
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed carbonylative synthesis of acrylamides from alkenyl thianthrenium salts
Ru-Han
A
ab,
Jiajun
Zhang
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
First published on 20th October 2025
Abstract
Covalent drugs have emerged as powerful tools in modern therapeutics, with acrylamide being one of the most widely used electrophilic warheads in recent years. Herein, we report a practical and selective strategy for synthesizing acrylamides via Pd-catalyzed carbonylative amidation of vinyl thianthrenium salts with arylamines. These salts serve as stable and safe surrogates for alkynes or activated alkenes. The reaction proceeds under mild conditions using only 0.1 mol% Pd catalyst and 1 atm of CO, delivering a broad range of N-aryl acrylamides in high yields (>90%) with excellent chemoselectivity and functional group tolerance. Furthermore, the method is readily scalable to the gram level without compromising efficiency. This work demonstrates the synthetic potential of vinyl thianthrenium salts in carbonylation chemistry and offers a general, robust, and scalable approach for the preparation of functionalized acrylamides under mild conditions.
Introduction
Acrylamide has emerged as a versatile and valuable structural motif in both medicinal chemistry and organic synthesis due to its broad reactivity and ability to form covalent bonds with biomolecular targets.1–4 In particular, its acrylamide moiety serves as an efficient electrophilic warhead, undergoing selective Michael addition with nucleophilic residues in proteins.5,6 Over the past decades, covalent inhibitors bearing acrylamide motifs have emerged as a central strategy in drug discovery (Fig. 1).7–10 Beyond pharmaceuticals, acrylamide is a key intermediate in organic synthesis, participating in various transformations and acrylamide monomers are also the fundamental building blocks of polyacrylamides, which are widely used in water treatment, enhanced oil recovery, and biomedical materials.11–16 Thus, the development of efficient synthetic methods for acrylamide derivatives is of significant research and practical value. | ||
| Fig. 1 Selected covalent drugs containing acrylamide warheads. | ||
Traditionally, acrylamides are prepared by nucleophilic condensation or substitution of amines with carboxylic acid derivatives in the presence of stoichiometric bases or coupling reagents;17–21 however, such protocols are often non-green, atom-inefficient, and occasionally suffer from chemoselectivity issues. The hydration of acrylonitrile remains the dominant industrial route,22–25 especially when combined with nitrile hydratase (NHase),25–29 which enables highly efficient production under mild conditions. In recent years, new methods for synthesizing acrylamide were reported,30–34 though they remain largely at the research stage with limited industrial implementation.
Carbon monoxide (CO) is one of the most atom-economical, inexpensive, and efficient C1 sources for constructing carbonyl-containing functionalities,35,36 including esters, amides, carboxylic acids, ketones, and acyl halides.37–40 Over the past several decades, CO has been extensively utilized in transition metal catalyzed carbonylation reactions, numerous studies have reported the synthesis of acrylamides via transition metal catalyzed carbonylation. Despite these advances, carbonylation reactions involving the simplest alkyne, acetylene, and N-nucleophiles remain relatively scarce. In 1953, Reppe et al. first reported the catalytic hydroamino–carbonylation of acetylene to yield acrylamides (Scheme 1, eqn (A)).41 However, this reaction was severely limited by the need for high temperatures (180 °C), high acetylene pressures (>12 bar), and highly toxic Ni(CO)4 catalysts. In 2024, Beller's group reported the first palladium-catalyzed hydroaminocarbonylation of acetylene, achieving moderate to excellent yields under milder conditions (Scheme 1, eqn (B)).42 Acrylamides can also be synthesized from activated alkenes,43,44 such as vinyl chloride or vinyl iodide, through carbonylation, thereby circumventing the use of highly hazardous acetylene gas and improving operational safety and scalability. In 2024, our group reported the first palladium-catalyzed carbonylation using 1,2-dichloroethane as the starting material,45 efficiently producing a range of acrylamides and acrylate ester derivatives, further avoiding the use of hazardous acetylene and vinyl chloride (Scheme 1, eqn (C)). Nonetheless, these methodologies still exhibit certain limitations concerning catalyst loading, operational safety, reaction selectivity and polymerization of the substrate. Therefore, there remains a strong demand for the development of greener, more efficient, and readily scalable synthetic strategies. On other hand, carbonylative transformation of thianthrenium salts has been recognized as an interesting topic and various procedures have been achieved.46 In our group, we also achieved the carbonylative reactions of aryl thianthrenium salts with arylboronic acids47 and terminal alkynes48 as the reaction partners. By using palladium as the catalyst, diaryl ketones, alkynones and furanones can be produced effectively. However, the carbonylative transformation with alkenyl thianthrenium salts has not been reported yet which can give different analogue of carbonyl-containing products. Under the above backgrounds, in this study, we developed a palladium-catalyzed carbonylative amidation using alkenyl thianthrenium salts as safer and more stable alternatives to alkynes or activated alkenes, allowing the synthesis of acrylamide derivatives from arylamines under mild conditions (Scheme 1, eqn (D)).
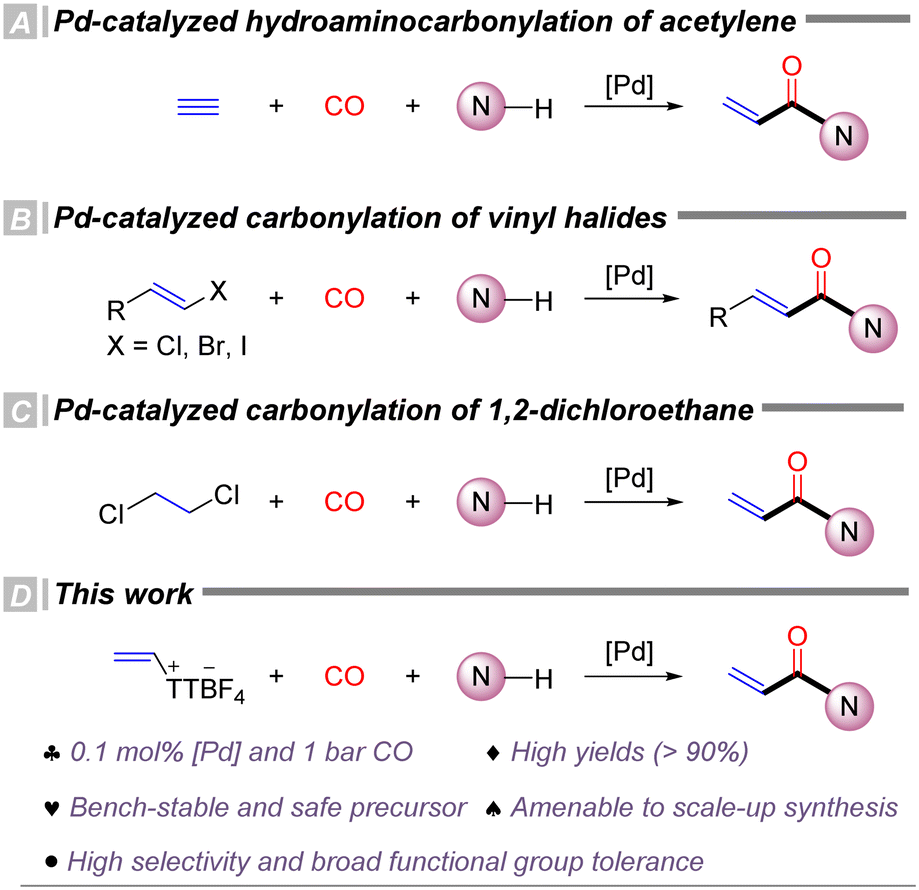 | ||
| Scheme 1 Studies on carbonylation for acrylamide synthesis. | ||
Results and discussion
We began our investigations by employing vinyl thianthrenium salt 1a as a surrogate for alkynes, using aniline as the nucleophile to construct the corresponding acrylamide. A systematic screening was conducted with respect to the catalyst and its loading, ligands, bases, solvents, and CO pressure to identify optimal conditions for the transformation (for details see, SI). These studies ultimately led to the establishment of an efficient catalytic system, which laid a solid foundation for subsequent substrate scope exploration. Initially, catalyst and ligand screenings revealed that palladium was essential for the reaction, as demonstrated by the complete failure of the transformation in its absence. Among various combinations, Pd(OAc)2 and xantphos proved to be the most effective, delivering the highest yield of 3aa. Solvents screening showed that MeCN was the most suitable, offering good solubility for both the palladium complex and organic substrates. In contrast, aromatic solvents (Table 1, entry 5) and highly polar solvents (Table 1, entry 9) gave inferior results. Interestingly, the reaction tolerated the presence of water (Table 1, entries 5, 6, and 8), and in some cases, water may even improve the reaction by enhancing base solubility.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] (mol%) | Ligand (mol%) | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (1.2 equiv.), base (2.0 equiv.), solvent (1.0 mL), CO (10 bar), 20 h. b The yields were determined by GC using n-hexadecane as the internal standard. c Pd(PPh3)4. d Isolated yield. e CO (1 bar). f Reaction time 10 h. g At room temperature. | |||||
| 1 | 4.0 | 6.0 | Na2CO3 | 1,4-Dioxane | 86 |
| 2c | 4.0 | 0 | Na2CO3 | 1,4-Dioxane | 84 |
| 3 | 4.0 | 6.0 | NaOAc | 1,4-Dioxane | 95 |
| 4 | 1.0 | 1.2 | NaOAc | MeCN | 95d |
| 5 | 1.0 | 1.2 | NaOAc | Tol. | 36 |
| 6 | 1.0 | 1.2 | NaOAc | Tol. + H2O | 69 |
| 7 | 1.0 | 1.2 | NaOAc | DMF | 13 |
| 8 | 1.0 | 1.2 | NaOAc | H2O | 35 |
| 9 | 1.0 | 1.2 | NaOAc | DMSO | n.d. |
| 10 | 1.0 | 1.2 | NaOH | MeCN | 19 |
| 11 | 1.0 | 1.2 | Li2CO3 | MeCN | 26 |
| 12 | 1.0 | 1.2 | Cs2CO3 | MeCN | 81 |
| 13 | 1.0 | 1.2 | — | MeCN | 65 |
| 14 | 1.0 | 1.2 | TEA | MeCN | 14 |
| 15e | 1.0 | 1.2 | NaOAc | MeCN | 97 |
| 16e | 0.1 | 0.12 | NaOAc | MeCN | 99 |
| 17e | 0.01 | 0.012 | NaOAc | MeCN | 42 |
| 18e | — | 0.12 | NaOAc | MeCN | n.d. |
| 19e,f | 0.1 | 0.12 | NaOAc | MeCN | 77 |
| 20e,g | 0.1 | 0.12 | NaOAc | MeCN | 72 |
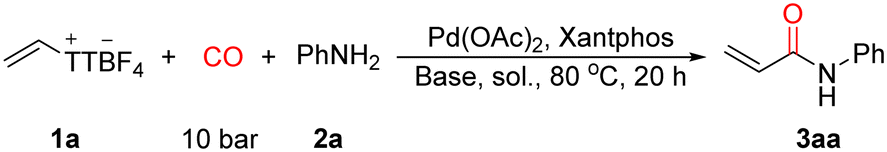
In terms of base selection, the reaction was found to be relatively base-tolerant, achieving 65% yield even under base-free conditions, suggesting that while a base is not strictly necessary, it can enhance efficiency. Among various bases evaluated, sodium acetate (NaOAc) emerged as the optimal choice, likely due to its balanced basicity and coordination ability, which may promote the Pd(II)/Pd(0) cycle or stabilize key intermediates. Strong bases (Table 1, entry 10) or organic bases (Table 1, entry 14) were detrimental to the reaction. We also evaluated the influence of CO pressure, reaction time and reaction temperature (Table 1, entries 15–20). While the initial optimization was performed under 10 bar of CO, we were pleased to find that the yield remained high even under 1 bar CO, significantly improving the operational safety and scalability of the protocol (Table 1, entry 15). Notably, the catalyst loading could be reduced from 4.0 mol% to as low as 0.1 mol% without compromising the yield, which even reached 99%, underscoring the high efficiency of the catalytic system (Table 1, entry 16). However, when the Pd loading was further decreased to 0.01 mol%, the yield dropped significantly, and prolonging the reaction time failed to improve the outcome (Table 1, entry 17), and no desired produced detectable in the absence of palladium (Table 1, entry 18). This may be attributed to a slower reaction rate under ultra-low Pd concentrations, leading to premature decomposition of the vinyl-TT salt before productive turnover. Moreover, the reaction maintained over 70% yield when either the temperature was lowered to room temperature or the reaction time was halved, further highlighting the high catalytic activity and practical potential of the system (Table 1, entries 19 and 20). In addition, Pd(PPh3)4 or Pd2dba3 can be applied as pre-catalyst as well and similar results can be obtained in the presence of xantphos. The yield of the desired product was formed in 81% in the presence of 1 equivalent of base. Decreased yields were obtained with shorten reaction time or decreased reaction temperature. Taken together, the optimized conditions were established as follows: Pd(OAc)2 (0.1 mol%), xantphos (0.12 mol%), NaOAc (2.0 equiv.), MeCN (1.0 mL) as solvent, 1 bar CO, at 80 °C for 20 h.
With the optimized conditions in hand, we next explored the substrate scope of this palladium catalyzed carbonylative amidation, focusing on both amines and alkenyl thianthrenium salts (Scheme 2). A wide range of arylamines proved compatible with this transformation, delivering the corresponding N-aryl acrylamides in generally excellent yields. Electron-donating substituents such as methyl, tert-butyl, methoxy, and acetoxy at the para- or meta-positions were well tolerated, affording products 3aa–3af and 3ap–3aq in 90–99% yields. These substituents typically enhance the nucleophilicity of anilines, facilitating the amidation and indicating that the reaction proceeds efficiently even with moderately nucleophilic amines. Moreover, substitution at different positions on the aryl ring, including para-, meta-, and even a sterically hindered group at ortho, did not significantly affect the yield, demonstrating the excellent steric tolerance of the catalytic system. Interestingly, when both amino and hydroxyl groups are present on the aromatic ring, the reaction proceeds selectively at the amino group to afford product 3ao. Electron-deficient arylamines bearing halogens, cyano, trifluoromethyl, or nitro groups also participated smoothly in the coupling, giving products 3ak–3an and 3au–3ax in 76–99% yields. Notably, strongly electron-withdrawing groups such as NO2 and CF3 were well tolerated without compromising yield, reflecting the high functional group compatibility and robustness of the catalytic system. The excellent chemoselectivity is further demonstrated by the fact that even in the presence of highly reactive iodide substituents, the reaction proceeded exclusively at the amino moiety with high efficiency, providing opportunities for further late-stage derivatization and cross-coupling. Heterocyclic arylamines such as 2-aminobenzofuran and fused aromatic systems like 2-naphthylamine also reacted smoothly, affording the desired products in over 90% yield, highlighting the method's potential utility for the construction of bioactive heterocyclic motifs. In contrast, when an alkynyl-substituted arylamine was employed, the desired product 3ay was not observed. Instead, the hydrolysis product 3aw was obtained in large quantities, indicating that 3ay is highly sensitive to trace amounts of water under the reaction conditions and readily undergoes hydrolysis. In addition to primary arylamines, we also evaluated the reactivity of tertiary amines. For instance, N-methylaniline delivered the corresponding product 4aa in excellent yield, whereas bulky tertiary amines such as diphenylamine failed to give any product (4ab), likely due to steric hindrance. Furthermore, aliphatic amines, alcohols, and phenols were not viable nucleophiles under these conditions, indicating that the protocol is highly selective for arylamines. In the case of aliphatic amine and benzyl amine, only the corresponding formamide compounds were detected. In the cases of 2-aminopyridine and 4-aminopyridine, the desired product was detected on trace amount. We further examined the generality of this reaction with respect to the alkenyl thianthrenium salt component. A series of alkenes bearing either aryl or alkyl side chains were successfully converted via their corresponding TT salts, giving products 3ba–3da in 88–99% yields. These results underscore the versatility of alkenyl thianthrenium salts as robust electrophilic surrogates in carbonylation chemistry.
 | ||
| Scheme 2 Substate scope of arylamines and alkenyl thianthrenium salts. Reaction conditions: 1a (0.1 mmol), 2a (1.2 equiv.), NaOAc (2.0 equiv.), MeCN (1.0 mL), CO (1 bar), 80 °C, 20 h, isolated yield. | ||
To gain some insights into the reaction mechanism, radical quenching experiments were conducted under standard conditions (Fig. 2A). The addition of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) significantly suppressed the formation of the desired amide product 3aa, resulting in only 19% yield. This result suggests the possible involvement of a radical intermediate, but the oxidation property of TEMPO should also be considered as it might oxidize phosphine or the in situ generated Pd(0) intermediate. The reaction proceeded smoothly, when BHT (2,6-di-tert-butyl-4-methylphenol) or DPE (1,1-diphenylethylene) was added, furnishing 3aa in 97% and 93% yields, respectively. These results imply that the process likely proceeds through a non-radical mechanism. In addition, a control experiment conducted in the absence of CO resulted in no product formation (Fig. 2B), highlighting the essential role of carbon monoxide in the reaction. To evaluate the practicality and scalability of the developed method, we performed a gram–scale reaction using 2.0 mmol of vinyl thianthrenium salt 1a (Fig. 2C). The reaction proceeded efficiently under the optimized conditions, affording the desired product 3aa in 96% isolated yield, demonstrating the excellent robustness and scalability of the protocol for potential synthetic applications.
 | ||
| Fig. 2 Control experiments and scale-up reaction. | ||
Based on our results and understanding, a plausible catalytic cycle for this carbonylative amidation is outlined in Scheme 3. The reaction is proposed to be initiated by oxidative addition of Pd(0) into the C–S bond of the vinyl thianthrenium salt, forming a vinyl-Pd(II) complex (B). The coordinating of CO with vinyl-Pd(II) complex (B) supposed to be important for its reactivity and stability which also explains no product was detected in the absence of CO in control experiment. Then, this intermediate undergoes migratory insertion of carbon monoxide to generate an acyl-Pd(II) species (C). Coordination and nucleophilic attack by the arylamine then affords intermediate D, which undergoes reductive elimination to deliver the final acrylamide product and regenerate the catalytically active Pd(0) species. This pathway is consistent with the requirement of CO for product formation and supports the critical role of TT+ as a leaving group in initiating the Pd-catalyzed cycle.
 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient and highly selective Pd-catalyzed carbonylative amidation of vinyl thianthrenium salts with arylamines under mild conditions, requiring only 0.1 mol% of catalyst and 1 bar of CO. This strategy enables the synthesis of a broad range of N-aryl acrylamide derivatives in excellent yields, typically exceeding 90%, and shows broad functional group compatibility. Importantly, vinyl thianthrenium salts serve as effective electrophilic surrogates for alkynes or activated alkenes, enabling carbonylative amidation under mild conditions and without the need for alkyne substrates. The method also demonstrates excellent scalability, affording the desired product in 96% yield on gram scale. This work highlights the synthetic potential of vinyl thianthrenium salts in carbonylation chemistry, and offers a practical and general approach for the synthesis of functionalized acrylamides under mild and scalable conditions.Author contributions
R. H. A. performed all the experiments and prepared the manuscript and SI. J. Z. prepared some substrates. X. F. W. conceived the project, supervised the research, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general comments, general procedure, analytic data, and NMR spectra. See DOI: https://doi.org/10.1039/d5sc06079f.Acknowledgements
The authors thank the National Key R&D Program of China (2023YFA1507500) and DICP for their financial support.Notes and references
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- P. Cohen, D. Cross and P. A. Jänne, Nat. Rev. Drug Discovery, 2021, 20, 551–569 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- D. Schaefer and X. Cheng, Pharmaceuticals, 2023, 16, 663 CrossRef CAS PubMed.
- L. Zheng, Y. Li, D. Wu, H. Xiao, S. Zheng, G. Wang and Q. Sun, MedComm: Oncol., 2023, 2, e56 Search PubMed.
- C. S. Lee, M. Milone and N. Seetharamu, OncoTargets Ther., 2021, 14, 4579–4597 CrossRef PubMed.
- F. Ran, Y. Liu, C. Wang, Z. Xu, Y. Zhang, Y. Liu, G. Zhao and Y. Ling, Eur. J. Med. Chem., 2022, 229, 114009 CrossRef CAS PubMed.
- X. Duan, N. Zheng, M. Li, G. Liu, X. Sun, Q. Wu and W. Song, Nat. Commun., 2022, 13, 4362 CrossRef CAS PubMed.
- D. Schaefer and X. Cheng, Pharmaceuticals, 2023, 16, 663 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2020, 80, 91–97 CrossRef CAS PubMed.
- Y. W. Huo, L. Yao, X. Qi and X. F. Wu, Org. Chem. Front., 2021, 8, 6974–6978 RSC.
- H. A. Blair, Drugs, 2023, 83, 1315–1321 CrossRef PubMed.
- S. Gou, Y. He, Y. Ma, S. Luo, Q. Zhang, D. Jing and Q. Guo, RSC Adv., 2015, 5, 51549–51558 RSC.
- W. de Cássia Novaes and A. Berg, Aesthetic Plast. Surg., 2003, 27, 376–380 CrossRef PubMed.
- S. Liang, Y. Liu, S. Hu, A. Shen, Q. Yu, H. Yan and M. Bai, Energies, 2019, 12, 562 CrossRef CAS.
- A. S. Fouda, E. M. Khalil, G. A. El-Mahdy, M. M. Shaban, A. S. Mohammed and N. A. Abdelsatar, Sci. Rep., 2023, 13, 3519 CrossRef CAS PubMed.
- X. Wang, J. Chem. Res., 2006, 37, 484–485 Search PubMed.
- Q. L. Luo, L. Lv, Y. Li, J. P. Tan, W. Nan and Q. Hui, Eur. J. Org Chem., 2011, 2011, 6916–6922 CrossRef CAS.
- Y. Abe and K. Emori, Org. Process Res. Dev., 2022, 26, 43–55 CrossRef CAS.
- L. Zhang, S. Bai and L. Zheng, Organometallics, 2023, 42, 2262–2268 CAS.
- M. Hossain, I. Habib, K. Singha and A. Kumar, Heliyon, 2024, 10, e23172 CrossRef CAS PubMed.
- T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS.
- S. Ichikawa, S. Miyazoe and O. Matsuoka, Chem. Lett., 2011, 40, 512–514 CrossRef CAS.
- C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed.
- D. Cirri, T. Marzo and A. Pratesi, Front. Chem., 2023, 11, 1253008 CrossRef CAS PubMed.
- H. Yamada and M. Kobayashi, Biosci. Biotechnol. Biochem., 1996, 60, 1391–1400 CrossRef CAS PubMed.
- S. Martinez, R. Wu, R. Sanishvili, D. Liu and R. Holz, J. Am. Chem. Soc., 2014, 136, 1186–1189 CrossRef CAS PubMed.
- Z. Cheng, Y. Xia and Z. Zhou, Front. Bioeng. Biotechnol., 2020, 8, 352 CrossRef PubMed.
- D. Ma, Z. Cheng and L. Peplowski, et. al. , Chem. Sci., 2022, 13, 8417–8428 RSC.
- F. Fouda, E. Khalil and G. El-Mahdy, et al. , Sci. Rep., 2023, 13, 3519 CrossRef PubMed.
- Q. L. Luo, L. Lv, Y. Li, J. P. Tan, W. Nan and Q. Hui, Eur. J. Org Chem., 2011, 2011, 6916–6922 CrossRef CAS.
- Z. J. Quan, H. D. Xia, Z. Zhang, Y. X. Da and X. C. Wang, Appl. Organomet. Chem., 2014, 28, 81–85 CrossRef CAS.
- F. Pandolfi, I. Chiarotto, L. Mattiello, R. Petrucci and M. Feroci, ChemistrySelect, 2019, 4, 12871–12874 CrossRef CAS.
- K. Yang, Y. Li, Z. Ma, L. Tang, Y. Yin, H. Zhang, Z. Li and X. Sun, Eur. J. Org Chem., 2019, 2019, 5812–5814 CrossRef CAS.
- (a) L. C. Wang, B. Chen and X. F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202203797 CrossRef CAS PubMed; (b) H.-Q. Geng, Y.-H. Zhao, P. Yang and X.-F. Wu, Chem. Sci., 2024, 15, 3996–4004 RSC; (c) H. Yang, Y. Wang, L.-C. Wang and X.-F. Wu, Chem. Sci., 2024, 15, 14304–14309 RSC; (d) X.-W. Gu, Y.-H. Zhao and X.-F. Wu, Chem. Sci., 2024, 15, 19970–19976 RSC; (e) Y. Wang, X. Qi, Z.-P. Bao and X.-F. Wu, Chem. Sci., 2025, 16, 9872–9880 RSC; (f) S. Shao, Y. Yuan, A. Schmoll and X.-F. Wu, Chem. Sci., 2025, 16, 10951–10956 RSC.
- Y. Wang, H. Yang and Y. Zheng, et al. , Nat. Catal., 2024, 7, 1065–1075 CrossRef CAS.
- L. C. Wang, B. Chen, Y. Zhang and X. F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207970 CrossRef CAS PubMed.
- R. R. Xu, D. Wen, X. Qi and X. F. Wu, Org. Biomol. Chem., 2022, 20, 2605–2608 RSC.
- F. P. Wu, Y. Yang, D. P. Fuentes and X. F. Wu, Chem, 2022, 8, 1982–1992 CAS.
- (a) R. H. A, Z. P. Bao, Y. W. Huo and X. F. Wu, Chem.–Asian J., 2024, 19, e202400892 CrossRef CAS PubMed; (b) H. Yang, Y. Wang, L.-C. Wang and X.-F. Wu, Chem. Sci., 2024, 15, 14304–14309 RSC; (c) X.-W. Gu, Y.-H. Zhao and X.-F. Wu, Chem. Sci., 2024, 15, 19970–19976 RSC; (d) Y. Wang, X. Qi, Z.-P. Bao and X.-F. Wu, Chem. Sci., 2025, 16, 9872–9880 RSC; (e) S. Shao, Y. Yuan, A. Schmoll and X.-F. Wu, Chem. Sci., 2025, 16, 10951–10956 RSC.
- W. Reppe, Adv. Cycloaddit., 1953, 582, 1–37 CAS.
- Z. Cao, Q. Wang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2024, 63, e202410597 CrossRef CAS PubMed.
- J. Eriksson, O. Åberg and B. Långström, Eur. J. Org Chem., 2007, 2007, 455–461 CrossRef.
- Y. Li, Y. Jing, Y. R. Shi, H. Li, M. G. Yang, Y. L. Kou and Q. W. Fan, Eur. J. Org Chem., 2024, 27, e202301255 CrossRef CAS.
- R. R. Xu, C. S. Kuai and X. F. Wu, Catal. Sci. Technol., 2024, 14, 6180–6185 RSC.
- (a) Y.-H. Zhao and X.-F. Wu, Chem.: Methods, 2025, e202500089 Search PubMed; (b) C. Chen, L.-Y. Ding, X.-X. Zhang, G.-S. Chen, Y.-P. Zhu, C. Ni and B. Zhu, Org. Lett., 2025, 27, 4915–4920 CrossRef CAS PubMed.
- J. Zhang and X.-F. Wu, Org. Lett., 2023, 25, 2162–2166 CrossRef CAS PubMed.
- (a) Y.-H. Zhao, X.-W. Gu and X.-F. Wu, Org. Lett., 2024, 26, 10702–10707 CrossRef CAS PubMed; (b) Y.-H. Zhao, X.-W. Gu and X.-F. Wu, Org. Lett., 2024, 26, 6507–6511 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |