBAF (0.6 mmol, 1.0 M in THF) and EtOH (1.0 mL) were added at 0 °C to rt for 4 h; Isolated yield; dr value was determined by crude 1H NMR; ee value was determined by chiral HPLC.
b −10 °C.
c
2 was added in four portions.
|
|---|
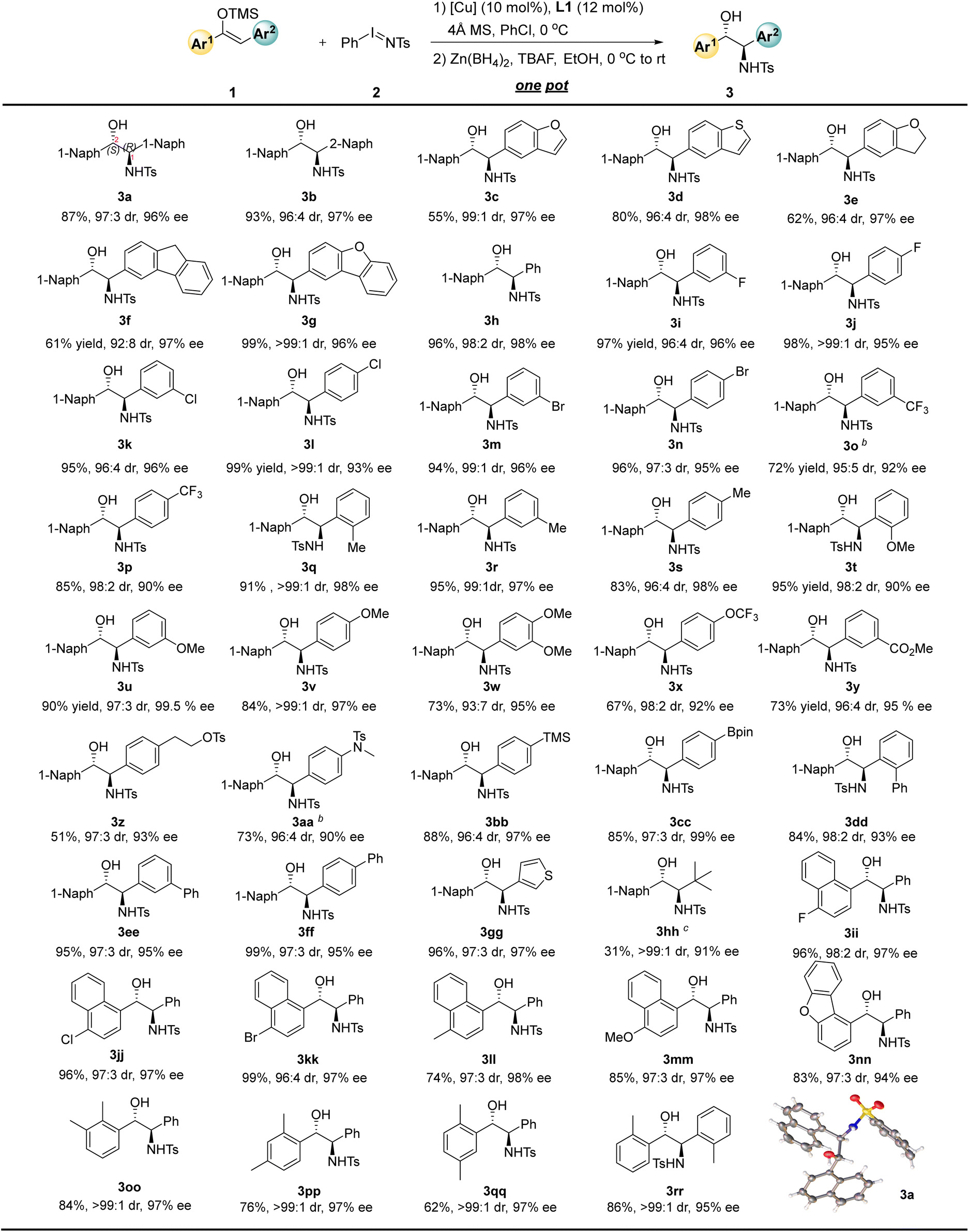
|
On the other hand, the Ar1 group can also be extended in a variety of ways. For instance, naphthyl substrates substituted with fluorine, chlorine, or bromine atoms afforded the corresponding amino alcohols 3ii–3kk in 96–99% yields with excellent diastereoselectivity and 97% ee. Naphthyl substrates with electron-donating substituents also performed well, giving 3ll and 3mm with 97–98% ee. Other more complex substituents were well compatible with the reaction, and the dibenzofuranyl-substituted product 3nn was obtained with 94% ee. This method also allowed for the convenient preparation of various dimethyl-substituted phenyl amino alcohols 3oo–3qq as well as 1,2-diaryl-substituted amino alcohol 3rr with 95–97% ee. Meanwhile, the absolute configuration of product 3a was confirmed to be (1R, 2S) by X-ray diffraction analysis of its single crystal.
To gain insights into the fluoride benefited reaction mechanism, density functional theory (DFT) calculations were performed on the pathway of the asymmetric aziridination reaction between copper nitrenes and silyl enol ethers, based on the reported computational literature studies (calculation details and the reaction potential energy surface calculations are provided in the SI).23 DFT calculations revealed that the initiating species of the reaction is PF63[Cu]NTs, corresponding to the structure where the triplet nitrene and ligand are coordinated to Cu+, respectively. During the reaction, for the major and minor enantiomers, the rate-determining step involves the formation of the first N–C bond via the transition states (TS) with the highest energy barrier, PF63TS1 (ΔG≠ = 8.4 kcal mol−1) and PF63TS'1 (ΔG≠ = 11.0 kcal mol−1), which thus serves as the chirality determining step. The calculated ΔΔG≠ for this step is 2.6 kcal mol−1, consistent with the experimental 98% ee (Fig. 2a).
 | ||
| Fig. 2 DFT-derived interactions in the enantioselective induction model. | ||
Notably, calculations indicated that in the initiating species PF63[Cu]NTs, one of the F atoms of the anion exhibit certain coordination interactions with Cu, with a Cu–F distance of 2.42 Å (significantly shorter than the sum of the van der Waals radii of Cu and F, 2.87 Å). Such secondary interactions enable the hexafluorophosphate ion to occupy the space above the PF63[Cu]NTs species, confining the Ts group to the region below the anion (Fig. 2a). The identification of these Cu–F secondary interactions corroborates the experimental observation that fluorinated counterions influence reaction activity and enantioselectivity. In the TS of the major enantiomer (PF63TS1), as the silyl enol ether substrate approaches the reaction center, the spatial confinement by the hexafluorophosphate ion induces a conformational change in the Ts group, facilitating its deformation and π–π stacking with the naphthyl ring of the silyl enol ether—this contributes to the lower energy of PF63TS1. As shown in Fig. 2a, the dihedral angle ∠N–S–C1–C2 (θ) changes from −91.7° in PF63[Cu]NTs to −75.3° in PF63TS1, corresponding to a 16.4° rotation of the benzene ring in the Ts group around the S–C1 bond. In contrast, no such π–π stacking is observed in PF63TS'1, and the variation in ∠θ is comparatively small.
The presence of the hexafluorophosphate ion also affects the conformation of the BOX ligand. IGMH (Independent Gradient Model based on Hirshfeld partition) analysis revealed two pairs of C⋯H weak interactions and two pairs of H⋯H weak attractive interactions between the benzene ring and the ligand in PF63TS1, which favor its lower energy barrier. In contrast, the weak interactions between the naphthyl ring and the ligand in PF63TS'1 are less pronounced. Based on the above IGMH analysis, the weak interaction sites in PF63TS1 are located at the ortho- and para-positions of the phenyl substituent. Thus, introducing an electron-donating substituent (e.g., methoxy) at the meta-position of the benzene ring is expected to enhance these electrostatic attractions, thereby lowering the energy barrier of PF63TS1 and increasing the energy barrier difference between PF63TS1 and PF63TS'1 (i.e., enhancing the ee value). Calculations showed that the energy barrier difference increases from 2.6 kcal mol−1 to 3.5 kcal mol−1, consistent with the experimental improvement in the ee value from 98% to 99.5% (Fig. 2b). Conversely, introducing an electron-withdrawing substituent (e.g., trifluoromethyl) at this position might have the opposite effect: the calculated energy barrier difference decreased to 2.0 kcal mol−1, and the experimental ee value dropped from 98% to 92% (Fig. 2b). These computational results are in good agreement with the experimental data, validating the reliability of the theoretical model.
This method provides a practical approach for the gram-scale synthesis of disubstituted chiral amino alcohols.
With 1,2-Dinaphthyl-substituted amino alcohol 3a, 1,2-diaryl-substituted amino alcohol 3rr, or the product 3h with two different substituents, the reaction can be successfully scaled up to the gram level, affording 79–94% yield with 3.42 g to 3.92 g (Fig. 3). Moreover, the resulting products can be isolated and purified by recrystallization to afford optically pure products with >99% ee and >99:1 dr. It is worth noting that these N-Ts protected amino alcohols can undergo mild and convenient deprotection under Red-Al conditions, thereby yielding the corresponding optically pure chiral amino alcohols 4a, 4rr, and 4h in 60–70% yields.
 | ||
| Fig. 3 Gram scale synthesis and transformation. | ||
Conclusions
In summary, we report a chiral copper-catalyzed two-step one-pot strategy for the synthesis of diaryl chiral vicinal amino alcohols with excellent diastereo- and enantioselectivity (up to >99:1 dr and 99.5% ee). A key innovation is the identification of fluorinated counteranions as critical regulators of reactivity and enantioselectivity—an underappreciated role in copper catalysis. DFT calculations revealed that secondary Cu–F interactions, coupled with the spatial confinement of hexafluorophosphate, fine-tune the catalytic chiral environment to enable precise stereocontrol, as validated by IGMH analysis. This method features broad substrate scope, gram-scale accessibility, and facile deprotection to free amino alcohols, offering a robust platform for accessing valuable chiral vicinal amino alcohols. Importantly, it highlights counterion engineering as a powerful tool for optimizing asymmetric catalytic performance.
Author contributions
L. W. conceived and directed the project, and prepared the manuscript; Y. Y. performed all the experiments and the experiment part of the SI; J.-L. W. performed the DFT calculation and the calculation part of the SI; S.-Y. Z. provided useful DFT calculation suggestions; Y. Z. supervised DFT calculations and the calculation part of the SI; X. L. and T. S. synthesized some substrates.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. Supplementary information: experimental procedures, spectroscopic data for new compounds, NMR, HRMS spectra and HPLC chromatograms, and CIF file of enantiopure product 3a. See DOI: https://doi.org/10.1039/d5sc06853c.CCDC 2478618 (3a) contains the supplementary crystallographic data for this paper.24
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91956103 and 22571088), the Science and Technology Commission of Shanghai Municipality (23JC1404500), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0610000) and the Fundamental Research Funds for the Central Universities. We also thank Dr Xiao-Li Zhao for X-ray crystal analysis.Notes and references
- (a) P. Gupta, and N. Mahajan, in Chemistry of Biologically Potent Natural Products and Synthetic Compounds, ed. Shahid-ul-Islam and J. A. Banday, Scrivener Publishing LLC, 2021 Search PubMed; (b) Y.-J. Cheng, L.-P. Zhao, L. Wang and Y. Tang, CCS Chem., 2023, 5, 124 CrossRef CAS; (c) P.-J. Li, P.-C. Yin, L. Wang and Y. Tang, CCS Chem., 2025 DOI:10.31635/ccschem.025.202506445.
- T. Hayashi, A. Katsumura, M. Konishi and M. Kumada, Asymmetric synthesis of 2-amino-1-arylethanols by catalytic asymmetric hydrogenation, Tetrahedron Lett., 1979, 20, 425–428 CrossRef.
- (a) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561–2576 CrossRef CAS; (b) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem.–Eur. J., 2011, 17, 58–76 CrossRef CAS PubMed; (c) B. N. Hemric, Org. Biomol. Chem., 2021, 19, 46–81 RSC; (d) X. Yan, W. Feng, J. X. Ng, J. Li, Y. Liu, B. Zhang, P.-L. Shao and Y. Zhao, Chem. Soc. Rev., 2025, 54, 7966–8018 RSC.
- (a) G. Li, H.-T. Chang and K. B. Sharpless, Angew. Chem., Int. Ed., 1996, 35, 451–454 CrossRef CAS; (b) P. O'Brien and K. B. Sharpless, Angew. Chem., Int. Ed., 1999, 38, 326–329 CrossRef.
- For recent leading examples on asymmetric hydrogenation, see: (a) J. Wang, X. Lin, P.-L. Shao, J. Song, J. Wen and X. Zhang, ACS Catal., 2021, 11, 12729–12735 CrossRef CAS; (b) Y. Xu, Y. Luo, J. Ye, Y. Deng, D. Liu and W. Zhang, J. Am. Chem. Soc., 2022, 144, 20078–20089 CrossRef CAS PubMed; (c) M. Zhang, T. Niu, M. Liang, F. Xu, Y. Du, H. Zhuang, R.-J. Song, H. Yang and Q. Yin, J. Am. Chem. Soc., 2025, 147, 18197–18207 CrossRef CAS PubMed.
- For recent leading examples on asymmetric reductive coupling, see: (a) C.-X. Ye, Y. Y. Melcamu, H.-H. Li, J.-T. Cheng, T.-T. Zhang, Y.-P. Ruan, X. Zheng, X. Lu and P.-Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef PubMed; (b) H. Hu and Z. Wang, J. Am. Chem. Soc., 2023, 145, 20775–20781 CrossRef CAS PubMed; (c) T. Bender and A. Fürstner, J. Am. Chem. Soc., 2024, 146, 33295–33301 CrossRef CAS; (d) Z. Chi, J.-B. Liao, X. Cheng, Z. Ye, W. Yuan, Y.-M. Lin and L. Gong, J. Am. Chem. Soc., 2024, 146, 10857–10867 CrossRef CAS PubMed; (e) J. Zhu, F. Rahim, P. Zhou, A. Zhang and S. J. Malcolmson, J. Am. Chem. Soc., 2024, 146, 20270–20278 CrossRef CAS PubMed.
- For recent leading examples on asymmetric radical addition, see: (a) J. Caner, A. Matsumoto and K. Maruoka, Chem. Sci., 2023, 14, 13879–13884 RSC; (b) H. Hu, Z. Shi, X. Guo, F.-H. Zhang and Z. Wang, J. Am. Chem. Soc., 2024, 146, 5316–5323 CrossRef CAS PubMed; (c) Z. Zhong, H. Wu, X. Chen, Y. Luo, L. Yang, X. Feng and X. Liu, J. Am. Chem. Soc., 2024, 146, 20401–20413 CrossRef CAS PubMed.
- For recent leading examples on miscellaneous strategies, see: (a) E. M. Mumford, B. N. Hemric and S. E. Denmark, J. Am. Chem. Soc., 2021, 143, 13408–13417 CrossRef CAS PubMed; (b) C. Hou, B. Peng, S. Ye, Z. Yin, J. Cao, X. Xiao and B. Zhao, Nat. Catal., 2022, 5, 1061–1068 CrossRef CAS; (c) B. G. Hejna, J. M. Ganley, H. Shao, H. Tian, J. D. Ellefsen, N. J. Fastuca, K. N. Houk, S. J. Miller and R. R. Knowles, J. Am. Chem. Soc., 2023, 145, 16118–16129 CrossRef CAS PubMed; (d) H. Yan, Q. Liao, Y. Chen, G. G. Gurzadyan, B. Lu, C. Wu and L. Shi, Angew. Chem., Int. Ed., 2023, 62, e202302483 CrossRef CAS PubMed; (e) A. Mondal and G. C. Fu, J. Am. Chem. Soc., 2025, 147, 10859–10863 CrossRef CAS PubMed.
- L. E. Martinez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef CAS.
- C. Schneider, A. R. Sreekanth and E. Mai, Angew. Chem., Int. Ed., 2004, 43, 5691–5694 CrossRef CAS PubMed.
- B. Gao, Y. Wen, Z. Yang, X. Huang, X. Liu and X. Feng, Adv. Synth. Catal., 2008, 350, 385–390 CrossRef CAS.
- G. L. Hamilton, T. Kanai and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 14984–14986 CrossRef CAS PubMed.
- M. R. Monaco, B. Poladura, M. Diazde Los Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed., 2014, 53, 7063–7067 CrossRef CAS PubMed.
- J. Li, Y. Liao, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 6672–6674 RSC.
- L. Wang, D. Yang, D. Li, H. Zhu, P. Wang, X. Liu, L. Bai and R. Wang, Adv. Synth. Catal., 2018, 360, 4491–4496 CrossRef CAS.
- (a) C. Wang and H. Yamamoto, Angew. Chem., Int. Ed., 2014, 53, 13920–13923 CrossRef CAS PubMed; (b) C. Wang and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 4308–4311 CrossRef CAS.
- J. Liu, Y.-Y. Du, Y.-S. He, Y. Liang, S.-Z. Liu, Y.-Y. Li and Y.-M. Cao, Chem. Sci., 2023, 14, 12152–12159 RSC.
- D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Org. Chem., 1991, 56, 6744–6746 CrossRef CAS.
- (a) W. Adam, K. J. Roschmann and C. R. Saha-Möller, Eur. J. Org Chem., 2000, 2000, 557–561 CrossRef; (b) J.-L. Liang, X.-Q. Yu and C.-M. Che, Chem. Commun., 2002, 124–125 RSC; (c) M. Anada, M. Tanaka, T. Washio, M. Yamawaki, T. Abe and S. Hashimoto, Org. Lett., 2007, 9, 4559–4562 CrossRef CAS PubMed; (d) M. Tanaka, Y. Kurosaki, T. Washio, M. Anada and S. Hashimoto, Tetrahedron Lett., 2007, 48, 8799–8802 CrossRef CAS; (e) M. Nakanishi, C. Minard, P. Retailleau, K. Cariou and R. H. Dodd, Org. Lett., 2011, 13, 5792–5795 CrossRef CAS PubMed; (f) K. Makino, Y. Kumagai, T. Yoshino, M. Kojima and S. Matsunaga, Org. Lett., 2023, 25, 3234–3238 CrossRef CAS PubMed.
- D. Sandoval, A. V. Samoshin and J. Read de Alaniz, Org. Lett., 2015, 17, 4514–4517 CrossRef PubMed.
- (a) B. Liu, P. Xie, J. Zhao, J. Wang, M. Wang, Y. Jiang, J. Chang and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8396–8400 CrossRef CAS PubMed; (b) R. Mi, Z. Ding, S. Yu, R. H. Crabtree and X. Li, J. Am. Chem. Soc., 2023, 145, 8150–8162 CrossRef CAS PubMed; (c) N. J. Hodson, S. Takano, A. Fanourakis and R. J. Phipps, J. Am. Chem. Soc., 2024, 146, 22629–22641 CrossRef CAS PubMed.
- (a) C.-T. Li, L.-G. Liu, J.-Z. Li, H.-X. Dong, R. A. Novikov, Z.-S. Wang, X. Hong, B. Zhou and L.-W. Ye, Nat. Commun., 2025, 16, 4107 CrossRef CAS PubMed; (b) S. Maurya, N. Navaneetha, P. Behera, J. B. Nanubolu, L. Roy and R. Chegondi, Angew. Chem., Int. Ed., 2025, 64, e202420106 CrossRef CAS PubMed; (c) K. Morisaki, Y. Furuki, R. Kousaka, S. Nagai, Y. Oonishi and Y. Sato, J. Am. Chem. Soc., 2025, 147, 12740–12748 CrossRef CAS PubMed; (d) C.-Y. Weng, L.-G. Liu, M. Sun, X. Lu, X. Hong, L.-W. Ye and B. Zhou, Angew. Chem., Int. Ed., 2025, 64, e202418254 CrossRef CAS PubMed; (e) X. Zhang, Y. Zhou, Z.-X. Yu, C.-H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2025, 64, e202420667 CrossRef CAS PubMed.
- (a) P. Brandt, M. J. Södergren, P. G. Andersson and P.-O. Norrby, J. Am. Chem. Soc., 2000, 122, 8013–8020 CrossRef CAS; (b) L. Maestre, W. M. C. Sameera, M. M. Díaz-Requejo, F. Maseras and P. J. Pérez, J. Am. Chem. Soc., 2013, 135, 1338–1348 CrossRef CAS PubMed; (c) M. R. Rodríguez, A. M Rodríguez, S. López-Resano, M. A. Pericàs, M. M. Díaz-Requejo, F. Maseras and P. J. Pérez, ACS Catal., 2023, 13, 706–713 CrossRef PubMed; (d) S.-Y. Zhang, Y. Zhao, L. Wang, Z.-P. Liu and Y. Tang, CCS Chem., 2025, 7, 3493–3506 CrossRef.
- CCDC 2478618, (3a): Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p66fk.
| This journal is © The Royal Society of Chemistry 2026 |