
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring the efficiency of synthetic routes and transformations using vectors derived from similarity and complexity
Samuel
Genheden
 a and
Gareth P.
Howell
*b
a and
Gareth P.
Howell
*b
aMolecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Pepparedsleden 1, SE-431 83 Mölndal, Sweden
bChemical Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield SK10 2NA, UK. E-mail: gareth.howell@astrazeneca.com
First published on 21st August 2025
Abstract
With the aim of providing new tools for the design and assessment of synthetic routes, we describe an approach that mimics human interpretation whilst being highly amenable to machine implementation. The representation of molecular structures as 2D-coordinates derived from molecular similarity and complexity allows individual transformations to be viewed as vectors (reactant to product) where the magnitude and direction of travel can be used to assess and quantify efficiency. Using a dataset comprising 640k literature syntheses and 2.4m reactions taken from six journals between 2000 and 2020, we show that vectors derived in this way follow logical patterns when grouped by reaction type. Similarly, complete synthetic routes can be visualised as sequences of head-to-tail vectors traversing the range between starting material and target, allowing the efficiency with which this range is covered to be quantified. Three applications of the methodology are demonstrated: a comparison of CASP performance between two versions of AiZynthFinder for generating synthetic routes to 100k ChEMBL targets, analysis of predicted routes to a specific target molecule and, finally, a perspective on how the efficiency of published synthetic routes has changed over the last two decades.
1. Introduction
The assessment and comparison of synthetic routes in organic chemistry can, after suitable training and experience, be readily accomplished by humans. Regardless of the criteria we are assessing against (cost, time, waste, etc.), someone “skilled in the art” can make a judgement as to whether the route represents a logical and efficient series of chemical transformations. This is typically achieved by considering the structural complexity of the target and assessing the number and type of transformations present, the order in which they are carried out and any reliance on protecting groups, auxiliaries, etc.To do the same assessment on hundreds or thousands of synthetic routes, our suitably trained chemist quickly becomes the rate-limiting step. If empirical information (e.g., yield or waste) is available, then automation is trivial. If such information is either unavailable or unreliable, for example at the route design stage, the task becomes significantly more challenging due to the sparsity of meaningful or generally accepted metrics.
Step count – either longest linear sequence (LLS) or total – is by far the most common gauge against which synthetic routes are assessed. It is easy to conceptualise, machine-interpretable and a reasonable predictor of the quantitative metrics we are ultimately interested in. If defined and counted consistently, it is a reliable tool for comparing synthetic routes to a specified target – with fewer steps usually being better. Unfortunately, step counting is subject to major inconsistencies within the synthetic-organic community. It is clear that step-counting should stop when the desired target structure is reached, but there is no accepted convention for when to begin. Typically, step-counting begins at the first material (working backwards) that can be purchased, regardless of cost or availability. Alternatively, counting begins at the first material whose synthesis has been reported and deemed “simple”. These approaches are practical, since exhaustively step-counting back to hydrocarbons and biomass feedstock is unrealistic. The result is a high degree of inconsistency, however, with unspecified steps upstream of the starting materials being unaccounted for.
Assessment of a synthetic route based on its constituent reaction types is often informative and can be automated using commercial software such as NameRxn1 or InfoChem.2 Certain reaction types, for example redox manipulations or functional group interconversions (FGI), can be penalised in favour of “constructive” steps where bonds present in the target skeleton are formed. This strategy remains challenging since the classification of reactions is prone to failure, particularly when considering novel or tandem/cascade transformations. Furthermore, the binary assignment of transformations as productive or non-productive is somewhat limiting for the purposes of comparison or ranking.
Other metrics, such as atom economy,3 step economy,4–6 redox economy,7 ideality8 and convergence9 have been reported to assess aspects of efficiency relating to synthetic routes. These concepts are all eminently logical and automatable, provided fully atom-mapped synthetic sequences are available (including reagents, which can be far from accurate);10 none are routinely used, however, when assessing or reporting synthetic routes. In computer-aided synthesis planning (CASP) and in the development of retrosynthesis analysis algorithms, there is no general agreement on how to assess route predictions, especially with respect to quality.11,12
With the above in mind, we have an interest in novel, automatable strategies for the assessment of synthetic routes, primarily at the route design stage, that are less reliant on step count and do not require empirical data (yield, etc.), atom mapping or reaction class assignment.
2. Results and discussion
2.1. Dataset compilation
We recently published an analysis of approximately 640![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 synthetic routes and 2.4 million reactions from the period 2000 to 2020.13 As sources, Angewandte Chemie International Edition,14The European Journal of Medicinal Chemistry,15The Journal of Medicinal Chemistry,16The Journal of Organic Chemistry,17Organic Letters18 and Organic Process Research and Development19 were used. Automated reaction classification gave a success rate of 68%.1 A wide range of trends were identified, describing various properties of synthetic routes, route targets and their constituent reactions. The same dataset was used here with some modifications. Routes where the starting material was found to be more complex than the route target (approx. 5% of routes) were removed. Routes leading to a target featuring a common protecting group were tagged and excluded from some of the analyses presented below.†
000 synthetic routes and 2.4 million reactions from the period 2000 to 2020.13 As sources, Angewandte Chemie International Edition,14The European Journal of Medicinal Chemistry,15The Journal of Medicinal Chemistry,16The Journal of Organic Chemistry,17Organic Letters18 and Organic Process Research and Development19 were used. Automated reaction classification gave a success rate of 68%.1 A wide range of trends were identified, describing various properties of synthetic routes, route targets and their constituent reactions. The same dataset was used here with some modifications. Routes where the starting material was found to be more complex than the route target (approx. 5% of routes) were removed. Routes leading to a target featuring a common protecting group were tagged and excluded from some of the analyses presented below.†
2.2. Similarity as a measure of route progression
We described the use of similarity (structural commonality between two molecules) to gauge the progress of a synthetic route in 2024.20 We would generally expect the starting material (sm) to be least similar to the route target and, as we move along the synthetic route, the reaction products (intermediates) should become increasingly similar to the route target; the final reaction in the route will necessarily deliver a product that is (identical to) the route target.To quantify similarity, we will use two approaches. Molecular fingerprints, as illustrated in Fig. 1, are widely used in cheminformatics for applications including structure activity relationship (SAR) analysis, virtual library screening and computer-aided synthesis planning (CASP). Amongst the various fingerprint types, Morgan fingerprints21 are popular for fast comparison of small molecules and can be easily generated from the Simplified Molecular Input Line Entry System (SMILES) strings of any pair of molecules using RDKit.22 As a mathematical measure of the similarity between two fingerprints, the Tanimoto coefficient is routinely used and yields values (SFP) between 0 (no similarity) and 1 (identical).23
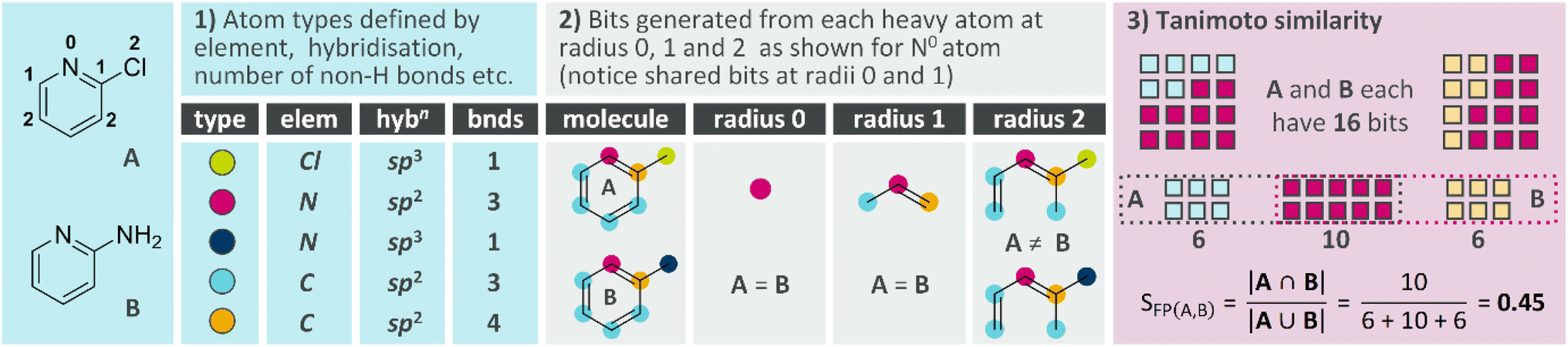 | ||
| Fig. 1 Morgan fingerprints and Tanimoto similarity between A and B. | ||
For comparison, we used an alternative measure of similarity: Maximum Common Edge Subgraph (MCES),24 which can also be generated using SMILES strings.22 This approach, summarised in Fig. 2, compares two molecular structures (graphs) and finds the largest fragment, or MCES, that is present in both. Tanimoto similarity was again used, this time to compare the number of atoms and bonds in the MCES with those in the two comparator molecules. This metric also yields values (SMCES) between 0 (no similarity) and 1 (identical).
 | ||
| Fig. 2 Maximum common edge subgraph (MCES) between A and B. | ||
For a given synthetic route, similarity measures (SFP, SMCES) between the target and all preceding intermediates can be generated, as shown in Fig. 3, for the synthesis of cell division cycle 25B (CDC25B) phosphatase inhibitor 6.25 The starting fingerprint similarity value (SFP 0.35) for naphthalene 1 is lower than the corresponding MCES similarity (SMCES 0.50). In step (a) (Boc-protection), there is a drop in both similarity metrics (ΔSFP, ΔSMCES = −0.07). This is logical since six heavy atoms (COC[CH3]3) have been introduced in an arrangement that does not feature in the target structure 6. In step (b) (C–N coupling), aniline 3 is added to give compound 4 which has significant positive impact on target similarity (ΔSFP = +0.12, ΔSMCES = +0.28). In the following Boc-deprotection step (c), positive changes are again observed (ΔSFP = +0.25, ΔSMCES = +0.21) since the previously added COC(CH3)3 fragment, which does not feature in target 6, is now being removed. The final transformation (d, ester hydrolysis/deprotection) shows a much lower change in MCES similarity (ΔSMCES = +0.08) compared to fingerprint similarity (ΔSFP = +0.36).
 | ||
| Fig. 3 Similarity (SFP and SMCES) changes during the synthesis of phosphatase inhibitor 6. | ||
As synthetic chemists, we would identify only one of the four transformations as being “productive” (step b); the other three protecting group manipulations would be considered non-ideal.8 The two similarity metrics both yield negative values (−ΔS) for the first step (a) and we would interpret this as non-productive since structurally, we are moving further away from our desired target. By this interpretation, however, both similarity metrics suggest steps (c and d), two deprotection operations, to be productive (+ΔS). In the case of SFP, both deprotections have a larger, positive ΔS value than the C–N coupling reaction (b).
We can obtain a more logical interpretation by adding a second descriptor to describe the magnitude of structural change taking place in each transformation. The changes in molecular weight for each step along the route might suffice here but, for our purposes of route assessment, we are interested in more than mass variation. Ideally, we are aiming to generate some measure of route efficiency related to cost, waste, time, etc. and, since this information is seldom available directly, we will use a molecular complexity metric as a surrogate. There is an important assumption to recognise here: we are assuming the “complexity” of a molecule is proportional to how easily it is obtained or synthesised, and therefore the implicit cost, time and waste. For the most part this seems reasonable in that “complex” molecules, where there is a variety of atom types, bond orders and ring systems are generally more challenging to obtain than “less complex” molecules. We must be mindful, however, that this assumption does not hold for complex molecules that are readily available (e.g., steroids, carbohydrates).
2.3. Similarity (S) and complexity (C) as Cartesian coordinates
There are numerous molecular complexity metrics that might be used here.26–32 We will demonstrate our approach using a path-based complexity metric, CM*,33 that we have recently shown to be useful as a predictor of process mass intensity (PMI).34 This metric, which can be easily generated from a SMILES string,22 does not make any special consideration of symmetry or chirality. For comparison, some of the analyses described below were repeated using spacial scores35 in place of CM*, leading to the same overall conclusions (see SI).In the same way that we have used similarity (SFP and SMCES) to measure progress along a synthetic route, we will use CM* to gauge the magnitude of structural change and show that similarity and complexity can operate in opposing directions, providing complementary information. After cleaning, the observed dataset values for similarity (SFP and SMCES) necessarily lie in the range 0 to 1 whilst the observed values for CM* vary between 3.5 and 12.0. To ensure equal weight is given to all metrics, CM* values were normalised (nC) to the range 0–1.‡
Still considering the synthesis of 6 (Fig. 3), two route plots can be generated using similarity (SFP and SMCES for comparison) and complexity as Cartesian co-ordinates, are shown in Fig. 4. Each transformation is now represented as a vector with magnitude and direction. There are various observations to be made here:
 | ||
| Fig. 4 Vector plots derived from similarity and complexity for the synthesis of phosphatase inhibitor 6 (Fig. 3). | ||
• The starting material must necessarily be to the left of the route target, since it must be less similar to the target than the target itself.
• The starting material is of lower complexity than the target (routes that do not satisfy this criterion were removed) and so the overall direction of travel from starting material to target will be in the direction +S (right) and +C (up).
• Transformations that are “productive” should therefore travel in the direction +S (right) and +C (up).
This 2D-coordinate system gives more information about the individual transformations than similarity alone. Step (a) (Boc-protection) is non-productive since complexity is added without an increase in target similarity, giving a vector that moves up and left. Step (b) (C–N coupling) is productive since it increases complexity and similarity toward that of the target (the vectors move up and right). The final deprotection operations (c and d) increase target similarity, as demonstrated in Fig. 3, but are associated with a decrease in complexity. The vectors move down and right which signifies “wasted” complexity (molecular structure that was not needed) and can be viewed as non-productive. Compared to similarity alone, the use of similarity and complexity gives a more logical assessment of the individual transformations in the route.
2.4. Vectors and transformation efficiency (ηT) by reaction class
Our next objective was to determine whether the basic trends identified above were consistent throughout the entire dataset, and whether certain reaction types result in vectors of a particular direction or magnitude. In Fig. 5, a general synthetic route is depicted leading from starting material A to target T, via intermediate B. Each individual step (AB, BT) is a vector with an associated change in similarity (ΔS) and complexity (ΔC), each of which can be positive or negative. The vector vmin represents the “ideal” route (if such a transformation could be achieved in a single step) leading directly from A to T. The magnitude of this line ‖vmin‖ represents the total amount of structural change or “work” that is required across the route. The “efficiency” (ηT), or contribution of each step to the overall route, is calculated as the scalar projection of its vector onto vmin.36 Efficiency values can be negative (which effectively lengthens vmin) leaving more work for the remaining steps in the route to achieve. | ||
| Fig. 5 Derivation of synthetic range (vmin), and transformation efficiency (ηT). | ||
Analysis of dataset reactions is shown in Fig. 6. The distributions of values for ΔSFP, ΔSMCES, ΔnC and ηT (derived from SMCES values) are grouped by ten main reaction super-classes,37 which do indeed display characteristic patterns.
 | ||
| Fig. 6 Distribution of similarity changes (left), complexity changes (centre) and efficiency values (right) for those 850k reactions where automatic reaction class assignment was successful. Whiskers show 5th and 95th percentiles, boxes show 25th, 50th and 75th percentiles. Note: to avoid influencing protection and deprotection classes, reactions from routes where the target features a common protecting group were not included (see SI). | ||
• Carbon–carbon bond forming reactions are predominantly associated with +ΔS and +ΔC changes, which indicates a productive reaction. The efficiency ηT values are the highest of all ten reaction super-classes.
• Heteroatom alkylation/arylation, heteroatom acylation and heterocycle formation show the same +ΔS and +ΔC mean values although the efficiency ηT values are slightly lower.
• Deprotection reactions yield −ΔnC values in conjunction with +ΔS values. The mean efficiency value ηT is positive but significantly less than for the four super-classes mentioned above.
• Redox, functional group interconversion (FGI) and addition (FGA) reactions show very low (close to zero) mean values for ΔS, ΔC and ηT.
• Protection reactions are unique (and in keeping with our earlier observations) in that they are associated with −ΔS and +ΔC values. This is the only reaction super-class with a negative mean ηT value.
Crucially, this definition of transformation efficiency ηT is not binary, in that reaction super-classes are not wholly designated as productive or non-productive. An acylation reaction, for example, can have low or negative ηT if the fragment being introduced represents a small proportion of the target structure (e.g., a methyl ester) or is not present at all in the target structure (e.g., a methyl ester that is functioning as a protecting group).
The distribution of ΔSMCES values is generally narrower than that of ΔSFP, particularly so for redox, FGI and FGA reactions. This is due to subtleties in the way that the fingerprint and MCES similarity algorithms operate. The fingerprint similarity approach is more sensitive to changes in bond order and can be significantly impacted by relatively small, single atom changes.38 This can be illustrated by examining how mean ΔS values change depending on proximity (in terms of step count) to the route target, as shown Fig. 7. The fingerprint based metric SFP is much more sensitive to the final changes taking place in the synthetic route (i.e., in the final step) than the alternative SMCES metric. It is possible to tune the way atom and bond types are distinguished in the fingerprint algorithm (or use a different fingerprint type), which would likely alleviate this problem, but we would suggest the SMCES metric to be more suitable and will use it from hereon.
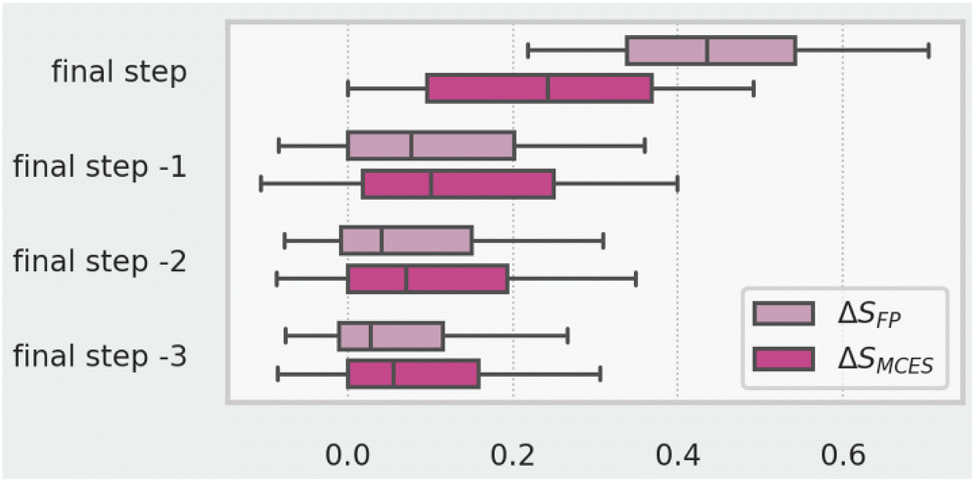 | ||
| Fig. 7 Comparison of similarity metrics vs. step location for 553k reactions. Whiskers show 5th and 95th percentiles, boxes show 25th, 50th and 75th percentiles. | ||
2.5. Route efficiency (ηR) and penalised route efficiency (ηPR)
We can apply the various measures described above to whole synthetic routes. The synthetic range (‖vmin‖) is useful for comparing routes that begin at differing starting materials but end at the same target. Step count is by far the most common metric for comparing routes but there are no specific rules for what designates an acceptable starting material; this frequently leads to unfair or inaccurate comparisons. The use of ‖vmin‖ allows us to quantify how different a starting material is from a route target in terms of similarity and complexity.Further to this, if we accept that (i) vmin represents the shortest possible route (a single transformation) from starting material to route target and (ii) productive transformations should move towards the route target when plotted on similarity, complexity axes, then we might assess the efficiency of a route using simple path comparison, as shown in Fig. 8.
 | ||
| Fig. 8 Route efficiency μR demonstrated with the synthesis of pyridol 12. Tabulated values show transformation efficiency ηT and magnitude ‖v‖ for each step. | ||
Pyridol 12 is synthesised in four steps from 7.39 The synthetic range of the route ‖vmin‖ is relatively large since the starting material is of lower complexity than the route target and bears limited similarity to it. The actual path of the route (a–d) is very close to the “ideal” path vmin (note the y-axis scale) and, as shown, a simple ratio of the magnitudes (lengths) of the ideal to actual path gives a measure of the efficiency ηR, where values can range from 0 (low efficiency) to 1 (perfect efficiency). This measure is both easy to conceptualise and there is no inherent need to examine or chemically interpret the actual transformations taking place. Routes that feature significant detours (e.g., protecting group manipulations, auxiliaries, complex leaving groups) are immediately obvious both mathematically and visually, as demonstrated in Fig. 9. To O-methylate 13 chemo- and regioselectively,40 global protection and deprotection was necessary, eventually yielding O-GlcNAcase inhibitor 18. The synthetic range of the route is very small (‖vmin‖ = 0.06) since little overall structural change is taking place whilst the actual path length (∑‖v‖) is very large due to the significant “detours”; the result is a low efficiency value (μR = 0.05).
 | ||
| Fig. 9 Protecting group manipulations leading to low route efficiency ηR as demonstrated by the synthesis of O-GlcNAcase inhibitor 18. Tabulated values show transformation efficiency ηT and magnitude ‖v‖ for each step. | ||
This definition of route efficiency is wholly agnostic towards step count: ηR is not influenced by the number of steps, only the direction in which they travel. Whilst this partially satisfies our original aim, it introduces a significant shortcoming: transformations with very small or zero magnitude (e.g., redox, FGI, FGA) or those which offer small, incremental progress in a direction similar to vmin are not penalised. Whilst we are trying to develop a theoretical analysis that is not governed by step count, we cannot ignore the fact that fewer steps is generally better. As shown in Fig. 10, it is possible for the real path of a route to be identical to vmin but also comprise low-efficiency transformations. Conversion of alcohol 19 to alkene 23 is achieved with very high stereoselectivity.41 In terms of introducing the heavy atoms present in the target skeleton, the entirety of the work is achieved in step (b) where iodide 20 is reacted with 1-trimethylsilylpropyne. Regarding similarity (SMCES) and complexity, steps a (FGI), c (alkyne migration) and d (hydrogenation) effect no change and display zero efficiency ηT values. The resulting route efficiency ηR is perfect, however, since our analysis ignores zero-magnitude vectors and effectively treats this as a single step transformation (19–23).
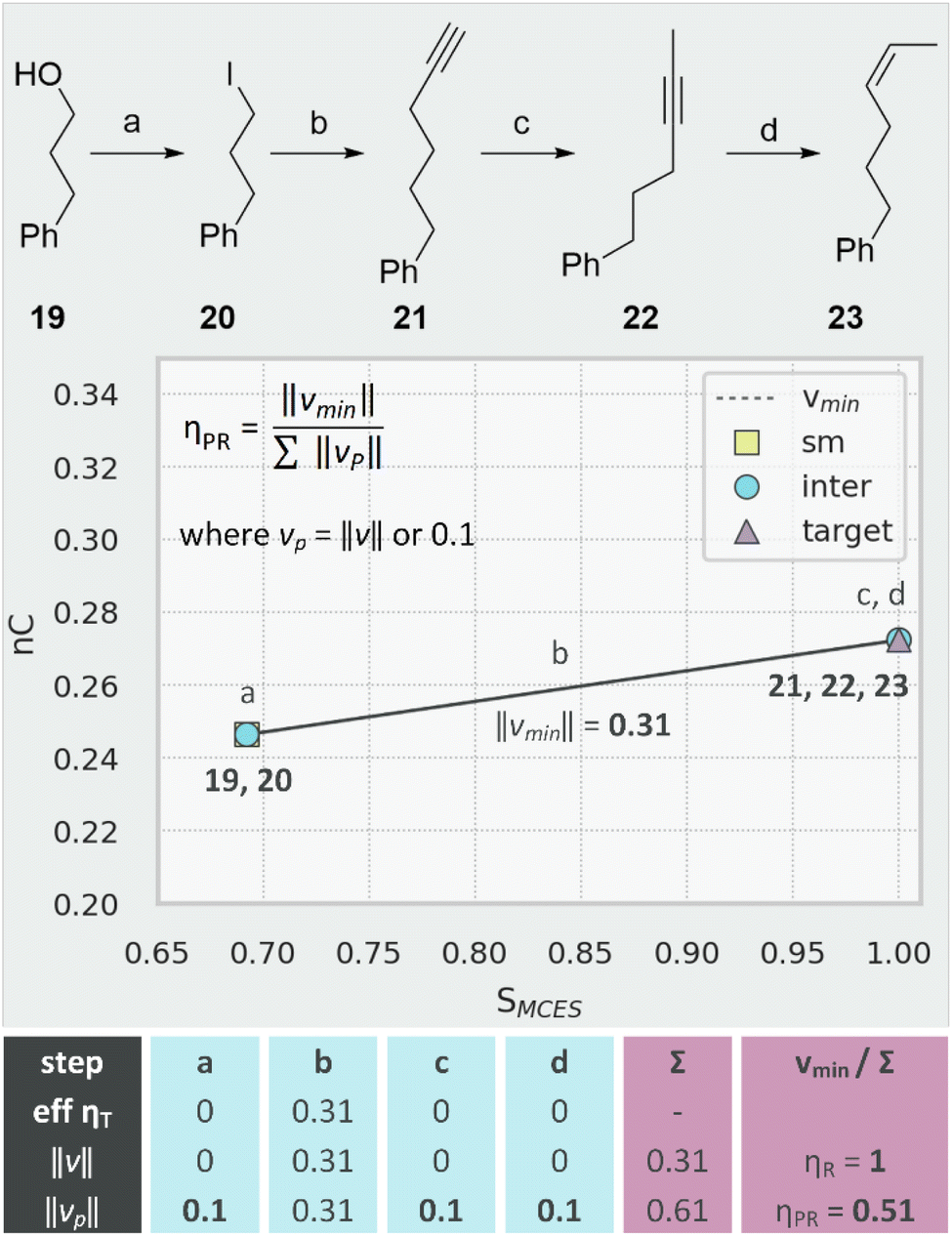 | ||
| Fig. 10 Penalised route efficiency ηPR demonstrated by the synthesis of 23. Tabulated values show transformation efficiency μT, magnitude ‖v‖ and penalised magnitude ‖vp‖ for each step. | ||
This shortcoming can be remedied by using a penalised function for route efficiency (ηPR) where the minimum path length for any single step is set to 0.1. As shown in Fig. 10, this significantly reduces the efficiency for the synthesis of 23 (ηR = 1, ηPR = 0.51) but would have much less impact on the route to 12 (Fig. 8, ηR = 0.94, ηPR = 0.84). The minimum value of 0.1 (a somewhat arbitrary value chosen by inspection of the data in Fig. 6) can, of course, be tuned to vary the extent to which low efficiency transformations are penalised. This penalised efficiency metric might be considered as an amalgam of atom economy3 and ideality8 since wasted molecular structure and inefficient transformations are disfavoured.
2.6. Convergent routes
Up to this point, we have only discussed linear routes with no parallel synthetic branches. The definition of a convergent route is again somewhat subjective since it depends on whether any fragment incoming to the LLS is designated as a starting material i.e., its synthesis is not included. Basic inspection of the dataset shows that many routes reported (or retrieved from Reaxys) as linear have complex incoming fragments part-way through the LLS and should therefore probably be designated as branched or convergent.Regardless of this inconsistency, Fig. 11 (left) shows that for all route lengths, convergent routes show higher penalised route efficiency values μPR than linear routes and (Fig. 11, right) coupling reactions occurring at branch points are significantly more efficient (ηT) than those occurring at non-branch points. This is logical since the branch point in a convergent route, where two significant skeletal fragments come together, is likely to feature a large increase in similarity and complexity. The same efficiency analysis we have described for the LLS could also be carried out for any parallel synthetic branches, between its starting material and the branch point, to give a second efficiency measure for the route. As a final, expected observation, Fig. 11 shows a steady decrease in route efficiency with route length.
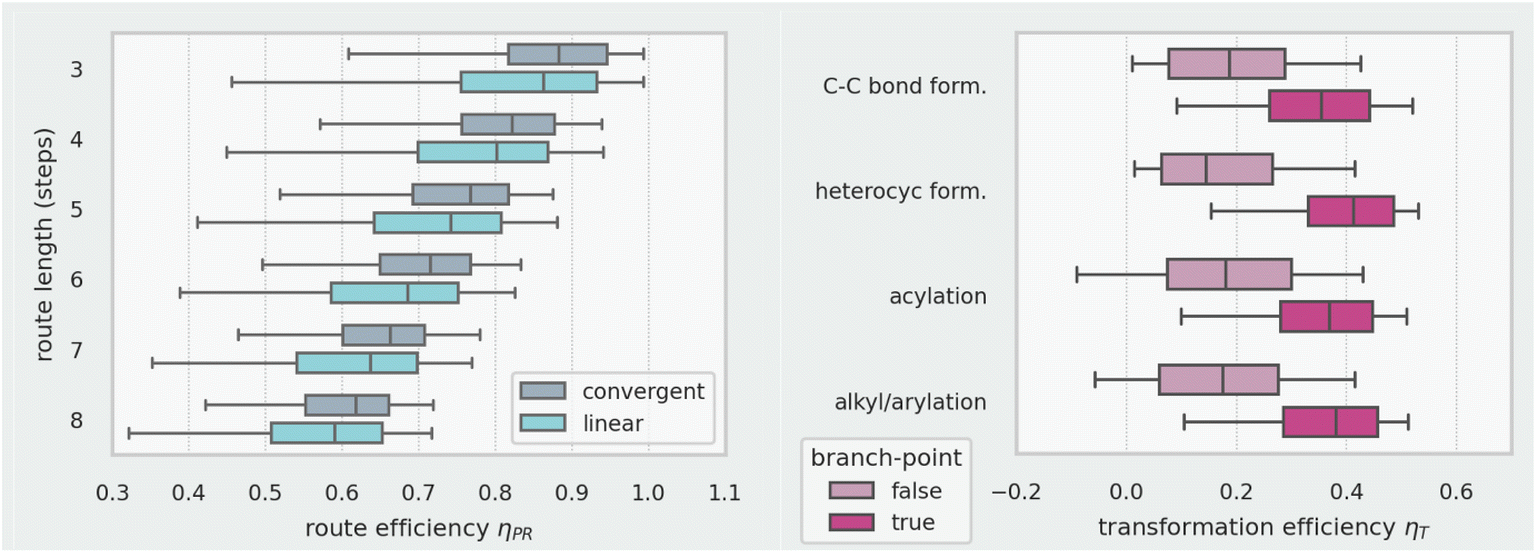 | ||
| Fig. 11 Penalised route efficiency ηPRversus route topography for 280k routes (left); transformation efficiency ηTversus reaction class (right) for 523k reactions. Whiskers show 5th and 95th percentiles, boxes show 25th, 50th and 75th percentiles. | ||
2.7. Analysis of retrosynthesis predictions
To demonstrate the utility of our methodology with CASP output, we took a set of 100000 molecules from ChEMBL42 and generated predicted synthetic routes using two versions of the AiZynthFinder software.§43,44 There was a slight difference in the success rate of each version: routes leading back to available starting materials (eMolecules)45 were found for 69% and 71% of the targets for AiZynthFinder versions v1 and v4 respectively. For 64% of the targets, routes were found using both software versions. The total number of routes identified for these 64400 common targets was 650000 for v1 and 867000 for v4.
As shown in Fig. 12 (top), if we consider only the best routes (one per target) by step count, there is negligible difference in the output of the two software versions. The output from the two software versions becomes distinguishable, however, using both synthetic range ‖vmin‖ (Fig. 12, middle) and penalised route efficiency ηPR (Fig. 12, bottom). The later version predicts routes that are, on average, of wider synthetic range. Since the collection of route targets is the same for both software versions, we can deduce that the route starting materials identified by version v4 are generally simpler and less similar to the route targets compared to version v1. The later version also predicts routes with slightly higher penalised route efficiency, which indicates a reduced reliance on non-productive transformations (redox, FGI, FGA, etc.) compared to version v1.
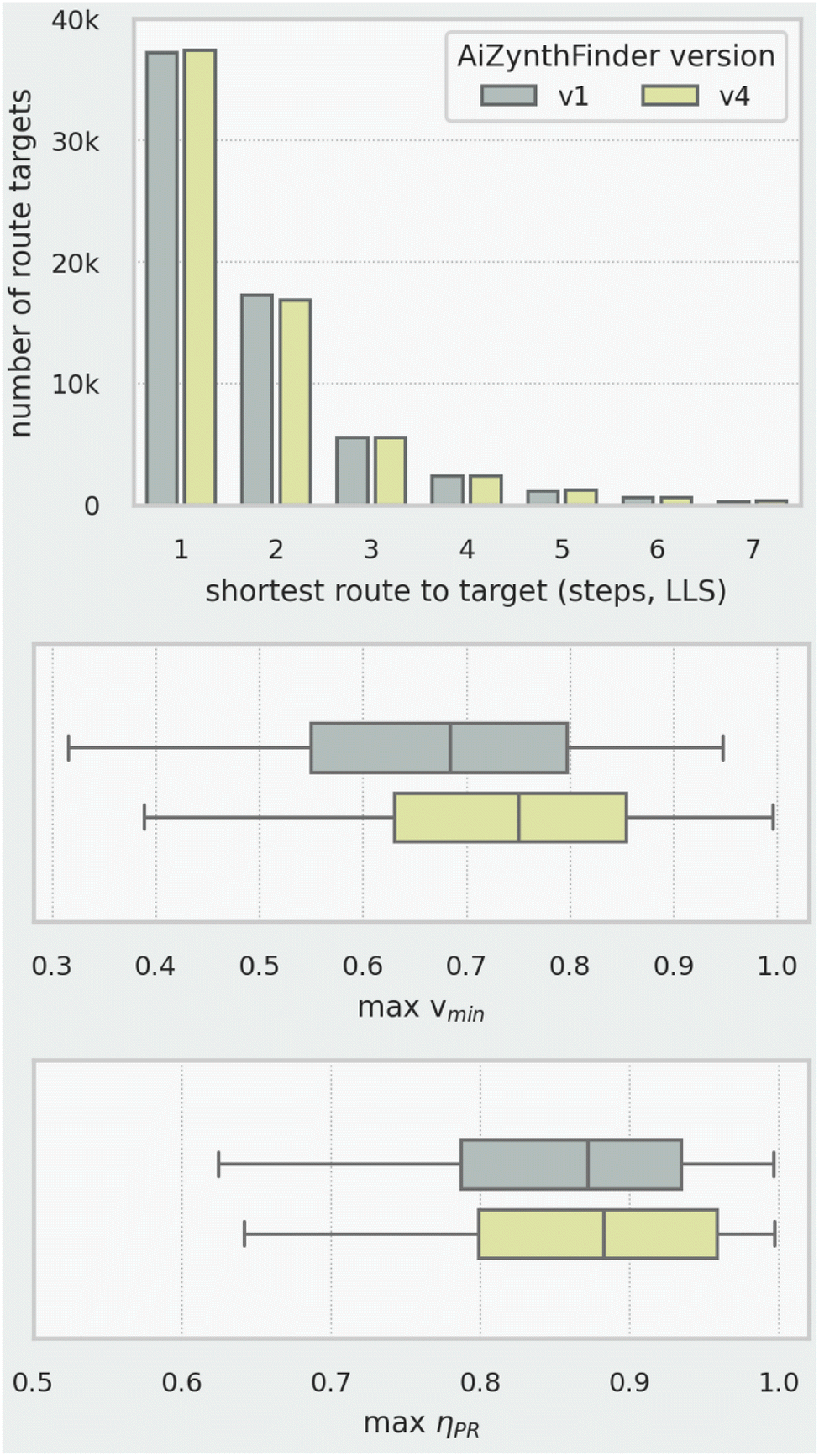 | ||
| Fig. 12 Comparison of best routes to 64k ChEMBL targets identified by AiZynthFinder: step count of shortest route per target (top), maximum synthetic range per target (middle) and maximum penalised route efficiency per target (bottom). | ||
We can also use this methodology to assess and rank CASP routes, using androgen receptor antagonist 26 from the ChEMBL dataset as an example.46 Construction of the central thiohydantoin unit is the main challenge and four predicted routes are shown in Fig. 13. Route A is the joint shortest (two steps) and the strategy here is to purchase the thiohydantoin core. The starting material is therefore complex and of very high similarity to the route target, meaning the synthetic range ‖vmin‖ is very small (0.04, notice the difference in scale for the vector plot of route A). The two proposed transformations are both of low efficiency (oxidation and FGI) resulting in low route efficiency (ηR = 0.36) and even lower penalised route efficiency (ηPR = 0.19).
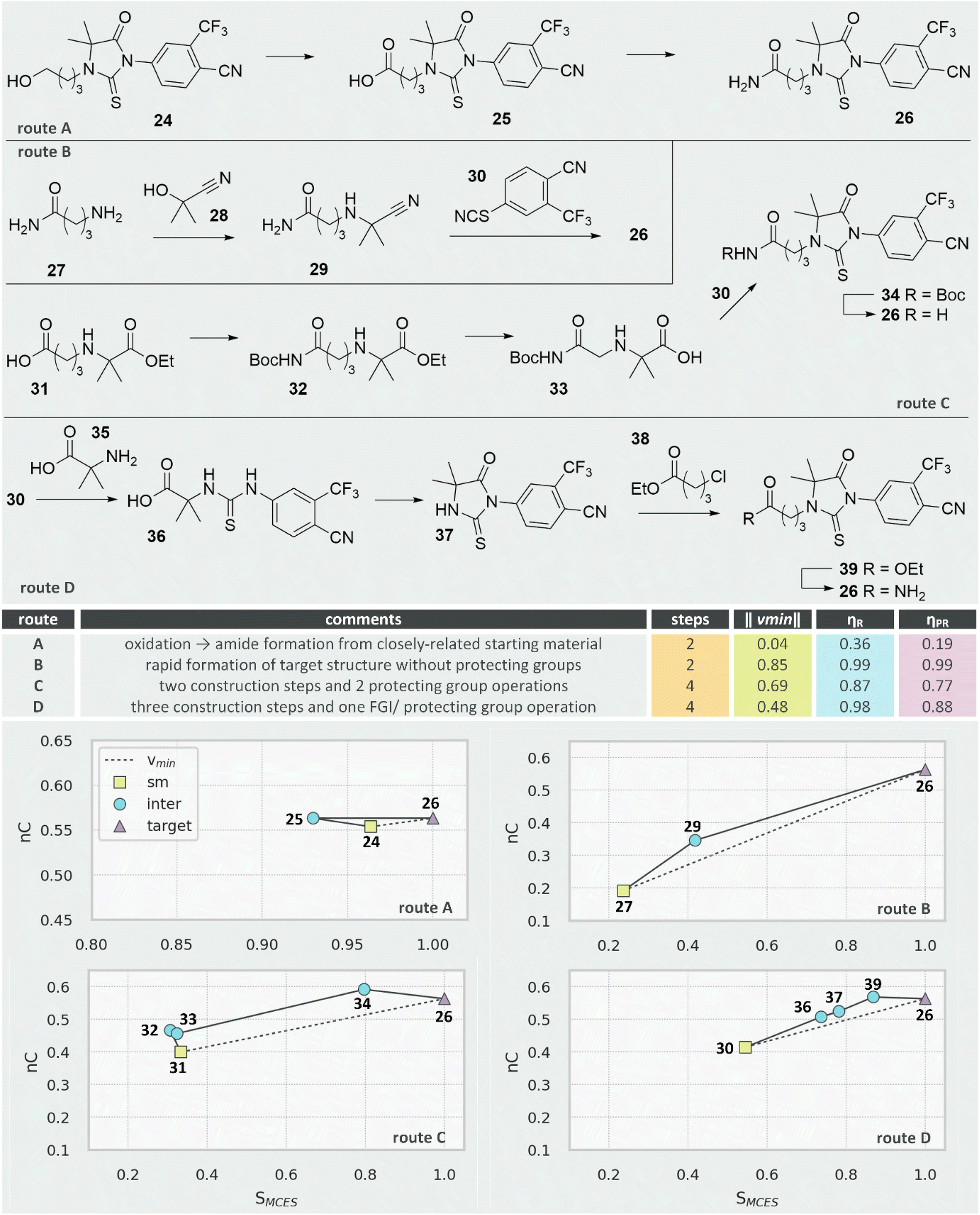 | ||
| Fig. 13 Comparison of routes to androgen receptor antagonist 26. | ||
Route B is also two steps but, in comparison to route A, covers a much wider synthetic range since the starting material 27 is much less complex and less similar to 26. Both transformations are of high efficiency and the resulting route efficiency is almost perfect (ηR = ηPR = 0.99). We might speculate on the viability of the thiohydantoin-forming step (29 + 30) or whether the primary amide might interfere but, if successful, route B would be a highly efficient synthesis.
The remaining two routes are both longer (4 steps). Route C has wider synthetic range ‖vmin‖ but comprises two protecting group manipulations resulting in a lower route efficiency (ηPR = 0.77). Route D has narrower synthetic range but, with three construction reactions and one FGI, has a higher route efficiency (ηPR = 0.88).
We propose that the overall analysis is in keeping with human assessment: route B would be the most efficient (if viable), routes C and D are less efficient (but perhaps more viable) and route A is unlikely to be of use (the cost and availability of starting material 24 is unlikely to be significantly different to that of target 26).
2.8. Analysis of synthetic routes between 2000 and 2020
As a final analysis, Fig. 14 shows how synthetic range (‖vmin‖) and penalised route efficiency (ηPR) have changed between 2000 and 2020. The steady upward trend in both suggests that, based on our analysis, the synthetic-organic community is making increasingly complex route targets from simpler starting materials and doing so with less wasted complexity and reliance on low efficiency reactions.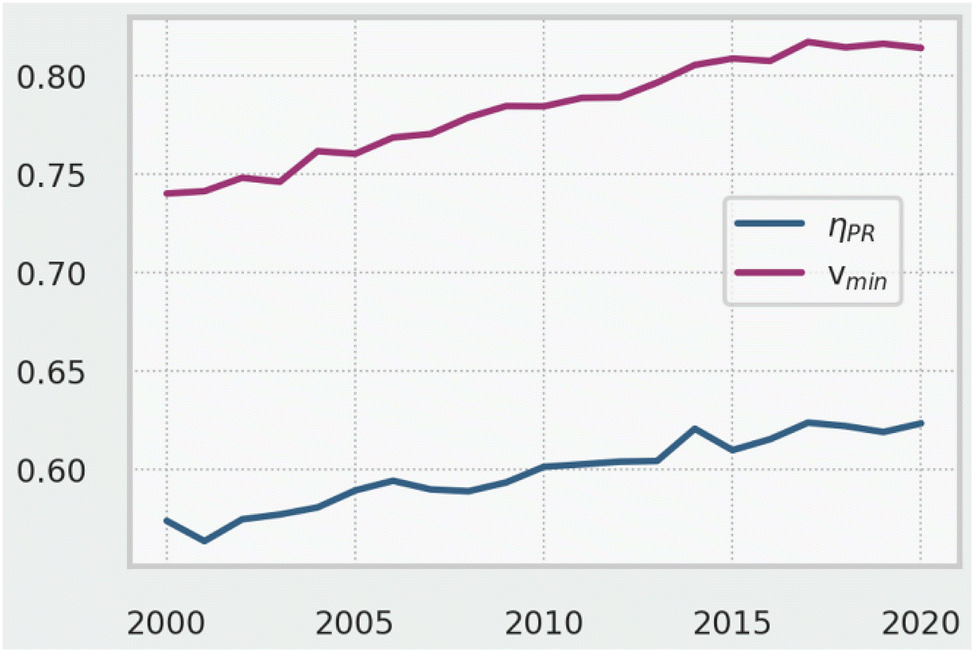 | ||
| Fig. 14 Mean penalised route efficiency ηPR for 654k routes between 2000 and 2020. | ||
3. Conclusion
The use of vectors comprising similarity and complexity components to describe synthetic transformations and routes has been shown to be useful for visualising and quantifying several qualities that we look for as organic chemists. Using a large dataset, we have demonstrated that the transformation efficiencies (ηT) associated with reaction super-classes follow logical trends. The specific similarity (Morgan fingerprint, MCES) and complexity metrics (CM*, SPS) we have used are easily interchangeable and alternative, or even custom-made fingerprints and/or complexity metrics, could be used, following the same principles. We have described how the synthetic range of a route can be represented as a vector and quantified using ‖vmin‖ to alleviate inconsistencies with step-counting and starting material designation. Penalised route efficiency (ηPR) has also been introduced to describe how effectively a given route traverses said range. This efficiency measure functions as an amalgam of atom economy and ideality.To demonstrate the use of this methodology, we have shown how (i) large sets of CASP output from differing sources can be rapidly compared and contrasted and (ii) the automated assessment of individual CASP-derived routes to a given synthetic target can be achieved in a similar way to human interpretation.
Our mathematical approach to route analysis is highly amenable to further analysis. The impact of ordering in the sequence of transformations along a synthetic route ought to be of interest. We would instinctively suggest that similarity and complexity values should continually increase from starting material to target such that wasted complexity (due to yield losses) is minimised. Similarly, we might expect low-efficiency transformations to be better situated at the start of a route and high-efficiency transformations towards the end; these properties could be assessed using rank correlation metrics (e.g., Pearson,47 Spearman48).
It should be stressed that the methodology described here, derived only from chemical structures and route topography is wholly theoretical and will always be inferior to real, empirical data such as cost, time and waste. Obtaining reliable empirical data for known transformations is problematic however and, in the case of unknown or theoretical transformations, Hendrickson's observation made in 1976 (ref. 9) (“when planning an organic synthesis it is presently impossible to predict the yields of individual reactions, or indeed even whether they will succeed or fail”) remains pertinent today. Thus, we believe this methodology will be useful wherever assessment of synthetic transformations and routes is required.
Prior submission to ChemRxiv
This manuscript is an extension and rework of a previous submission (https://doi.org/10.26434/chemrxiv-2024-nbx35-v2);20 some sections of text have been reused.Author contributions
SG: data curation, software and writing – review & editing. GPH: conceptualisation, analysis, methodology, software and writing – original draft.Conflicts of interest
For the duration of this study, GPH and SG were both employees of and shareholders in AstraZeneca, who funded the research.Abbreviations
| CASP | Computer aided synthesis planning |
| ECFP4 | Enhanced connectivity fingerprint (diameter 4) |
| FGA | Functional group addition |
| FGI | Functional group interconversion |
| LLS | Longest linear sequence |
| MCES | Maximum common edge subgraph |
| PMI | Process mass intensity |
| SAR | Structure activity relationship |
| SMILES | Simplified molecular-input line-entry system |
Data availability
We are unable to provide the main Reaxys dataset used for most of this study as it is a paid product. As an alternative, we have provided (via a link in the Jupyter notebook) a dataset of 457k routes taken from the patent literature.Also provided are the python scripts and an accompanying Jupyter notebook that allows the user to search, retrieve data and generate vector plots for individual routes within the patent dataset.
• Vectors_scripts.py: Python file containing all the necessary code (except the file below) to convert the patents datafile into a useable dataframe with the necessary metrics.
• proudfoot_complexity.py: Python file containing the necessary code for generating CM* complexity metrics.
• Vectors_Notebook.ipynb: Jupyter notebook containing instructions on how to process data, search, retrieve route information and generate vector plots.
Complementary analyses using alternative complexity metrics and the code files mentioned above is available. See DOI: https://doi.org/10.1039/d5sc06089c.
Notes and references
- https://www.nextmovesoftware.com/namerxn.html, accessed December 2024.
- H. Kraut, J. Eiblmaier, G. Grethe, P. Löw, H. Matuszczyk and H. Saller, Algorithm for Reaction Classification, J. Chem. Inf. Model., 2013, 53(11), 2884–2895, DOI:10.1021/ci400442f.
- B. Trost, The Atom Economy—A Search for Synthetic Efficiency, Science, 1991, 254(5037), 1471–1477, DOI:10.1126/science.1962206.
- P. A. Wender, M. P. Croatt and B. Witulski, New Reactions and Step Economy: The Total Synthesis of (±)-Salsolene Oxide Based on the Type II Transition Metal-Catalyzed Intramolecular [4+4] Cycloaddition, Tetrahedron, 2006, 62(32), 7505–7511, DOI:10.1016/j.tet.2006.02.085.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Function-Oriented Synthesis, Step Economy, and Drug Design, Acc. Chem. Res., 2008, 41(1), 40–49, DOI:10.1021/ar700155p.
- P. A. Wender and B. L. Miller, Synthesis at the Molecular Frontier, Nature, 2009, 460(7252), 197–201, DOI:10.1038/460197a.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Redox Economy in Organic Synthesis, Angew. Chem., Int. Ed., 2009, 48(16), 2854–2867, DOI:10.1002/anie.200806086.
- T. Gaich and P. S. Baran, Aiming for the Ideal Synthesis, J. Org. Chem., 2010, 75(14), 4657–4673, DOI:10.1021/jo1006812.
- J. B. Hendrickson, Systematic Synthesis Design. 6. Yield Analysis and Convergency, J. Am. Chem. Soc., 1977, 99(16), 5439–5450, DOI:10.1021/ja00458a035.
- A. Lin, N. Dyubankova, T. I. Madzhidov, R. I. Nugmanov, J. Verhoeven, T. R. Gimadiev, V. A. Afonina, Z. Ibragimova, A. Rakhimbekova, P. Sidorov, A. Gedich, R. Suleymanov, R. Mukhametgaleev, J. Wegner, H. Ceulemans and A. Varnek, Atom-to-Atom Mapping: A Benchmarking Study of Popular Mapping Algorithms and Consensus Strategies, Mol. Inf., 2022, 41(4), 2100138, DOI:10.1002/minf.202100138.
- S. Genheden and E. Bjerrum, PaRoutes: Towards a Framework for Benchmarking Retrosynthesis Route Predictions, Digital Discovery, 2022, 1(4), 527–539, 10.1039/D2DD00015F.
- K. Maziarz, A. Tripp, G. Liu, M. Stanley, S. Xie, P. Gaiński, P. Seidl and M. H. S. Segler, Re-Evaluating Retrosynthesis Algorithms with Syntheseus, Faraday Discuss., 2025, 256, 568–586, 10.1039/D4FD00093E.
- S. Genheden and G. P. Howell, An Analysis of Published Synthetic Routes, Route Targets, and Reaction Types (2000–2020), Org. Process Res. Dev., 2024, 28(12), 4225–4239, DOI:10.1021/acs.oprd.4c00389.
- https://onlinelibrary.wiley.com/journal/15213773, accessed March 2025.
- https://www.sciencedirect.com/journal/european-journal-of-medicinal-chemistry, accessed March 2025.
- https://pubs.acs.org/journal/jmcmar, accessed March 2025.
- https://pubs.acs.org/journal/joceah, accessed March 2025.
- https://pubs.acs.org/journal/orlef7, accessed March 2025.
- https://pubs.acs.org/journal/oprdfk, accessed March 2025.
- G. Howell and S. Genheden, Synthetic Route Design & Assessment Using Vectors Derived from Similarity and Complexity, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-nbx35-v2.
- H. L. Morgan, The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service, J. Chem. Doc., 1965, 5(2), 107–113, DOI:10.1021/c160017a018.
- https://www.rdkit.org/, accessed March 2025.
- D. Bajusz, A. Rácz and K. Héberger, Why Is Tanimoto Index an Appropriate Choice for Fingerprint-Based Similarity Calculations?, J. Cheminf., 2015, 7(1), 20, DOI:10.1186/s13321-015-0069-3.
- J. W. Raymond and P. Willett, Maximum Common Subgraph Isomorphism Algorithms for the Matching of Chemical Structures, J. Comput.-Aided Mol. Des., 2002, 16(7), 521–533, DOI:10.1023/A:1021271615909.
- C. Cerchia, R. Nasso, M. Mori, S. Villa, A. Gelain, A. Capasso, F. Aliotta, M. Simonetti, R. Rullo, M. Masullo, E. De Vendittis, M. R. Ruocco and A. Lavecchia, Discovery of Novel Naphthylphenylketone and Naphthylphenylamine Derivatives as Cell Division Cycle 25B (CDC25B) Phosphatase Inhibitors: Design, Synthesis, Inhibition Mechanism, and in Vitro Efficacy against Melanoma Cell Lines, J. Med. Chem., 2019, 62(15), 7089–7110, DOI:10.1021/acs.jmedchem.9b00632.
- S. H. Bertz, The First General Index of Molecular Complexity, J. Am. Chem. Soc., 1981, 103(12), 3599–3601, DOI:10.1021/ja00402a071.
- J. B. Hendrickson, P. Huang and A. G. Toczko, Molecular Complexity: A Simplified Formula Adapted to Individual Atoms, J. Chem. Inf. Comput. Sci., 1987, 27(2), 63–67, DOI:10.1021/ci00054a004.
- J. Li and M. D. Eastgate, Current Complexity: A Tool for Assessing the Complexity of Organic Molecules, Org. Biomol. Chem., 2015, 13(26), 7164–7176, 10.1039/C5OB00709G.
- R. P. Sheridan, N. Zorn, E. C. Sherer, L.-C. Campeau, C. Chang, J. Cumming, M. L. Maddess, P. G. Nantermet, C. J. Sinz and P. D. O'Shea, Modeling a Crowdsourced Definition of Molecular Complexity, J. Chem. Inf. Model., 2014, 54(6), 1604–1616, DOI:10.1021/ci5001778.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, SCScore: Synthetic Complexity Learned from a Reaction Corpus, J. Chem. Inf. Model., 2018, 58(2), 252–261, DOI:10.1021/acs.jcim.7b00622.
- A. S. Tyrin, D. A. Boiko, N. I. Kolomoets and V. P. Ananikov, Digitization of Molecular Complexity with Machine Learning, Chem. Sci., 2025, 16(16), 6895–6908, 10.1039/D4SC07320G.
- T. Böttcher, An Additive Definition of Molecular Complexity, J. Chem. Inf. Model., 2016, 56(3), 462–470, DOI:10.1021/acs.jcim.5b00723.
- J. R. A. Proudfoot, Path Based Approach to Assessing Molecular Complexity, Bioorg. Med. Chem. Lett., 2017, 27(9), 2014–2017, DOI:10.1016/j.bmcl.2017.03.008.
- L. Angelini, C. E. Coomber, G. P. Howell, G. Karageorgis and B. A. Taylor, Cumulative Complexity Meta-Metrics as an Efficiency Measure and Predictor of Process Mass Intensity (PMI) during Synthetic Route Design, Green Chem., 2023, 25(14), 5543–5556, 10.1039/D3GC00878A.
- A. Krzyzanowski, A. Pahl, M. Grigalunas and H. Waldmann, Spacial Score – A Comprehensive Topological Indicator for Small-Molecule Complexity, J. Med. Chem., 2023, 66(18), 12739–12750, DOI:10.1021/acs.jmedchem.3c00689.
- https://en.wikipedia.org/wiki/Scalar_projection, accessed March 2025.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules, Org. Biomol. Chem., 2006, 4(12), 2337–2347, 10.1039/B602413K.
- https://greglandrum.github.io/rdkit-blog/posts/2024-05-14-similarity-and-centrality.html, accessed March 2025.
- S. Wang, Y.-Q. Guo, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, K2CO3-Mediated Cyclization and Rearrangement of γ,δ-Alkynyl Oximes To Form Pyridols, Org. Lett., 2017, 19(7), 1574–1577, DOI:10.1021/acs.orglett.7b00389.
- H. G. Selnick, J. F. Hess, C. Tang, K. Liu, J. B. Schachter, J. E. Ballard, J. Marcus, D. J. Klein, X. Wang, M. Pearson, M. J. Savage, R. Kaul, T.-S. Li, D. J. Vocadlo, Y. Zhou, Y. Zhu, C. Mu, Y. Wang, Z. Wei, C. Bai, J. L. Duffy and E. J. McEachern, Discovery of MK-8719, a Potent O-GlcNAcase Inhibitor as a Potential Treatment for Tauopathies, J. Med. Chem., 2019, 62(22), 10062–10097, DOI:10.1021/acs.jmedchem.9b01090.
- X. Liu, B. Liu and Q. Liu, Migratory Hydrogenation of Terminal Alkynes by Base/Cobalt Relay Catalysis, Angew. Chem., Int. Ed., 2020, 59(17), 6750–6755, DOI:10.1002/anie.201916014.
- https://www.ebi.ac.uk/chembl/, accessed April 2025.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, AiZynthFinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning, J. Cheminf., 2020, 12(1), 70, DOI:10.1186/s13321-020-00472-1.
- L. Saigiridharan, A. K. Hassen, H. Lai, P. Torren-Peraire, O. Engkvist and S. Genheden, AiZynthFinder 4.0: Developments Based on Learnings from 3 Years of Industrial Application, J. Cheminf., 2024, 16(1), 57, DOI:10.1186/s13321-024-00860-x.
- https://www.emolecules.com/, accessed April 2025.
- H. Yoshino, H. Sato, K. Tachibana, T. Shiraishi, M. Nakamura, M. Ohta, N. Ishikura, M. Nagamuta, E. Onuma, T. Nakagawa, S. Arai, K.-H. Ahn, K.-Y. Jung and H. Kawata, Structure–Activity Relationships of Bioisosteric Replacement of the Carboxylic Acid in Novel Androgen Receptor Pure Antagonists, Bioorg. Med. Chem., 2010, 18(9), 3159–3168, DOI:10.1016/j.bmc.2010.03.036.
- https://en.wikipedia.org/wiki/Pearson_correlation_coefficient, accessed January 2025.
- https://en.wikipedia.org/wiki/Spearman%27s_rank_correlation_coefficient, accessed January 2025.
Footnotes |
| † A list of protecting groups with structures is provided in the SI. |
| ‡ For application with other datasets, we would suggest using nC values of 0 and 1 respectively for compounds where CM* is <3.5 or >12.0. |
| § Version v1 corresponds to the first public version of AiZynthFinder with models trained on USPTO data in 2019, whereas version v4 corresponds to the latest major release of AiZynthFinder with models trained on USPTO data in 2022. |
| This journal is © The Royal Society of Chemistry 2025 |