
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater drives sequential breakdown of dynamic nanodomains in deep eutectic electrolytes
Tubai Chowdhury ab,
Athira Babuab,
Sreekumar Kurungot*ab,
Rajib Biswas*c,
Sapna Ravindranathan*bd and
Sayan Bagchi*ab
ab,
Athira Babuab,
Sreekumar Kurungot*ab,
Rajib Biswas*c,
Sapna Ravindranathan*bd and
Sayan Bagchi*ab
aPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune 411008, India. E-mail: k.sreekumar.ncl@csir.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India. E-mail: s.bagchi.ncl@csir.res.in
cDepartment of Chemistry, Indian Institute of Technology Tirupati, Yerpedu, Tirupati, Andhra Pradesh 517619, India. E-mail: rajib@iittp.ac.in
dCentral NMR Facility, CSIR – National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411008, India. E-mail: s.ravindranathan.ncl@csir.res.in
First published on 3rd December 2025
Abstract
The rational design of functional materials hinges on understanding molecular interactions in complex hydrogen-bonded liquids like deep eutectic electrolytes (DEEs), where molecular structure governs ionic conductivity and electrochemical stability. Hydration levels critically influence these properties, yet the underlying mechanisms remain elusive, hindering systematic design. Using multidimensional NMR, 2D infrared spectroscopy, and molecular dynamics simulations, we studied choline chloride-malic acid DEEs at varying hydration levels. We show a sequential, component-specific breakdown of molecular nanodomains that overturns dilution models. This process proceeds in distinct stages: water first disrupts ionic domains at moderate hydration levels, while hydrogen-bonded organic networks persist until higher water content. These insights enabled us to design a DEE with enhanced electrolyte performance, achieving ionic conductivity of 13.0 mS cm−1 and stable cycling over 1000 cycles while suppressing parasitic reactions. This work demonstrates how fundamental molecular insights can resolve critical bottlenecks in sustainable technology development, enabling systematic engineering of nanostructured liquids for energy storage, catalysis, and sustainable chemistry applications.
Introduction
The rational design of functional materials increasingly depends on understanding and controlling structure across multiple length scales, from molecular interactions to bulk properties. This challenge is particularly acute for complex, hydrogen-bonded liquids that exhibit hierarchical organization, where small changes in composition can dramatically alter macroscopic behavior through cascading structural reorganization. Deep eutectic solvents (DESs), formed from hydrogen bond donors (HBDs) and acceptors (HBAs), represent a paradigmatic example of such systems, with their unique combination of ionic and molecular components creating nanostructured liquids with tunable properties.1,2Deep eutectic electrolytes (DEEs), the electrochemically active variants of DESs, have emerged as promising candidates for next-generation energy storage systems due to their wide electrochemical stability windows, thermal stability, and environmental compatibility.3,4 However, pristine DEEs typically suffer from high viscosities and limited ionic conductivities that severely constrain their practical applications.5 The controlled addition of water has become a widely adopted strategy to modulate these properties, often leading to dramatic improvements in ionic transport while maintaining advantageous characteristics like reduced flammability and enhanced safety compared to conventional organic electrolytes.5–8
Despite the widespread use of hydrated DEEs, the molecular-level mechanisms by which water remodels their nanostructure remain poorly understood.6,7 This knowledge gap represents a fundamental limitation in the rational design of these materials. Previous experimental studies have largely focused on bulk property correlations,9–12 while computational investigations have been limited by the challenge of simulating these complex, multi-component systems across relevant length and time scales.13–15
The few mechanistic studies available, notably neutron scattering work by Hammond and coworkers on the choline chloride-malic acid (ChCl-MA) system, established that water is sequestered within the existing ionic network at very low hydration levels (≤2 water equivalents) without significant structural disruption.16 However, the structural evolution at higher, performance-relevant hydration levels, along with the specific molecular pathways of network breakdown, remained largely unexplored.
The absence of a detailed mechanistic understanding has prevented the development of predictive frameworks for hydrated DEE design. Current approaches rely heavily on empirical screening, limiting the ability to rationally optimize these systems for specific applications.17 This is particularly problematic for emerging technologies like aqueous metal-ion batteries, where electrolyte performance depends critically on the delicate balance between ionic conductivity, electrochemical stability, and suppression of parasitic reactions, all of which are intimately connected to molecular-level structure and dynamics.18,19
Recent advances in multidimensional spectroscopy and molecular simulation provide unprecedented opportunities to bridge the gap between molecular structure and macroscopic function in complex liquids.20–24 Two-dimensional nuclear magnetic resonance (2D NMR) spectroscopy can directly probe intermolecular interactions and network connectivity,25,26 while 2D infrared (2D IR) spectroscopy reveals hydrogen bond (HB) dynamics on ultrafast timescales.20,27–31 When combined with modern molecular dynamics simulations employing validated force fields, these techniques enable comprehensive characterization of structure and dynamics across multiple length and time scales.32,33
Here, we combine this suite of advanced techniques to elucidate the molecular mechanism of hydration-driven network disruption in a prototypical ChCl-MA DEE. By systematically varying water content from 0 to 15 equivalents, we uncover a hierarchical sequence of nanodomain breakdown: water preferentially solvates chloride anions, disrupting choline chloride-rich networks at low hydration, while malic acid-rich domains persist until high water content. Around a critical hydration level (∼10w), water disrupts the malic-rich networks and bridges both choline and malic acid components, resulting in a reorganized solvation network that maximizes ion transport while suppressing parasitic water-splitting reactions. This molecular-level insight enables rational optimization of the electrolyte for zinc-ion battery applications, demonstrating stable cycling performance that directly correlates with the identified optimal nanostructure. By providing direct, experimental observation of sequential nanodomain breakdown in DESs, this work establishes a generalizable framework for engineering nanostructured liquids with predictable, application-specific properties.
Results and discussion
Choline chloride and malic acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio form a unique eutectic system characterized by an exceptionally high viscosity (14000 mPa s at 298 K). This extreme viscosity results from the strong hydrogen bonding network facilitated by malic acid's carboxylic acid and branched hydroxyl groups, which enhance intermolecular interactions with the choline cation and chloride anion.16 The robust network in ChCl-MA makes it an ideal model system for investigating how controlled hydration can systematically remodel nanostructure and tune macroscopic properties for electrolyte applications.
:1 molar ratio form a unique eutectic system characterized by an exceptionally high viscosity (14000 mPa s at 298 K). This extreme viscosity results from the strong hydrogen bonding network facilitated by malic acid's carboxylic acid and branched hydroxyl groups, which enhance intermolecular interactions with the choline cation and chloride anion.16 The robust network in ChCl-MA makes it an ideal model system for investigating how controlled hydration can systematically remodel nanostructure and tune macroscopic properties for electrolyte applications.
Sequential disruption of dynamic nanodomains
The pristine ChCl-MA DES exhibits a strong hydrogen bond network, evidenced by broad 1H NMR resonances of both aliphatic and hydroxyl protons, reflecting severely restricted molecular mobility (Fig. 1a). Upon incremental water addition, pronounced narrowing of these resonances occurs, with the exchange-averaged hydroxyl signal approaching the resonance of pure water at 4.77 ppm (Fig. S1). This progressive line narrowing and upfield shift of the hydroxyl resonance provide direct evidence for HB network disruption and the emergence of enhanced molecular dynamics upon water addition.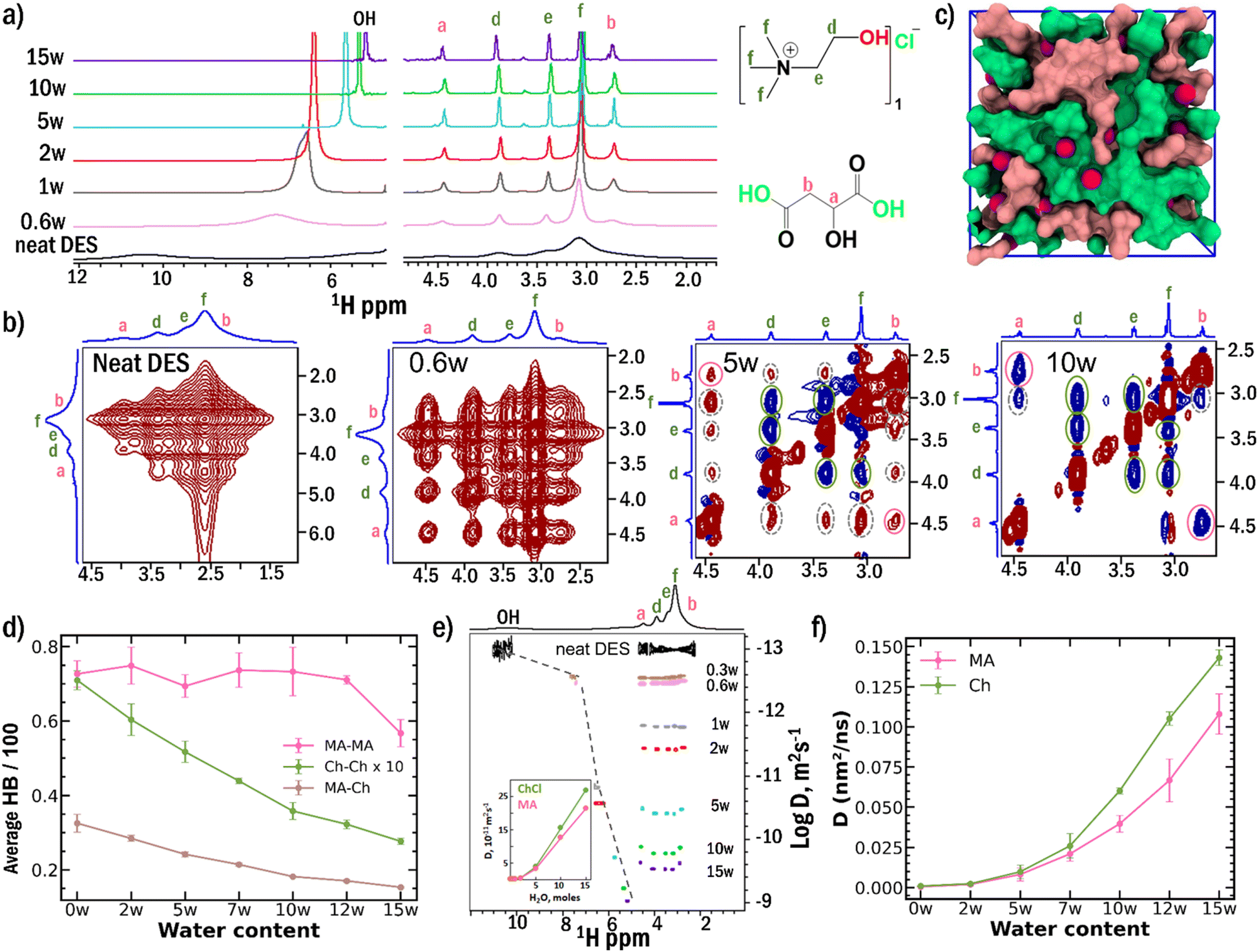 | ||
| Fig. 1 (a) 1H NMR spectra recorded on a 500 MHz spectrometer at 298 K, showing progressive line narrowing with increasing water content, providing direct evidence for hydrogen-bond network disruption and enhanced molecular dynamics. (b) 2D NOESY cross-peak evolution in the aliphatic region demonstrating hierarchical breakdown: choline network disruption at 5w (cross-peak inversion from positive to negative) while MA domains persist (positive cross-peaks remain). Spectra are shown for neat DES, 0.6w, 5w, and 10w hydration levels. Positive and negative contours are indicated in red and blue, respectively. (c) MD simulation snapshot of neat DES: heterogeneous nanostructure is dominated by choline chloride-rich (green with chloride ions shown in magenta) and malic acid-rich domains (pink) (d) MD-calculated hydrogen bond populations revealing component-specific breakdown: Ch–Ch interactions decrease steadily, while MA–MA bonds remain almost constant till 12w with a nominal fluctuation and decrease after that. Ch–Ch data are multiplied by a factor of 10 for clarity. (e) Experimental PFG-NMR and estimated diffusion coefficients (inset) showing component-specific mobility enhancement. Clear divergence emerges at 5w and above: choline diffuses faster than malic acid despite similar molecular weights, indicating sequential liberation from the hydrogen-bond network. (f) MD-calculated diffusion coefficients corroborate the experimental trend. | ||
The hierarchical nature of this HB network breakdown is revealed through 2D NOESY NMR spectroscopy (Fig. 1b). In the neat system, broad, intense positive cross-peaks connecting all aliphatic (non-exchanging) proton pairs indicate slow dynamics (ω0τ ≫ 1, ω0 = Larmor frequency and τ = rotational correlation time) within a rigid, cooperative HB network.26 The addition of 0.6 moles of water leads to significant signal narrowing due to increased mobility as water penetrates the network; however, strong inter- and intra-component positive cross-peaks persist, indicating that the fundamental network topology remains intact.
A critical transition occurs at 5w hydration: choline–choline cross-peaks invert from positive (red) to negative (blue) signals, marking the onset of choline network disruption and a transition toward fast dynamics (ω0τc ≪ 1, see SI, Section 2, Note S1). Remarkably, cross-peaks between malic acid proton pairs and ChCl-MA protons remain positive (red) at 5w, providing direct experimental evidence that malic acid-rich domains persist with restricted mobility even after choline network breakdown. Significant nanodomain disruption occurs only at around 10w, where all cross-peaks become negative (blue) and inter-component cross-peaks weaken significantly. Results of ROESY experiments further confirm that the observed inter-component cross peaks arise due to the spatial proximity of ChCl and MA in the HB networks and that inter-component interactions persist beyond hydration levels of 10w (Fig. S2, SI Section 2, Note S1). The heteronuclear NOE cross peaks observed between 13C and 1H in HOESY experiments corroborate the conclusions from NOESY and ROESY spectra (Fig. S3, see SI Section 2, Note S2).
Molecular dynamics simulations show a heterogeneous nanostructure that is dominated by choline chloride rich and malic acid rich domains (Fig. 1c). Further, simulations provide quantitative atomistic validation of this sequential breakdown mechanism (Fig. 1d). The calculated HB populations (see SI Section 2, Note S3) show that Ch–Ch and MA–Ch HB interactions decrease steadily with hydration (Tables S2 and S3, see SI Section 2, Note S4), while MA–MA HB population remains almost constant till 12w, and decrease after that (Table S4). These trends in HB populations provide molecular-level insight into the experimental observation: as the choline network is disrupted, malic acid molecules reorganize into more extensive, resilient self-associated domains before eventual breakdown by excess water.
Pulsed field gradient NMR diffusion measurements reveal component-specific changes in translational dynamics that directly reflect the sequential breakdown mechanism (Fig. 1e, S4, S5 and Table S5).34–37 In neat DES, choline chloride and malic acid exhibit nearly identical diffusion coefficients (∼1 × 10−13 m2 s−1), consistent with a tightly bound, cooperative network with strong intercomponent interactions and restricted mobility. At 5w and above, a clear divergence emerges: choline diffuses faster than malic acid despite similar molecular weights, indicating component-specific liberation from the hydrogen-bond network. MD simulations quantitatively reproduce this differential mobility, with calculated diffusion coefficients showing excellent qualitative agreement with experimental trends (Fig. 1f).
Chloride anion solvation as the primary disruption trigger
The preferential solvation of chloride anions emerges as the fundamental mechanism driving sequential network breakdown. 35Cl NMR spectroscopy provides direct experimental evidence for this process (Fig. 2a). The Cl− resonance exhibits pronounced narrowing as water content increases from 5w to 15w, with the full width at half maximum decreasing from 5095 Hz to 426 Hz, reflecting a transition from a highly anisotropic, restricted environment to a more symmetric and mobile solvation shell. Below 5w, the Cl− resonance remains extremely broad and barely observable due to strong quadrupolar interactions in an asymmetric environment, consistent with limited water access and strong choline-Cl− interactions.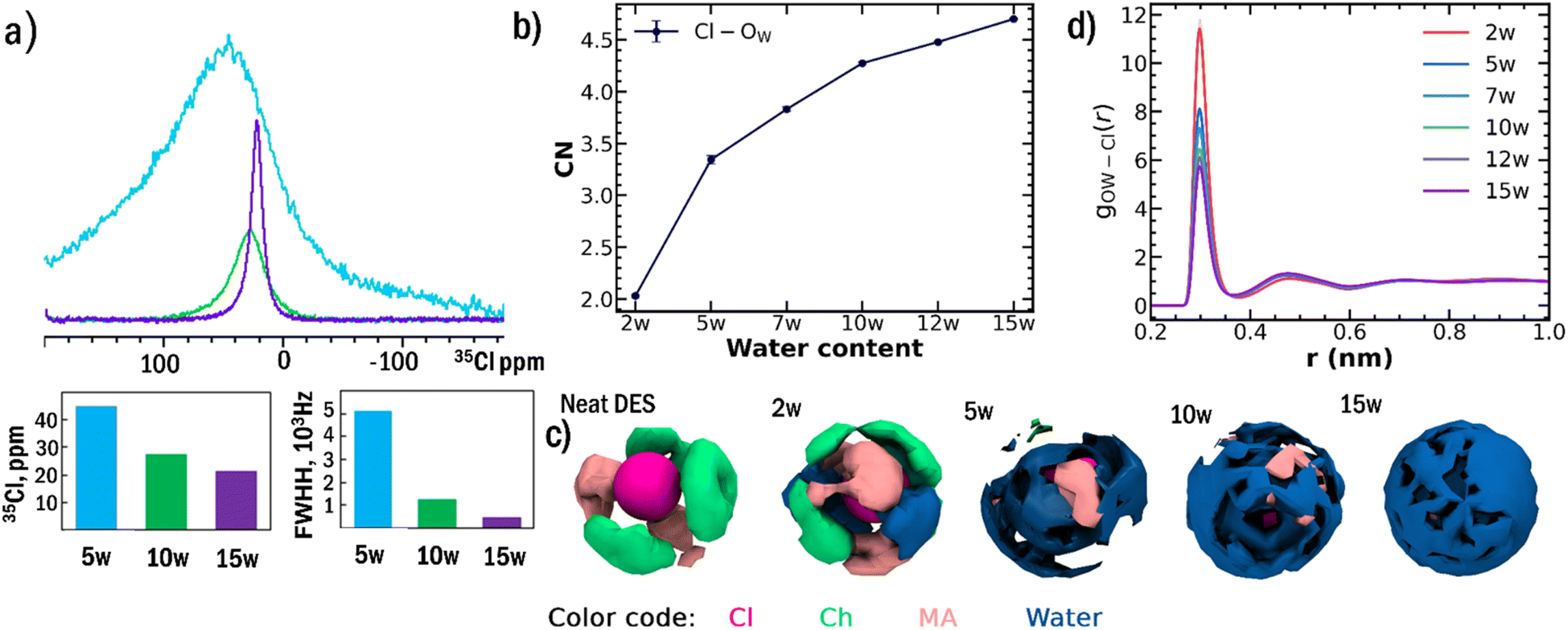 | ||
| Fig. 2 (a) 35Cl NMR spectra of ChCl-MA at 298 K, showing dramatic line narrowing from 5095 Hz (5w) to 426 Hz (15w), indicating enhanced Cl− mobility due to progressive hydration shell formation. Peak positions and line widths are displayed in bar plots. Below 5w, resonances remain extremely broad due to strong quadrupolar interactions in asymmetric environments. (b) MD-calculated water-Cl− coordination numbers increasing systematically from 1.99 (2w) to 4.35 (10w), demonstrating progressive formation of well-defined chloride hydration shells. (c) Spatial distribution functions reveal preferential water positioning around chloride anions rather than random distribution throughout the DES matrix, providing direct computational evidence for a selective solvation mechanism. (d) Radial distribution functions of water oxygen (Ow) and chloride ions showing decreasing peak intensities with concomitant increases in coordination number. This peak height reduction reflects increased mobility of water molecules in chloride solvation shells rather than decreased water density, corroborating the 35Cl NMR line narrowing observations. | ||
MD simulations provide atomistic details of this solvation process (Fig. 2b–d).13,38,39 The average water Cl− coordination number increases systematically from 2.0 at 2w to 4.3 at 10w (Table S6). The systematic increase in Cl-Ow coordination number upon hydration reflects the preferential solvation of chloride anions by water molecules. The radial distribution functions show decreasing peak intensities with concomitant increases in coordination number as water content rises. Importantly, this decrease in peak height does not indicate fewer water molecules around chloride anions but rather reflects increased mobility of water molecules preferentially located in the chloride solvation shell, corroborating the decrease in FWHM in 35Cl NMR. Spatial distribution functions confirm that water exhibits strong positional preferences around chloride anions even at moderate hydration levels, providing direct computational evidence for the selective solvation mechanism.
DFT calculations of isolated pair interactions confirmed that the driving force for this preferential solvation is thermodynamic, with the Cl−-water interaction energy (−14.38 kcal mol−1) being significantly stronger than the MA-water interaction (−12.28 kcal mol−1). The DFT results (Table S7) are consistent with MD-derived coordination trends (Fig. 2b) and 35Cl NMR line-narrowing. The energetic preference (2.10 kcal mol−1) supports the experimental observation that water partitions into chlorine-rich sites, stabilizing chloride hydration shells and destabilizing choline-Cl− networks at low hydration.
This selective solvation disrupts the choline-Cl− interactions that stabilize choline-rich nanodomains, providing a molecular explanation for the NOESY cross-peak inversion observed experimentally at 5w. The preferential anion solvation is thermodynamically driven by the high charge density of Cl− compared to the organic components. This selectivity explains why the initial stages of hydration (≤2w) cause minimal network disruption, as water molecules are sequestered around chloride anions without significantly perturbing the overall hydrogen-bonding topology, consistent with previous neutron scattering observations.16
Hydrogen bond network dynamics and spectroscopic correlation
2D IR spectroscopy using SCN− as a vibrational reporter reveals the dynamic evolution of the hydrogen bond network (Fig. 3a and b, see SI Section 4, Note S5).27–29,40–42 In neat DES, the HB dynamics are extremely slow, with a correlation time (τc) of ∼221 ps, reflecting a highly viscous, extensively hydrogen-bonded network. Upon addition of a small amount of water (2w), τc decreases only slightly to ∼216 ps, indicating that initial water incorporation does not significantly disrupt the HB network. Beyond 2w, a dramatic hydration-dependent acceleration is observed: the correlation time (τc) decreases from 221 ps in neat DES to ∼44 ps at 10w (Table S8), reflecting the transition from a highly viscous, extensively hydrogen-bonded network to a more fluid, dynamically exchanging system. This sharp decrease corresponds to the regime where water preferentially solvates chloride anions, destabilizing choline-rich domains and facilitating the breakdown of the extended HB network, as corroborated by 2D NOESY NMR and MD simulations. Beyond 10w, the rate of τc decreases considerably, with τc reaching ∼18 ps at 15w as the system gradually approaches bulk water behavior (∼2 ps), though it remains distinct from pure water due to persistent structural influences from choline and malic acid. This delineates three distinct regimes: minimal disruption at low hydration (≤2w, r1), rapid network breakdown between 2w and 10w (r2), and gradual transition toward water-like dynamics at higher hydration (r3, Fig. 3c). | ||
| Fig. 3 (a) 2D IR spectra of SCN− vibrational reporter in ChCl-MA DES showing evolution with waiting time (Tw) at key hydration levels. Spectra recorded at 1 ps and 5 ps waiting times, with Tw increasing from top to bottom. (b) Experimental frequency–frequency correlation function (FFCF, details in SI, Section 1) decay curves demonstrating dramatic hydration-dependent acceleration, reflecting transition from a highly viscous, extensively hydrogen-bonded network to a fluid, dynamically exchanging system. (c) Three distinct regimes were identified based on the timescales: minimal disruption (≤2w, r1), rapid breakdown (2w–10w, r2), and water-like dynamics (>10w, r3). (d) MD-derived intermittent hydrogen bond autocorrelation functions for MA–MA, MA–Ch, and Ch–Ch interactions showing accelerated decay with increasing hydration. Calculated correlation times exhibit excellent qualitative agreement with experimental FFCF measurements. (e) Anisotropy decay measurements of SCN− at 5w and 10w hydration levels. Faster anisotropy decay at 10w confirms that preferential solvation of the anion facilitates anion rotation within the evolving network structure. | ||
To isolate individual component contributions, binary solutions of 5w-choline chloride and 5w-malic acid were analyzed separately (Fig. S6 and Table S9). The faster dynamics in malic acid solutions (τc ∼6 ps) compared to choline chloride solutions (τc ∼24 ps) confirm that the sequential breakdown observed in the eutectic mixture reflects intrinsic differences in component-water interactions rather than cooperative effects.
The MD-derived hydrogen bond correlation functions provide quantitative validation of these experimental observations (Fig. 3d).27 Intermittent hydrogen bond autocorrelation functions for MA–MA, MA–Ch, and Ch–Ch interactions all show accelerated decay with increasing hydration. The calculated correlation times (τ3, Tables S10–S12) exhibit excellent qualitative agreement with the 2D IR decay times, decreasing from >1000 ps at 0w to ∼100 ps at 10w. It should be noted that MD-calculated hydrogen bond dynamics in viscous ionic liquids and DESs are generally slower than the frequency fluctuations obtained from 2D IR, as the latter involves faster reversible environmental fluctuations that do not result in complete hydrogen bond breaking, whereas MD intermittent analysis specifically tracks full hydrogen bond dissociation and reformation events.32,33
Polarization-selective pump-probe anisotropy measurements further corroborate the hydration-induced acceleration of molecular reorientation (Fig. 3e). The anisotropy decay of SCN− becomes markedly faster with increasing water content, confirming that anion solvation facilitates rotational motion within the evolving network structure (Table S13). This result supports the increasingly mobile environment around chloride anions upon hydration, as evidenced by the 35Cl NMR line narrowing and the formation of dynamic hydration shells observed in MD simulations.
Structure–property-function integration and rational design
The molecular-level picture of sequential nanodomain disruption correlates directly with macroscopic property evolution. Differential scanning calorimetry (DSC) reveals that the glass transition temperature initially increases with hydration up to 5w, consistent with network reorganization rather than simple dilution (Fig. 4a and Table S14). Beyond 5w, the disappearance of Tg and emergence of endothermic transitions signal major structural reorganization coinciding with the experimentally observed nanodomain breakdown.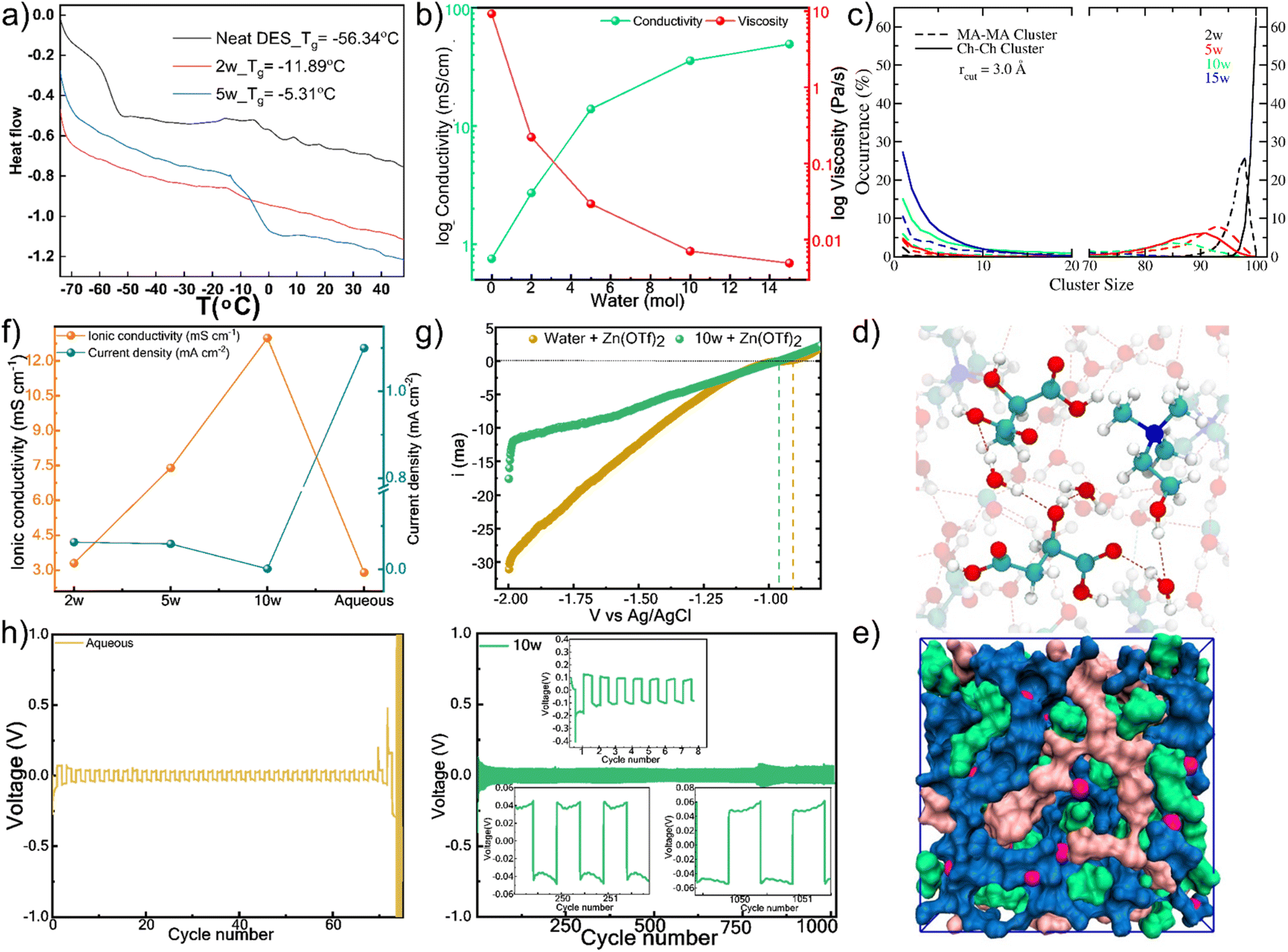 | ||
| Fig. 4 Structure–property-function correlations. (a) Glass transition temperature evolution showing distinct structural reorganization regimes: initial increase up to 5w reflecting network reorganization rather than simple dilution, followed by Tg disappearance beyond 5w signalling major structural transformation coinciding with observed nanodomain breakdown. (b) Macroscopic property evolution demonstrating direct consequences of network disruption: monotonic changes in conductivity and viscosity with diminishing rates of change per water increment, reflecting transition from structured, heterogeneous liquid to homogeneous, water-dominated fluid. (c) MD cluster analysis providing quantitative insight into network fragmentation: probability of largest Ch–Ch clusters and MA–MA clusters drops sharply beyond 10w, representing percolation-like breakdown where networks fragment into isolated clusters rather than gradual erosion. (d) Molecular snapshot of 10w showing the breakdown of DES nano-domains and formation of water-bridged molecular conformations. (e) Molecular snapshot of 10w showing the formation of water channels. (f) Electrochemical impedance spectroscopy in SS‖SS showing ionic conductivity maximum of 13.0 mS cm−1 at 10w hydration, linear polarization resistance (LPR) analysis in Zn‖Zn symmetric cells shows that the corrosion current density is minimized at the 10w hydration level. (g) Linear sweep voltammetry profile demonstrating hydrogen evolution reaction suppression: onset potential shifts negatively by 50 mV compared to aqueous electrolytes, with current density at −2.0 V reduced by 43% due to a unique water-sequestering nanostructure at optimal hydration. (h) Galvanostatic Zn plating/stripping analysis in Zn‖Zn symmetric cells showing stable cycling over 1000 cycles at 1.0 mA cm−2 with low overpotential, directly correlating superior performance with rationally identified optimal nanostructure at 10w hydration level. | ||
Viscosity and proton conductivity measurements demonstrate the macroscopic consequences of network disruption (Fig. 4b and Table S14).43 While both properties change monotonically with hydration, their rates of change diminish with successive water additions, reflecting the transition from structured, heterogeneous liquid to homogeneous, water-dominated fluid. Analysis of the electric double layer capacitance further confirms this evolution, showing a systematic increase in interfacial charge storage capability with water content (Fig. S7), consistent with the formation of a more diffuse and polarizable interface. Temperature-dependent viscosity measurements (Fig. S8 and Table S15) show systematic decreases in activation energy with hydration, confirming that water dismantles energetic barriers to ion transport by disrupting the extended hydrogen-bond network.
MD cluster analysis reveals the nanodomains as dynamic, fluctuating entities rather than static phase-separated domains (Fig. S9). Analysis of different trajectory segments showed temporal variations in cluster size distributions, confirming their transient nature. The cluster sizes for MA–MA, MA–Ch, and Ch–Ch are significantly reduced beyond 10w, along with a concurrent sharp increase in water–water (w–w) cluster size beyond 10w, indicating the formation of more bulk-like water domains (Fig. 4c and S10). This data, supported by the weakening of inter-component cross-peaks in ROESY spectra (Fig. S2), indicates a structural transformation. The system evolves into a state where the DES components form smaller, solvated clusters embedded within a percolating, yet still constrained, water network (Fig. 4d, e and S11).
The microstructural evolution from segregated nanodomains to a homogeneous network fundamentally reshapes the electrode–electrolyte interface. At low hydration, the persistent domain structure creates a rigid interface that restricts ion rearrangement, while at the optimal 10w hydration, the breakdown of domains enables formation of a fluid, dynamic interface where water bridges facilitate ion transport while structurally constraining water molecules away from the electrode surface. This interfacial transformation is quantified by electric double layer capacitance measurements (Fig. S7), which show systematically increasing charge storage capability with hydration, reflecting the transition from a compact to a diffuse, polarizable interface that correlates with the observed electrochemical performance. The fundamental understanding of sequential nanodomain disruption enables rational design of high-performance electrolytes for ZIB. DEE offers a promising approach to enhance the stability and performance of zinc-ion batteries (ZIBs) by suppressing water-induced side reactions and supporting highly reversible zinc (Zn) plating and stripping by modulating the solvation sphere. Ionic conductivity in aqueous electrolytes may be limited due to factors such as Zn2+ aggregation, insufficient ion solvation, and clustering. In contrast, the DEE, with precisely controlled water content, facilitates the regulation of ionic conductivity. Electrochemical impedance spectroscopy on electrolytes containing 0.7 M Zn(OTf)2 reveals that ionic conductivity increases from 3.3 mS cm−1 (2w) to 7.4 mS cm−1 (5w), reaching a maximum of 13.0 mS cm−1 at 10w hydration (Fig. 4f), where both primary nanodomains are disrupted but the system retains sufficient structure to create optimal ion transport pathways.44 Linear polarization resistance (LPR) analysis (Fig. 4f) in Zn‖Zn symmetric cells shows that the corrosion current density is minimized at the 10w hydration level, reaching a value of 0.0015 mA cm−2.
The optimized nanostructure simultaneously suppresses parasitic side reactions. Linear sweep voltammetry (Fig. 4g) shows the hydrogen evolution reaction onset potential shifts negatively by 50 mV compared to aqueous electrolytes, with current density at −2.0 V reduced by 43%.45 The exceptional HER suppression at 10w hydration stems from both reduced water activity and modified Zn2+ solvation structure. MD simulations reveal that at 10w hydration, Zn2+ exhibits a mixed coordination environment with 4–5 water molecules and 1–2 anions (Cl− and/or OTf−) in the primary solvation shell (Fig. S12), representing a significant departure from the conventional [Zn(H2O)6]2+ complex in aqueous electrolytes. This modified coordination, combined with the constrained water network, reduces the availability of readily reducible water molecules at the electrode–electrolyte interface. Plating-stripping analysis in the Zn‖Zn symmetric cells is used to analyze the reversibility and stability of Zn dissolution-deposition in aqueous electrolyte and DES (Fig. 4h). A stable plating-stripping for over 1000 h at 1.0 mA cm−2 with low over potential highlights the ability of DES in suppressing Zn dendrite formation and interfacial stability.46
To validate the practical implications of the identified optimal hydration state, we assembled Zn‖MnO2 full cells with electrolytes at key hydration levels (5w, 10w, and 15w). Cyclic voltammetry (Fig. 5a) carried out at 1 mV s−1 reveals distinct redox behavior dependent on water content, with the 10w electrolyte exhibiting the most pronounced and symmetric peaks, indicating optimal Zn2+ intercalation/deintercalation kinetics with minimal polarization. In contrast, the 5w electrolyte shows sluggish kinetics due to high viscosity, while the 15w electrolyte exhibits weakened peak definition due to excessive dilution of the coordination network. Galvanostatic charge–discharge measurements (Fig. 5b) at 0.1 A g−1 corroborate these findings, with the 10w electrolyte delivering the highest specific capacity and superior cycling stability. These results demonstrate that the nanostructural optimization at 10w hydration, which enables balanced ion transport and stable interfacial chemistry in symmetric cells, directly translates to enhanced performance in practical full-cell configurations.
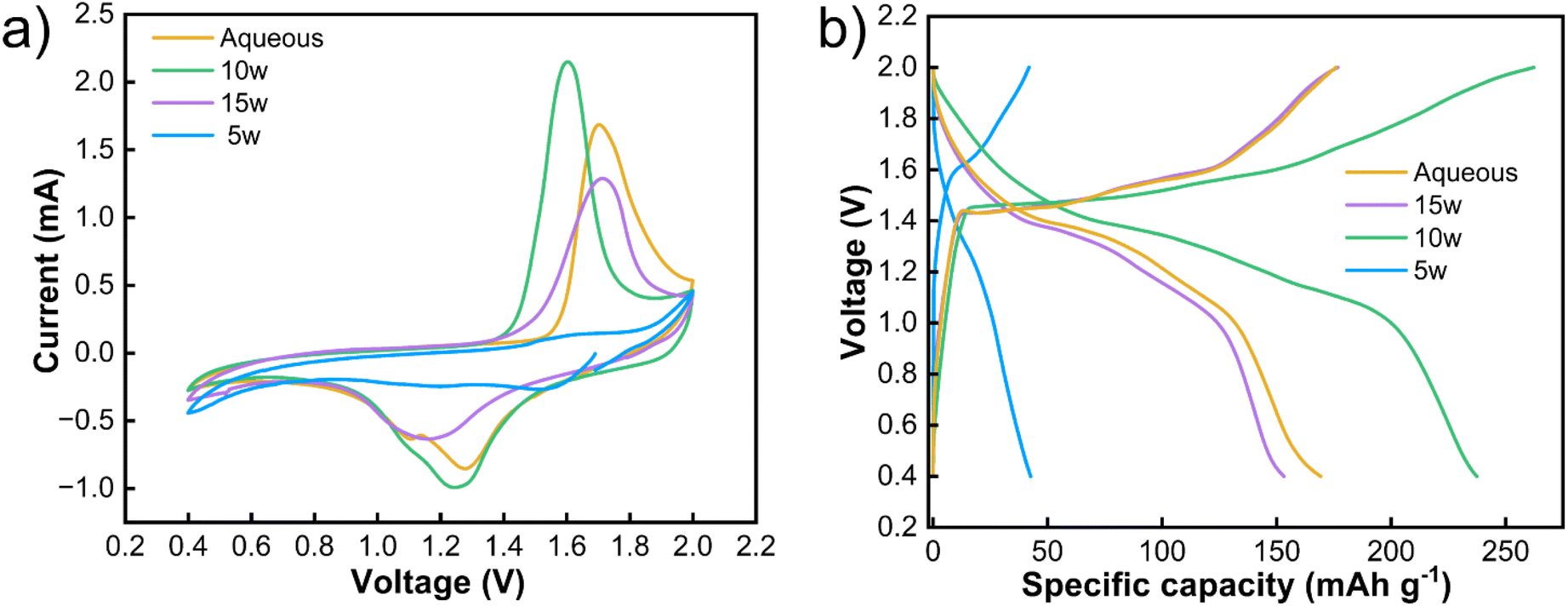 | ||
| Fig. 5 (a) The cyclic voltammograms (CV) at 1 mV s−1 and (b) galvanostatic charge discharge (GCD) profiles of the Zn‖MnO2 full cells employing deep eutectic electrolytes (DEEs) with varying hydration levels at 0.1 A g−1. | ||
The molecular origin of this superior performance lies in the unique water-bridged structure at 10w revealed by MD simulations. Water forms continuous but constrained channels that facilitate ion transport while avoiding bulk water regions that promote side reactions.47 While the percolating water network at 10w facilitates ion conduction, further hydration leads to bulk-like water clusters that increase water activity and promote HER, thereby degrading electrochemical performance. This represents an optimal balance between ionic mobility and structural integrity that cannot be achieved through empirical optimization alone.
Conclusion
This study establishes a comprehensive, molecular-level framework for understanding and engineering the properties of deep eutectic electrolytes through controlled hydration. By integrating multidimensional spectroscopy and molecular simulation, we provide direct experimental evidence for a sequential, component-specific breakdown of transient clusters in ChCl-MA DES. This process is initiated by the thermodynamically favored preferential solvation of chloride anions, which disrupts choline-rich domains at moderate hydration (∼5w), followed by the breakdown of more resilient malic acid-rich networks at higher water content (>10w).The sequential breakdown mechanism represents a fundamental advance in understanding hydration effects in nanostructured liquids. Unlike simple dilution models, our results reveal that water acts as a selective structural disruptor, with the sequence and energetics of breakdown determined by the relative binding affinities of water for different DES components.48 This insight explains the nonlinear property evolution commonly observed in hydrated DESs and provides a predictive framework for rational design.
These nanostructural transformations correlate directly with macroscopic property evolution, culminating in an optimal state around 10w hydration where ionic conductivity is maximized while maintaining structural features that suppress parasitic electrochemical reactions. We demonstrated the practical significance of this fundamental understanding by engineering a high-performance zinc-ion battery electrolyte. At the 10w optimum, the unique water-bridged network simultaneously maximizes ionic transport and minimizes hydrogen evolution, enabling stable long-term cycling that directly results from the rationally identified nanostructure.
The broader implications of this work extend well beyond the specific ChCl-MA system studied here. The sequential breakdown framework provides a generalizable approach for understanding hydration effects in the vast chemical space of DESs and related nanostructured liquids.49,50 We hypothesize that the specific sequence and transition points of nanodomain disruption will be systematically tunable based on the chemical nature of the DES components. For example, replacing chloride with larger, less electronegative halides should predictably shift the onset of network breakdown by altering water-anion binding energies. Similarly, employing hydrogen bond donors with different association strengths than malic acid should modulate the resilience of HBD-rich nanodomains.
Future work systematically mapping these chemical variations will validate the broader applicability of our mechanistic model and establish it as a predictive tool for materials design. The integration of advanced spectroscopic techniques with molecular simulation demonstrated here provides a powerful platform for such investigations. Ultimately, this work establishes clear structure–property-function relationships that enable predictive design of next-generation electrolytes and other functional nanostructured liquids for applications spanning energy storage, catalysis, separation science, and sustainable chemistry.
The sequential breakdown mechanism revealed here represents a new paradigm for understanding and controlling the properties of complex hydrogen-bonded liquids. By moving beyond empirical correlations to mechanistic understanding, this framework opens pathways for rational engineering of nanostructured materials with precisely tailored properties for emerging technological applications.
Author contributions
TC prepared the samples, performed 2D IR experiments and data analysis, bulk property measurements, conducted the literature review, and contributed to project design. AB performed electrochemical experiments and data analysis. RB performed theoretical calculations and analysis. SK supervised electrochemical measurements and data analysis. SR performed NMR experiments, data analysis, and contributed to project design. SB conceptualized and designed the project, supervised experiments and data analysis, and provided overall coordination of the multidisciplinary project components. TC and AB drafted the initial manuscript, which was refined to its final form after discussions and contributions from all authors.Conflicts of interest
The authors declare no conflict of interest.Data availability
Supplementary information (SI): materials and methods, additional spectroscopic data, molecular dynamics simulation details, tables of diffusion coefficients, FFCF decay parameters, anisotropy decay parameters, thermal properties, electrochemical data, and hydrogen bond correlation functions. See DOI: https://doi.org/10.1039/d5sc06819c.Acknowledgements
We thank the CSIR-National Chemical Laboratory for providing research facilities. T. C. and A. B. acknowledge CSIR for research fellowships. S. B. acknowledges CSIR-NCL and ANRF, India (Grant No. CRG/2022/004454), for the financial support. We acknowledge the National Supercomputing Mission (NSM) for providing computing resources of ‘PARAM Brahma’ at IISER Pune and ‘PARAM Seva’ at IIT Hyderabad, which are implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India. R. B. acknowledges IIT Madras for partial computational support via the AQUA HPC facility.References
- E. L. Smith, A. P. Abbott and K. S. Ryder, Deep Eutectic Solvents (DESs) and Their Applications, Chem. Rev., 2014, 114(21), 11060–11082 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Deep Eutectic Solvents: A Review of Fundamentals and Applications, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- W. Xiong, X. Zhang, Z. Tu, X. Hu and Y. Wu, Novel Deep Eutectic Electrolyte Induced by Na···N Interactions for Sodium Batteries, Ind. Eng. Chem. Res., 2023, 62, 51–61 CrossRef CAS.
- W. Wang, M. M. Sabugaa, S. Chandra, Y. P. Asmara, B. A. Alreda, N. Ulloa, Y. Elmasry and M. M. Kadhim, Choline chloride-based deep eutectic solvents as electrolytes for wide temperature range supercapacitors, J. Energy Storage, 2023, 71, 108141 CrossRef.
- S. Azmi, M. F. Koudahi and E. Frackowiak, Reline deep eutectic solvent as a green electrolyte for electrochemical energy storage applications, Energy Environ. Sci., 2022, 15, 1156–1171 RSC.
- P. Luo, Y. Wang, W. Zhang, Z. Huang, F. Chao, Y. Yuan, Y. Wang, Y. He, G. Yu, D. Zhu, Z. Wang, H. Tang and Q. An, Water-Deficient Interface Induced via Hydrated Eutectic Electrolyte with Restrictive Water to Achieve High-Performance Aqueous Zinc Metal Batteries, Small, 2025, 21, 2410946 CrossRef CAS PubMed.
- M. Sun, K. Wan, Y. Huang, H. Yang, X. Zhou, C. Yan, J. Zhou and T. Qian, Playing with Water Molecules: “Repulsing” or “Trapping” to Exclude Water-Induced Side Reactions on Zn Metal Anode, Adv. Funct. Mater., 2025, 35, 2417890 CrossRef CAS.
- Q. Liu, J. Li, D. Xing, Y. Zhou and F. Yan, Ternary Eutectic Electrolyte for Flexible Wide-Temperature Zinc-Ion Batteries from −20 °C to 70 °C, Angew. Chem., Int. Ed., 2025, 64, e202414728 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, The Effect of Water upon Deep Eutectic Solvent Nanostructure: An Unusual Transition from Ionic Mixture to Aqueous Solution, Angew. Chem., Int. Ed., 2017, 56(33), 9782–9785 CrossRef CAS PubMed.
- T. Zhekenov, N. Toksanbayev, Z. Kazakbayeva, D. Shah and F. S. Mjalli, Formation of type III Deep Eutectic Solvents and effect of water on their intermolecular interactions, Fluid Phase Equilib., 2017, 441, 43–48 CrossRef CAS.
- H. Srinivasan, V. K. Sharma and S. Mitra, Can the microscopic and macroscopic transport phenomena in deep eutectic solvents be reconciled?, Phys. Chem. Chem. Phys., 2021, 23(40), 22854–22873 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Liquid structure of the choline chloride-urea deep eutectic solvent (reline) from neutron diffraction and atomistic modelling, Green Chem., 2016, 18, 2736–2744 RSC.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Doubly ionic hydrogen bond interactions within the choline chloride–urea deep eutectic solvent, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- S. L. Perkins, P. Painter and C. M. Colina, Experimental and Computational Studies of Choline Chloride-Based Deep Eutectic Solvents, J. Chem. Eng. Data, 2014, 59, 3652–3662 CrossRef CAS.
- H. de Araujo Chagas, G. Colherinhas and E. E. Fileti, Unveiling the Potential of Natural Deep Eutectic Solvents in Electrochemical Energy Storage Applications, ACS Phys. Chem. Au, 2025 DOI:10.1021/acsphyschemau.5c00063.
- O. S. Hammond, D. T. Bowron, A. J. Jackson, T. Arnold, A. Sanchez-Fernandez, N. Tsapatsaris, V. Garcia Sakai and K. J. Edler, Resilience of Malic Acid Natural Deep Eutectic Solvent Nanostructure to Solidification and Hydration, J. Phys. Chem. B, 2017, 121(31), 7473–7483 CrossRef CAS PubMed.
- X. Lin, G. Zhou, M. J. Robson, J. Yu, S. C. T. Kwok and F. Ciucci, Hydrated Deep Eutectic Electrolytes for High-Performance Zn-Ion Batteries Capable of Low-Temperature Operation, Adv. Funct. Mater., 2022, 32, 2109322 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, Cation-Deficient Spinel ZnMn2O4 Cathode in Zn(CF3SO3)2 Electrolyte for Rechargeable Aqueous Zn-Ion Battery, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- J. Pan, A. P. Charnay, W. Zheng and M. D. Fayer, Revealing Lithium Ion Transport Mechanisms and Solvation Structures in Carbonate Electrolytes, J. Am. Chem. Soc., 2024, 146, 35329–35338 CrossRef CAS PubMed.
- J. Pan, K. A. Carter-Fenk, S. T. Hung, N. Dao, J. N. S. Smith and M. D. Fayer, Dynamics of Deep Eutectic Mixtures of Tetraethylammonium Halides/Ethylene Glycol Investigated with Ultrafast Infrared Spectroscopy, J. Phys. Chem. B, 2025, 129, 2718–2729 CrossRef CAS PubMed.
- K. Kwak, J. Jeon, S. Y. Chun and M. Cho, Ion Networks in Water-based Li-ion Battery Electrolytes, Acc. Chem. Res., 2025, 58, 199–207 CrossRef CAS PubMed.
- J. Jeon, H. Lee, J.-H. Choi and M. Cho, Modeling and Simulation of Concentrated Aqueous Solutions of LiTFSI for Battery Applications, J. Phys. Chem. C, 2020, 124, 11790–11799 CrossRef CAS.
- S. Y. Chun, J. W. Shim, K. Kwak and M. Cho, Molecular Photothermal Effect on the 2D-IR Spectroscopy of Acetonitrile-Based Li-Ion Battery Electrolytes, J. Phys. Chem. Lett., 2024, 15, 7302–7311 CrossRef CAS PubMed.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, John Wiley & Sons, Chichester, England, 2008, pp. 752 Search PubMed.
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer, M. Rance, N. J. Skelton, Protein NMR spectroscopy: Principles and practice, Academic Press, 2nd edn, Burlington, 2007, pp. 912 Search PubMed.
- T. Chowdhury, S. Chatterjee, S. H. Deshmukh and S. Bagchi, A Systematic Study on the Role of Hydrogen Bond Donors in Dictating the Dynamics of Choline-Based Deep Eutectic Solvents, J. Phys. Chem. B, 2023, 127(33), 7299–7308 CrossRef CAS PubMed.
- S. Chatterjee, S. H. Deshmukh, T. Chowdhury and S. Bagchi, Viscosity effects on the dynamics of diols and diol-based deep eutectic solvents, Photochem. Photobiol., 2024, 100, 946–955 CrossRef CAS PubMed.
- S. Chatterjee, T. Chowdhury and S. Bagchi, Solvation Dynamics and Microheterogeneity in Deep Eutectic Solvents, J. Phys. Chem. B, 2024, 128(51), 12669–12684 CrossRef CAS PubMed.
- S. S. Sakpal, S. H. Deshmukh, S. Chatterjee, D. Ghosh and S. Bagchi, Transition of a Deep Eutectic Solution to Aqueous Solution: A Dynamical Perspective of the Dissolved Solute, J. Phys. Chem. Lett., 2021, 12, 8784–8789 CrossRef CAS PubMed.
- M. D. Fayer, Dynamics of Water Interacting with Interfaces, Molecules, and Ions, Acc. Chem. Res., 2012, 45(1), 3–14 CrossRef CAS PubMed.
- S. A. Yamada, W. H. Thompson and M. D. Fayer, Water-anion hydrogen bonding dynamics: Ultrafast IR experiments and simulations, J. Chem. Phys., 2017, 146(23), 234501 CrossRef PubMed.
- J. B. Asbury, T. Steinel, C. Stromberg, S. A. Corcelli, C. P. Lawrence, J. L. Skinner and M. D. Fayer, Water Dynamics: Vibrational Echo Correlation Spectroscopy and Comparison to Molecular Dynamics Simulations, J. Phys. Chem., 2004, 108(7), 1107–1119 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- S. J. Gibbs and C. S. Johnson, A PFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents, J. Magn. Reson., 1991, 93, 395–402 Search PubMed.
- T. Brand, E. J. Cabrita and S. Berger, Intermolecular interaction as investigated by NOE and diffusion studies, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 159–196 CrossRef CAS.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Molecular motion and ion diffusion in choline chloride based deep eutectic solvents studied by 1H pulsed field gradient NMR spectroscopy, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- S. Kaur, A. Malik and H. K. Kashyap, Anatomy of Microscopic Structure of Ethaline Deep Eutectic Solvent Decoded through Molecular Dynamics Simulations, J. Phys. Chem. B, 2019, 123, 8291–8299 CrossRef CAS PubMed.
- S. Spittle, D. Poe, B. Doherty, C. Kolodziej, L. Heroux, M. A. Haque, H. Squire, T. Cosby, Y. Zhang, C. Fraenza, S. Bhattacharyya, M. Tyagi, J. Peng, R. A. Elgammal, T. Zawodzinski, M. Tuckerman, S. Greenbaum, B. Gurkan, C. Burda, M. Dadmun, E. J. Maginn and J. Sangoro, Evolution of microscopic heterogeneity and dynamics in choline chloride-based deep eutectic solvents, Nat. Commun., 2022, 13(1), 219 CrossRef CAS PubMed.
- S. Chatterjee, T. Chowdhury and S. Bagchi, Does variation in composition affect dynamics when approaching the eutectic composition?, J. Chem. Phys., 2023, 158(11), 114203 CrossRef CAS PubMed.
- Y. Cui, K. D. Fulfer, J. Ma, T. K. Weldeghiorghis and D. G. Kuroda, Solvation dynamics of an ionic probe in choline chloride-based deep eutectic solvents, Phys. Chem. Chem. Phys., 2016, 18(46), 31471–31479 RSC.
- T. Chowdhury, A. Pathania, S. Ravindranathan and S. Bagchi, Probing solvent fluctuations in deep eutectic solvents: Influence of probe charge and nano-domain localization, J. Chem. Phys., 2025, 163(4), 044506 CrossRef CAS PubMed.
- H. Kivelä, M. Salomäki, P. Vainikka, E. Mäkilä, F. Poletti, S. Ruggeri, F. Terzi and J. Lukkari, Effect of Water on a Hydrophobic Deep Eutectic Solvent, J. Phys. Chem. B, 2022, 126, 513–527 CrossRef PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Recent Advances in Zn-Ion Batteries, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- K. Roy, A. Rana, T. K. Ghosh, J. N. Heil, S. Roy, K. J. Vannoy, B. M. Tackett, M. Chen and J. E. Dick, How Solvation Energetics Dampen the Hydrogen Evolution Reaction to Maximize Zinc Anode Stability, Adv. Energy Mater., 2024, 14, 2303998 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Highly reversible zinc metal anode for aqueous batteries, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- M. McEldrew, Z. A. H. Goodwin, A. A. Kornyshev and M. Z. Bazant, Theory of the Double Layer in Water-in-Salt Electrolytes, J. Phys. Chem. Lett., 2018, 9, 5840–5846 CrossRef CAS PubMed.
- Y. Marcus, Preferential solvation in mixed solvents. Part 11. Eight additional completely miscible aqueous co-solvent binary mixtures and the relationship between the volume-corrected preferential solvation parameters and the structures of the co-solvents, Phys. Chem. Chem. Phys., 2002, 4(18), 4462–4471 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, Natural Deep Eutectic Solvents – Solvents for the 21st Century, ACS Sustain. Chem. Eng., 2014, 2(5), 1063–1071 CrossRef CAS.
- T. El Achkar, H. Greige-Gerges and S. Fourmentin, Basics and properties of deep eutectic solvents: a review, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |